

Abstract
Congenital chloride-losing diarrhea (CLD)
is a genetic disorder causing watery stool and dehydration. Mutations in
SLC26A3 (solute carrier 26 family member 3), which
functions as a coupled
Cl-/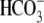 exchanger, cause
CLD. SLC26A3 is a membrane protein predicted to contain 12
transmembrane-spanning α-helices and a C-terminal STAS
(sulfate transporters and
anti-sigma-factor) domain homologous to the bacterial
anti-sigma-factor antagonists. The STAS domain is required for SLC26A3
Cl-/
exchanger, cause
CLD. SLC26A3 is a membrane protein predicted to contain 12
transmembrane-spanning α-helices and a C-terminal STAS
(sulfate transporters and
anti-sigma-factor) domain homologous to the bacterial
anti-sigma-factor antagonists. The STAS domain is required for SLC26A3
Cl-/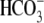 exchange function
and for the activation of cystic fibrosis transmembrane conductance regulator
by SLC26A3. Here we investigate the molecular mechanism(s) by which four
CLD-causing mutations (ΔY526/7, I544N, I675/6ins, and G702Tins) in the
STAS domain lead to disease. In a heterologous mammalian expression system
biochemical, immunohistochemical, and ion transport experiments suggest that
the four CLD mutations cause SLC26A3 transporter misfolding and/or
mistrafficking. Expression studies with the isolated STAS domain suggest that
the I675/6ins and G702Tins mutations disrupt the STAS domain directly, whereas
limited proteolysis experiments suggest that the ΔY526/7 and I544N
mutations affect a later step in the folding and/or trafficking pathway. The
data suggest that these CLD-causing mutations cause disease by at least two
distinct molecular mechanisms, both ultimately leading to loss of functional
protein at the plasma membrane.
exchange function
and for the activation of cystic fibrosis transmembrane conductance regulator
by SLC26A3. Here we investigate the molecular mechanism(s) by which four
CLD-causing mutations (ΔY526/7, I544N, I675/6ins, and G702Tins) in the
STAS domain lead to disease. In a heterologous mammalian expression system
biochemical, immunohistochemical, and ion transport experiments suggest that
the four CLD mutations cause SLC26A3 transporter misfolding and/or
mistrafficking. Expression studies with the isolated STAS domain suggest that
the I675/6ins and G702Tins mutations disrupt the STAS domain directly, whereas
limited proteolysis experiments suggest that the ΔY526/7 and I544N
mutations affect a later step in the folding and/or trafficking pathway. The
data suggest that these CLD-causing mutations cause disease by at least two
distinct molecular mechanisms, both ultimately leading to loss of functional
protein at the plasma membrane.
The SLC26A (solute carrier family 26) is a
recently described protein family containing 10 human genes
(1-3).
These genes encode membrane proteins that are predicted to contain between 10
and 14 transmembrane-spanning α-helices and a C-terminal domain
(4-13).
The family members have varied tissue distributions, some being expressed in
most organs and others with more restricted tissue expression patterns
(1,
4,
6,
12,
14-23).
The SLC26A proteins function as anion exchangers or Cl- channels in
the luminal membrane of epithelial cells, transport solutes, including
oxalate, 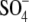 , I-,
Cl-,
, I-,
Cl-, 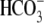 ,
,
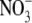 , SCN-, OH-,
and thus are important in a number of physiological processes
(9,
19,
20,
23-28).
, SCN-, OH-,
and thus are important in a number of physiological processes
(9,
19,
20,
23-28).
SLC26A3 was first identified as a gene that was
down-regulated in human colon adenocarcinomas
and subsequently named DRA
(29). Thus far,
SLC26A3 has not been conclusively demonstrated to be a tumor
suppressor gene (30,
31), but it has been
unequivocally demonstrated to be a
Cl-/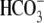 exchanger with a
2:1 transport stoichiometry
(27,
30-33).
The transport function of SLC26A3 is thought to play an important role in
Cl- absorption and
exchanger with a
2:1 transport stoichiometry
(27,
30-33).
The transport function of SLC26A3 is thought to play an important role in
Cl- absorption and 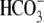 secretion in the colon and perhaps the pancreas
(4,
9,
30-33).
secretion in the colon and perhaps the pancreas
(4,
9,
30-33).
The highly conserved C-terminal domain of the SLC26A proteins has been
demonstrated to be important for proper ion transport activity
(29,
32-36).
This C-terminal domain is homologous to the SpoIIAA family of
anti-sigma-factor antagonists
(37), which controls bacterial
sporulation through a protein-protein interaction with the anti-sigma-factor
(38-40),
an interaction that is regulated by phosphorylation
(41-44).
Because the SLC26A proteins were originally predicted to be sulfate
transporters, their C-terminally conserved domain was named STAS for
sulfate transporters and
anti-sigma-factor antagonists
(37). Similar to the
anti-sigma-factor antagonists, phosphorylation regulates the interaction
between the SLC26A3 STAS domain and the R-domain of
CFTR2
(33). The STAS and R-domain
interaction coordinately activates Cl- channel activity of CFTR and
Cl-/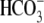 exchange activity
of SLC26A3 (33) as required
for epithelial function.
exchange activity
of SLC26A3 (33) as required
for epithelial function.
Mutations in four of the SLC26A transporters have been linked with human diseases (45-51). Mutations in SLC26A2 (also known as DTDST) have been linked to various chondro-dysplasias that cause skeletal defects, including clubbed feet, cleft palate, and short limbed dwarfism (47, 48, 51). Mutations in the SLC26A4 gene (also known as Pendrin or PDS) have been linked to Pendred syndrome, which is the most common form of syndromic deafness (45, 46, 50), whereas mutations in the SLC26A5 gene (also known as Prestin) have been linked to non-syndromic deafness (49). Mutations found in the SLC26A3 gene have been linked to congenital chloride-losing diarrhea (CLD) (4), a disease in which patients suffer from watery diarrhea containing elevated Cl- concentrations that can prove fatal if left untreated (52). Clinical management of the disease includes increased dietary uptake of water, KCl, and NaCl (52, 53). A recent report also suggests butyrate is effective in controlling the diarrhea (54).
Currently, over 30 mutations in SLC26A3 have been linked to CLD (8, 14, 52). Four of these mutations are missense, deletion, or insertion mutations that reside in the STAS domain of SLC26A3 (ΔY526/7, I544N, I675/6ins and G702T + GFE-VKIQNF insertion (G702Tins)). The I544N and I675/6ins mutations have been demonstrated to cause a loss of measurable transport activity of the human and mouse SLC26A3 protein, respectively (32-34); however, the mechanism by which these mutations undermine function is unknown. The effects of the ΔY526/7 and G702Tins mutations on SLC26A3 protein function and folding are also unknown. To elucidate the molecular mechanism(s) by which these CLD-causing mutations give rise to disease, we functionally, biochemically, and cell biologically characterized the wild type and mutant transporters. In addition, we monitored the effect of the CLD-causing mutations on the isolated STAS domain. These data suggest distinct mechanisms by which the CLD-causing mutations lead to disease.
EXPERIMENTAL PROCEDURES
Plasmid Construction—Mammalian expression constructs for full-length human SLC26A3 protein (gi:4557534) were constructed using standard molecular biology techniques and cloned into the BglII and NotI restriction sites of the pCMV-Myc vector (Clontech) resulting in a protein construct with a N-terminal Myc tag. Site-directed mutagenesis was performed using PfuTurbo DNA polymerase (Stratagene) to generate the in-frame deletion ΔY526/7, I544N, I675/6ins, and G702Tins mutant constructs. Constructs for mammalian cell expression of human SLC26A3 STAS domain constructs were generated with domain boundaries encompassing residues 509-741 and were cloned into the BglII and NotI sites of the pCMV-Myc vector, whereas nine different human STAS domain constructs for bacterial expression were cloned into the BamHI site of the Smt3-pET28 vector using the combination of three N-terminal domain boundaries (amino acids 503, 510, and 525) and three C-terminal domain boundaries (720, 741, and 764) (55). All of these constructs were N-terminal fusions with the His6-Smt3 protein and were verified by DNA sequencing.
Cl-/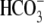 Exchange Activity—HeLa cells grown on glass coverslips were
transfected with the appropriate SLC26A3 plasmid along with a green
fluorescent protein expression plasmid. 3 μl of FuGENE 6 (Roche Applied
Science) was used per transfection, and the cells were used for
Cl-/
Exchange Activity—HeLa cells grown on glass coverslips were
transfected with the appropriate SLC26A3 plasmid along with a green
fluorescent protein expression plasmid. 3 μl of FuGENE 6 (Roche Applied
Science) was used per transfection, and the cells were used for
Cl-/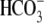 exchange
measurements 48 h post-transfection. Green fluorescent protein-positive cells
were selected on a fluorescence microscope (Nikon) and incubated with 10
μm
2′,7′-bis-(2-carboxyethyl)-5-(and-6)-carboxyfluorescein,
acetoxymethyl ester (BCECF-AM) (Molecular Probes), and 0.2% Pluronic F-127
(Molecular Probes) for 10 min in HEPES buffer (10 mm HEPES, pH 7.4,
140 mm sodium chloride, 5 mm potassium chloride, 1
mm magnesium chloride, 1 mm calcium chloride, 10
mm glucose, osmolarity 310 mosm). Cells were perfused
with HEPES buffer until a steady base line was achieved, and then cells were
perfused with
exchange
measurements 48 h post-transfection. Green fluorescent protein-positive cells
were selected on a fluorescence microscope (Nikon) and incubated with 10
μm
2′,7′-bis-(2-carboxyethyl)-5-(and-6)-carboxyfluorescein,
acetoxymethyl ester (BCECF-AM) (Molecular Probes), and 0.2% Pluronic F-127
(Molecular Probes) for 10 min in HEPES buffer (10 mm HEPES, pH 7.4,
140 mm sodium chloride, 5 mm potassium chloride, 1
mm magnesium chloride, 1 mm calcium chloride, 10
mm glucose, osmolarity 310 mosm). Cells were perfused
with HEPES buffer until a steady base line was achieved, and then cells were
perfused with 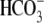 buffer for 3 min
(2.5 mm HEPES, pH 7.4, 120 mm sodium chloride, 5
mm potassium chloride, 1 mm magnesium chloride,
1mm calcium chloride, 10 mm glucose, 260
mosm, then 25 mm of sodium bicarbonate was added).
Subsequently the cells were perfused with Cl--free buffer for 3 min
(2.5 mm HEPES, pH 7.4, 120 mm sodium gluconate, 5
mm potassium gluconate, 1 mm magnesium sulfate, 1
mm cyclamic acid, 10 mm glucose, 260 mosm,
then 25 mm sodium bicarbonate was added), followed by a 3-min
perfusion with
buffer for 3 min
(2.5 mm HEPES, pH 7.4, 120 mm sodium chloride, 5
mm potassium chloride, 1 mm magnesium chloride,
1mm calcium chloride, 10 mm glucose, 260
mosm, then 25 mm of sodium bicarbonate was added).
Subsequently the cells were perfused with Cl--free buffer for 3 min
(2.5 mm HEPES, pH 7.4, 120 mm sodium gluconate, 5
mm potassium gluconate, 1 mm magnesium sulfate, 1
mm cyclamic acid, 10 mm glucose, 260 mosm,
then 25 mm sodium bicarbonate was added), followed by a 3-min
perfusion with 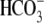 buffer. Finally,
the cells were perfused with HEPES buffer for 3 min. 5% carbon dioxide gas was
bubbled through buffers containing bicarbonate throughout the experiment. The
change in the fluorescence excitation ratio of BCECF-AM was monitored at 490
nm/440 nm with the emission at 510 nm. pHi was calibrated using
three standard buffers containing 10 mm HEPES, 140 mm
potassium chloride, 1mm magnesium chloride, 1 mm calcium
chloride, 10 mm glucose, and 5 μm nigericin at pH
6.65, 7.4, and 8.15.
buffer. Finally,
the cells were perfused with HEPES buffer for 3 min. 5% carbon dioxide gas was
bubbled through buffers containing bicarbonate throughout the experiment. The
change in the fluorescence excitation ratio of BCECF-AM was monitored at 490
nm/440 nm with the emission at 510 nm. pHi was calibrated using
three standard buffers containing 10 mm HEPES, 140 mm
potassium chloride, 1mm magnesium chloride, 1 mm calcium
chloride, 10 mm glucose, and 5 μm nigericin at pH
6.65, 7.4, and 8.15.
Immunofluorescence—HeLa and COS-7 cells were grown on glass coverslips (Fisher) in 35-mm dishes using Dulbecco's modified Eagle's medium (Invitrogen) containing 10% calf serum (Gemini) and supplemented with penicillin and streptomycin at 37 or 30 °C in a humidified incubator with 5% carbon dioxide. HeLa cells were transfected with 1 μg of wild type or mutant SLC26A3 pDNA using FuGENE 6 following the manufacturer's protocol. 48 h post-transfection cells were washed twice with PBS and fixed in 3.7% formaldehyde for 20 min and subsequently washed twice for 5 min in PBS. Cells were permeabilized with 0.1% Triton X-100 in PBS for 10 min at room temperature followed by two washes with PBS. The permeabilized cells were incubated for 20 min in blocking solution (1.3% fish skin gelatin and 1% bovine serum albumin in PBS) followed by a 1-h incubation with a 1:100 dilution of rabbit anti-Grp78 (Bip) (StressGen), 1:500 anti-c-Myc (Santa Cruz Biotechnology), and a 1:500 phalloidin conjugated to tetramethylrhodamine isothiocyanate (TRITC) (Sigma) in blocking solution. The cells were subsequently washed twice with PBS for 5 min and then incubated with a 1:100 dilution of the anti-rabbit antibody conjugated to cyanine-5.18 (cy5) (Jackson ImmunoResearch) and a 1:500 dilution of anti-mouse antibody conjugated to fluorescein isothiocyanate (Jackson ImmunoResearch) in blocking solution. Cells were washed and mounted using gel mount (Biomedia). Staining cells with the STAS domain antibody required a change of secondary antibody species, but the same protocol was used as described above. A 1:500 dilution of the STAS domain antibody was used along with a 1:100 dilution of mouse anti-Grp78 (BiP) (StressGen). The secondary antibodies were a 1:500 dilution of anti-rabbit antibody conjugated to (fluorescein isothiocyanate) (Jackson ImmunoResearch) and a 1:100 dilution of the anti-mouse antibody conjugated to (cy5) (Jackson ImmunoResearch). All stained cells were visualized and images collected using a Zeiss confocal microscope and Zeiss LSM 510 software version 2.8.
Glycosylation Assay—COS-7 cells were grown in Dulbecco's modified Eagle's medium (Invitrogen) containing 10% calf serum (Gemini) supplemented with penicillin and streptomycin at 37 or 30 °C in a humidified incubator with 5% CO2. 35-mm dishes of COS-7 cells were transfected with 1 μg of wild type or mutant SLC26A3 pDNA using FuGENE 6 (Roche Applied Science) following the manufacturer's guidelines. 48 h post-transfection, the cells were washed twice with 0.5 ml of TBS and then lysed in 0.5 ml of RIPA buffer (20 mm Tris, pH 8.0, 140 mm sodium chloride, 0.1% SDS, 0.5% deoxycholic acid, 1% polyoxyethylene(2) isooctylphenyl ether, and protease inhibitors (Roche Applied Science)) on ice for 30 min. The lysates were centrifuged at 12,000 × g for 5 min, and then 100 μl of lysate was incubated at 37 °C for 1 h with 500 units of EndoH (New England Biolabs), 500 units of PNGaseF (New England Biolabs), or no enzyme. The proteins were separated by SDS-PAGE and transferred to PVDF membrane for Western blot analysis.
STAS Solubility Assay—To aid in the soluble expression and purification of the human SLC26A3 STAS domain, the yeast small ubiquitin-related modifier homolog Smt3 was fused with the STAS domain N terminus (55). The wild type or mutant His6-Smt3-SLC26A3 STAS domain plasmids were transformed into Escherichia coli BL21 Codon Plus RIL cells (Stratagene) and grown at 37 °C in LB (Qbiogene) containing 50 μg/liter kanamycin and 34 μg/liter chloramphenicol to an A600 of 1.0. The cells were induced with 750 μm isopropyl β-d-1-thiogalactopyranoside (Research Products International) and shifted to 15 °C for overnight expression. The following day A600 measurements were taken and used to ensure that the same number of cells were pelleted at 4 °C at 3000 × g. The cell pellets were then resuspended in 20 ml of lysing buffer (20 mm Tris, pH 7.9, 200 mm sodium chloride, 1 mm 2-mercaptoethanol) and lysed via sonication with a Branford 450 sonicator on power level 4, duty cycle 60%, three times for 30 s. The resulting lysates were cleared via a 40,000 × g centrifugation step at 4 °C for 30 min. Equal volumes of cell lysates were separated by SDS-PAGE and stained with Coomassie Blue or transferred for Western blot analysis.
HEK 293 cells were transiently transfected with wild type and mutant pCMV-Myc SLC26A3 STAS domains and incubated at 37 °C for 48 h and subsequently scraped off the plate in 0.5 ml of lysing buffer. The cells were lysed by sonication and centrifuged at 60,000 × g for 30 min at 4 °C. Equivalent amounts of protein were loaded on the gel as determined by a Bradford assay.
Western Blotting—SDS-polyacrylamide gels for Western blots were transferred to PVDF membrane (Millipore) using the wet transfer method in Tris/glycine transfer buffer containing 10% methanol. Membranes were blocked using 5% milk (Bio-Rad) in TBS containing 0.05% Tween 20 (Sigma), and primary and secondary antibody incubations were performed in blocking buffer. Primary antibody dilutions were as follows: a 1:1000 dilution of the anti-His6 (Novagen), a 1:3000 dilution of the anti-c-Myc (Santa Cruz Biotechnology), and a 1:5000 dilution of the anti-STAS antibody. Secondary antibody dilutions were as follows: 1:10,000 of the anti-mouse horseradish peroxidase (Jackson ImmunoResearch) and 1:10,000 of the anti-rabbit horseradish peroxidase (Jackson ImmunoResearch). ECL Plus (Amersham Biosciences) or SuperSignal West Dura Extended Duration (Pierce) detection reagent was used following the manufacturer's guidelines and visualized using the STORM 840 (Amersham Biosciences) or Hyblot CL autoradiography film (Denville Scientific).
STAS Expression and Purification—Five liters of E. coli BL21 Codon Plus RIL cells (Stratagene) containing the wild type or mutant Smt3-STAS domain plasmids were grown at 37 °C in LB containing 50 μg/liter kanamycin and 34 μg/liter chloramphenicol to an A600 of 0.8. The cells were induced with 750 μm isopropyl β-d-1-thiogalactopyranoside (Research Products International) and shifted to 15 °C for overnight expression. The following day the cells were pelleted at 3000 × g and resuspended in 70 ml of lysing buffer. The cells were lysed via sonication and the resulting lysates cleared by centrifugation at 40,000 × g. Standard nickel chromatography was performed on the resulting supernatants using a 10-ml bed volume of resin (Novagen). The equilibration/binding buffer (buffer A) contained 20 mm Tris, pH 7.9, 200 mm sodium chloride, 1 mm 2-mercaptoethanol, and the column was washed using buffer A containing 40 mm imidazole, and the protein was eluted from the column using buffer A containing 400 mm imidazole. Fractions containing the Smt3-STAS fusion protein were pooled together and digested overnight with a 1:1000 dilution of the Ulp1 protease at 4 °C (55).
Ammonium sulfate (750 mm) was added to the cleaved protein solution, then run through a 0.22-μm filter (Nalgene), and then subjected to butyl hydrophobic interaction chromatography (Amersham Biosciences). The equilibration/binding buffer contained 20 mm sodium phosphate, pH 5.9, 750 mm ammonium sulfate, 1 mm DTT, and the elution buffer contained 20 mm sodium phosphate, pH 6.2, 1 mm DTT. Fractions containing the STAS protein were pooled together, and 350 mm sodium chloride was added to the solution. The protein was concentrated to 2 ml using an Amicon Ultra-concentrator (5,000 molecular weight cutoff) at 4 °C in a swinging bucket rotor, run through a 0.22-μm filter (Nalgene), and run over a HiPrep 16/60 Sephacryl S-200 column (Amersham Biosciences) in 20 mm sodium phosphate, pH 6.2, 350 mm sodium chloride, 1 mm DTT. Monodisperse STAS domain-containing fractions were pooled and concentrated as needed. The molecular weight of the STAS domain was estimated by the elution profile of protein standards and is consistent with a monomer.
Circular Dichroism—Circular dichroism spectra were measured using an AVIV 62DS spectrophotometer with a 0.1-cm quartz cuvette. The STAS CD spectra and thermal denaturation curves were collected at 10 μm in 20 mm sodium phosphate, pH 6.2, 350 mm sodium chloride, 1 mm DTT.
NMR Spectroscopy—Uniformly and isotopically enriched 15N wild type and ΔY526/7 SLC26A3 STAS domain were expressed in M9 minimal media with [15N]ammonium chloride as the nitrogen source, supplemented with 5% uniformly 15N-labeled BioExpress cell growth media (Cambridge Isotope Laboratories). The proteins were purified as described above with the only change being that the protein was eluted off of the gel filtration column in 50 mm potassium phosphate, pH 6.8, 400 mm potassium chloride, 100 mm arginine, 2% glucose, 2 mm DTT. TROSY-HSQC spectra (56) of 90 μm purified wild type and ΔY526/7 STAS domains were collected using a 600-MHz Varian Inova spectrometer with a pulsed-field gradient, triple resonance cold probe at 30 °C for 12 h each. Spectra were processed with NMRPipe (57) and displayed with NMRView (58).
STAS Domain Antibody Production—Recombinantly expressed and purified SLC26A3 STAS domain protein (described above) was used as the antigen for rabbit polyclonal antibody production. Briefly, 5 mg of purified protein was shipped to Proteintech Group Inc. for injection of two rabbits following their standard 102-day protocol. Two production bleeds and a final bleed from each animal were tested for their reactivity to the STAS domain protein, and all six antiserum samples were found to detect the STAS domain protein in a similar fashion. The STAS domain antibody has been shown to cross-react with mouse SLC26A3, SLC26A6, SLC26A7, and SLC26A9 proteins as well as with human SLC26A6 and SLC26A11 proteins. The ability of the antisera to recognize other SLC26A transporters has not been evaluated.
Tryptic Digests—Transiently transfected HEK 293 cells were allowed to express the wild type and mutant SLC26A3 proteins for 48 h at which time they were washed twice with TBS and scraped off the plate in 500 μl of 10 mm HEPES, pH 7.4, 200 mm sucrose. The cells were sonicated twice for 20 s on the lowest power setting and placed on ice. The resulting lysates were incubated in the presence of increasing concentrations of trypsin (0.3, 1, 3, 10, 30, 100, and 300 μg/ml) at room temperature for 12 min. The digestions were stopped with SDS sample buffer, and the digested protein was separated by SDS-PAGE and then transferred to PVDF membrane. The Western blot was performed as described above using the anti-STAS antibody, and densitometry was performed on representative Western blots using the ImageJ program.
RESULTS
A number of structural models have been suggested for the SLC26A transporters, which differ in the number of transmembrane segments, their topology, the location of the N and C termini, and the boundaries of the STAS domain. Although the vast majority of SLC26A topology predictions suggest these proteins contain an even number of transmembrane spans with their N and C termini located in the cytosol (1, 4, 5, 7-13, 18, 59-61), some topology models have suggested an odd number of transmembrane spans (22, 62), or that the N and C termini are extracellularly located (63). To identify the most appropriate topology model for SLC26A3, the location of the N termini and the conserved C-terminal STAS domain were determined (Fig. 1A). Immunofluorescence experiments were performed with HeLa cells transfected with empty vector, the SLC26A3 STAS domain, or the complete SLC26A3 transporter. Cells were then permeabilized by incubation with Triton X-100 or left untreated to determine the topology. N-terminal Myc tag or C-terminal STAS domain staining was only observed in permeabilized cells (Fig. 1, B and C), indicating that both termini are cytosolic. However, these experiments cannot rule out the possibility that the detergent treatment exposed extracellular epitopes that are inaccessible to the antibodies in the absence of detergent.
FIGURE 1.

SLC26A3 structural model. A, schematic model of a predicted
SLC26A3 structure, which includes 12 transmembrane-spanning α-helices
and a C-terminally conserved STAS domain. The experimentally determined STAS
domain boundaries are shown along with the predicted location of the
glycosylation site in the second extracellular loop between transmembrane
helices 3 and 4. SLC26A3 has been demonstrated to be an anion exchanger with a
2Cl-/1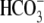 stoichiometry,
and the chemical equation showing how
stoichiometry,
and the chemical equation showing how
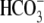 can affect cellular pH is shown.
B, HeLa cells transfected with empty vector, wild type STAS domain,
or wild type SLC26A3 are shown in the presence or absence of Triton X-100
permeabilization. The cells were stained with phalloidin (red),
anti-BIP antibody (blue), and anti-c-Myc antibody (green).
C, HeLa cells transfected with empty vector, wild type STAS domain,
or wild type SLC26A3 are shown in the presence or absence of Triton X-100
permeabilization. The cells were stained with phalloidin (red),
anti-BIP antibody (blue), and STAS domain antibody (green).
Scale bar is 50 μm.
can affect cellular pH is shown.
B, HeLa cells transfected with empty vector, wild type STAS domain,
or wild type SLC26A3 are shown in the presence or absence of Triton X-100
permeabilization. The cells were stained with phalloidin (red),
anti-BIP antibody (blue), and anti-c-Myc antibody (green).
C, HeLa cells transfected with empty vector, wild type STAS domain,
or wild type SLC26A3 are shown in the presence or absence of Triton X-100
permeabilization. The cells were stained with phalloidin (red),
anti-BIP antibody (blue), and STAS domain antibody (green).
Scale bar is 50 μm.
To further characterize the SLC26A3 protein, we decided to identify the domain boundaries of the STAS domain. As an initial step in determining the domain boundaries, a STAS domain multiple sequence alignment was generated (Fig. 2). From the alignment it is immediately apparent that the SLC26A STAS domains contain a large insertion of sequence compared with their structural homologs, the SpoIIAA proteins. The sequence in the SLC26A STAS domains not found in the SpoIIAA proteins is designated as the intervening sequence (IVS). The first 20 amino acids of the IVS show limited conservation among the SLC26A STAS domains, and a secondary structure prediction of these residues suggests that a portion of this sequence is α-helical in nature. The remaining residues in the IVS show no sequence conservation in the various SLC26A transporters, and secondary structure predictions of this region suggest they are largely unstructured.
FIGURE 2.

SLC26A3 STAS alignment. The C-terminal STAS domains of 10 SLC26A transporters from Homo sapiens were aligned with 12 Sultr family of sulfate transporters from A. thaliana, four SpoIIAA proteins from Bacillus sphaericus, Bacillus subtilis, Geobacillus stearothermophilus, and Thermotoga maritima and two predicted sulfate transporters from Desulfovibrio vulgaris Hildenborough and Aquifex aeolicus VF5. The alignment was generated using T-coffee and then shaded using the Boxshade program. The block arrows and tubes below the alignment represent the α-helices and β-strands found in the three-dimensional structures of the SpoIIAA proteins (44). The line above the alignment denotes the IVS, which is defined as the large sequence insertion in the STAS domains not present in the SpoIIAA homologs. The small filled arrows above the alignment indicate the various N- and C-terminal boundaries used to screen for the optimal STAS domain boundaries. The asterisk indicates the position of known CLD-causing mutations found in the SLC26A3 STAS domain and the ρ symbol denotes the location of the conserved Ser residue in the SpoIIAA proteins, which is phosphorylated (70). Residues shaded in red represent the carbonic anhydrase II-binding site as described by Alvarez et al. (67), whereas residues shaded in yellow indicate the residues which form the binding interface with SpoIIAB (71).
The poor sequence conservation between the SLC26A proteins and the SpoIIAA proteins makes it difficult to predict the N- and C-terminal boundaries of the SLC26A3 STAS domain. To identify the N- and C-terminal boundaries of the STAS domain, a matrix of three N-terminal boundaries (amino acids 503, 510, and 525) and three C-terminal boundaries (amino acids 720, 741, and 764) was used to produce nine possible SLC26A3 STAS constructs. All were expressed in E. coli, and the three constructs ending in amino acid 720 were insoluble after cell lysis and a 40,000 × g centrifugation step (data not shown). The remaining six constructs had greater than 50% of the total expressed protein in the supernatant fraction. These six were evaluated for secondary structure, thermal stability, and their ability to bind to the R-domain of CFTR (supplemental Fig. 1). Based on these data (see below), the STAS domain construct with domain boundaries of 510-741 was selected for further study.
With the topology of SLC26A3 elucidated and the STAS domain boundaries
identified, we set out to understand how mutations in the cytosolic STAS
domain of SLC26A3 lead to disease. We began by measuring chloride-bicarbonate
exchange (Cl-/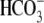 ) activity
of the wild type and mutant transporters in HeLa cells using the pH-sensitive
fluorescent dye BCECF-AM as described previously
(64). Briefly, BCECF
fluorescence is monitored upon the addition or removal of extracellular
Cl- in the presence of
) activity
of the wild type and mutant transporters in HeLa cells using the pH-sensitive
fluorescent dye BCECF-AM as described previously
(64). Briefly, BCECF
fluorescence is monitored upon the addition or removal of extracellular
Cl- in the presence of 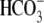 .
As extracellular Cl- is removed, intracellular Cl- exits
the cell. When Cl- ions leave the cell through the
Cl-/
.
As extracellular Cl- is removed, intracellular Cl- exits
the cell. When Cl- ions leave the cell through the
Cl-/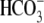 exchanger SLC26A3,
exchanger SLC26A3,
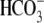 ions are transported into the
cell.
ions are transported into the
cell. 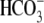 can react with an
intracellular proton to form H2CO3, which can then be
converted to H2O and CO2 by carbonic anhydrase
effectively increasing the intracellular pH
(Fig. 1A). Using this
assay Cl-/
can react with an
intracellular proton to form H2CO3, which can then be
converted to H2O and CO2 by carbonic anhydrase
effectively increasing the intracellular pH
(Fig. 1A). Using this
assay Cl-/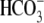 exchange
activity was observed for the wild type and ΔY526/7 SLC26A3 proteins
(Fig. 3A), with the
exchange rate of the ΔY526/7 mutant reduced by 46% relative to wild type
(Fig. 3B). No
significant Cl-/
exchange
activity was observed for the wild type and ΔY526/7 SLC26A3 proteins
(Fig. 3A), with the
exchange rate of the ΔY526/7 mutant reduced by 46% relative to wild type
(Fig. 3B). No
significant Cl-/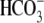 exchange activity was observed for the I544N, I675/6ins and G702Tins mutant
transporters (Fig. 3, A and
B).
exchange activity was observed for the I544N, I675/6ins and G702Tins mutant
transporters (Fig. 3, A and
B).
FIGURE 3.

SLC26A3
Cl-/ exchange
activity. A, representative
Cl-/
exchange
activity. A, representative
Cl-/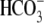 exchange activity
measurements are shown recorded from HeLa cells transiently transfected with
wild type or mutant (ΔY526/7, I544N, I675/6ins, G702Tins) SLC26A3 along
with cells transfected with empty vector. HeLa cells were loaded with
BCECF-AM, and ratiometric fluorescence excitation measurements were recorded
while continuously perfusing the indicated buffer over the cells. B,
Cl-/
exchange activity
measurements are shown recorded from HeLa cells transiently transfected with
wild type or mutant (ΔY526/7, I544N, I675/6ins, G702Tins) SLC26A3 along
with cells transfected with empty vector. HeLa cells were loaded with
BCECF-AM, and ratiometric fluorescence excitation measurements were recorded
while continuously perfusing the indicated buffer over the cells. B,
Cl-/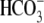 exchange rate
between the
exchange rate
between the 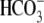 buffer and
Cl- free buffer transition for the indicated proteins. The number
of experiments used in the quantitation is indicated below the bar
graph, and error bars represent the mean ± S.E.
buffer and
Cl- free buffer transition for the indicated proteins. The number
of experiments used in the quantitation is indicated below the bar
graph, and error bars represent the mean ± S.E.
Two simple models can account for the reduced function of the mutant transporters as follows: 1) effects on the mechanochemistry of the transporter or 2) effects on protein folding and or trafficking. To distinguish between these two possibilities, biochemical experiments were performed on transiently transfected COS-7 cells to monitor the glycosylation state of the protein as a marker of movement through the secretory system. Transfected COS-7 cells expressing the wild type or mutant transporters for 48 h at 37 °C were lysed in RIPA buffer and incubated with either EndoH or PNGaseF enzyme or left untreated (Fig. 4A). The Western blot revealed two bands for the untreated wild type transporter with the higher molecular weight species being the predominant band. Upon treatment of the wild type lysates with EndoH, the migration of the lower molecular weight species was accelerated, indicating it was core-glycosylated and integrated into the ER membrane. Upon PNGaseF treatment of the wild type lysates, migration of both bands shifted to this lowest molecular weight species, consistent with the higher molecular weight species being a complexly glycosylated SLC26A3 protein that has trafficked to the Golgi apparatus or beyond. The bulk of the ΔY526/7 SLC26A3 protein was core-glycosylated with only a small amount of complexly glycosylated protein. The I544N, I675/6ins, and G702Tins mutants were all core-glycosylated but contained no observable complexly glycosylated protein, suggesting that these CLD-causing mutations give rise to the disease by preventing proper trafficking to the plasma membrane. In addition, it is worth noting that at steady state, the expression levels of the I675/6ins and G702Tins mutant SLC26A3 proteins are consistently reduced compared with the wild type, ΔY526/7, and I544N mutant transporters consistent with their recognition by proteolytic systems of the cell.
FIGURE 4.

SLC26A3 subcellular localization. A, lysates of COS-7 cells transiently transfected with wild type or mutant SLC26A3 vectors were treated with the endoglycosidase enzymes EndoH or PNGaseF and SLC26A3 detected by Western blot with the anti-c-Myc antibody. Molecular mass markers in kDa are indicated to the right of the blot. B, HeLa cells transiently transfected with the wild type or mutant SLC26A3 proteins were used for immunofluorescence imaging as described under the “Experimental Procedures.” Filamentous actin is stained red; Bip (an ER marker) is stained blue, and Myc (SLC26A3) is stained green. The panels are representative images of the various subcellular locations of the SLC26A3 protein. Two sets of ΔY526/7 images are shown highlighting representative plasma membrane or intracellular localization. The scale bar represents 50 μm.
The cellular location of the wild type and mutant SLC26A3 transporters was also determined by performing immunofluorescence experiments. In HeLa cells expressing SLC26A3, the wild type transporter is found in the plasma membrane and in the ER (Fig. 4B). Cells transfected with the ΔY526/7 transporter showed two distinct populations of cells. One population of cells showed the ΔY526/7 transporter at the plasma membrane (Fig. 4B, row 3), and the other showed ER retention (Fig. 4B, row 2). The I544N, I675/6ins, and G702Tins mutant transporters all show ER localization (Fig. 4B, rows 4-6). Identical results were observed in the COS-7 cells (data not shown). To exclude any possible epitope effects that may contribute to the observed subcellular localization of the wild type and mutant transporters, we also performed immunofluorescence experiments using the anti-STAS domain antibody. The staining pattern for the SLC26A3 transporters using the anti-STAS domain antibody was indistinguishable compared with the anti-c-Myc antibody in both HeLa and COS-7 cells (data not shown).
In an attempt to rescue the CLD-causing mutations that affect proper
trafficking out of the ER, we reduced the expression temperature from 37 to 30
°C and monitored the amount of the complexly glycosylated protein
produced. Transiently transfected COS-7 cells were incubated at 37 °C for
24 h and then shifted to 30 °C for an additional 24 h. The manipulation
led to a substantial increase in the amount of the complexly glycosylated
ΔY526/7 mutant transporter, whereas a decrease was observed in the
amount of complexly glycosylated wild type protein
(Fig. 5A compared with
Fig. 4A). In contrast
to the ΔY526/7 transporter, the I544N, I675/6ins, or G702Tins
transporters were not complexly glycosylated upon low temperature treatment
(Fig. 5A).
Immunofluorescence experiments were also performed to assess the subcellular
localization of the wild type and mutant transporters when expressed at 30
°C (Fig. 5B).
Consistent with the biochemical data, the vast majority of ΔY526/7
mutant transporter was found to be localized to the plasma membrane at 30
°C, highlighting the temperature sensitivity of this mutant. The increase
in plasma membrane expression at low temperature was not observed for the
I544N, I675/6ins, and G702Tins mutants, which all show ER localization and
vacuolation. To further substantiate this finding and to determine whether the
plasma membrane targeted ΔY526/7 is active,
Cl-/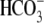 exchange activity
of the wild type and ΔY526/7 mutant transporters was measured
(Fig. 5C). By contrast
to the functional measurements made at 37 °C, the
Cl-/
exchange activity
of the wild type and ΔY526/7 mutant transporters was measured
(Fig. 5C). By contrast
to the functional measurements made at 37 °C, the
Cl-/ exchange rate of
the ΔY526/7 mutant transporter at 30 °C approached that of wild type
(Fig. 5D).
exchange rate of
the ΔY526/7 mutant transporter at 30 °C approached that of wild type
(Fig. 5D).
FIGURE 5.

Low temperature rescue of ΔY526/7 SLC26A3 at 30 °C.
A, Western blot of COS-7 cells expressing the wild type and mutant
SLC26A3 proteins after 24 h at 37 °C followed by 24 h of 30 °C
treatment. The protein samples in each lane are as follows: lane 1,
empty vector; lane 2, wild type SLC26A3; lane 3,
ΔY526/7 SLC26A3; lane 4, I544N SLC26A3; lane 5,
I675/6ins SLC26A3; lane 6, G702Tins SLC26A3. The molecular mass
markers to the left of the blot are measured in kDa. B,
representative immunofluorescent images of HeLa cells transiently transfected
with the wild type or mutant SLC26A3 proteins. Filamentous actin staining is
colored red; Bip staining is colored blue; Myc staining is
colored green; and the scale bar represents 50
μm. C, representative
Cl-/ exchange activity
at 30 °C is shown for HeLa cells transiently transfected with the
indicated vectors. The HeLa cells were initially grown and transfected at 37
°C, and then 24 h post-transfection the cells were shifted to 30 °C
for an additional 24 h before
Cl-/
exchange activity
at 30 °C is shown for HeLa cells transiently transfected with the
indicated vectors. The HeLa cells were initially grown and transfected at 37
°C, and then 24 h post-transfection the cells were shifted to 30 °C
for an additional 24 h before
Cl-/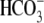 exchange
measurements were made. D, transition between the
exchange
measurements were made. D, transition between the
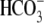 and Cl--free buffers
was used to calculate the
Cl-/
and Cl--free buffers
was used to calculate the
Cl-/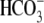 exchange rate of
HeLa cells transfected with the indicated plasmids. The number of experiments
used in the quantification is indicated below the bar graph,
and the error bars represent the mean ± S.E.
exchange rate of
HeLa cells transfected with the indicated plasmids. The number of experiments
used in the quantification is indicated below the bar graph,
and the error bars represent the mean ± S.E.
The reduced ability of the ΔY526/7 mutant SLC26A3 transporter to traffic to the plasma membrane, and the lack of plasma membrane trafficking of the I544N, I675/6ins, and G702Tins mutants, suggested that these mutations may be causing the transporters to misfold. To investigate the possibility that these mutations specifically disrupt folding of the STAS domain, the wild type and mutant STAS domains were recombinantly expressed to study their biochemical and biophysical properties.
Using the SLC26A3 STAS domain boundaries of 510-741, we created Smt3-STAS fusions of the wild type and mutant proteins and assessed their ability to fold by monitoring solubility in E. coli. The wild type, ΔY526/7, and I544N STAS domains were found to be solubly expressed in E. coli, whereas the I675/6ins and G702Tins SLC26A3 STAS domains were found to be produced in an insoluble form (Fig. 6A). A similar pattern of solubility/folding was observed when the STAS domains were produced in HEK 293 (Fig. 6B) and COS-7 cells (data not shown), suggesting that the effects of the mutations on STAS domain folding are independent of the cell line used.
FIGURE 6.

STAS domain folding in cells. A, wild type and mutant SLC26A3 STAS domains were expressed as Smt3 fusions in E. coli, and the amount of solubly expressed STAS domain in the crude lysates was assessed by Western blot analysis or Coomassie staining. Approximately 40 μg of crude lysate was loaded in each lane: lane 1, wild type STAS; lane 2, ΔY526/7 STAS; lane 3, I544N STAS; lane 4, I675/6ins STAS; lane 5, G702Tins STAS. B, wild type and mutant SLC26A3 STAS domains were expressed as Myc-tagged fusions HEK 293 cells, and the amount of solubly expressed STAS domain protein was assessed by Western blot analysis. The samples are as follows: lane 1, untransfected; lane 2, wild type STAS; lane 3, ΔY526/7 STAS; lane 4, I544N STAS; lane 5, I675/6ins STAS; lane 6, G702Tins STAS. The molecular mass markers to the left of the blots are measured in kDa.
The solubly expressed wild type, ΔY526/7, and I544N STAS domains were purified (Fig. 7A), and biophysical analyses using NMR and CD spectroscopies were performed (Fig. 7, B-D). The TROSY-HSQC spectrum of the wild type SLC26A3 STAS domain shows nitrogen-proton cross-peaks with chemical shift dispersions typical of a well folded protein, with peaks in the center of the spectrum likely from flexible loops, termini, or the IVS (Fig. 7B, black peaks). A TROSY-HSQC spectrum of the ΔY526/7 STAS domain (Fig. 7B, red peaks) overlays well with the wild type spectrum with only minor differences, consistent with absence of peaks and local chemical shift changes near the site of the mutation. In addition to the TROSY-HSQC spectra, CD spectra were collected for the wild type, ΔY526/7, and I544N STAS domains. The CD spectra for the wild type and two mutant STAS domains are indistinguishable from one another and indicate that the STAS domain is a mixed α/β protein, consistent with the predicted structure of the STAS domain (Fig. 7C). Thermal stability measurements were made of the wild type, ΔY526/7, and I544N STAS domains by monitoring the loss of molar ellipticity at 222 nm as the temperature was increased from 20 to 80 °C. No significant differences were observed between the wild type, ΔY526/7, and I544N STAS domain denaturation curves with all three proteins having melting temperature (Tm) values of 53 °C (Fig. 7D).
FIGURE 7.

SLC26A3 STAS structure and stability. A, representative Coomassie-stained gels of purified STAS domain protein is shown. B, NMRTROSY-HSQC spectra of purified 15N-labeled wild type and ΔY526/7 SLC26A3 STAS domain (black and red peaks, respectively) indicating well folded proteins. C, circular dichroism spectra of the wild type, ΔY526/7, and I544N SLC26A3 STAS domains. The spectra are averages of at least three experiments from at least two separate protein preparations. D, mean residue molar ellipticity at 222 nm of the wild type, ΔY526/7 and I544N SLC26A3 STAS domains as a function of temperature from 20 to 80 °C. The data presented are averaged data from at least three experiments from at least two independent protein preparations.
Although the ΔY526/7 and I544N mutations cause a partial or complete loss of plasma membrane trafficking of the transporter, no measurable differences in the domain itself were observed. By inference, the ΔY526/7 and I544N mutations may affect the ability of the STAS domain to interact with other regions of the transporter. To test this, the conformation of SLC26A3 was assessed by limited proteolysis of the wild type and mutant transporters with trypsin. Increasing concentrations of trypsin were incubated with the lysates at room temperature for 12 min, and the reactions were stopped by the addition of sample buffer. Tryptic fragments were detected via Western blot analysis using the STAS domain antibody, which detect the full-length protein and three predominant proteolytic fragments labeled 1-3 (Fig. 8A). Consistent with their STAS domains being misfolded and more readily degraded, the I675/6ins and G702Tins transporters are more susceptible to proteolysis than are the wild type, ΔY526/7, and I544N transporters. More subtle conformational differences between the wild type and the ΔY526/7 and I544N transporters are also observed. Quantification of the degradation patterns of the full-length transporters (Fig. 8B) reveal measurable differences between the wild type and the I544N transporters at the trypsin concentrations encompassing 3-100 μg/ml (p values lower than 0.05), where no significant differences are observed between the wild type and ΔY526/7 transporters. The most profound difference between the wild type, ΔY526/7, and I544N transporters is in the production of proteolytic fragment 1 (Fig. 8D). These data taken together suggest that the ΔY526/7 and I544N mutations may cause conformational changes in the transporter because of altered domain-domain interactions.
FIGURE 8.

Trypsin digest of SLC26A3. A, representative Western blots of SLC26A3 tryptic digests are shown with ∼125 μg of protein loaded in each lane. Triangles indicate the full-length (FL) protein and consistently observed tryptic fragments (1-3). B, quantification of the loss of the wild type, ΔY526/7, and I544N SLC26A3 full-length (FL) protein species at various trypsin concentrations. Each experiment was quantified and normalized independently of one another, and the means ± S.D. were calculated from all experiments quantified (wild type n = 5, ΔY527/6 n = 5, I544N n = 7). C, quantification of tryptic fragment 3 from the wild type, ΔY526/7, and I544N SLC26A3 trypsin digest experiments. The relative amounts of tryptic fragment 3 have been normalized to the most prevalent band within each individual experiment, and means ± S.D. were calculated from all experiments quantified (wild type n = 5, ΔY527/6 n = 5, I544N n = 7). D, quantification of tryptic fragment 1 from the wild type, ΔY526/7, and I544N SLC26A3 trypsin digest experiments. The amount of tryptic fragment 1 in the 300 μg/ml trypsin lane was quantified via densitometry and then normalized to the average amount of wild type tryptic fragment. The means ± S.D. are calculated from all experiments quantified (wild type n = 5, ΔY527/6 n = 6, I544N n = 7).
DISCUSSION
Multiple topology models have been presented for the SLC26A family, with
the most frequently published model having the N and C termini located in the
cytosol (1,
4,
5,
7-13,
18,
59-61).
The experimental topology data presented here is the first to be reported for
SLC26A3 and supports a model with intracellularly located N and C termini.
Consistent with our results, some previous studies on SLC26A5 and SLC26A6 have
indicated that the N and C termini of these proteins are located in the
cytosol (7,
10,
13,
61). The location of the N
terminus is important as previous reports have reported it to be
proteolytically cleaved in vivo
(65,
66), raising the possibility
that proteolytic cleavage may regulate transporter function. However, in this
study the same banding pattern was observed in our Western blots regardless of
whether an N- or C-terminal antibody was used (anti-c-Myc or anti-STAS domain;
data not shown), suggesting that under our experimental conditions SLC26A3 is
not undergoing any significant proteolysis. In addition, localizing the
SLC26A3 STAS domain to the cytosol is consistent with the previously described
model for CFTR activation by SLC26A3
(33). Indeed, establishing
that the STAS domain of SLC26A3 is in the same subcellular compartment as the
R-domain of CFTR is central to this model for
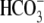 secretion.
secretion.
The boundaries of the cytosolic STAS domain have not yet been clearly identified; however, it must follow the final transmembrane segment of SLC26A3 that has been predicted to end between amino acids 494 and 497 (8, 59). Thus, the SLC26A3 STAS domain is likely to begin somewhere after amino acid 500. The absence of an SLC26A STAS domain structure and the lack of strong sequence identity between the SLC26A STAS domains and SpoIIAA have made identifying the precise beginning of the domain difficult. In our expression studies the three N-terminal boundaries tested all resulted in soluble protein indicating that the N terminus of the STAS domain may be at or C-terminal to amino acid 525. However, in our R-domain binding assay, the N-terminal boundary of amino acid 510 resulted in more consistent binding that we interpret to indicate that the extra 15 amino acids on the N terminus may aid in the binding interaction with the R-domain, suggesting that the functional N terminus is somewhere between amino acids 510 and 525. Constructs with a C-terminal boundary of amino acid 720 produced an insoluble STAS domain, whereas constructs ending in amino acid 741 were soluble. We interpret these data to indicate that the C terminus of the STAS domain must be beyond amino acid 720 but likely not past amino acid 741. In addition to SLC26A3, the boundaries of a STAS domain homolog from Aquifex aeolicus VF5 (gi:15606490) were screened with similar results to those for SLC26A3. The optimal domain boundaries for soluble protein expression were amino acids 450-588 (analogous to amino acids 525-739 of SLC26A3) with an N-terminal boundary of amino acid 459 (analogous to amino acid 534 in SLC26A3) or a C-terminal boundary of 575 (analogous to amino acid 728 of SLC26A3) rendering the expressed protein insoluble (data not shown). In addition, a previous report used amino acid 497 as the N-terminal boundary for the SLC26A6 STAS domain, which is analogous to SLC26A3 amino acid 513 in SLC26A3 (67). However, in this study the C-terminal boundary of SLC26A6 was defined to be amino acid 633, which from our multiple sequence alignment is predicted to be near the end of the IVS, suggesting that this construct is truncated at the C terminus (67). This study provides a systematic analysis of STAS domain boundaries and should aid in understanding the structural and functional properties of this domain.
The functional, glycosylation, and immunofluorescence data presented
suggest that the I544N, I675/6ins, and G702Tins mutations result in CLD
because of ER retention and the loss of functional protein at the plasma
membrane. This is a common disease-causing mechanism in the SLC26A protein
family as a number of mutations in SLC26A2 and SLC26A4 have shown ER retention
when monitored by immunofluorescence
(11,
32,
48,
68). It is interesting to note
that the equivalent I675/6ins mutation in mouse SLC26A3 results in an inactive
transporter that is properly trafficked to the plasma membrane
(32), suggesting that this
mutation differentially affects the human and mouse transporters. The
disease-causing mechanism of the ΔY526/7 transporter is less clear
because it retains some
Cl-/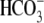 exchange activity,
and hence some functional protein at the plasma membrane. By lowering the
expression temperature the amount of functional, plasma membrane-localized,
and complexly glycosylated ΔY526/7 mutant transporter approached wild
type levels. These data suggest that the ΔY526/7 mutation is extremely
sensitive to its cellular environment and is possibly a less severe mutation
than the I544N, I675/6ins or G702Tins mutations. It should be noted that the
ΔY526/7 mutant was identified in a patient who had the severe I675/6ins
mutation on the other SLC26A3 allele
(69).
exchange activity,
and hence some functional protein at the plasma membrane. By lowering the
expression temperature the amount of functional, plasma membrane-localized,
and complexly glycosylated ΔY526/7 mutant transporter approached wild
type levels. These data suggest that the ΔY526/7 mutation is extremely
sensitive to its cellular environment and is possibly a less severe mutation
than the I544N, I675/6ins or G702Tins mutations. It should be noted that the
ΔY526/7 mutant was identified in a patient who had the severe I675/6ins
mutation on the other SLC26A3 allele
(69).
Although the major mechanism of disease seems to be ER retention, the CLD mutations have differential effects on the STAS domain itself. The I675/6ins and G702Tins mutations seem to prevent the STAS domain from folding as evident by the lack of soluble expression. We interpret the lack of solubly expressed protein to indicate protein misfolding. In contrast to the I675/6ins and G702Tins mutations, the ΔY526/7 and I544N mutations are solubly produced and do not cause dramatic changes in the measured biochemical or biophysical properties of the STAS domain. Nevertheless, the quality control machinery of the cell clearly recognizes the ΔY526/7 and I544N mutant transporters as different from the wild type transporter as evident by the mistrafficking of these proteins. It is possible that the ΔY526/7 and I544N mutations disrupt an interaction between the STAS domain and other regions of the SLC26A3 transporter that results in the recognition of this conformation as non-native by the cell. The trypsin proteolysis experiments revealed different conformations for the mutant SLC26A3 transporters compared with the wild type, consistent with the hypothesis that the ΔY526/7 and I544N mutations disrupt domain-domain interactions.
A recent study on an SLC26A homolog from Arabidopsis thaliana also supports the hypothesis that the STAS domain may be involved in a domain-domain interaction. Shibagaki and Grossman (36) performed random mutagenesis on the Sultr1.2 STAS domain from A. thaliana and identified a number of residues that are critical for proper sulfate transport and trafficking. Three of the residues identified by Shibagaki and Grossman (36) (Tyr-523, Ala-540, and Tyr-542) are at positions similar to two of the CLD-causing mutations (ΔY526/7 and I544N) in SLC26A3. The Y523H, A540S, and Y542C Sultr1.2 mutants were all expressed and found to be in the plasma membrane, with the Y523H and Y542C mutations resulting in inactive transporters and the A540S mutant reducing transport activity. Shibagaki and Grossman (36) mapped their mutants onto the SpoIIAA structure and found that many of their mutants, including Y523H, A540S and Y542C, cluster to the SpoIIAA-SpoIIAB binding interface. As a result, Shibagaki and Grossman (36) speculated that molecular interactions on this surface may be important for proper Sultr1.2 transport activity. Because SLC26A3 and Sultr1.2 are homologous, it is logical to think that the ΔY526/7 and I544N mutations may also disrupt an important domain-domain interaction; however, in contrast to the Sultr1.2 mutants, the CLD-causing mutations in SLC26A3 inhibit proper folding.
Taken together, our data suggest a model by which the CLD-causing mutations in the STAS domain of SLC26A3 result in disease by causing a loss in wild type levels of functional protein at the plasma membrane (Fig. 9). The loss of functional transporters at the membrane is caused by at least two distinct mechanisms, both of which result in transporter mistrafficking and ER retention. The I675/6ins and G702Tins mutations cause the STAS domain to misfold, which prevents the mutant transporters from reaching the native state. In contrast, the ΔY526/7 and I544N mutations do not cause the STAS domain to misfold, but rather likely disrupt important intramolecular interactions critical to forming a well folded and functional transporter. However, we cannot rule out the possibility that additional intermolecular interactions critical for proper folding are also being disrupted by these two mutations. These mechanisms have important implications for potential therapeutics, as with the former case directed therapy would require stabilization of the STAS domain itself, whereas the latter would require stabilization of a domain-domain interaction.
FIGURE 9.

Model for the folding and trafficking of wild type and mutant SLC26A3. A protein folding and trafficking model for the wild type and mutant SLC26A3 proteins is presented with proper folding of both the STAS domain and the transmembrane spans being required for trafficking from the ER to the plasma membrane. The circle labeled STAS denotes properly folded STAS domain, whereas the squiggly line represents improperly folded STAS domain. The vertical rectangles represent properly folded and assembled transmembrane spans, whereas the skewed rectangles symbolize non-native transmembrane structure. The horizontal black lines represent the ER and plasma membranes, and the branched structures on the transmembrane domain represent the glycosylation state of the protein. The wild type transporter and a portion of the ΔY526/7 transporters can adopt the appropriate structure that is recognized by the quality control machinery of the ER as native, and is then allowed to proceed through the secretory pathway where it is complexly glycosylated and eventually reaches the plasma membrane (PM). A portion of the ΔY526/7 transporters are retained in the ER and not trafficked to the plasma membrane. This misprocessing is more profound for the I544N, I675/6ins, and G702Tins mutant transporters. The large and small font type for the ΔY526/7 labels represents the larger and smaller population of cells that have native-like or non-native-like transporter. This conceptual model has no information regarding the order by which the STAS and transmembrane domains fold and are assembled in the ER, rather it highlights the different mechanisms by which the STAS domain mutations bring about protein misfolding and the absence of active protein at the plasma membrane.
Supplementary Material
This work was supported by National Institutes of Health Grants NIDDK 49835 (to P. J. T.), DE12309, and DK38938 (to S. M.), Canadian Institutes of Health Research Grant 145629 (to J. D. F.-K.), and National Institutes of Health Training Grant GM-08203 (to M. R. D.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The on-line version of this article (available at http://www.jbc.org) contains supplemental Fig. 1.
Footnotes
The abbreviations used are: CFTR, cystic fibrosis transmembrane conductance regulator; CLD, congenital chloride-losing diarrhea; BCECF-AM, 2′,7′-bis-(2-carboxyethyl)-5-(and-6)-carboxyfluorescein, acetoxymethyl ester; TRITC, tetramethylrhodamine isothiocyanate; EndoH, endoglycosidase H; PNGaseF, peptide N-glycosidase F; PVDF, polyvinylidene fluoride; DTT, dithiothreitol; TROSY, transverse relaxation optimized spectroscopy; HSQC, heteronuclear single quantum coherence; IVS, intervening sequence; ER, endoplasmic reticulum; PBS, phosphate-buffered saline; TBS, Tris-buffered saline.
References
- 1.Lohi, H., Kujala, M., Kerkela, E., Saarialho-Kere, U., Kestila, M., and Kere, J. (2000) Genomics 70 102-112 [DOI] [PubMed] [Google Scholar]
- 2.Mount, D. B., and Romero, M. F. (2004) Pfluegers Arch. 447 710-721 [DOI] [PubMed] [Google Scholar]
- 3.Wilkinson, D. J., Strong, T. V., Mansoura, M. K., Wood, D. L., Smith, S. S., Collins, F. S., and Dawson, D. C. (1997) Am. J. Physiol. 273 L127-L133 [DOI] [PubMed] [Google Scholar]
- 4.Hoglund, P., Haila, S., Socha, J., Tomaszewski, L., Saarialho-Kere, U., Karjalainen-Lindsberg, M. L., Airola, K., Holmberg, C., de la Chapelle, A., and Kere, J. (1996) Nat. Genet. 14 316-319 [DOI] [PubMed] [Google Scholar]
- 5.Kere, J., Lohi, H., and Hoglund, P. (1999) Am. J. Physiol. 276 G7-G13 [DOI] [PubMed] [Google Scholar]
- 6.Lohi, H., Kujala, M., Makela, S., Lehtonen, E., Kestila, M., Saarialho-Kere, U., Markovich, D., and Kere, J. (2002) J. Biol. Chem. 277 14246-14254 [DOI] [PubMed] [Google Scholar]
- 7.Lohi, H., Lamprecht, G., Markovich, D., Heil, A., Kujala, M., Seidler, U., and Kere, J. (2003) Am. J. Physiol. 284 C769-C779 [DOI] [PubMed] [Google Scholar]
- 8.Makela, S., Kere, J., Holmberg, C., and Hoglund, P. (2002) Hum. Mutat. 20 425-438 [DOI] [PubMed] [Google Scholar]
- 9.Moseley, R. H., Hoglund, P., Wu, G. D., Silberg, D. G., Haila, S., de la Chapelle, A., Holmberg, C., and Kere, J. (1999) Am. J. Physiol. 276 G185-G192 [DOI] [PubMed] [Google Scholar]
- 10.Navaratnam, D., Bai, J. P., Samaranayake, H., and Santos-Sacchi, J. (2005) Biophys. J. 89 3345-3352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Taylor, J. P., Metcalfe, R. A., Watson, P. F., Weetman, A. P., and Trembath, R. C. (2002) J. Clin. Endocrinol. Metab. 87 1778-1784 [DOI] [PubMed] [Google Scholar]
- 12.Toure, A., Morin, L., Pineau, C., Becq, F., Dorseuil, O., and Gacon, G. (2001) J. Biol. Chem. 276 20309-20315 [DOI] [PubMed] [Google Scholar]
- 13.Zheng, J., Long, K. B., Shen, W., Madison, L. D., and Dallos, P. (2001) Neuroreport 12 1929-1935 [DOI] [PubMed] [Google Scholar]
- 14.Dawson, P. A., and Markovich, D. (2005) Curr. Med. Chem. 12 385-396 [DOI] [PubMed] [Google Scholar]
- 15.Haila, S., Hastbacka, J., Bohling, T., Karjalainen-Lindsberg, M. L., Kere, J., and Saarialho-Kere, U. (2001) J. Histochem. Cytochem. 49 973-982 [DOI] [PubMed] [Google Scholar]
- 16.Jacob, P., Rossmann, H., Lamprecht, G., Kretz, A., Neff, C., Lin-Wu, E., Gregor, M., Groneberg, D. A., Kere, J., and Seidler, U. (2002) Gastroenterology 122 709-724 [DOI] [PubMed] [Google Scholar]
- 17.Knauf, F., Yang, C. L., Thomson, R. B., Mentone, S. A., Giebisch, G., and Aronson, P. S. (2001) Proc. Natl. Acad. Sci. U. S. A. 98 9425-9430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Royaux, I. E., Suzuki, K., Mori, A., Katoh, R., Everett, L. A., Kohn, L. D., and Green, E. D. (2000) Endocrinology 141 839-845 [DOI] [PubMed] [Google Scholar]
- 19.Satoh, H., Susaki, M., Shukunami, C., Iyama, K., Negoro, T., and Hiraki, Y. (1998) J. Biol. Chem. 273 12307-12315 [DOI] [PubMed] [Google Scholar]
- 20.Soleimani, M., Greeley, T., Petrovic, S., Wang, Z., Amlal, H., Kopp, P., and Burnham, C. E. (2001) Am. J. Physiol. 280 F356-F364 [DOI] [PubMed] [Google Scholar]
- 21.Vincourt, J. B., Jullien, D., Kossida, S., Amalric, F., and Girard, J. P. (2002) Genomics 79 249-256 [DOI] [PubMed] [Google Scholar]
- 22.Waldegger, S., Moschen, I., Ramirez, A., Smith, R. J., Ayadi, H., Lang, F., and Kubisch, C. (2001) Genomics 72 43-50 [DOI] [PubMed] [Google Scholar]
- 23.Xie, Q., Welch, R., Mercado, A., Romero, M. F., and Mount, D. B. (2002) Am. J. Physiol. 283 F826-F838 [DOI] [PubMed] [Google Scholar]
- 24.Jiang, Z., Grichtchenko, I. I., Boron, W. F., and Aronson, P. S. (2002) J. Biol. Chem. 277 33963-33967 [DOI] [PubMed] [Google Scholar]
- 25.Karniski, L. P., Lotscher, M., Fucentese, M., Hilfiker, H., Biber, J., and Murer, H. (1998) Am. J. Physiol. 275 F79-F87 [DOI] [PubMed] [Google Scholar]
- 26.Scott, D. A., and Karniski, L. P. (2000) Am. J. Physiol. 278 C207-C211 [DOI] [PubMed] [Google Scholar]
- 27.Shcheynikov, N., Wang, Y., Park, M., Ko, S. B., Dorwart, M., Naruse, S., Thomas, P. J., and Muallem, S. (2006) J. Gen. Physiol. 127 511-524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kim, K. H., Shcheynikov, N., Wang, Y., and Muallem, S. (2005) J. Biol. Chem. 280 6463-6470 [DOI] [PubMed] [Google Scholar]
- 29.Rouached, H., Berthomieu, P., El Kassis, E., Cathala, N., Catherinot, V., Labesse, G., Davidian, J. C., and Fourcroy, P. (2005) J. Biol. Chem. 280 15976-15983 [DOI] [PubMed] [Google Scholar]
- 30.Chapman, J. M., Knoepp, S. M., Byeon, M. K., Henderson, K. W., and Schweinfest, C. W. (2002) Cancer Res. 62 5083-5088 [PubMed] [Google Scholar]
- 31.Hemminki, A., Hoglund, P., Pukkala, E., Salovaara, R., Jarvinen, H., Norio, R., and Aaltonen, L. A. (1998) Oncogene 16 681-684 [DOI] [PubMed] [Google Scholar]
- 32.Ko, S. B., Shcheynikov, N., Choi, J. Y., Luo, X., Ishibashi, K., Thomas, P. J., Kim, J. Y., Kim, K. H., Lee, M. G., Naruse, S., and Muallem, S. (2002) EMBO J. 21 5662-5672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ko, S. B., Zeng, W., Dorwart, M. R., Luo, X., Kim, K. H., Millen, L., Goto, H., Naruse, S., Soyombo, A., Thomas, P. J., and Muallem, S. (2004) Nat. Cell Biol. 6 343-350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chernova, M. N., Jiang, L., Shmukler, B. E., Schweinfest, C. W., Blanco, P., Freedman, S. D., Stewart, A. K., and Alper, S. L. (2003) J. Physiol. (Lond.) 549 3-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shibagaki, N., and Grossman, A. R. (2004) J. Biol. Chem. 279 30791-30799 [DOI] [PubMed] [Google Scholar]
- 36.Shibagaki, N., and Grossman, A. R. (2006) J. Biol. Chem. 281 22964-22973 [DOI] [PubMed] [Google Scholar]
- 37.Aravind, L., and Koonin, E. V. (2000) Curr. Biol. 10 R53-R55 [DOI] [PubMed] [Google Scholar]
- 38.Duncan, L., Alper, S., and Losick, R. (1996) J. Mol. Biol. 260 147-164 [DOI] [PubMed] [Google Scholar]
- 39.Duncan, L., and Losick, R. (1993) Proc. Natl. Acad. Sci. U. S. A. 90 2325-2329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Garsin, D. A., Paskowitz, D. M., Duncan, L., and Losick, R. (1998) J. Mol. Biol. 284 557-568 [DOI] [PubMed] [Google Scholar]
- 41.Clarkson, J., Campbell, I. D., and Yudkin, M. D. (2003) Biochem. J. 372 113-119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kovacs, H., Comfort, D., Lord, M., Campbell, I. D., and Yudkin, M. D. (1998) Proc. Natl. Acad. Sci. U. S. A. 95 5067-5071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Magnin, T., Lord, M., Errington, J., and Yudkin, M. D. (1996) Mol. Microbiol. 19 901-907 [DOI] [PubMed] [Google Scholar]
- 44.Seavers, P. R., Lewis, R. J., Brannigan, J. A., Verschueren, K. H., Murshudov, G. N., and Wilkinson, A. J. (2001) Structure (Lond.) 9 605-614 [DOI] [PubMed] [Google Scholar]
- 45.Campbell, C., Cucci, R. A., Prasad, S., Green, G. E., Edeal, J. B., Galer, C. E., Karniski, L. P., Sheffield, V. C., and Smith, R. J. (2001) Hum. Mutat. 17 403-411 [DOI] [PubMed] [Google Scholar]
- 46.Everett, L. A., Glaser, B., Beck, J. C., Idol, J. R., Buchs, A., Heyman, M., Adawi, F., Hazani, E., Nassir, E., Baxevanis, A. D., Sheffield, V. C., and Green, E. D. (1997) Nat. Genet. 17 411-422 [DOI] [PubMed] [Google Scholar]
- 47.Karniski, L. P. (2001) Hum. Mol. Genet. 10 1485-1490 [DOI] [PubMed] [Google Scholar]
- 48.Karniski, L. P. (2004) Hum. Mol. Genet. 13 2165-2171 [DOI] [PubMed] [Google Scholar]
- 49.Liu, X. Z., Ouyang, X. M., Xia, X. J., Zheng, J., Pandya, A., Li, F., Du, L. L., Welch, K. O., Petit, C., Smith, R. J., Webb, B. T., Yan, D., Arnos, K. S., Corey, D., Dallos, P., Nance, W. E., and Chen, Z. Y. (2003) Hum. Mol. Genet. 12 1155-1162 [DOI] [PubMed] [Google Scholar]
- 50.Park, H. J., Shaukat, S., Liu, X. Z., Hahn, S. H., Naz, S., Ghosh, M., Kim, H. N., Moon, S. K., Abe, S., Tukamoto, K., Riazuddin, S., Kabra, M., Erdenetungalag, R., Radnaabazar, J., Khan, S., Pandya, A., Usami, S. I., Nance, W. E., Wilcox, E. R., Riazuddin, S., and Griffith, A. J. (2003) J. Med. Genet. 40 242-248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rossi, A., and Superti-Furga, A. (2001) Hum. Mutat. 17 159-171 [DOI] [PubMed] [Google Scholar]
- 52.Holmberg, C. (1986) Clin. Gastroenterol. 15 583-602 [PubMed] [Google Scholar]
- 53.Hihnala, S., Hoglund, P., Lammi, L., Kokkonen, J., Ormala, T., and Holmberg, C. (2006) J. Pediatr. Gastroenterol. Nutr. 42 369-375 [DOI] [PubMed] [Google Scholar]
- 54.Canani, R. B., Terrin, G., Cirillo, P., Castaldo, G., Salvatore, F., Cardillo, G., Coruzzo, A., and Troncone, R. (2004) Gastroenterology 127 630-634 [DOI] [PubMed] [Google Scholar]
- 55.Mossessova, E., and Lima, C. D. (2000) Mol. Cell 5 865-876 [DOI] [PubMed] [Google Scholar]
- 56.Pervushin, K., Riek, R., Wider, G., and Wuthrich, K. (1997) Proc. Natl. Acad. Sci. U. S. A. 94 12366-12371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Delaglio, F., Grzesiek, S., Vuister, G. W., Zhu, G., Pfeifer, J., and Bax, A. (1995) J. Biomol. NMR 6 277-293 [DOI] [PubMed] [Google Scholar]
- 58.Johnson, B. A., and Blevins, R. A. (1994) J. Biomol. NMR 4 603-614 [DOI] [PubMed] [Google Scholar]
- 59.Byeon, M. K., Westerman, M. A., Maroulakou, I. G., Henderson, K. W., Suster, S., Zhang, X. K., Papas, T. S., Vesely, J., Willingham, M. C., Green, J. E., and Schweinfest, C. W. (1996) Oncogene 12 387-396 [PubMed] [Google Scholar]
- 60.Deak, L., Zheng, J., Orem, A., Du, G. G., Aguinaga, S., Matsuda, K., and Dallos, P. (2005) J. Physiol. (Lond.) 563 483-496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ludwig, J., Oliver, D., Frank, G., Klocker, N., Gummer, A. W., and Fakler, B. (2001) Proc. Natl. Acad. Sci. U. S. A. 98 4178-4183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Vincourt, J. B., Jullien, D., Amalric, F., and Girard, J. P. (2003) FASEB J. 17 890-892 [DOI] [PubMed] [Google Scholar]
- 63.Girard, J. P., Baekkevold, E. S., Feliu, J., Brandtzaeg, P., and Amalric, F. (1999) Proc. Natl. Acad. Sci. U. S. A 96 12772-12777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Park, M., Ko, S. B., Choi, J. Y., Muallem, G., Thomas, P. J., Pushkin, A., Lee, M. S., Kim, J. Y., Lee, M. G., Muallem, S., and Kurtz, I. (2002) J. Biol. Chem. 277 50503-50509 [DOI] [PubMed] [Google Scholar]
- 65.Byeon, M. K., Frankel, A., Papas, T. S., Henderson, K. W., and Schweinfest, C. W. (1998) Protein Expression Purif. 12 67-74 [DOI] [PubMed] [Google Scholar]
- 66.Lohi, H., Makela, S., Pulkkinen, K., Hoglund, P., Karjalainen-Lindsberg, M. L., Puolakkainen, P., and Kere, J. (2002) Am. J. Physiol. 283 G567-G575 [DOI] [PubMed] [Google Scholar]
- 67.Alvarez, B. V., Vilas, G. L., and Casey, J. R. (2005) EMBO J. 24 2499-2511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Rotman-Pikielny, P., Hirschberg, K., Maruvada, P., Suzuki, K., Royaux, I. E., Green, E. D., Kohn, L. D., Lippincott-Schwartz, J., and Yen, P. M. (2002) Hum. Mol. Genet. 11 2625-2633 [DOI] [PubMed] [Google Scholar]
- 69.Hoglund, P., Auranen, M., Socha, J., Popinska, K., Nazer, H., Rajaram, U., Al Sanie, A., Al-Ghanim, M., Holmberg, C., de la Chapelle, A., and Kere, J. (1998) Am. J. Hum. Genet. 63 760-768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Najafi, S. M., Willis, A. C., and Yudkin, M. D. (1995) J. Bacteriol. 177 2912-2913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Masuda, S., Murakami, K. S., Wang, S., Anders Olson, C., Donigian, J., Leon, F., Darst, S. A., and Campbell, E. A. (2004) J. Mol. Biol. 340 941-956 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.