

Abstract
The cytoplasmic domain of LRP1 contains two NPXY motifs that have been shown to interact with signaling proteins. In previous work, we showed that Tyr4507 in the distal NPXY motif is phosphorylated by v-Src, whereas denaturation of the protein was required for phosphorylation of Tyr4473 in the membraneproximal NPXY motif. Amide H/D exchange studies reveal that the distal NPXY motif is fully solvent-exposed, whereas the proximal one is not. Phosphopeptide mapping combined with in vitro and in vivo kinase experiments show that Tyr4473 can be phosphorylated, but only if Tyr4507 is phosphorylated or substituted with glutamic acid. Amide H/D exchange experiments indicate that solvent accessibility increases across the entire LRP1 cytoplasmic region upon phosphorylation at Tyr4507; in particular the NPXY4473 motif becomes much more exposed. This differential phosphorylation is functionally relevant: binding of Snx17, which is known to bind at the proximal NPXY motif, is inhibited by phosphorylation at Tyr4473. Conversely, Shp2 binds most strongly when both of the NPXY motifs in LRP1 are phosphorylated.
The low density lipoprotein receptor-related protein LRP1 is an endocytic receptor involved in the uptake of a diverse range of ligands including lipoproteins, proteases and protease-inhibitor complexes, matrix proteins, and growth factors (1). LRP1 is presented on the cell surface as a two-chain molecule, which consists of a 515-kDa α chain and a 85-kDa β chain that are noncovalently associated (2). The α chain is made up of ligand-binding repeats organized in four domains, each resembling the extracellular domain of the low density lipoprotein receptor. The ligand-binding repeats have been shown to bind up to 30 different extracellular ligands, many of which are found in the proteinaceous plaques present in the brains of Alzheimer disease patients (3). The LRP1 cytoplasmic domain interacts with FE65, providing a direct link between LRP1 and the amyloid precursor protein (APP)2 (4, 5).
The β chain is composed of epidermal growth factor-like repeats, a single transmembrane domain, and a 100-amino acid cytoplasmic region with two NPXY motifs (NPXY4473 and NPXY4507) (2), which were thought to mediate interactions with the receptor internalization machinery. Mutational studies identified the YXXL motif (which overlaps with Tyr4507) as critical for LRP1 receptor endocytosis (6). Recently, the membrane proximal NPXY motif has been identified as the binding site for Snx17 (sorting nexin 17), and knock-in experiments in mice showed a strong phenotype upon mutation of this site (7, 8). Mutation of Tyr4473 to alanine, which abolishes Snx17 binding, resulted in impaired receptor recycling and reduced amounts of the mature form of LRP1 on the cell surface (7).
The cytoplasmic domain of LRP1 also binds the adapter protein, Shc, and this result provided the first hint of a signaling role for LRP1 (9). Phosphorylation of Tyr4507, which occurs in PDGF-stimulated or v-Src-transformed cells, is required for binding of Shc (9–11). Recently, LRP1 was shown to bind JIP, a scaffold protein involved in Jun N-terminal kinase activation (12, 13). The distal NPXY4507 motif was also identified as the primary site of binding to DAB1 (Disabled-1), an adaptor protein involved in neuronal path finding during the development of the central nervous system (4). Interactions between LRP1 and the engulfment adapter protein Gulp also require the distal NPXY4507 motif (14). Deletion of both NPXY motifs was required to abrogate binding of the neuronal adaptor protein, FE65. Finally, mutations at the distal NPXY4507 motif affected processing of APP (5, 15).
The distal NPXY4507 motif can be phosphorylated in v-Src-transformed cells (10, 16) and following stimulation of cells with PDGF-BB (10, 16). It is surprising that phosphorylation of only the distal of the two NPXY motifs has been observed, because the amino acid sequences N-terminal to Tyr4473 and Tyr4507 are very similar (9, 16). This raises the question as to why v-Src phosphorylation of Tyr4473 was not observed. One possible explanation is that the NPXY4473 site is bound by a protein that prevents access to the kinase. If this is the case, in vitro phosphorylation of the purified cytoplasmic tail should result in phosphorylation of this site. A second possibility is that the protein is somehow structured around the NPXY4473 site so as to prevent phosphorylation. We report here that the proximal NPXY4473 motif of LRP1 can be tyrosine-phosphorylated. However, our observations strongly suggest that Tyr4473 can only become phosphorylated after Tyr4507 is phosphorylated. We further show by in vivo and in vitro experiments that phosphorylation of Tyr4507 increases the solvent accessibility of much of the cytoplasmic tail, providing an explanation as to why Tyr4473 may only be accessible after Tyr4507 is phosphorylated. Finally, we show that Snx17 binding at the proximal NPXY4473 as well as Shp2 binding are regulated by phosphorylation.
EXPERIMENTAL PROCEDURES
Materials—HEK293 cells were grown in Dulbecco-Vogt's modified Eagle's medium containing 10% fetal bovine serum. 11H4 hybridoma cells (ATCC, Manassas, VA) were grown in Iscove's modified Dulbecco's medium containing 10% fetal bovine serum and 25 mm l-glutamine. Anti-phosphotyrosine monoclonal antibody G410, anti-HA monoclonal antibody 12CA5, and horseradish peroxidase goat anti-mouse serum, and protein G-agarose were purchased from Upstate Biotechnology, Inc. (Lake Placid, NY). Anti-Myc antibodies, α-cyano-4-hydroxycinnamic acid, and protein A-Sepharose were obtained from Sigma. Anti-His antibodies were purchased from Qiagen. Glutathione-Sepharose was purchased from GE Healthcare. Pepsin beads were obtained from Pierce. Recombinant Src kinase domain and YopH phosphatase plasmids were a kind gift from Dr. John Kuriyan (University of California, Berkeley, CA).
Bacterial Protein Expression and Purification—DNA corresponding to the LRP1 C-terminal residues 4444–4544 was cloned into pGEX 4T2. Mutations at Tyr4473 and Tyr4507 were introduced using a Stratagene site-directed mutagenesis kit, and mutants were sequenced to ensure fidelity. Glutathione S-transferase (GST) fusion proteins were expressed in pLysS cells and induced with 0.1 mm isopropyl thiogalactoside overnight at room temperature. The cells were collected by centrifugation, resuspended in lysis buffer (50 mm Tris-HCl, pH 7.4, 150 mm NaCl (TBS), 1 mm benzamidine, 1 mm dithiothreitol, 1 μg/ml leupeptin, 1 μg/ml aprotinin), sonicated on ice, and centrifuged for 30 min at 10,000 rpm. The supernatant was incubated with 10 ml of glutathione-Sepharose for 1 h at 4°C, and the beads were washed exhaustively with TBS. Fusion proteins were eluted with 10 mm reduced glutathione in TBS, pH 7.4, dialyzed overnight in TBS at 4 °C, and quantified by BCA assay (Pierce). Maintaining the LRP1-CT as a fusion with GST reduced aggregation and improved stability. Recombinant v-Src kinase domain was expressed and purified as described (17). Shp2SH21,2, (1–222), Shp2SH21(1–109), and Shp2SH22(109–228) were produced as His-tagged ubiquitin fusion proteins in Escherichia coli BL21(DE3). Expression was induced overnight with 0.1 mm isopropyl thiogalactoside. The cells were pelleted and sonicated, and the extract was cleared by centrifugation. The proteins were captured on 5 ml of nickel-nitrilotriacetic acid resin (Qiagen), eluted with 250 mm imidazole in TBS, pH 7.4, and further purified by size exclusion chromatography (S-75 column in 50 mm Tris and 150 mm NaCl, pH 7.4).
Phosphopeptide Mapping—To map the location of phosphorylation sites in LRP1, 1 μg of wild type and mutant GST-LRP1-CT fusion proteins were incubated with 1 μl of v-Src kinase in 25 μl of kinase buffer (100 mm Tris, pH 7.4, 10 mm MnCl2, 10 mm MgCl2, 1 mm dithiothreitol, 25 μCi of [γ-32P]ATP) for 15 min at 37 °C. The reaction was quenched, and samples were resolved by SDS-PAGE. Phosphorylated proteins were localized by autoradiography, isolated from the gel, and digested with trypsin (18). The phosphopeptides were separated in two dimensions on a TLC plate by electrophoresis at pH 1.9 for 35 min and by chromatography in isobutyric acid buffer. Phosphopeptides were visualized by autoradiography. Amino acid analysis was performed as described before (18).
Hydrogen/Deuterium Exchange Mass Spectrometry—Purified GST-LRP1 or full-length v-Src (Upstate Biotechnology, Inc.)-phosphorylated GST-LRP1 (400 μg, in TBS, pH 7.4) was bound to glutathione-Sepharose and cleaved with thrombin for 1 h at room temperature. Free LRP1-CT was collected by centrifugation and used immediately. Triplicate samples (2 μl) of free LRP1-CT were mixed with 20 μl of 2H2O-TBS for 30 s to 10 min at room temperature. After quenching by the addition of chilled 0.1% trifluoroacetic acid (adjusted with 2% trifluoroacetic acid so that the final pH of mixed solution was 2.2), the mixture was added to prewashed (25 μl of beads washed twice with 0.1% trifluoroacetic acid) pepsin beads and incubated for 10 min at 0 °C. The supernatant was collected by centrifugation and immediately frozen. Peptides produced by pepsin digests were identified as described previously (19). Matrix-assisted laser desorption ionization time-of-flight mass spectrometry was used to monitor deuterium incorporation into the amide positions of the LRP1-CT. The samples were thawed and mixed with α-cyano-4-hydroxycinnamic acid matrix, and the spectra were acquired on a Voyager DE-STR instrument (Applied Biosystems, Foster City, CA) exactly as previously described (19). The centroids of the individual peptides were calculated using the CAPP software program. Backbone deuteration is reported for each peptide as previously described (20).
To directly compare the amount of deuterium incorporation into the phospho-versus non-phospho-LRP1, an experiment was performed in which 14N-LRP1 and 15N-phospho-LRP1 were compared in the same sample. 15N-Labeled GST-LRP1-CT was isolated from cells grown in M9 minimal medium containing 15N-NH4Cl. Labeled protein was then phosphorylated with full-length v-Src (Upstate Biotechnology, Inc.). Equal amounts of 14N-GST-LRP1-CT and 15N-GST-LRP1-CT-P were bound to glutathione-Sepharose beads and mixed together. The beads were washed and incubated with thrombin as before, and free LRP1 was collected by centrifugation. Free protein was then used immediately for H/D experiments as above.
Circular Dichromism and NMR—Freshly prepared LRP1-CT and phospho-LRP1 were concentrated to 63 μm in 25 mm Tris, 50 mm NaCl, pH 7.4, and tested for structure of the LRP1-CT by CD and NMR as described previously (21).
In Vitro Binding Experiments—HEK293 cells transiently expressing Snx17 (1–470) were lysed in HUNT buffer (7) and incubated with wild type and mutant GST-LRP1-CT fusion proteins bound to glutathione-Sepharose beads for 2 h at 4°C while rocking. The beads were washed, and bound proteins were detected by Western blotting. In other experiments, recombinant His-tagged (ubiquitin)-Shp2 fusion proteins were incubated with wild type and mutant GST-LRP1-CT fusion proteins immobilized on Glutathione-Sepharose, and bound proteins were identified by immunoblotting.
Transient Transfections—pcDNA3.1-based constructs containing N-terminally Myc-tagged LRP1 β chain (Myc-LRP1β), v-Src, N-terminally HA-tagged Snx-17, or N-terminally HA-tagged Shp2 deletion mutants were expressed by transient transfection into HEK293 cells as described before (10). Shp2 mutants Shp2Δcterm (1–531), Shp2Δnterm (SH22-PTP) (109–598), Shp2(SH21,2) (1–222), Shp2 (PTP, 228–598), and Shp2 Y2F (1–598, Y542F/Y580F) were generated using a Stratagene site-directed mutagenesis kit, and the mutants were sequenced to ensure fidelity.
Immunoprecipitation and Immunoblotting—HEK293 cells were lysed in PLC lysis buffer (50 mm HEPES, pH 7.5, 150 mm NaCl, 10% glycerol, 0.1% Triton X-100, 1.5 mm MgCl2, 1 mm EDTA, 10 mm NaP2O7, 100 mm NaF, 1 mm Na3VO4) (9) 48 h after transfection, and incubated with primary antibodies for 1 h at 4 °C followed by incubation with 10 μl of protein G or protein A for 1 h at 4 °C on rocker. The beads were washed four times with PLC lysis buffer, and the proteins were resolved by SDS-PAGE. The proteins were transferred to nitrocellulose membranes by semi-dry blotting. The blots were incubated for 1 h at room temperature with 1% casein in TBS with 0.1% Tween 20 (TBST) and incubated with primary antibodies diluted in blocking buffer. The blots were washed twice for 10 min with TBST and twice with TBS for 10 min and then incubated with a 1:5000 dilution of horseradish peroxidase goat anti-mouse or horseradish peroxidase-protein A in TBST for 30 min at room temperature; the blots were washed as before, and the proteins were visualized by ECL (22). The anti-LRP1, anti-HA, anti-His, anti-phosphotyrosine, and anti-Myc antibodies were used at 1:100, 1:20, 0.1 μg/ml, 1:10 and 1:1000, respectively.
RESULTS
Tyrosine Phosphorylation of LRP1 NPXY4473 Motif Requires Phosphorylation or a Phosphomimic at NPXY4507—LRP1 contains two NPXY motifs in its cytoplasmic domain. It was previously shown that Tyr4507 in the distal NPXY4507 motif of LRP1 can be phosphorylated in response to stimulation with PDGF-BB and v-Src-transformed cells (10, 16). We also showed that Tyr4473 in the proximal NPXY motif could be phosphorylated in vitro if the LRP1 protein was first denatured (10). Endogenous LRP1 isolated from v-Src-transformed fibroblasts migrates in multiple bands, and upon vanadate/H2O2 treatment the band migration is most consistent with two phosphorylation events (Fig. 1A). However, the putative doubly phosphorylated species was only observed in the vanadate-H2O2-treated cells. To ascertain whether Tyr4473 was the second site of phosphorylation and to understand why it appeared to be less readily phosphorylated, we carried out in vitro kinase reactions with recombinant v-Src using a purified GST fusion protein containing the cytoplasmic domain of LRP1 (GST-LRP1-CT) and [γ-32P]ATP as substrates. In a time course experiment, the wild type fusion protein continued to incorporate 32P for at least an hour, as did the Y4473F mutant (Fig. 1B). In contrast, the Y4507F and the Y2F (Y4473F,Y4507F) mutant fusion proteins were not phosphorylated at all by recombinant v-Src, but the mutant in which Tyr4507 was replaced by glutamic acid was able to be phosphorylated (Fig. 1B). Quantitative analysis showed that the wild type GST-LRP1-CT incorporated approximately twice the amount of 32P as either the Y4473F or the Y4507E mutant. This suggests that in the wild type protein both NPXY motifs are subject to phosphorylation, whereas only one of these motifs is phosphorylated in either the Y4473F or the Y4507 mutant protein. Phosphorylation of the single mutants appeared to lag at early times compared with the wild type protein (Fig. 1C). Importantly, substitution of Tyr4507 with Phe completely abrogated all phosphorylation of the LRP1 cytoplasmic region, whereas substitution of Tyr4507 with Glu promoted phosphorylation, suggesting that phosphorylation of Tyr4473 depends on phosphorylation of Tyr4507.
FIGURE 1.
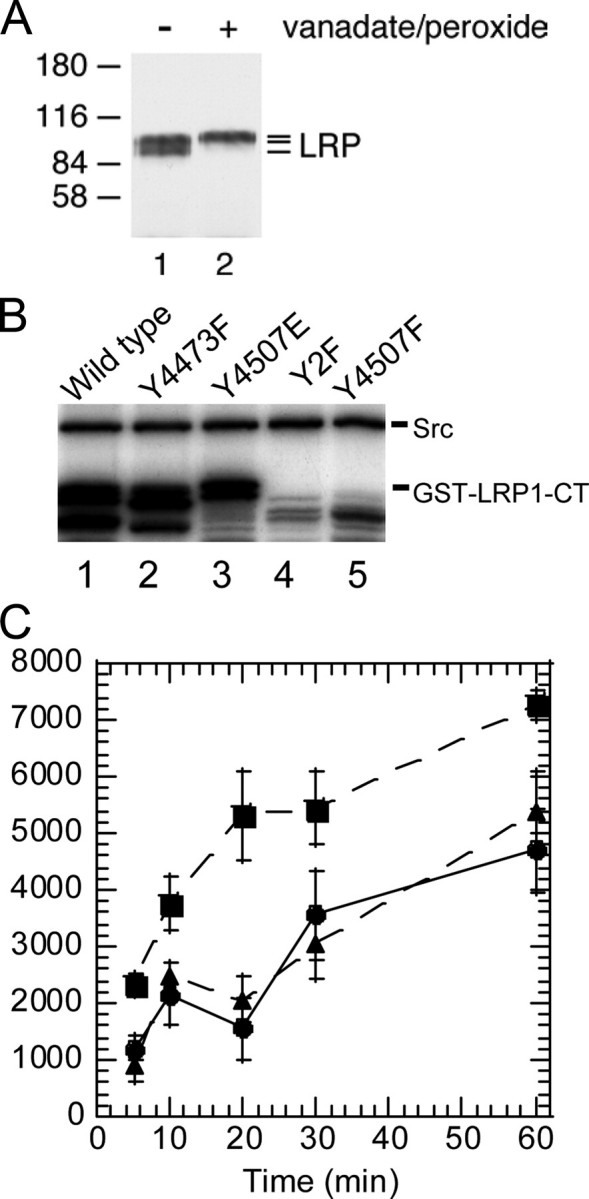
Phosphorylation of GST-LRP1-CT constructs. A, anti-LRP1 immunoprecipitates from control and vanadate/H2O2-treated v-Src-transformed fibroblasts cells were analyzed by immunoblotting with anti-LRP1 antibodies. The two bands observed for LRP1 in the absence of vanadate/H2O2 treatment (lane 1) both migrate lower than that observed after treatment (lane 2). B, GST-LRP1-CT fusion proteins were incubated with recombinant v-Src and [γ-32P]ATP. Samples were removed at 5, 10, 20, 30, and 60 min. The top band corresponds to v-Src autophosphorylation. Lanes 4 and 5 show the lack of phosphorylation of GST-LRP1-CT (Y4507F) and GST-LRP1-CT (Y2F). C, densitometry of phosphorylated bands of GST-LRP1-CT (▪), GST-LRP1-CT (Y4473F) (•), and GST-LRP1-CT (Y4507E) (▴) over 1 h.
Phosphopeptide Mapping Identifies Both NPXY Motifs in LRP1 as Sites of Phosphorylation—To confirm the identity of the sites of phosphorylation in LRP-CT, phosphorylated proteins from in vitro kinase reactions were analyzed by phosphopeptide mapping. Wild type GST-LRP1-CT phosphorylated with full-length v-Src yielded two major 32P-containing peptides that were identified by comparison of maps obtained from wild type and mutant proteins (Fig. 2). This analysis indicates that the main phosphopeptide seen on maps of the wild type protein represents the Tyr4507 phosphorylation site, whereas the minor peptide represents the Tyr4473 phosphorylation site (Fig. 2, B and C). The relative intensities of the phosphopeptides indicated that the majority of phosphorylation was found on Tyr4507, whereas Tyr4473 had lower intensity corresponding to a lower level of phosphorylation at this site. Possible phosphorylation of the other two tyrosine residues in the LRP1-CT was ruled out by use of the Y2F mutant control, which did not incorporate any label. The results from this mapping experiment confirmed that phosphorylation of Tyr4473 only occurs following phosphorylation of Tyr4507 or upon substitution of Tyr4507 with glutamate.
FIGURE 2.
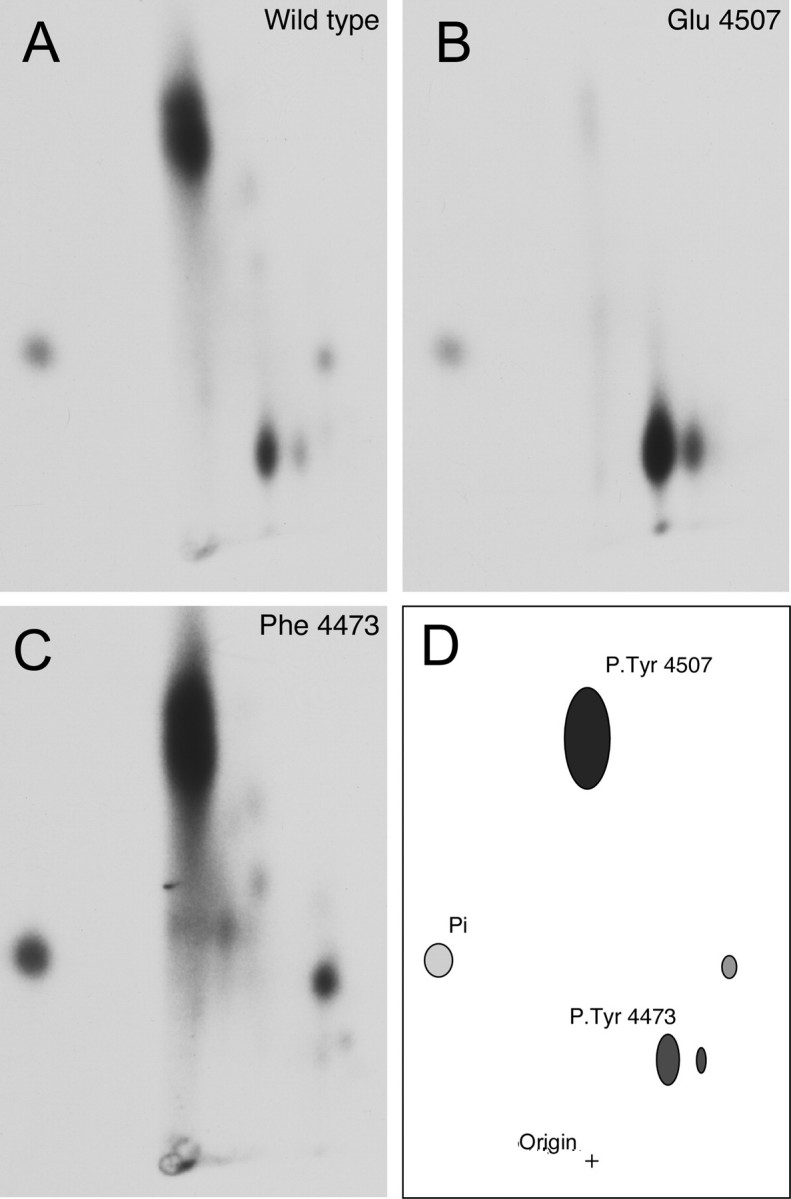
Phosphopeptide mapping of the GST-LRP1-CT. GST-LRP1-CT fusion proteins were incubated with [γ-32P]ATP and full-length v-Src followed by SDS-PAGE and autoradiography. Phosphorylated LRP1 was excised from the gel and digested with trypsin, and the resulting peptide mixture was resolved in two dimensions by electrophoresis and chromatography. A, wild type GST-LRP1-CT, yielded two major 32P-phospho-peptides. B, Y4507E GST-LRP1-CT yielded one 32P-phospho-peptide. C, Y4473F GST-LRP1-CT yielded one 32P-phospho-peptide. D, schematic map of the 32P-phosphopeptides derived from the tryptic digest of phospho-GST-LRP1-CT.
Phosphorylation of NPXY4473 in Vivo—To test whether Tyr4473 can also be phosphorylated in vivo, Myc-tagged LRP1-β chain (Myc-LRP1β) and v-Src were coexpressed by transient transfection in HEK293 cells (10, 16). Myc-LRP1β was isolated by anti-Myc immunoprecipitation and probed for tyrosine phosphorylation. In the presence of v-Src, phosphorylation of the wild type protein as well as the Y4473F and Y4507E mutants was observed (Fig. 3). The highest level of phosphorylation was seen in the wild type protein, followed by the Y4473F mutant, and the Y4507E gave a weak but detectable signal. These results confirm that Tyr4473 can be phosphorylated in vivo. The Y4507E Myc-LRP1β appeared to be a better kinase substrate then the Y4507F form, in agreement with the in vitro results. The observation that the wild type protein was phosphorylated to a higher level than the Y4473F mutant again indicates that phosphorylation of the Tyr4473 residue by v-Src is occurring in these cells. Given that Tyr4473 can be phosphorylated in vitro and in vivo but that it requires prior phosphorylation at Tyr4507 or substitution with glutamic acid, we surmised that the cytoplasmic domain might have structural constraints preventing phosphorylation by v-Src but that phosphorylation or the presence of a negative charge at Tyr4507 might relieve these constraints.
FIGURE 3.

Phosphorylation of Tyr4473in vivo. Wild type (WT) or mutant myc-LRP1β was expressed in the absence (lanes 1, 3, 5, 7, and 9) or presence (lanes 2, 4, 6, 8, 10, and 11) of v-Src. Myc (LRP1β) immunoprecipitations (IP) were analyzed by anti-phosphotyrosine (top panel) or anti-LRP1 (bottom panel) immunoblotting. v-Src phosphorylated wild type (lane 2) and Y4473F (lane 4) but not Y4507F (lane 6) or Y2F (lane 8), indicating that Tyr4507 is readily phosphorylated by v-Src. v-Src did not phosphorylate Y4507F (lane 6) but did phosphorylate Y4507E (lane 10), indicating that Tyr4473 can be phosphorylated when a negative charge is present at 4507. WB, Western blot.
Structural Characterization of the LRP1 Cytoplasmic Region—Having confirmed the dual tyrosine phosphorylation of LRP1, we sought to determine whether a negative charge at Tyr4507 might lead to a change in accessibility of Tyr4473. The cytoplasmic tail is 100 amino acid residues long and has no significant predicted structure according to homology or sequence predictors. The CD spectrum of the untagged LRP1-CT suggested that it consists primarily of random coil secondary structure (Fig. 4). Additional CD spectra collected on the GST-LRP1-CT as compared with GST alone and an NMR HSQC spectrum of 15N-labeled GST-LRP1 all showed only random coil secondary structure (data not shown).
FIGURE 4.

CD Spectrum of free LRP1-CT. GST-LRP1-CT bound to glutathione-Sepharose was cleaved in solution with thrombin (1:50) in TBS, pH 7.4, 2.5 mm CaCl2. The free LRP1-CT was rapidly concentrated by microcolumn centrifugation before analysis. The sample conditions were 63 μm LRP1-CT in 25 mm Tris, 50 mm NaCl, 25 °C.
Hydrogen/Deuterium Exchange of Wild Type and Phosphorylated LRP1 Cytoplasmic Region Shows Increased Solvent Accessibility in the Phosphorylated Form—To determine whether the LRP1 cytoplasmic region was at all structured and whether phosphorylation affected the structure, we measured the amide hydrogen/deuterium exchange by mass spectrometry. This experiment is ideal for probing weakly structured regions of proteins (23). The extent of exchange depends on a number of factors including whether or not the amide is sequestered from solvent because it is in a folded part of the molecule and/or is participating in a hydrogen bond. The experiment involved incubating freshly prepared LRP1-CT or phospho-LRP1-CT in buffered 2H2O, quenching the reaction by lowering the pH and temperature, and digesting the protein with pepsin. The resulting peptides were analyzed by mass spectrometry. Peptides covering 82% of the protein were identified, including those that contain the two NPXY motifs (Fig. 5A). Phosphorylation of Tyr4507 causes the peptides covering the NPXY4507 motif to disappear from the spectrum. Surprisingly, much of the phosphorylated LRP1-CT became more deuterated than the unphosphorylated protein (Fig. 5, B–E). The difference was quantitatively significant at all time points measured (Table 1 gives the 2-min data). To control for the possibility of a systematic difference between the samples that may lead to such an overall difference in deuteration, a sample of the phosphorylated protein that was 15N-labeled was prepared and compared with unphosphorylated protein that was 14N-labeled in the same reaction mixture (data not shown). This analysis confirmed the global differences in solvent accessibility between the unphosphorylated and phosphorylated proteins.
FIGURE 5.

Increased deuterium exchange of LRP1-CT upon phosphorylation. A, pepsin cleavage of LRP1-CT resulted in nine peptides covering 82% of the protein sequence. Tyrosine residues in the context of NPXY motifs are marked in bold. B, the N terminus of LRP1-CT (residues 4442–4452), which was represented by the peptide of mass 1249.72, showed higher deuteration in the phosphorylated form. i, the peptide before deuteration of LRP1-CT; ii, the peptide after LRP1-CT was deuterated for 30 s; iii, the peptide after phospho-LRP1-CT was deuterated for 30 s. C, residues 4453–4465 of LRP1-CT (the peptide of mass 1491.66) under the same conditions as in B. D, residues 4466–4475 of LRP1-CT, which includes NPXY4473 (peptide of mass 1151.57) under the same conditions as in B. E, residues 4492–4507 of LRP1-CT, which include NPXY4507 (peptide of mass 1838.89), under the same conditions as in B except iii is not shown because the intensity of the peptide signal decreased markedly because of phosphorylation. The data for the C-terminal region is not shown.
TABLE 1.
Summary of hydrogen/deuterium exchange in the LRP1 cytoplasmic domain
| Peptide (N to C termini) | Coverage region | Number of amides | Deuteration in LRP1 | Deuteration in LRP1-P (Tyr(P)4507) | Deuteration in LRP1 Y4507E |
|---|---|---|---|---|---|
| 1249.72 | 4442-4452 | 10 | 7.5 ± 0.1 | 8.4 ± 0.2 | 9.7 ± 0.1 |
| 1377.61 | 4452-4464 | 11 | 7.3 ± 0.1 | 9.0 ± 0.3 | 9.1 ± 0.3 |
| 1491.64 | 4453-4465 | 12 | 7.8 ± 0.2 | 9.7 ± 0.3 | 10.1 ± 0.5 |
| 1151.57 | 4466-4475 | 8 | 4.9 ± 0.2 | 6.5 ± 0.1 | 7.9 ± 0.5 |
| 1800.80 | 4465-4480 | 14 | 8.1 ± NAa | 10.4 ± 0.04 | NA |
| 1691.83 | 4493-4507 | 12 | 8.6 ± 0.1 | NA | NA |
| 1838.89 | 4492-4507 | 12 | 11.3 ± 0.1 | NA | NA |
| 1704.74 | 4511-4526 | 15 | 7.1 ± NA | 9.6 ± 0.1 | 10.6 ± NA |
| 1965.03 | 4527-4544 | 15 | 8.6 ± 0.5 | 12.2 ± 0.1 | 11.0 ± 0.1 |
NA, not applicable.
The changes in solvent accessibility that occurred upon phosphorylation led us to ask whether the same changes could be induced simply by the presence of a negative charge at the NPXY4507 site. To this end, we repeated the H/D exchange experiment with the Y4507E mutant. The overall deuterium incorporation of Y4507E mutant was higher than both the wild type and phosphorylated protein. The phospho-LRP1 was only approximately 30% phosphorylated by full-length v-Src, and only Tyr4507 and not Tyr4473 was phosphorylated as seen by the decreased intensity of peptide spanning residues 4492–4507 because of phosphorylation, whereas the peptide spanning 4466–4475 did not decrease in intensity.
The Effect of LRP1 Tyrosine Phosphorylation on Interaction with Snx17—Sorting nexin protein Snx17 was recently shown to bind to the NPXY4473 motif of LRP1 (7). This prompted us to investigate whether tyrosine phosphorylation affects the binding of Snx17 to LRP1. To do so, unphosphorylated and phosphorylated GST-LRP1-CT fusion proteins immobilized on glutathione-Sepharose were incubated with lysates of HEK293 cells containing HA-tagged Snx17, and bound proteins were analyzed by anti-HA immunoblotting. By using a series of GST-LRP1-CT mutants, we determined that Snx17 prefers to bind to the LRP1-CT when Tyr4473 is either not phosphorylated or is substituted with Phe. Phosphorylation of the wild type protein (at both Tyr4507 and Tyr4473) abolished binding, but phosphorylation of the LRP1-CT at Tyr4507 or substitution of Tyr4507 with glutamic acid in the context of Y4473F did not affect binding of Snx17. Importantly, phosphorylation of the Y4507E mutant, which is only phosphorylated at Tyr4473, also abolished binding of Snx17 (Fig. 6A). To confirm this result in vivo, HA-Snx17 was expressed in HEK 293 cells together with wild type or mutant mycLRP1β, in the presence or absence of v-Src. HA immunoprecipitates were probed for the presence of Myc-LRP1β. Again, in all cases, phosphorylation of Tyr4473 blocked binding (Fig. 6B). These results suggest strongly that the interaction between LRP1 and Snx17 is inhibited by tyrosine phosphorylation at Tyr4473.
FIGURE 6.
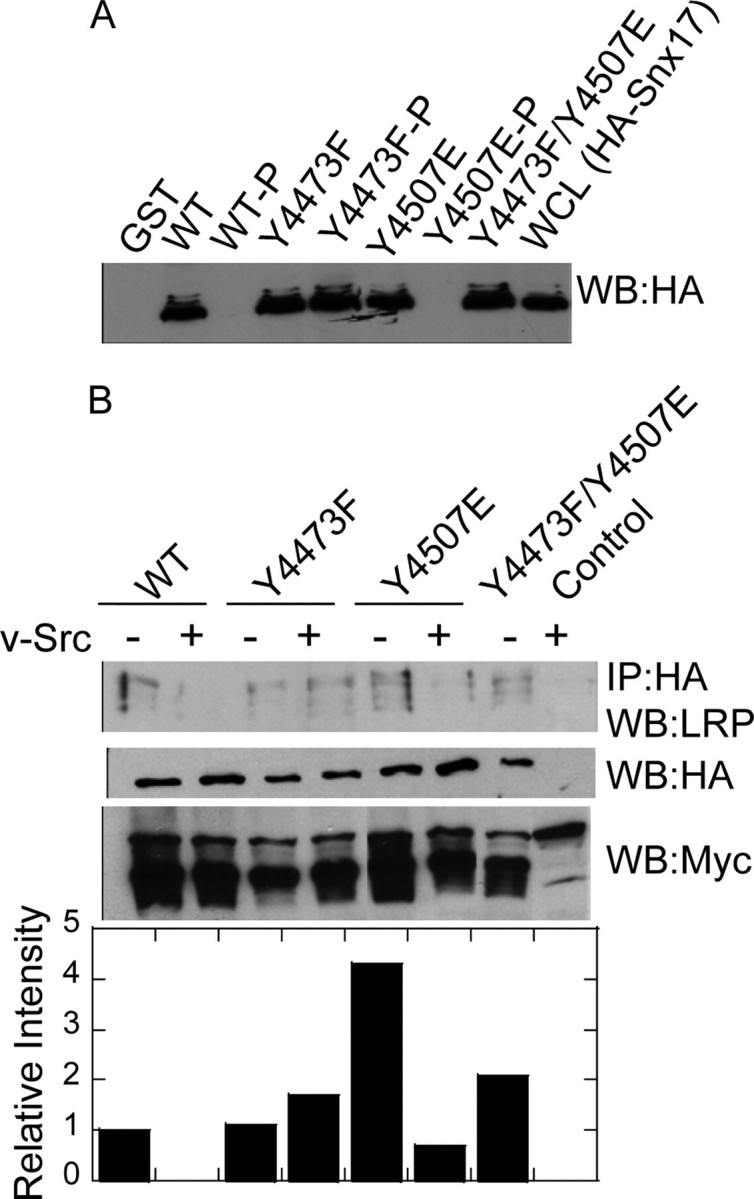
Phosphorylation at Tyr4473 abolishes the LRP1-Snx17 interaction. A, unphosphorylated and phosphorylated GST-LRP1-CT fusion proteins immobilized on Sepharose beads were incubated with lysates of HEK293 cells expressing HA-tagged Snx17. Bound proteins were analyzed by anti-HA immunoblotting. B, wild type (WT) or mutant myc-LRP1β was expressed together with HA-Snx17 in the presence or absence of v-Src. Anti-Myc immunoprecipitations (IP) were probed for the presence of Snx17 by anti-HA immunoblotting. As controls for expression, whole cell lysates were analyzed by anti-HA and anti-Myc immunoblotting. WB, Western blot.
Characterization of LRP1 Binding to Shp2—Shp2 contains two SH2 domains that are known to bind to phosphorylated NPXY motifs (24). We recently showed that Shp2 binds strongly to peptides corresponding to the sequence containing Tyr4507.3 To determine how Shp2 interacts with LRP1, we constructed a series of Shp2 deletion mutants (Fig. 7A). The HA-tagged Shp2 proteins were coexpressed with Myc-LRP1β and v-Src in HEK293 cells, and HA immunoprecipitates were probed for the presence of Myc-LRP1β. We found that only deletion of both SH2 domains from Shp2 (Fig. 7B, lane 1) completely abrogated binding to LRP1. A Shp2 mutant containing only one SH2 domain showed strongly reduced binding to Myc-LRP1β (Fig. 7B, lane 3). To test whether both SH2 domains can bind simultaneously if both NPXY motifs are phosphorylated, we made use of the phosphorylation of Tyr4473 in the context of the Y4507E mutant. Control and phosphorylated GST-LRP1-CT fusion proteins immobilized on glutathione-Sepharose were mixed with Shp2 deletion mutants expressed as His-tagged ubiquitin fusion proteins. Either SH2 domain could bind to either of the phosphorylated NPXY motifs (Fig. 8A). The N-terminal SH2 domain bound better to LRP1-CT phosphorylated on Tyr4507 than to LRP1-CT phosphorylated on Tyr4473, whereas the C-terminal SH2 domain bound equally well to either one of the phosphorylated NPXY motifs. Comparison of Shp2 binding to Myc-LRP1β showed a large decrease in binding upon mutation of Y4473. We did not observe Shp2 binding to the Y4507E mutant in vivo. This may have been because the amount of Tyr4473 phosphorylation is low in this mutant or because expression levels were low (Fig. 8B). In in vitro experiments using the extremely active recombinant v-Src, we were able to observe binding of the LRP1-CT, in which only Tyr4473 is phosphorylated (Fig. 8C). Taken together, the data suggest that binding is strongest when both SH2 domains of Shp2 can engage LRP1 at both phosphorylated NPXY motifs.
FIGURE 7.

Shp2 domains responsible for LRP1 binding. Myc-LRP1β was expressed together with v-Src and various Shp2 deletion mutants (A). HA immunoprecipitates (IP) were probed for the presence of LRP1 by anti-Myc immunoblotting (B). As a control for expression of myc-LRP1β and HA-Shp2, whole cell lysates were probed by anti-Myc and anti-HA immunoblotting. WB, Western blot.
FIGURE 8.
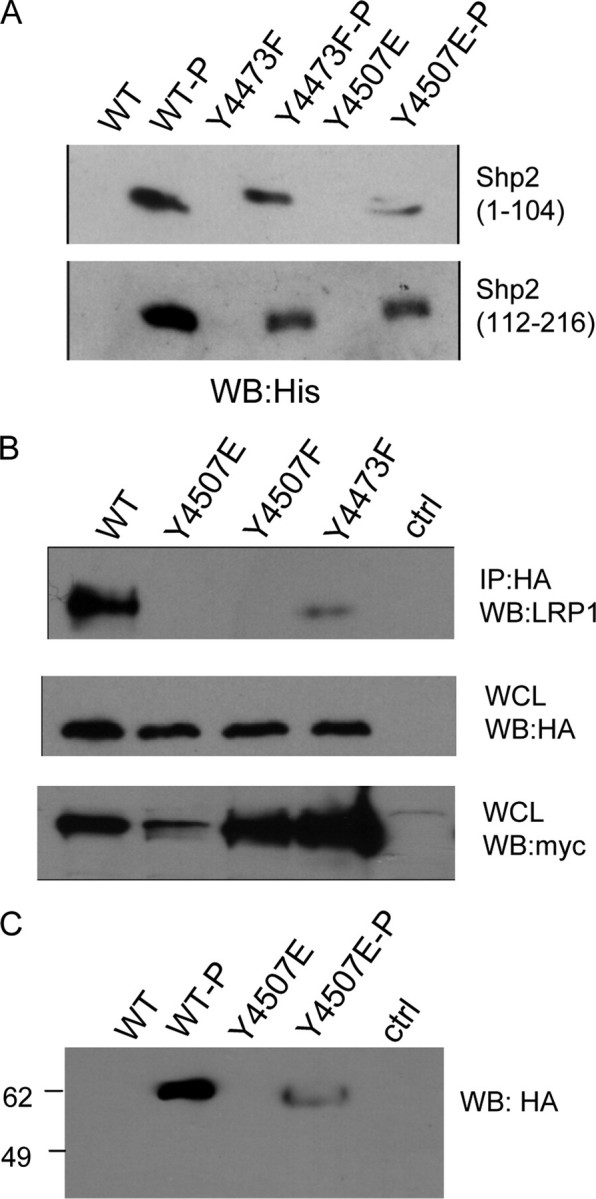
Phosphorylation dependence of LRP1-Shp2 interaction. A, unphosphorylated and phosphorylated GST-LRP1-CT fusion proteins immobilized on Sepharose beads were incubated with recombinant His-tagged Shp2 SH2 domains. Bound proteins were visualized by anti-His immunoblotting. Binding is always phosphorylation-dependent. Either of the phosphorylation sites can act as binding sites, and both Shp2 SH2 domains can bind. B, HA-Shp2, v-Src, and various Myc-LRP1β constructs were expressed in HEK293 cells. HA (Shp2) immunoprecipitations (IP) were probed for the presence of LRP. Untransfected cells were analyzed in parallel. Even though the expression of the LRP phenylalanine mutants is much higher, the amount bound to Shp2 is still much lower compared with wild type (WT). C, Shp2 interacts with LRP1-CT when only Tyr4473 is phosphorylated in vitro (lane 4). Binding is still stronger to the dually phosphorylated protein (lane 2). WB, Western blot; WCL, whole cell lysate.
DISCUSSION
Implications of Phosphorylation at the Two NPXY Motifs in LRP1—The LRP1-CT contains two NPXY motifs. The distal NPXY4507 motif has been shown to bind signaling proteins (10, 16, 25), and the proximal NPXY4473 motif has been shown to be involved in receptor recycling and demonstrated a strong phenotype in knock-in mice (7, 8). Binding of Shc and Dab1 has been localized to the NPXY4507 motif and a few surrounding residues using peptides as the pull-down bait (4, 9). Phosphorylation at Tyr4507 is required for the binding of Shc in vitro and in vivo (10). We previously reported that Tyr4507 is the principle phosphorylation site on LRP1 because tyrosine phosphorylation in vitro and in vivo was blocked by mutation of this residue (10). Phosphopeptide mapping studies reported here show that LRP1 can be phosphorylated on two tyrosines that are represented by two phosphopeptides on the peptide map. Substitution of Tyr4473 with Phe results in the disappearance the phosphopeptide that represents the Tyr4473 phosphorylation site. Strikingly, substitution of Tyr4507 with Phe results in disappearance of both phosphopeptides. Mutation of Tyr4507 to glutamate blocks phosphorylation on Tyr4507 but allows phosphorylation on Tyr4473. These results are consistent with a model in which the LRP1 can be phosphorylated at both NPXY motifs, but phosphorylation of Tyr4507, or mutation to glutamate, is a prerequisite for phosphorylation of Tyr4473.
Phosphorylation at Tyr4507 Opens the Structure of LRP1-CT near Tyr4473—Amide exchange followed by mass spectrometry revealed different solvent accessibility of the two NPXY motifs in LRP1. Whereas the distal NPXY4507 region was the most solvent-accessible region in the LRP1-CT, the proximal NPXY4473 appeared at least partly sequestered from solvent. Phosphorylation at Tyr4507 caused a significant increase in solvent accessibility of the region around Tyr4473. Thus, in the unphosphorylated LRP1-CT, the proximal NPXY4473 motif is sequestered from solvent and unavailable for phosphorylation by v-Src. However, when Tyr4507 becomes phosphorylated, the protein backbone near the proximal NPXY4473 motif becomes more exposed. This experiment helps to define a mechanism in which the increase in solvent accessibility is likely to also increase accessibility to v-Src. Notably, the increases both in phosphorylation and in solvent accessibility are also seen in the Y4507E mutant. The observation that phosphorylation of one NPXY motif changes the reactivity of the other is, as far as we know, novel.
LRP1 is one of many receptors that have short cytoplasmic domains. Similar to the APP-CT, structure prediction programs, CD, and NMR generally predict that these domains are mainly random coil (26). The APP-CT is approximately half the size of the LRP1-CT and contains a single NPXY motif (NPTY687) that forms a type I reverse turn (26). Phosphorylation at Thr668 in APP-CT was shown to alter the cis-trans ratio of Pro669 located in a second type 1 reverse turn comprised of residues TPEE671 some 15 amino acids upstream from the NPXY. No long distance structural changes were seen near the NPXY motif upon phosphorylation of Thr668 (26, 27). In contrast, phosphorylation of LRP1-CT at the distal NPXY4507 caused structural changes more than 30 residues away at the proximal NPXY4473. Our results are consistent with the possibility that the LRP1-proximal NPXY4473 is present within a type I reverse turn similar to that seen in APP-CT because a tight turn would reduce solvent accessibility and may restrict access to the v-Src active site. Introduction of a phosphate at Tyr4507 or replacement of Tyr4507 with a negatively charged glutamic acid appears to disrupt the turn and open up the Tyr4473 region both to backbone exchange with solvent and to phosphorylation.
Phosphorylation at Tyr4473 Abolishes Binding of Snx17—Binding of Snx17 aids in the proper recycling of LRP1, and mutation of the Tyr4473-binding site and small interfering RNA knockdown of Snx17 expression in U87 cells led to a decrease in the distribution of LRP1 on the cell surface (7). It has also been reported that Tyr4473 is necessary for correct basolateral sorting of LRP1 in polarized cells (28). Snx17 interacts with the LRP1-CT at the NPXY4473, but what effect phosphorylation of LRP1 would have on the interaction was not determined (7). Our results conclusively show that Snx17 binding is inhibited by phosphorylation of the LRP1-CT at Tyr4473. Phosphorylation at Tyr4507 had no effect on binding, hinting that Snx17 does not discriminate between the more solvent-accessible and less solvent-accessible conformations of the LRP1-CT. These results suggest an interplay between control of the phosphorylation state of the LRP1-CT and receptor sorting/recycling. Others have proposed that alteration of LRP1 recycling and/or sorting may affect APP processing and Aβ peptide production (15), and our results suggest a mechanism by which phosphorylation of the LRP1-CT may be important in this control.
Phosphorylation at Both NPXY Motifs Potentiates Binding of Shp2—We show here for the first time that neuronal Shp2 associates directly with the LRP1-CT. Both SH2 domains of Shp2 can bind to tyrosine-phosphorylated GST-LRP1-CT. Deletion of the N-terminal SH2 domain of Shp2 greatly weakened binding, recapitulating other experiments in Xenopus with similar Shp2 truncations that also found a greater influence of the N-SH2 domain in responding to basic fibroblast growth factor (29). Binding was tightest when both SH2 domains of Shp2 bound to the dually phosphorylated LRP1-CT. This first report of Shp2 interaction with LRP1 may provide a clue as to how maintenance of vascular wall integrity depends, at least in part, on the ability of LRP1 to regulate the activity of PDGFRβ (Ref. 30 and references therein). It is known that Shp2 is recruited to the PDGF receptor upon receptor stimulation by PDGF-BB (31) and that PDGF stimulation of cells also results in tyrosine phosphorylation of LRP1 and possibly formation of an LRP1-PDGF receptor heterodimer (16, 32). Studies conflict as to whether Shp2 inactivation is necessary for PDGF signaling to occur or whether Shp2 activity contributes to this process (33, 34). Thus, one might speculate that binding of Shp2 to LRP1 may control its activity toward PDGFRβ. Depending on the localization and amount of expression of LRP1, activity of Shp2 toward PDGFRβ and/or its downstream targets may increase or decrease. Further experiments are required to ascertain the exact role of these proteins in PDGFRβ signaling processes in different cellular contexts.
This work was supported, in whole or in part, by National Institutes of Health Grants AG025343 and CA78629. This work was also supported by NIH Training Grant T32-DK07233 (to G. N. B.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
The abbreviations used are: APP, amyloid precursor protein; PDGF, platelet-derived growth factor; PDGFR, PDGF receptor; HA, hemagglutinin; GST, glutathione S-transferase; TBS, Tris-buffered saline; Y2F, Y4473F,Y4507F.
G. N. Betts, J. W. Torpey, H. Barnes, M. Guttman, D. W. Peterson, J. Lew, P. van der Geer, and E. A. Komives, manuscript submitted.
References
- 1.Strickland, D. K., and Ranganathan, S. (2003) J. Thromb. Haemostasis 1 1663–1670 [DOI] [PubMed] [Google Scholar]
- 2.Herz, J., Hamann, U., Rogne, S., Myklebost, O., Gausepohl, H., and Stanley, K. K. (1988) EMBO J. 7 4119–4127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Herz, J., and Strickland, D. K. (2001) J. Clin. Investig. 108 779–784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Trommsdorff, M., Borg, J. P., Margolis, B., and Herz, J. (1998) J. Biol. Chem. 273 33556–33560 [DOI] [PubMed] [Google Scholar]
- 5.Pietrzik, C. U., Yoon, I. S., Jaeger, S., Busse, T., Weggen, S., and Koo, E. H. (2004) J. Neurosci. 24 4259–4265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li, Y., Marzolo, M. P., van Kerkhof, P., Strous, G. J., and Bu, G. (2000) J. Biol. Chem. 275 17187–17194 [DOI] [PubMed] [Google Scholar]
- 7.van Kerkhof, P., Lee, J., McCormick, L., Tetrault, E., Lu, W., Schoenfish, M., Oorschot, V., Strous, G. J., Klumperman, J., and Bu, G. (2005) EMBO J. 24 2851–2861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Roebroek, A. J., Reekmans, S., Lauwers, A., Feyaerts, N., Smeijers, L., and Hartmann, D. (2006) Mol. Cell Biol. 26 605–616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Barnes, H., Larsen, B., Tyers, M., and van Der Geer, P. (2001) J. Biol. Chem. 276 19119–19125 [DOI] [PubMed] [Google Scholar]
- 10.Barnes, H., Ackermann, E. J., and van der Geer, P. (2003) Oncogene 22 3589–3597 [DOI] [PubMed] [Google Scholar]
- 11.Boucher, P., Liu, P., Gotthardt, M., Hiesberger, T., Anderson, R. G., and Herz, J. (2002) J. Biol. Chem. 277 15507–15513 [DOI] [PubMed] [Google Scholar]
- 12.Gotthardt, M., Trommsdorff, M., Nevitt, M. F., Shelton, J., Richardson, J. A., Stockinger, W., Nimpf, J., and Herz, J. (2000) J. Biol. Chem. 275 25616–25624 [DOI] [PubMed] [Google Scholar]
- 13.Lutz, C., Nimpf, J., Jenny, M., Boecklinger, K., Enzinger, C., Utermann, G., Baier-Bitterlich, G., and Baier, G. (2002) J. Biol. Chem. 277 43143–43151 [DOI] [PubMed] [Google Scholar]
- 14.Goto, J. J., and Tanzi, R. E. (2002) J. Mol. Neurosci. 19 37–41 [DOI] [PubMed] [Google Scholar]
- 15.Pietrzik, C. U., Busse, T., Merriam, D. E., Weggen, S., and Koo, E. H. (2002) EMBO J. 21 5691–5700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Loukinova, E., Ranganathan, S., Kuznetsov, S., Gorlatova, N., Migliorini, M. M., Loukinov, D., Ulery, P. G., Mikhailenko, I., Lawrence, D. A., and Strickland, D. K. (2002) J. Biol. Chem. 277 15499–15506 [DOI] [PubMed] [Google Scholar]
- 17.Seeliger, M. A., Young, M., Henderson, M. N., Pellicena, P., King, D. S., Falick, A. M., and Kuriyan, J. (2005) Protein Sci. 14 3135–3139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.van der Geer, P., and Hunter, T. (1994) Electrophoresis 15 544–554 [DOI] [PubMed] [Google Scholar]
- 19.Mandell, J. G., Falick, A. M., and Komives, E. A. (1998) Proc. Natl. Acad. Sci. U. S. A. 95 14705–14710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hughes, C. A., Mandell, J. G., Anand, G. S., Stock, A. M., and Komives, E. A. (2001) J. Mol. Biol. 307 967–976 [DOI] [PubMed] [Google Scholar]
- 21.Ferreiro, D. U., Cervantes, C. F., Truhlar, S. M., Cho, S. S., Wolynes, P. G., and Komives, E. A. (2007) J. Mol. Biol. 365 1201–1216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Thorpe, G. H., and Kricka, L. J. (1986) Methods Enzymol. 133 331–353 [DOI] [PubMed] [Google Scholar]
- 23.Truhlar, S. M. E., Croy, C. H., Torpey, J. W., Koeppe, J. R., and Komives, E. A. (2006) J. Am. Soc. Mass Spectrom. 17 1490–1497 [DOI] [PubMed] [Google Scholar]
- 24.Songyang, Z., Shoelson, S. E., McGlade, J., Olivier, P., Pawson, T., Bustelo, X. R., Barbacid, M., Sabe, H., Hanafusa, H., Yi, T., Ren, R., Baltimore, D., Ratnofsky, S., Feldman, R. A., and Cantley, L. C. (1994) Mol. Cell Biol. 14 2777–2785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Swertfeger, D. K., Bu, G., and Hui, D. Y. (2002) J. Biol. Chem. 277 4141–4146 [DOI] [PubMed] [Google Scholar]
- 26.Kroenke, C. D., Ziemnicka-Kotula, D., Xu, J., Kotula, L., and Palmer, A. G., III (1997) Biochemistry 36 8145–8152 [DOI] [PubMed] [Google Scholar]
- 27.Ramelot, T. A., and Nicholson, L. K. (2001) J. Mol. Biol. 307 871–884 [DOI] [PubMed] [Google Scholar]
- 28.Marzolo, M. P., Yuseff, M. I., Retamal, C., Donoso, M., Ezquer, F., Farfan, P., Li, Y., and Bu, G. (2003) Traffic 4 273–288 [DOI] [PubMed] [Google Scholar]
- 29.O'Reilly, A. M., and Neel, B. G. (1998) Mol. Cell Biol. 18 161–177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Boucher, P., Li, W.-P., Matz, R. L., Takayama, Y., Auwerx, J., Anderson, R. G. W., and Herz, J. (2007) PLoS ONE 2 e448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Klinghoffer, R. A., and Kazlauskas, A. (1995) J. Biol. Chem. 270 22208–22217 [DOI] [PubMed] [Google Scholar]
- 32.Newton, C. S., Loukinova, E., Mikhailenko, I., Ranganathan, S., Gao, Y., Haudenschild, C., and Strickland, D. K. (2005) J. Biol. Chem. 280 27872–27878 [DOI] [PubMed] [Google Scholar]
- 33.Meng, T. C., Fukada, T., and Tonks, N. K. (2002) Mol. Cell 9 387–399 [DOI] [PubMed] [Google Scholar]
- 34.Araki, T., Nawa, H., and Neel, B. G. (2003) J. Biol. Chem. 278 41677–41684 [DOI] [PubMed] [Google Scholar]