

Abstract
The Haemophilus influenzae HMW1 adhesin is secreted via the two-partner secretion pathway and requires HMW1B for translocation across the outer membrane. HMW1B belongs to the Omp85-TpsB superfamily of transporters and consists of two structural domains, a C-terminal transmembrane β-barrel and an N-terminal periplasmic domain. We investigated the electrophysiological properties of the purified full-length HMW1B and the C-terminal domain using planar lipid bilayers. Both the full-length and the truncated proteins formed conductive pores with a low open probability, two well defined conductance states, and other substates. The kinetic patterns of the two conductance states were distinct, with rapid and frequent transitions to the small conductance state and occasional and more prolonged openings to the large conductance state. The channel formed by the full-length HMW1B showed selectivity for cations, which decreased when measured at pH 5.2, suggesting the presence of acidic residues in the pore. The C-terminal domain of HMW1B was less stable and required reconstitution into liposomes prior to insertion in the bilayer. It formed a channel of smaller conductance but a similar gating pattern as the full-length protein, demonstrating the ability of the last 312 C-terminal amino acids to form a pore and suggesting that the periplasmic domain is not involved in occluding the pore, nor in controlling the inherent basal kinetics of the channel. The HMW1 pro-piece containing the secretion domain, although binding to the channel with high affinity, did not induce channel opening.
Secretion of proteins in the external medium from the cytosol of a Gram-negative bacterium is a complex mechanism. Because of the compartmented structure of the cell envelope, a periplasm limited by the inner membrane and the outer membrane, secreted proteins may need to undergo two translocation processes. Seven main types of mechanisms exist to translocate proteins through the cell envelope (1–6). Some of these pathways export the protein directly from the cytosol to the extracellular milieu, whereas others include a periplasmic intermediate step. In this case, independent translocation pathways exist through the inner membrane and comprise the Sec pathway and the TAT pathway (7). The type V secretion pathway is one of those two-step processes, where proteins are first exported to the periplasm in a Sec-dependent manner (5). The second step involves the translocation through the outer membrane of a passenger peptide through a transporter. In many cases, the passenger and the transporter domains belong to the same polypeptide, thus defining an auto-transporter type of mechanism. The two-partner secretion pathway is distinct in that the secreted exoprotein (TpsA) and its cognate transporter protein (TpsB) are translated as two separate proteins but are coded on the same operon or the same locus. Each TpsB transporter is devoted to the translocation of one specific TpsA. For example, the Haemophilus influenzae HMW1/HMW1B pair represents a prototype two-partner secretion pathway where HMW1 is the transported exoprotein (TpsA) and HMW1B is the outer membrane translocator (TpsB) (8).
The TpsB proteins belong to a large family of polypeptide-transporting or -assembling proteins that also includes Bordetella pertussis FhaC, Neisseria meningitidis Omp85, Escherichia coli YaeT, Toc 75 of chloroplasts, and Sam50-Tob55 of mitochondria (9, 10). The proteins of this superfamily are characterized by a C-terminal transmembrane β-barrel and the presence of one to five polypeptide transport-associated (POTRA)2 domains in their N-terminal periplasmic region, which are believed to be involved in the interaction with the substrate protein (11, 12). For TpsBs the pore formed by the β-barrel is postulated to be used for the translocation of the specific TpsA protein; the function of the β-barrel in Omp85 is not yet well defined (13, 14), although it has been shown to form ion-conductive pores in artificial bilayers (15, 16). Recently, the structure of FhaC has been solved, giving new insight into the organization of TpsB proteins. The FhaC barrel is composed of 16 β strands and is partially plugged by an N-terminal α-helix and the inwardly folded extracellular loop L6 (13). Two POTRA domains are found in a periplasmic region between the N-terminal α-helix and the β-barrel. FhaC has also been characterized in electrophysiology and found to have a conductance of 1200 pS in 1 m KCl (17, 18), a value comparable with a single OmpF porin monomer in these conditions (19). Deletion of the POTRA domains abrogated secretion but had no effect on the channel properties (13, 18). Deletion of L6 also abolished secretion, despite the fact that it appears to make a wider pore based on increased antibiotic sensitivity and FhaC-ΔL6 structure predicted in silico (13). Based on the FhaC structure and functional experiments, Clantin et al. (13) propose a model of FHA transport whereby FHA first interacts with the POTRA domains of FhaC via its N-terminal secretion domain, which has the resulting effect of displacing L6 of FhaC and allowing FHA to penetrate in the pore as a β-hairpin.
The crystal structure of HMW1B is unknown, but a recently published topology study predicts a protein with multiple domains (20). By using epitope tagging and accessibility to engineered cysteines, St. Geme and co-workers (21) showed that the N terminus is surface-exposed and followed by a large periplasmic domain and then finally by a C-terminal domain consisting of a β-barrel with 10–13 transmembrane β strands. Liposome swelling assays suggested that HMW1B and the C-terminal domain have pore forming activity. The measurement of the swelling rate performed with a range of different size sugars gave an estimation of the size of the pore around 2.7 nm. The specific activities are much lower than for OmpF, suggesting that the pores are either of smaller size than the general diffusion porins (which is unlikely based on the estimated pore size) or remain mainly closed (21). The integrity of the periplasmic domain was shown to be essential for secretion, and thus this region is postulated to participate in substrate recognition and/or interaction (20).
HMW1B appears to exist as an oligomer, but the size of the oligomer is still controversial. A tetrameric organization of HMW1B was first proposed by St. Geme and co-workers in 2004 (21). This was supported by direct observation with electron microscopy as well as by the retention time in the size exclusion column during purification and by migration on nondenaturating gels. The protein used for these studies was produced from a histidine affinity tag-tagged construct. More recently, similar experiments conducted with an untagged HMW1B construct suggest that HMW1B rather forms dimers (22) in a manner reminiscent of other polypeptide-transporting proteins, such as PapC (23) and the TIM and TOM complexes of mitochondrial inner and outer membranes, respectively (24, 25).
In this paper, we report the first electrophysiological characterization of HMW1B. We show that this protein is able to form conductive pores with a low open probability, in agreement with liposome swelling assays (20). Moreover, we demonstrate the pore forming ability of the C-terminal domain of HMW1B (HMW1B234–545) and found that the absence of the periplasmic domain has a minimal effect on the conductance and the kinetic behavior of HMW1B. We also show that the N-terminal domain of the HMW1 substrate (HMW1 pro-piece) harboring the secretion domain has the ability to bind to the HMW1B channel with high affinity, although it does not induce channel opening.
EXPERIMENTAL PROCEDURES
Strains and Protein Purification—HMW1B, HMW1B234–545, and the HMW1 pro-piece (HMW1-PP, residues 69–441, harboring the HMW1 secretion domain) were purified as described previously (20, 21, 26) with modifications. The strains used in this study were the porin-less E. coli BL21(DE3)omp8 derivative (27) harboring either plasmid pHAT:HMW1B (21) or pHAT: HMW1B234–545 (20) and BL21(DE3) harboring pGEX:HMW69–441 (26). The cells were grown overnight at 37 °C in 4 liters of Luria-Bertani medium supplemented with 100 μg/ml ampicillin and harvested by centrifugation at 6,000 × g for 20 min. The cell pellets were resuspended in 25 ml of lysis buffer (20 mm HEPES/EDTA-free pro-tease inhibitor mixture; Roche Applied Science) and disrupted by sonication. The membranes were collected by centrifugation at 40,000 × g for 40 min. After selective solubilization of inner membranes by Sarkosyl (Sigma), outer membranes were recovered by a spin at 40,000 × g for 1 h. Outer membrane proteins were then solubilized in 25 ml of buffer A (20 mm HEPES, pH 8.0, 150 mm NaCl, 1% Elugent; CalBiochem) by vigorously stirring for 1 h. Following centrifugation at 40,000 × g for 30 min, solubilized proteins were collected and applied to a 5-ml nickel-nitrilotriacetic acid superflow column (Qiagen) equilibrated with buffer B (20 mm HEPES, pH 8.0, 150 mm NaCl, 0.1% dodecyl maltoside; CalBiochem). Bound proteins were eluted with an imidazole linear gradient using buffer B containing 0.5 m imidazole. The pooled fractions were further loaded on a 5-ml HiTrap-Q column and/or 5-ml HiTrap-Heparin column (GE Healthcare) equilibrated with buffer C (20 mm HEPES, pH 8.0, 50 mm NaCl, 0.1% dodecyl maltoside). The proteins were further enriched in the flow-through, whereas minor contaminant proteins were bound to the column. Finally, the proteins were concentrated using a spin concentrator (VivaSpin) and applied to a Sephacryl S200 (or S300) 16/60 column (GE Healthcare) equilibrated with buffer D (20 mm HEPES, pH 8.0, 150 mm NaCl, 0.1% dodecyl maltoside). Peak fractions were pooled and concentrated to ∼4 mg/ml. Alternatively, during the final concentration step, the protein samples were subjected to buffer exchange by a quick dilution with buffer E (20 mm HEPES, pH 8.0, 150 mm NaCl, 0.1% octyl-β-d-glucoside; Anatrace) and concentrated to ∼4 mg/ml. An N-terminal sequencing (Midwest Analytical, Inc.) confirmed HMW1B without any detectable contaminants. The results of the size exclusion chromatography (supplemental Fig. S1) confirm that both full-length and C-terminal domain HMW1B eluted as tetramers, as described previously (21). An SDS-PAGE of full-length HMW1B (supplemental Fig. S1) shows the heat modifiability of the protein, as reported (21), confirming the retention of the native structure during purification. Note that, unlike porins, oligomers are not observed in the SDS-PAGE even when the sample has not been heated to 100 °C.
Channel Reconstitution—Channel activity was measured in planar lipid bilayers, as described (28, 29) using a 0.01-mm-thick Teflon film in which a ∼100-μm hole has been formed and pretreated with a solution of 1% of hexadecane (TCI America) in pentane (Burdick & Jackson). The chambers were both filled with 1.5 ml of the appropriate buffer, and 5 μl of a 5 mg ·ml–1 diphytanoyl phosphatidylcholine (Avanti Polar Lipids) solution diluted in pentane was added on each side. After evaporation of the solvent (∼5 min), the buffer on one side only of the Teflon film was lowered and raised back, creating a single lipid bilayer membrane over the hole, as witnessed by an increase in resistance. For the full-length HMW1B, once the membrane was formed and stable, 200 ng of the protein (5 μl of the protein diluted 1:100 in the appropriate buffer containing 1% of N-octyl-oligo-oxyethylene (Axxora)) were added to one chamber only, thus defined as the cis side. A single insertion usually occurs within 10 min of stirring. HMW1B234–545 was reluctant to insert in this manner, and the protein had to be reconstituted first into soybean phospholipids (l-α-phosphatidylcholine Type II-S; Sigma) at a protein to lipid ratio of 1:200 to 1:125 (w/w); then 5 μl of the reconstituted proteoliposomes were added to the cis side. The preparation of proteoliposomes was done as described (30), except that the dehydration-rehydration steps were omitted, and proteoliposomes were added to the bilayer chamber after resuspension in MOPS buffer. Insertions are seen within 10 min of stirring.
Electrophysiological Recordings and Data Analysis—The currents were recorded with an Axopatch 1D amplifier (Axon Instruments) using a CV-4B headstage. The current was filtered at 1 KHz, digitized at 1.25-ms sampling intervals (ITC-18, Instrutech), and stored on a computer using the Acquire software (Bruxton). The analysis and reading of the traces were performed with the Clampfit 9.0.1.07 program from Axon Instruments. The voltage was always applied on the cis side, with the trans side grounded. All of the buffers used in electrophysiology contained 5 mm of HEPES (pH 7.2) or 5 mm MES (pH 5.2) and various concentrations of KCl. The pH was adjusted by the addition of KOH or HCl. To measure reversal potentials, the insertions were first obtained in symmetrical conditions of 500 mm KCl buffer. The salt concentration was raised on the cis side by the addition of 750 μl of a 3 m KCl solution in the appropriate buffer, and the same volume of 500 mm KCl buffer was added on the trans side. This created a salt gradient of 1500 mm KCl on the cis side and 500 mm on the trans side. In some experiments, purified HMW1-PP (26) containing the secretion domain (residues 69–441) was added to either side of a bilayer containing HMW1B (up to 20 μm final concentration) or on both sides of the bilayer (5 μm final concentration).
Solid Phase Binding Assay—The interaction between HMW1B and HMW1-PP was tested using an enzyme-linked immunosorbent assay-based protein binding assay. Reacti-Bind glutathione-coated plate wells (Pierce) were incubated with 1 μg of GST:HMW1-PP in 100 μl of PBS-T buffer (PBS buffer + 0.02% (v/v) Tween 20) overnight at 4 °C. After washing the wells with 200 μl of PBS-T buffer, the unreacted material was blocked with 150 μl of PBS-T buffer containing 1% (w/v) bovine serum albumin for 2 h at 4°C. After washing the wells three times with PBS-T buffer, varying amounts of pHAT: HMW1B in PBS-T buffer (100 μl/well) were added and incubated for 2 h at 4°C. After several washings with PBS-T buffer, 100 μl of HisProbe-HRP (Pierce) were added (1:2,000 dilution in PBS-T) and incubated for 1 h at 4°C. Following extensive washings with PBS-T, 200 μl of Sigma FAST OPD substrate (Sigma) was added to wells and incubated in the dark for 30 min at room temperature. The reaction was terminated by the addition of 50 μl of 3 n HCl, and the plates were read at 492 nm using Multiskan EX microplate photometer (Thermo Scientific).
RESULTS
HMW1B Forms a Very Dynamic Channel in Bilayers—Liposome swelling assays have suggested that HMW1B could form a pore. However, because of the low rate of translocation of arab-inose, the channel formed by HMW1B was thought to be either in a nonfunctional conformation or closed most of the time (21). Using the planar lipid bilayer technique, we have been able to record the electrophysiological activity of HMW1B and confirmed that HMW1B has pore forming activity. The traces presented in Fig. 1A were recorded at different voltages from the same inserted protein in a 1 m KCl buffer. HMW1B displays frequent transient transitions to various open levels. Despite a highly dynamic behavior, the channel has a relatively low probability of being in the open state (Po is ∼0.1 at +50 and –50 mV for this representative experiment). When the pore is in a closed state (marked with level C in Fig. 1A), the current is close to 0 pA, demonstrating a full and tight closure of the channel, as well as the absence of a leak at the protein/membrane interface. Moreover, the very low level of noise observed when HMW1B is closed confirms the stability of the membrane after protein insertion.
FIGURE 1.

HMW1B forms ion-conductive pores. A, representative current traces of HMW1B obtained from the same single inserted protein at the indicated voltages. The current level corresponding to the closed channel is marked by C. The arrows show the direction of openings (upward and downward deflections at positive and negative voltages, respectively). B, HMW1B displays two main levels of conductance, labeled S and L and indicated by the tick marks. The bottom trace shows 2 s of the upper trace (marked by the thick horizontal bar) on an expanded time scale to highlight the details of the two types of openings. The voltage was +50 mV. C, amplitude histogram from the whole trace showed in B. C, closed; S, small conductance state; L, large conductance state.
As seen on the enlarged segment of the trace in Fig. 1B, two types of openings can be defined: openings of small conductance (marked with S) with a current of ∼17 pA at +50 mV and openings of larger conductance (marked with L) with a current of ∼70 pA. The amplitude histogram obtained from the entire trace displays three main peaks corresponding to the three states that are most often visited by the channel. The peak corresponding to the closed state has the highest amplitude because HMW1B spends most of the time in a closed conformation. It is also somewhat asymmetric and cannot be fitted with a Gaussian distribution. Indeed, although the noise level is very low when the channel is in the fully closed state, the lopsided distribution of the first peak indicates that the channel frequently opens to intermediate ill-defined levels, smaller than the S level. The second peak, corresponding to the S openings, can be distinguished as a hump on the edge of the first peak. The third peak corresponds to the L level and displays an asymmetrical distribution too, which can be explained by the constant flickering between the L open level and various ill-defined lower conductance states, as seen on the enlarged trace.
The opening kinetics is highly variable; in some experiments, the channel displays few transitions to the large conductance state; in other experiments, the channel is much less active altogether. Still, the overall fingerprint of the HMW1B emerges as that of a channel fluctuating between two main conductance states. There does not appear to be any dependence on the magnitude of the voltage, although the behavior seems dependent on the polarity of the voltage, as observed in Fig. 1A. In this experiment, fewer openings of large conductance are monitored at positive voltages than at negative voltages. Interestingly, more S openings are observed in positive voltages too. The polarity of the voltage dependence is not preserved from one experiment to the other (i.e. with different insertions), implying that the protein could insert in both orientations or that the polarity of the asymmetrical voltage dependence is variable. The possibility that the protein could insert in either direction is unlikely, because it would require that the large hydrophilic N-terminal domain translocate through the lipid bilayer as well.
Biophysical Properties of HMW1B—Because of the variability in kinetic behavior and the high level of flickering of the channel, the frequency of channel openings could not be measured reliably. The sizes of the openings, on the other hand, are consistent parameters. In 45 experiments performed with three different preparations of HMW1B, we have never observed an opening state of higher amplitude than the L level, and thus we assume that the L level corresponds to the full open state of HMW1B in these conditions. The current of the large conductance state was measured from different inserted proteins and plotted versus the applied voltage. The channel has ohmic behavior from –150 mV to +150 mV. The measurement of the openings becomes unreliable over ±150 mV because of the high instability of the channel at higher voltages. A linear fit of the current-voltage plot gives a conductance of 1417 pS (Fig. 2A) for the large conductance state.
FIGURE 2.
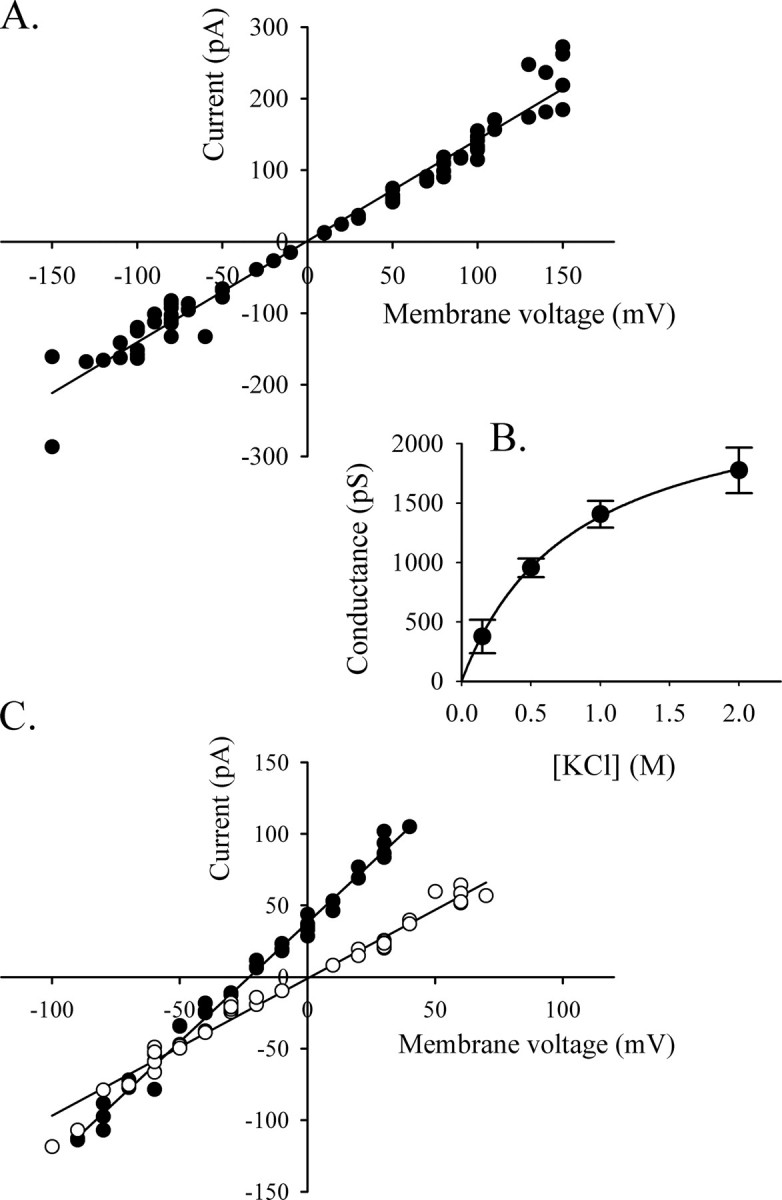
Conductance and selectivity of HMW1B. A, current-voltage (I/V) curve of the L conductance in 1 m KCl (pH 7.2). The current was measured from nine independent experiments at different voltages and plotted on the ordinate. The best fitted line was calculated with Sigmaplot, and the slope gives a conductance of 1417 pS. B, the current of the L conductance saturates with increasing salt concentration. The conductance was calculated from the I/V curves from at least three independent inserted proteins in each condition. The data represent the average conductance ± S.E. of the L openings. The fit to the Michaelis-Menten equation gave the following parameters: Gmax = 2505 pS, and Km = 808 mm. C, the reversal potential of HMW1B was measured from the L openings in the presence of a salt gradient of 1500/500 mm KCl (cis/trans)(closed circles, n = 3). The observed Erev of –22 mV in these conditions corresponds to a PK/PCl of 11.3. The open circles show the I/V relationship in symmetrical conditions of 500/500 mm KCl (n = 4).
The majority of the experiments were performed in a 1 m KCl buffer. Openings of large conductance were also observed at lower or higher salt concentrations, and the conductance values were calculated from I/V curves and plotted versus salt concentration (Fig. 2B). Above 1 m KCl the conductance starts to saturate, indicating weak ion binding (31). By fitting the data to a Michaelis-Menten equation, we were able to estimate the maximal conductance of the channel to be Gmax = 2505 ± 60 pS and the Km to be 808 ± 45 mm.
The measurement of current in asymmetrical buffer conditions allows the calculation of the reversal potential of the channel, which relates to selectivity. For these experiments, the trans compartment contained a 500 mm KCl buffer, and the cis chamber was filled with a 1.5 m KCl buffer. In these conditions, the reversal potential measured for HMW1B is –22 mV (Fig. 2C). The permeability ratio calculated by the Goldman-Hodgkin-Katz equation (31) is PK/PCl = 11.2, indicating cation selectivity. The cation selectivity of large channels such as porins or polypeptide-transporting channels (32–34) is usually due to the presence of titrable negative charges at the constriction zone. If this is the case, loss of cation selectivity is expected upon titration of the negative charges at acidic pH. This assumption was confirmed by the measurement of the selectivity at lower pH. Indeed, at pH 5.2, the reversal potential decreased to –17 mV, corresponding to a PK/PCl of 4.7 (Fig. 3C). Another effect of acidic pH on the pore was to increase the conductance of the openings. The L conductance is increased 1.5-fold (2115 pS), as calculated from the I/V curve shown in Fig. 3C (symmetrical conditions).
FIGURE 3.

Acidic pH affects HMW1B conductance and selectivity. A and B, representative segments obtained from the same inserted protein at the indicated voltages and at pH 5.2 show that the two main conductance levels are still observed, although the channel is slightly noisier than at neutral pH. The trace between traces A and B shows 2 s of the +50 mV trace on an expanded time scale (scale bar between A and B). The bottom scale bar refers to the top trace in A and the trace in B. C, current-voltage relationship for the L openings at pH 5.2 in symmetric (○, 1 m KCl on each side) and asymmetric (•, 1500/500 mm KCl cis/trans) conditions. The reversal potential drops to –17 mV at pH 5.2, reflecting a PK/PCl of 4.7, whereas the conductance calculated from the I/V curve in symmetric conditions increases to 2115 pS.
Initial attempts at modulating channel activity were unsuccessful. Neither membrane potential, acidic pH, nor changes in ionic strength produced changes in channel open probability or in the overall activity pattern. The addition of purified HMW1-PP containing the secretion domain was also ineffective. The channel activity was unchanged, therefore ruling out either channel activation alone or channel activation in combination with block because of the transported peptide. The secretion domain alone is competent for translocation in vivo, as demonstrated in a previous study (26) where HMW1-PP could be recovered from cell supernatants when co-expressed with HMW1B. In addition, here we show that the purified HMW1-PP used in bilayer studies does indeed bind to HMW1B. An enzyme-linked immunosorbent assay-based protein binding assay shows that HMW1B binds GST:HMW1-PP with a Kd of 125.8 ± 14.4 nm (supplemental Fig. S2A) and that this binding can be competed by HMW1-PP (not fused to GST, i.e. the material used in bilayer experiments) with an IC50 of 125.5 ± 12.5 nm (supplemental Fig. S2B). This high affinity of HMW1-PP for HMW1B supports the contention that the HMW1-PP is present in a folded state, as shown by x-ray crystallography (26) and in a biochemically relevant conformation. Because the bilayer experiments were performed with HMW1-PP concentrations that ranged up to 100 times Kd, it appears that the N-terminal domain of HMW1 alone can bind to the channel but does not have the ability to or is not sufficient to trigger channel opening in vitro.
Pore Properties of the C-terminal Domain HMW1B234–545—The domain analysis performed by St. Geme co-workers (20) suggests that the truncated mutant HMW1B234–545 constitutes a C-terminal domain containing 10 membrane-spanning β-strands involved in the formation of the pore. We decided to test this prediction by checking the behavior of this mutant in electrophysiology. The HMW1B234–545 truncated protein appears less stable than the full-length protein during purification and did not insert in the bilayer. To overcome the insertion problem, we had to first reconstitute the proteins in liposomes (see “Experimental Procedures”). Once inserted, HMW1B234–545 showed pore activity with a behavior similar to the one recorded for the full-length protein (Fig. 4). The pore is closed most of the time and displays two types of openings with a small and a large conductance. A current-voltage plot gave a value of 973 pS in 1 m KCl for the large conductance. This value is slightly smaller than that of the full-length protein, either because of some underestimation in the measurement caused by the high level of flickering activity or because the β-barrel domain of the full-length protein includes a greater number of β-strands than in the truncated construct. In any case, these results demonstrate that the last 312 amino acids of the protein are sufficient to form a pore and that the periplasmic domain is not involved in the formation of the channel. The unchanged activity and the decreased conductance of HMW1B234–545 suggest that the removal of the N-terminal domain has not increased the open probability or the size of the pore and therefore rule out a role for this domain in the obstruction of the pore.
FIGURE 4.

The C-terminal domain HMW1B234–545 also forms pores in bilayers. Representative traces from the same inserted protein at the indicated voltages show that the overall behavior is very similar to that of the full-length protein, and two main conductance levels are also seen. The expanded trace shows 1 s of the –50 mV trace marked by a thick horizontal line. The size of the L openings calculated from the I/V curve is 973 pS. C, closed.
DISCUSSION
All of the bacterial secretion systems in Gram-negative bacteria include an outer membrane component used for transport and/or assembly of the secreted polypeptide. With the exception of the capsule polysaccharide export machinery whose outer membrane translocon comprises a novel α-helical barrel (35), outer membrane translocator proteins or domains characterized to date appear to be β-barrel channel-like conduits, presumably used for the transport of unfolded, or in some cases folded, polypeptides (13, 36–38). Only a very small number of them have been subjected to electrophysiological analysis (17, 39–41), and, along with polypeptide-import machineries of chloroplasts and mitochondria (24, 33, 42, 43), they display spontaneous kinetic transitions between open and closed states, and many of them have conductance properties typical of fairly large channels.
Here we provide the first description of the pore properties of HMW1B by electrophysiology. Unlike FhaC, a TpsB transporter whose channel activity was initially reported in 1999 (17), HMW1B is thought to belong to the oligomeric branch of the Omp85/TpsB superfamily of transporters, although the native oligomerization state, dimeric or tetrameric, is still unclear (21, 22). In the experiments reported here, HMW1B is purified as a tetramer and displays two main conductances of 1417 pS (L) and 343 pS (S) in 1 m KCl. Although the small conductance is about a quarter of the size of the large one, our experiments do not allow us to conclude whether the S conductance corresponds to the opening of a single monomer, whereas the L conductance represents the full opening of the four monomers in concert. Such a high level of cooperativity has not been reported among outer membrane channels. Thus, these two conductance levels might simply reflect two states of a single pore. Indeed, even monomeric pores such as FhaC display subconductance states (17).
The channel properties of HMW1B have similarities with those of FhaC, the other TpsB that has been analyzed by electrophysiology. Both channels have similar conductances in 1 m KCl (∼1400 pS for HMW1B and 1200 pS for FhaC (18)). We have not been able to achieve multiple insertions to obtain macroscopic current information, which, in the case of FhaC, showed activation of the channel at potentials more negative than –60 mV (18). However, we also observed a polarity-dependent behavior in HMW1B. In addition, a greatly increased activity occurred at potentials larger than ±100 mV, leading to noisy and unstable traces (data not shown). A similar behavior was also described for FhaC (18). We observed that the C-terminal β-barrel domain of HMW1B was sufficient to form ion-conducting pores, although the channel appears less stable, as was found in FhaC (18). Although substates were reported for FhaC, examination of the published traces reveals that the FhaC channel seems to show a more homogenous behavior with mostly transitions to the 1200 pS state (17, 18). On the contrary, a hallmark of the HMW1B channel kinetic behavior is the presence of two well defined conductance states that may relate to the oligomeric nature of the channel.
The topology model presented by St. Geme and colleagues predicts a C-terminal 10 β-stranded barrel (20). Would such architecture allow for the formation of a channel with an observed conductance of ∼1400 pS in 1 m KCl? It is difficult to establish a strict correlation between the number of β-strands, the size of the pore and the conductance. A survey of the literature shows that barrels with fewer or with more β-strands than the predicted 10 for HMW1B form pores with smaller conductance values than what we report here. For example, the conductance of the eight β-stranded N-terminal domain barrel of OmpA is ∼110 pS in 1 m KCl (44). The conductance appears to increase for pores containing a larger number of β strands (for example, 800 pS for the monomeric OmpG (14 strands) (45) or 1200 pS for FhaC (16 strands) (18)), but one has to exercise caution because the conductance is not only determined by the pore diameter but also by the charges and the electric field within the pore as well as by the access resistance of the pore (46). In addition, we must keep in mind that the inward folding of extracellular loops or other domains may constrict the pore. For instance, other barrels made of 10 β-strands include the N. meningitidis adhesin OpcA (47) and the E. coli OmpT protease (48), both of which appear to have occluded pores. The proposed topology must also be taken with caution, because the C-terminal barrel domain may include more than 10 β-strands, as stated by the authors themselves (20). In fact, the smaller conductance (973 pS) we obtain for the C-terminal domain HMW1B234–545 may be due to truncation within the actual β-barrel domain, because the deleted construct was made on the basis of the 10-β strand putative topology.
The concentration dependence of the L conductance yielded saturation kinetics parameters (Km = 808 mm, Gmax = 2.5 nS) that are in the range of those typically observed for large channels with weak affinity for ions. For example, the values for the chloroplast import channel Toc75 were Km = 2.5 m and Gmax = 6.2 nS (33). The fact that HMW1B is likely to form a large channel is also suggested by its permeability to maltodextrins (21), although we were unable to demonstrate interruption of ion flux through HMW1B because of flux of maltohexaose (but this could be due to a very low affinity of the sugar for the pore and thus a short residency time). The cation selectivity of the channel is not in disagreement with a large channel architecture, because permeability ratios for potassium to chloride (PK/PCl) in a similar range have been reported for other large channels, such as the chloroplast Toc75 (PK/PCl = 14.3) (33), the Vibrio cholerae porin OmpU (PK/PCl = 13.8) (32) and E. coli TolC (PK/PCl = 16.5) (39).
Experiments done at different pH values can provide biophysical insights into the mechanism of permeation and pore function, even if pH may not be a physiologically relevant parameter in the context of secretion of HMW1 adhesin. Although one cannot dismiss the possibility that protons affect the conformation or geometry of the pore, pH effects are often ascribed to protonation of key pore residues, leading to changes in the distribution of charges inside the pore (34). These electrostatic effects can be responsible for changes in selectivity. As shown for other large cation-selective channels, the reduction in the number of negative charges inside the pore caused by the titration of the acidic residues results in decreased cation selectivity. For example, the permeability ratio PK/PCl of 14.3 of the chloroplast import protein Toc75 decreases 2-fold between pH 7.0 and 5.0 (33). The mild cation selectivity of OmpF (PK/PCl of 4) can even be turned into anion selectivity in strongly acidic solutions (34). Here we find that the cation selectivity of HMW1B (PK/PCl of 11.2) is considerably reduced at pH 5.2 and propose that the inner channel wall displays acidic residues at the constriction zone.
So far, we have not been able to promote channel opening by varying physico-chemical parameters such as voltage, pH, and ionic strength, nor by providing HMW1-PP containing the secretion domain (up to 20 μm) to either side of a reconstituted pore. However, we have demonstrated that the HMW1-PP binds to the HMW1B channel with high affinity. Therefore, assuming that the channel is activated by the substrate that it translocates, it appears that the N-terminal secretion domain alone is not able or sufficient to open the channel, despite its ability to interact with the channel. The high affinity of HMW1-PP for HMW1B is consistent with its role in specific recognition of HMW1 by HMW1B, as clearly demonstrated by experiments with chimera where HMW1-PP fused to the Hia adhesin passenger domain could be secreted when co-expressed with HMW1B but not with the Hia translocator (21). The work presented here suggests a possible model for the functional interaction between HMW1B and its substrate, whereby the secretion domain is involved in the recognition event, but not in channel activation, a process that may require either the whole polypeptide or another unidentified domain. It is also possible that additional unidentified factors not provided in the bilayer system are in fact required and that the reconstituted system used here does not fully reproduce the in vivo situation, despite the fact that the translocator is active and able to bind the HMW1 pro-piece. Unfortunately there are limitations in testing the activation of the channel by the whole HMW1 polypeptide, because the latter, a large polypeptide of over 1400 amino acids, is rather insoluble. Note that, to our knowledge, the ligand activation of other secretion systems, such as FhaC or Omp85, has also not been reported in the electrophysiological analysis of such channels to date (13, 15–17).
The fact that removing the entire N-terminal domain did not increase the conductance or open probability of the pore supports the idea that this region is not involved in plugging the channel. Nor does it control the flickering between the S and L conductance states. This is in contrast with the effect of the N-terminal domain of a cyanobacterial Omp85, which introduces a great amount of flickering and additional subconductance states (15). In FhaC, the N-terminal region contains the H1 α-helix, which runs down the inner channel wall, and two POTRA domains. Deletion of each of these regions had minor effects on channel behavior (13). On the other hand, deletion of the inwardly folded L6 loop of FhaC affected the conductance of the channel and increased the antibiotic sensitivity of cells expressing such mutated protein, suggesting that L6 dictates the architecture of the channel (13). Molecular dynamics simulations have suggested that the movement of the pore constricting loop L2 in the N. meningitidis adhesin OpcA can lead to channel opening (49). Long loops such as these exist in the topology prediction model of HMW1B (20). Because of the similarity between HMW1B and FhaC, it is tempting to assume that the closed state of HMW1B is also maintained by an inwardly folded loop.
Supplementary Material
Acknowledgments
We are grateful to Joseph St. Geme III for the HMW1B full-length and C-terminal domain constructs.
This work was supported, in whole or in part, by National Institutes of Health Grant AI068943 (to H.-J. Y.). This work was also supported by Grant E-1616 from the Welch Foundation (to H.-J. Y.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1 and S2.
Footnotes
The abbreviations used are: POTRA domain, polypeptide transport-associated domain; MOPS, 4-morpholinepropanesulfonic acid; MES, 4-morpholineethanesulfonic acid; GST, glutathione S-transferase; PBS, phosphate-buffered saline.
References
- 1.Kostakioti, M., Newman, C. L., Thanassi, D. G., and Stathopoulos, C. (2005) J. Bacteriol. 187 4306–4314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Thanassi, D. G., Stathopoulos, C., Karkal, A., and Li, H. (2005) Mol. Membr. Biol. 22 63–72 [DOI] [PubMed] [Google Scholar]
- 3.Mota, L. J., Sorg, I., and Cornelis, G. R. (2005) FEMS Microbiol. Lett. 252 1–10 [DOI] [PubMed] [Google Scholar]
- 4.Cascales, E., and Christie, P. J. (2003) Nat. Rev. Microbiol. 1 137–149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Desvaux, M., Parham, N. J., and Henderson, I. R. (2004) Curr. Issues Mol. Biol. 6 111–124 [PubMed] [Google Scholar]
- 6.Pukatzki, S., Ma, A. T., Sturtevant, D., Krastins, B., Sarracino, D., Nelson, W. C., Heidelberg, J. F., and Mekalanos, J. J. (2006) Proc. Natl. Acad. Sci. U. S. A. 103 1528–1533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Holland, I. B. (2004) Biochim. Biophys. Acta 1694 5–16 [DOI] [PubMed] [Google Scholar]
- 8.St. Geme, J. W., III, and Grass, S. (1998) Mol. Microbiol. 27 617–630 [DOI] [PubMed] [Google Scholar]
- 9.Gentle, I. E., Burri, L., and Lithgow, T. (2005) Mol. Microbiol. 58 1216–1225 [DOI] [PubMed] [Google Scholar]
- 10.Moslavac, S., Mirus, O., Bredemeier, R., Soll, J., von Haeseler, A., and Schleiff, E. (2005) FEBS J. 272 1367–1378 [DOI] [PubMed] [Google Scholar]
- 11.Sanchez-Pulido, L., Devos, D., Genevrois, S., Vicente, M., and Valencia, A. (2003) Trends Biochem. Sci. 28 523–526 [DOI] [PubMed] [Google Scholar]
- 12.Habib, S. J., Waizenegger, T., Niewienda, A., Paschen, S. A., Neupert, W., and Rapaport, D. (2007) J. Cell Biol. 176 77–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Clantin, B., Delattre, A. S., Rucktooa, P., Saint, N., Meli, A. C., Locht, C., Jacob-Dubuisson, F., and Villeret, V. (2007) Science 317 957–961 [DOI] [PubMed] [Google Scholar]
- 14.Tommassen, J. (2007) Science 317 903–904 [DOI] [PubMed] [Google Scholar]
- 15.Bredemeier, R., Schlegel, T., Ertel, F., Vojta, A., Borissenko, L., Bohnsack, M. T., Groll, M., von Haeseler, A., and Schleiff, E. (2007) J. Biol. Chem. 282 1882–1890 [DOI] [PubMed] [Google Scholar]
- 16.Stegmeier, J. F., and Andersen, C. (2006) J. Biochem. (Tokyo) 140 275–283 [DOI] [PubMed] [Google Scholar]
- 17.Jacob-Dubuisson, F., El-Hamel, C., Saint, N., Guedin, S., Willery, E., Molle, G., and Locht, C. (1999) J. Biol. Chem. 274 37731–37735 [DOI] [PubMed] [Google Scholar]
- 18.Meli, A. C., Hodak, H., Clantin, B., Locht, C., Molle, G., Jacob-Dubuisson, F., and Saint, N. (2006) J. Biol. Chem. 281 158–166 [DOI] [PubMed] [Google Scholar]
- 19.Basle, A., Iyer, R., and Delcour, A. H. (2004) Biochim. Biophys. Acta 1664 100–107 [DOI] [PubMed] [Google Scholar]
- 20.Surana, N. K., Buscher, A. Z., Hardy, G. G., Grass, S., Kehl-Fie, T., and St. Geme, J. W., III (2006) J. Biol. Chem. 281 18051–18058 [DOI] [PubMed] [Google Scholar]
- 21.Surana, N. K., Grass, S., Hardy, G. G., Li, H., Thanassi, D. G., and St. Geme, J. W., III (2004) Proc. Natl. Acad. Sci. U. S. A. 101 14497–14502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li, H., Grass, S., Wang, T., Liu, T., and St. Geme, J. W., III (2007) J. Bacteriol. 189 7497–7502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li, H., Qian, L., Chen, Z., Thibault, D., Liu, G., Liu, T., and Thanassi, D. G. (2004) J. Mol. Biol. 344 1397–1407 [DOI] [PubMed] [Google Scholar]
- 24.Rehling, P., Model, K., Brandner, K., Kovermann, P., Sickmann, A., Meyer, H. E., Kuhlbrandt, W., Wagner, R., Truscott, K. N., and Pfanner, N. (2003) Science 299 1747–1751 [DOI] [PubMed] [Google Scholar]
- 25.Ahting, U., Thun, C., Hegerl, R., Typke, D., Nargang, F. E., Neupert, W., and Nussberger, S. (1999) J. Cell Biol. 147 959–968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yeo, H. J., Yokoyama, T., Walkiewicz, K., Kim, Y., Grass, S., and St. Geme, J. W., III (2007) J. Biol. Chem. 282 31076–31084 [DOI] [PubMed] [Google Scholar]
- 27.Prilipov, A., Phale, P. S., Van Gelder, P., Rosenbusch, J. P., and Koebnik, R. (1998) FEMS Microbiol. Lett. 163 65–72 [DOI] [PubMed] [Google Scholar]
- 28.Rostovtseva, T. K., Nestorovich, E. M., and Bezrukov, S. M. (2002) Biophys. J. 82 160–169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Duret, G., Simonet, V., and Delcour, A. H. (2007) Channels 1 70–79 [DOI] [PubMed] [Google Scholar]
- 30.Delcour, A. H., Martinac, B., Adler, J., and Kung, C. (1989) J. Membr. Biol. 112 267–275 [DOI] [PubMed] [Google Scholar]
- 31.Hille, B. (2001) Ion Channels of Excitable Membranes, 3rd Ed., Sinauer, Sunderland, MA
- 32.Simonet, V. C., Basle, A., Klose, K. E., and Delcour, A. H. (2003) J. Biol. Chem. 278 17539–17545 [DOI] [PubMed] [Google Scholar]
- 33.Hinnah, S. C., Wagner, R., Sveshnikova, N., Harrer, R., and Soll, J. (2002) Biophys. J. 83 899–911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nestorovich, E. M., Rostovtseva, T. K., and Bezrukov, S. M. (2003) Biophys. J. 85 3718–3729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dong, C., Beis, K., Nesper, J., Brunkan-Lamontagne, A. L., Clarke, B. R., Whitfield, C., and Naismith, J. H. (2006) Nature 444 226–229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Koronakis, V., Sharff, A., Koronakis, E., Luisi, B., and Hughes, C. (2000) Nature 405 914–919 [DOI] [PubMed] [Google Scholar]
- 37.Oomen, C. J., van Ulsen, P., van Gelder, P., Feijen, M., Tommassen, J., and Gros, P. (2004) EMBO J. 23 1257–1266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Thanassi, D. G., Stathopoulos, C., Dodson, K., Geiger, D., and Hultgren, S. J. (2002) J. Bacteriol. 184 6260–6269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Andersen, C., Hughes, C., and Koronakis, V. (2002) J. Membr. Biol. 185 83–92 [DOI] [PubMed] [Google Scholar]
- 40.Burghout, P., van Boxtel, R., Van Gelder, P., Ringler, P., Muller, S. A., Tommassen, J., and Koster, M. (2004) J. Bacteriol. 186 4645–4654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nouwen, N., Ranson, N., Saibil, H., Wolpensinger, B., Engel, A., Ghazi, A., and Pugsley, A. P. (1999) Proc. Natl. Acad. Sci. U. S. A. 96 8173–8177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Goetze, T. A., Philippar, K., Ilkavets, I., Soll, J., and Wagner, R. (2006) J. Biol. Chem. 281 17989–17998 [DOI] [PubMed] [Google Scholar]
- 43.Hill, K., Model, K., Ryan, M. T., Dietmeier, K., Martin, F., Wagner, R., and Pfanner, N. (1998) Nature 395 516–521 [DOI] [PubMed] [Google Scholar]
- 44.Saint, N., El Hamel, C., De, E., and Molle, G. (2000) FEMS Microbiol. Lett. 190 261–265 [DOI] [PubMed] [Google Scholar]
- 45.Conlan, S., Zhang, Y., Cheley, S., and Bayley, H. (2000) Biochemistry 39 11845–11854 [DOI] [PubMed] [Google Scholar]
- 46.Van Gelder, P., Dumas, F., and Winterhalter, M. (2000) Biophys. Chem. 85 153–167 [DOI] [PubMed] [Google Scholar]
- 47.Prince, S. M., Achtman, M., and Derrick, J. P. (2002) Proc. Natl. Acad. Sci. U. S. A. 99 3417–3421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Vandeputte-Rutten, L., Kramer, R. A., Kroon, J., Dekker, N., Egmond, M. R., and Gros, P. (2001) EMBO J. 20 5033–5039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bond, P. J., Derrick, J. P., and Sansom, M. S. (2007) Biophys. J. 92 L23–L25 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.