

Abstract
Chromosome translocations in the common epithelial cancers are abundant, yet little is known about them. They have been thought to be almost all unbalanced and therefore dismissed as mostly mediating tumour suppressor loss. We present a comprehensive analysis by array painting of the chromosome translocations of breast cancer cell lines HCC1806, HCC1187 and ZR-75-30. In array painting, chromosomes are isolated by flow cytometry, amplified and hybridized to DNA microarrays. A total of 200 breakpoints were identified and all were mapped to 1Mb resolution on BAC arrays, then 40 selected breakpoints, including all balanced breakpoints, were further mapped on tiling-path BAC arrays or to around 2kb resolution using oligonucleotide arrays. Many more of the translocations were balanced at 1Mb resolution than expected, either reciprocal (eight in total) or balanced for at least one participating chromosome (19 paired breakpoints). Secondly, many of the breakpoints were at genes that are plausible targets of oncogenic translocation, including balanced breaks at CTCF, EP300/p300, and FOXP4. Two gene fusions were demonstrated, TAX1BP1-AHCY and RIF1-PKD1L1. Our results support the idea that chromosome rearrangements may play an important role in common epithelial cancers such as breast cancer.
Keywords: breast cancer, chromosome rearrangements, microarrays
Introduction
A major mechanism of gene alteration in cancer is genome rearrangement, such as chromosome translocation and inversion, which can result in gene fusion, promoter insertion or gene inactivation. As is well known in haematopoietic tumours and sarcomas, translocations and inversions can have powerful oncogenic effects on specific genes and play a central role in cancer development (Rowley, 1998). In the past there has been an implicit assumption that such rearrangements are not significant players in the common epithelial cancers (e.g. Vogelstein & Kinzler, 2004), but this view is now being challenged. Most provocatively, about 70% of prostate cancers have gene fusions analogous to those found in leukaemias and sarcomas such as fusions of transcription factors of the ETS family to the TMPRSS2 gene as a result of translocation or deletion (Tomlins et al 2005; Mehra et al, 2007). In breast cancer, we have described translocations of the NRG1/heregulin gene in 6% primary cases (Huang et al, 2004), and Soda et al (2007) have described fusions of ALK in 7% lung cancers. Other approaches also suggest that fusion transcripts are present in epithelial cancers (Hahn et al 2004, Volik et al, 2003). It has recently been argued that the low number of known gene fusions in epithelial cancers could simply reflect lack of data (Mitelman et al, 2004).
The study of translocations in epithelial cancers has been limited by technology (Mitelman, 2000). It is difficult to get useful numbers of metaphase spreads from tumour tissue for cytogenetic approaches, and primary cultures of tumours may be overgrown by premalignant and normal cells (Persson et al, 1999). Chromosome analysis therefore has to be done on cell lines. Even when chromosome spreads can be made, the karyotypes are mostly too complex for classical cytogenetic analysis. 24-colour chromosome painting—spectral karyotyping (SKY) or ‘M-FISH’ analysis—helps, but it gives no direct information about breakpoints (see e.g. Adeyinka et al, 2000; Davidson et al, 2000). There are not yet any established DNA-based methods to detect chromosome rearrangements - in particular, recent high-throughput screens for sequence change (e.g. Davies et al, 2002; Sjoblom et al, 2006) were unable to detect rearrangements, since the screens rely on being able to amplify sequencing targets by PCR, and translocation breaks will not be amplified. Array comparative genomic hybridisation (array-CGH) can in principle identify the breakpoints of unbalanced translocations as changes in genomic copy number, but cannot detect balanced translocations.
The distinction between balanced and unbalanced translocations has been considered an important issue (Dutrillaux, 1995; Mitelman, 2000). Balanced translocations are more likely to have effects at their breakpoints, whereas unbalanced translocations result in gain or loss of material—in particular, loss of tumour suppressor genes—so that the breakpoints themselves may not be important. While the familiar gene-fusing translocations in haemopoietic disease are usually balanced, translocations in common epithelial cancers are said to be mostly, though not exclusively, unbalanced (Dutrillaux, 1995; Davidson et al, 2000; Abdel-Rahman et al, 2001; Roschke et al, 2003).
As we show here, ‘array painting’ is a powerful advance in the analysis of the complex chromosome rearrangements of cancers. Introduced in the context of constitutional chromosome abnormalities (Fiegler et al, 2003b; Gribble et al 2007), it is a logical extension of reverse chromosome painting, which we previously applied to three breast cancer cell lines (Morris et al, 1997). In both approaches, individual chromosomes are purified using a fluorescence-activated cell sorter and the DNA amplified and hybridized to normal genomic DNA—to normal metaphase chromosomes in reverse painting and to DNA microarrays in array painting. Array painting gives a resolution only limited by the arrays used. It can be used to map all translocation breakpoints, including those of balanced translocations, and it identifies which chromosome fragments are joined, which is important in the context of looking for fusion genes.
We report here a comprehensive analysis by array painting of the chromosome translocations of three breast cancer cell lines. We analysed all chromosomes to 1Mb resolution, then analysed all balanced breakpoints and a number of unbalanced breakpoints to higher resolution by using either chromosome ‘tiling-path’ arrays or custom oligonucleotide arrays, since the breakpoints of balanced rearrangements have a higher probability of targeting genes.
Results
We determined the complete karyotypes of 3 breast cancer cell lines (Table 1). In total 200 breakpoints were identified and mapped to at least 1Mb resolution by array painting: we flow-sorted all chromosomes, amplified the chromosome DNA, and hybridized it to genomic DNA microarrays (Supplementary Table 1). The DNA of almost all chromosomes was also hybridized to normal metaphase chromosome spreads: although not strictly necessary, this identified independently the chromosome fractions sorted. Figure 1 and Supplementary Figures 1 and 2 show flow karyotypes of the cell lines. Almost all chromosomes were resolved into distinct fractions—‘peaks’ on the cytometry plot—except normal chromosomes 9 to 12 which are always superimposed. Where chromosomes did sort together, SKY data allowed us to identify which chromosomes were present in almost all cases. The chromosomes identified corresponded closely to the karyotypes of the three lines obtained by SKY (Davidson et al, 2000; M. Grigorova and PAWE, unpublished; shown at http://www.path.cam.ac.uk/~pawefish/). Minor discrepancies reflected the limitations of SKY analysis, principally the misclassification of small fragments. All were clarified by conventional chromosome painting. A summary of all abnormal chromosomes in the three cell lines is shown in Figure 2.
Table 1.
Chromosome rearrangements of breast cancer cell lines HCC1187 (a), HCC1806 (b) and ZR-75-30 (c) as determined by array painting
| a | |||
|---|---|---|---|
| HCC1187 | |||
| Peak1 | Chromosome2 | Peak1 | Chromosome2 |
| A | der(1)(6pter->6p21.1::1p35.2->1q21.3::8p22->8pter)*3 | T | der(19)t(2;19)(p16;p13.3)* |
| B | 4 | U | del(18)(q21.2)4 |
| C | 3 | V | der(2;5) t(2;5)(p10;p10)del(2)(p16p25.1)* |
| D | der(X)(6pter->6p21.1::1p35->1p21.3::Xp11.22->Xqter) | W | 14 |
| E | der(8)(1q10->1q21.3::8p22->8q22.2::1p31.1->1pter)* | X | 15 |
| F | 5 | Y | 16 |
| G | der(20)t(2;20)(q10;q11.21)* | der(20)t(14;20)({14qter->14q24.3:}{20pter->20qter})$ | |
| H | der(8)t(1;8)(p31.1;q22.2)* | Z | 17 |
| I | 6 | a | 18 |
| J | der(1)t(1;8)(p13;q22.2)* | b | der(13)t(10;13)(p12;q21.31) * |
| K | 7 | c | i(13q)del(13)(q10q31) |
| L | X | d | 20 |
| M | del(7)(q36.1)3 | e | 19 |
| N | 8 | f | 21 |
| der(10)(13qter->13q21::10p12->10q23.1::19q13.41->19qter)*3 | g | 21 | |
| O | der(11)t(11;12)(p15.4;p11.22)del(11)(q13.5q21) | h | 22 |
| P | der(19)t(2;19)(p10;p13.3)* | i | der(19)t(1;19)(p36.22;q13.1)3 |
| Q | 9,10,11,12 | j | der(?)({20pter->20p13:}{:13q31.1->13qter})3 |
| R | der(11)t(11;16)(p15.3;q22.1)del(11)(q13.5q21)*3 | k | trc(1;X;1)(1qter->1p11::Xp21.3->Xq25::1p11->1qter) |
| S | der(16)(16pter->16q22.1::11p15.3->11p15.4::12p11.22->12pter)* | ||
| b | |||
|---|---|---|---|
| HCC1806 | |||
| Peak1 | Chromosome2 | Peak1 | Chromosome2 |
| A | der(2)(5pter->5p10::2p23.3->2q32.2::5p10->5pter)5 | S | del(11)(q23.1q23.3)3 |
| B | der(4)(1qter->1q10::6p22.3->6p21.1::4p15.33->4qter) * 3 | T | del(8)(p22p12) |
| C | der(5)t(1;5)(p10;q10) * | U | der(11)t(11;13;11;13;11;13)8 |
| D | 1 | V | der(10)t(6;10)(p22.3;p11.22)* |
| E | der(4)t(4;6)(p15.33;p21.1) * | W | der(15)(15p?->15q21.1:{:19q31.1-> 19qter}:10p12.31->10pter)3,7,9,12 |
| F | der(6)t(1;6)(q24.3;p21.1)* | X | der(17)({17pter->17qter}:3q26.32->3qter)$ |
| G | der(7)t(2;7)(q23.3;p12.3)del(7)(q36.1q36.2)* | Y | der(22)t(12;22)(q13.2;q13.2) * |
| der(14)(2qter->2q31::14p?->14qter)6+ | Z | der(14)t(6;14)(p22;p11.2)* | |
| H | 5 | a | 13 |
| I | 8 | b | 15 |
| der(3)t(3;16)(p21.1;p12.1)*3 | c | der(19)(1pter->1p36:{:19p13-> 19q13.2:}:8q22.2->8qter)#,3,9 |
|
| J | der(12)t(12;13)(p13.31;q12)del(13)(q14.11q32.3) | d | 16 |
| K | der(11)t(2;11)(p24.3;p15.4)del(11)(p12p10)3 | e | 17 |
| L | der(7)(8qter->8q23.3:{:7p15.2->7qtel}:17q23.2->17qtel)*7 | f | der(5)t(5;10)(p10;p10)* |
| der(16)(11qter->11q13.1::3pter->3p21.2::16p12->16qter)*7 | g | 18 | |
| i(10q) | h | der(21)t(3;21)(p21.31;p?)+ | |
| M | X | i | der(20)(10pter->10p12.31: {:15q14->15q21.1:}:20p12->20q11.2::7p15.2->7pter)*3,10 |
| der(20)(3qter->3q10::20p11.2->20q11.2::7p15.2->7pter)*3 | der(12)t(2;12)(p21;p10) | ||
| N | der(6)t(4;6)(p15.33;p21.1) * | j | del(6)(q10qter) |
| O | der(3;20)t(3;20)(q10;?)# | k | der(12)t(12;22)(q13.2;q13.2)*3,13 |
| P | 9 | l | der(21)t(21;22)(p?;p?)+ |
| der(2)(13qtel->13q32.3::13q14.11->13q12.13:{:11q13.5-> 11q14:}:2p11.2->2q23.3::7p12.3->7pter)* |
m | 20 | |
| Q | der(17)(3qter->3q26.32:{17pter->17q24.31:}:15q21.3->15qter)7 | n | 19 |
| R | der(9)t(9;12)(p21.1;q23) | o | der(19)t(18;19)(p10;q12?)9 |
| c | |||
|---|---|---|---|
| ZR-75-30 | |||
| Peak1 | Chromosome2 | Peak1 | Chromosome2 |
| A | dic(1;1)(1qter->1q10:{8q?;17q?}n:1q10->1qter)11 | Q | 8 |
| B | der(5)dup(5)(p)({5pter->5qter}{:13q21->13q33:}:7q11.22->7qter)* | R | der(7)t(5;7)(q21;q11.22)*3 |
| C | der(1;14)(1qter->1q10:{8q?;17q?}n:14q10->14qter)11 | S | 9 |
| D | der(14;20)(20pter->20p10::14q10->14q?:{8q?;17q?}n:14q?->14qter)11 | T | 10,11,12 |
| E | 2 | U | der(14;20)t(14;20)(q10;p10) |
| F | 1 | V | 15 |
| der(14)(8qter->8q11::8p11->8p11:{:17q25->17q21:}:14p10->14qter)11 | W | der(21?)({:21p10?->21qter}{:13q10?->13q21:})*+ | |
| G | der(1)t(1;21)(p35;q11)del(1)(p21p13)del(21)(q21q22) | X | 16 |
| H | i(13)(q10) | Y | 17 |
| I | 4 | Z | 18 |
| der(5)dup(5)(p)( {5pter->5q14:} {:13q21->13qter})* | a | del(6)(q13) | |
| J | 3 | b | del(11)(q13.5) |
| K | der(11)t(10;11)(q11.21;q24) * | c | del(16)(q22)3 |
| L | der(7)(7pter->7q21:{8q?;17q?}n:14q32->14qter) | d | 20 |
| M | 5 | e | del(19)(p11)3,4 |
| N | 6 | f | 19 |
| O | 7 | g | der(10)t(10;11)(q11.21;q24) * |
| P | X | h | 22 |
Asterisks mark translocations that include a balanced breakpoint, which is shown in bold type.
Structural rearrangements as determined by array painting. Breaks are given by banding position for identification: bands are deduced from array painting, not cytogenetic analysis. Notation is essentially according to ISCN, using the extended notation for complex translocations. For example, der(7)t(2;7)(q23.3;p12.3)del(7)(q36.1q36.2) indicates ‘derivative of chromosome 7 which is a translocation with chromosome 2, the breakpoints being at q23.3 on chromosome 2 and p12.3 on chromosome 7, and there is also a deletion on the chromosome 7 between q36.1 and q36.2’. As the chromosome is a der(7), i.e. the centromere belongs to chromosome 7, the notation implies that the majority of chromosome 7 is present, but chromosome 2 is only present distal to the breakpoint. In extended notation this is der(7)(2qter->2q23.3::7p12.3->7q36.1::7q36.2->7qter). The order of fragments in complex translocations was deduced from SKY and in come cases FISH. The orientation of fragments was generally deduced by assuming that telomeres were provided by telomeric ends of fragments, and in complex rearrangements by comparison with similar or reciprocal junctions. Where the orientation was not known the fragment is enclosed in curly brackets {}. Mapped breakpoint intervals are provided in Table 2 and Supplementary Tables 1 and 2. A ? is given in some instances where the breakpoint position was unclear. This was either:
due to a lack of BAC clones on the p-arm of the acrocentric chromosomes;
due to no obvious break on the chromosome despite its presence in the translocation (confirmed by FISH), suggesting possible fusion at the telomere;
or due to ‘noise’ in the array data at the breakpoint.
There is one der(?) in HCC1187 where it was unclear which chromosome provided the centromere.
Part or all of rearrangement not detected in the SKY data (http://www.path.cam.ac.uk/~pawefish/) because the pieces of chromosome involved were too small to be resolved or were misclassified, but was verified by FISH. In the SKY analysis of HCC1187, a dmin classifying as chromosome 22 had also been tentatively identified but it would have been too small to appear in the flow karyotype.
Based on their position in the flow karyotype these deleted chromosomes are expected to contain a duplication in addition to the deletion.
Hybridization suggests part of 2p is duplicated.
Chromosome fraction Peak G of HCC1806 was a rare example where two chromosomes that sorted together had fragments from the same normal chromosome, chromosome 2.
There are 4 examples of 3-way translocations where only one break was detected by array painting, suggesting possible telomere fusion. The orientation of the middle fragment was not in general determined.
Breakpoint positions for the 11 and 13 breaks are given in Supplementary Table 1. Banding is not given here as the arrangement of the different pieces of 11 and 13 have not been clarified by FISH.
Chromosome 19 breaks not well resolved on array and order of fragments not confirmed.
Chromosome painting confirms order and chromosome origin of fragments. Probably the chromosome identified as der(19)t(7;19;10) in SKY.
ZR-75-30 has multiple small fragments of 17, some interspersed with fragments of 8, which were not well resolved by the 1Mb array.
SKY suggests a possible isodicentric chromosome 15. This can not be detected by array painting.
The der(12)t(12;22), which was uncertain from SKY data was confirmed to be present in all cells.
Figure 1.

Flow karyotype of chromosomes from the breast cancer cell line HCC1187.
The axes show fluorescence intensities of Hoechst 33258 and Chromomycin A3 DNA staining on an arbitrary scale. The chromosome fractions or ‘peaks’ are designated A to k. Peak k was off the top of the scale at the gain shown: it contains the largest chromosome, approximately 1.5 times the size of the chromosome in peak A.
Figure 2.

Summary of chromosome segments comprising the abnormal chromosomes in breast cancer cell lines, HCC1187, HCC1806 and ZR-75-30. Ideograms of chromosomes 1 to 22 and X are shown. To the right of each ideogram are lines representing the segments that constitute abnormal chromosomes and their approximate breakpoints in each of the three lines, HCC1187 (1187), HCC1806 (1806) and ZR-75-30 (ZR). The precise breakpoints are given in Supplementary Tables 1 and 2. Peak letters are shown at the top of each segment and correspond to the peak letters given in figure 1 and supplementary figures 1 and 2. Balanced breakpoints are indicated*. Normal copies of chromosomes are not shown.
All chromosome fractions were first hybridized to 1Mb-resolution BAC arrays, with amplified normal whole-genome DNA as reference (Figure 3 and Supplementary Table 1). Regions of the genome present in the sorted chromosome gave much stronger hybridisation signals than regions absent, in almost all cases clearly identifying the genome content of the chromosomes and the breakpoints (Figure 3). Crosshybridisation between large segmental duplications was occasionally seen, notably on proximal 10q, but mostly was weaker than authentic hybridisation.
Figure 3.

Example hybridisations of chromosomes from reciprocal translocations to arrays of various resolutions. (a - c) Balanced chromosome 8 breakpoints in HCC1187. (a) Array CGH of genomic DNA from HCC1187 on 1Mb array, showing fluorescence log2 ratios for clones on chromosome 8: note no copy number changes are detected, showing that all chromosome 8 rearrangements are balanced. (b) Hybridisation of flow-sorted chromosomes der(8)t(1;8)t(1;8) (peak E) (open circles) and der(1)t(1;8) (peak J) (closed circles) to a 1Mb BAC array, showing fluorescence log2 ratios for chromosome 8. A change in the log2 ratio from ∼0 to greater than 2.5 represents the 8q22.2 translocation breakpoint, indicated with a vertical line. (c) Hybridisation of sorted chromosomes der(8)t(1;8)t(1;8) (peak E) (open circles) and der(1)t(1;6)t(1;8) (closed circles) to an 8p tiling-path BAC array shows the balanced 8p22 breakpoint. (d - f) Examples of balanced breakpoints in HCC1806. (d) Hybridisation of sorted chromosomes der(4)t(4;6) (peak E) (open circles) and der(6)t(4;6) (peak N) (closed circles) to a chromosome 6 tiling path BAC array. (e) Hybridisation of sorted chromosome der(20)t(3;20;7) (peak M) to a custom Nimblegen oligonucleotide array. The breakpoint in TAX1BP1 on chromosome 7 is indicated with a black circle. (f) Hybridisation of sorted chromosome der(12)t(12;22) (peak k) to a custom Nimblegen oligonucleotide array. The breakpoint in P300 on chromosome 22 is indicated with a black circle. Nimblegen data shown is averaged over 300 bp.
Many translocations were balanced
Definitions of ‘balanced’ rearrangements vary. The definition we adopted of a balanced rearrangement was pragmatic, because we wanted to use it to identify rearrangements whose genetic consequences, for a given chromosome, should be confined to the breakpoint region. Reciprocal translocations are the simplest kind of balanced rearrangement. By ‘balanced’ for a given chromosome, we mean that there is no net gain or loss of sequence at the resolution of the 1Mb arrays. This is a realistic definition, since many near-balanced translocations have additional rearrangements such as deletions, insertions and inversions around the translocation breakpoints (e.g. Gribble et al, 2005; Sinclair et al, 2000; for references see Pole et al, 2006). For some of the balanced translocations we found that the two products were present in a different number of copies (e.g. for the t(10;13) of HCC1187, SKY analysis shows three copies of the derivative 13 chromosome but only one copy of the derivative 10), but since we assume this reflects numerical changes subsequent to the translocation we consider this unimportant.
The array hybridizations showed that many translocations were balanced for at least one of the participating chromosomes (Figure 3; marked in bold in Table 1). For example, in HCC1187, two chromosome 8 translocation breakpoints were identified, one at 8p22 and one at 8q22.2, and in both cases hybridization of the separated chromosomes to arrays showed that the translocations were balanced for chromosome 8, at the resolution of the arrays (Figures 3b and 3c). Array CGH using the 1Mb array also showed no copy number step at the breakpoints confirming that the breaks were balanced (Figure 3a). In some cases, the breaks on the partner chromosome were also balanced, forming a reciprocal translocation, but in several cases they were not. For example, the balanced breaks on 8q22.2 were joined to chromosome 1 breaks at 1p31.1 (in peaks E and H, Table 1) and 1p13 (in peak J), with net loss of 31 Mb of chromosome 1. More unexpected were the junctions of the balanced breaks on 8p22 (peaks A and E, Table 1): both were joined to chromosome 1 at 1q21, but instead of the chromosome 1 fragments being balanced, both the chromosome 8 breaks were joined to similar fragments of chromosome 1, extending from 1q21 towards the centromere. Other balanced breaks appeared to be joined to unrelated chromosomes, e.g. the 7p15.2 breaks in HCC1806. Conventional chromosome painting confirmed that the balanced fragments were on the expected chromosomes (Supplementary Figure 3). Overall, 26% (53/200) of breaks were balanced —17/53 (32%) in HCC1187, 27/93 (29%) in HCC1806, and 9/54 (17%) in ZR-75-30.
Among the balanced translocations there were eight reciprocal translocations in the three lines, allowing for some loss or duplication at the breakpoint. The straightforward reciprocal translocations were the t(10;13) in HCC1187; the t(4;6), t(12;22) and the t(2;7) in HCC1806, the latter consisting of the der(7)t(2;7) (peak G) and the der(2)t(13;11;2;7) (peak P); and the t(10;11) in ZR-75-30. In HCC1187, the complex rearrangement of 11, 12 and 16 is reciprocal, comprising a der(11)t(11;16)(p15.3;q22.1)del(11)(q13.5q21) and a der(16)t(11;16)(p15.3;q22.1)t(11;12)(p15.4;p11.22). HCC1187 also has a translocation between chromosomes 11 and 12 with the same 11q deletion, der(11)t(11;12)(p15.4;p11.22)del(11)(q13.5q21), so the reciprocal translocation appears to have been formed between another copy of this and a normal chromosome 16, with the break on 11 slightly proximal to the junction between 11 and 12. The t(3;16) in HCC1806, in which the chromosome 3 break is balanced and the 16 break is close to balanced, with a duplication of around 1Mb at the breakpoint, is likely to be a reciprocal rearrangement. Finally, the t(1;8) in HCC1187, which was expected to be a reciprocal translocation from SKY analysis, can be considered reciprocal with a very large deletion on chromosome 1p, as discussed above.
A comparison of array painting results and 1Mb array CGH data for the three cell lines showed no copy number change where balanced breaks were expected, unless the two products of the translocation were present in different copy numbers. The positions of unbalanced breakpoints matched precisely, although the signal to noise ratio was much weaker by array CGH.
Three breakpoints were recurrent at 1Mb resolution: all three lines had breaks at 8q22; HCC1187 and ZR-75-30 both had breaks at 16q22 and 13q21.31.
High resolution analysis of balanced breaks
We then mapped to higher resolution all the balanced breaks, together with some of the unbalanced breaks that were on the same chromosomes, by hybridizing at least one of the product chromosomes to either a chromosome-specific ‘tiling-path’ BAC array, i.e. an array of BACs at around 150-200 kb intervals or a custom oligonucleotide array designed to span the break, with oligonucleotides spaced at an average of about 120 to 170 bp (Figure 3e and 3f, Table 2, Supplementary Tables 1 and 2).
Table 2.
High resolution mapping of balanced breakpoints and associated genes in breast cancer cell lines HCC1187, HCC1806 and ZR-75-30
| Cell line | Breakpoint | Chromosome fraction mapped (Peak Letter)1 | Chromosome fraction containing reciprocal break (Peak letter)2 | Gene at breakpoint3 | Breakpoint interval (Mb)4 | Mapping method5 |
|---|---|---|---|---|---|---|
| HCC1187 | 2p16 | T | V | PSME4 | Chr2: 54.050- 54.053 | O |
| 8p22 | E | A* | SGCZ | Chr8: 14.5890-14.5917 | O, T8 | |
| 8p22 | A | E* | SGCZ | Chr8: 14.3315-14.4659 | T8 | |
| 8q22.2 | E | J* | RGS22 | Chr8: 101.059276 | O, C | |
| 10p12.2 | N | b | PIP5K2A | Chr10: 22.830-22.833, 22.863-22.866, 22.872-22.8758 | O, T10 | |
| 10p12.2 | b | N | none | Chr10: 22.49-23.02 | T10 | |
| 11p15.3 | R | S | AMPD3 | Chr11: 10.435-10.438 | O | |
| 13q21.31 | N | b | none | Chr13: 58.2726-58.2733 | O | |
| 16q22.1 | S | R | CTCF | Chr16: 66.1975-66.2120 | O | |
| 16q22.1 | R | S | 11 kb 5′ of CTCF | Chr16: 66.1419-66.1422 | O | |
| HCC1806 | 2q23.3 | P | G | RIF1 | Chr2: 152.0170-152.0176 | O |
| 3p21.1 | L | I | 25kb 3′ of PRKCD. Between PRKCD and TKT | Chr3: 53.2171 - 53.2274 | O | |
| 4p15.33 | E | N* | within apparent alt 3′ end of RAB28 (BE501997) | Chr4: 12.9048-12.9068 | O | |
| 4p15.33 | N | E*, B* | within apparent alt 3′ end of RAB28 (BE501997) | Chr4: 12.9057-12.9059 | O | |
| 6p22.3 | B | V, Z | ∼100kb 3′ of HDGFL1 | Chr6: 23.05-24.49 | T6 | |
| 6p22.3 | V | B | none | Chr6: 22.82-23.00 | T6 | |
| 6p21.1 | N | B, E | FOXP4 | Chr6: 41.649-41.655 | O, T6 | |
| 6p21.1 | E | N | 16kb 3′ of FOXP4 | Chr6: 41.694 - 41.696 | O | |
| 6p21.1 | B | N | ∼16kb 3′ of FOXP4 | Chr6: 41.59-41.79 | T6 | |
| 7p15.2 | L | M, i | ∼1 kb 5′ of HIBADH. Between TAX1BP1 and HIBADH | Chr7: 27.6695 - 27.6715 | O, T7 | |
| 7p15.2 | M | L | TAX1BP1 | Chr7: 27.752-27.7543 | O, T7 | |
| 7p12.3 | P | G* | PKD1L1 | Chr7: 47.8685-47.8688 | O, T7 | |
| 7p12.3 | G | P* | PKD1L1 | Chr7: 47.80-47.91 | T7 | |
| 12q13.2 | k | Y | ANKRD52 | Chr12: 54.928-54.930 | O | |
| 22q13.2 | k | Y | P300 | Chr22: 39.8895-39.8915 | O, T22 | |
| 22q13.2 | Y | K | P300, L3MBTL2, RANGAP1, ZC3H7B | Chr22: 39.90-40.09 | T22 | |
| ZR-75-30 | 7q11.22 | B | R* | AUTS2 | Chr7: 69.28-69.34 | T7 |
| 7q11.2 | R | B* | AUTS2 | Chr7: 69.34-69.43 | T7 | |
| 10q11.2 | K | g | Undetermined to gene level6 | Chr10: 45.322-45.661 | T10 | |
| 10q11.2 | g | K | Undetermined to gene level6 | Chr10: 45.661-45.980 | T10 | |
| 11q24.2 | g | K | PUS3/HYLS17 | Chr11: 125.26-125.28 | P | |
| 13q21.31 | I | W | none | Chr13: 56.686-56.688 | O |
Balanced breakpoints are breakpoints of translocations that are balanced to 1Mb resolution, for the chromosome broken. (High resolution mapping of some unbalanced breaks is given in Supplementary Table 2). Each balanced breakpoint is present on (at least) two chromosomes; in most cases the breakpoint was mapped precisely on only one of them. Centromeric breaks have not been included.
Peak letters assigned to chromosome fractions in the flow karyotypes in Figure 1 and Supplementary Figures 1 and 2.
High resolution mapping was not performed on both products of all the balanced breaks. In general, the break was not precisely balanced below 1Mb, therefore the same gene was not always broken. Where it was established that the same gene was broken on both partners the peak letter is marked with an asterix*.
Gene disrupted by the translocation. Where the breakpoint interval included several genes they are listed.
Positions in Mb on NCBI Build 36 (UCSC Genome Browser hg18). Where a single position is given the precise breakpoint has been defined by sequencing. Breakpoint positions determined by BAC arrays are given as the midpoints of the BACs.
O, High density oligonucleotide array; T6, T7, etc, tiling-path BAC array for chromosomes 6, 7, etc respectively; C, genomic breakpoint cloned and sequenced; P, breakpoint fine mapped by PCR.
The exact break remains undetermined due to segmental duplications in this region preventing further analysis.
Two genes span this breakpoint, transcribed in opposite orientations.
Complex translocation with multiple breakpoints. The balanced breakpoint appears to be at 22.83 Mb, with PIP5K2A affected by these additional rearrangements.
In addition, some breakpoints were confirmed by FISH with fosmids or BACs, or by PCR to detect presence or absence of sequences on sorted chromosomes (McMullan et al, 2002), as listed in Supplementary Table 1. For example, the breakpoint at CTCF on chromosome 16 in HCC1187 was mapped by FISH using BACs followed by fosmids, and PCR (Supplementary figure 4). Notably, fosmids W12-1762O11 and WI2-1996C17 were both positive on both products of the translocation, as were PCRs on the sorted chromosomes using primer pairs between 66.152 and 66.162 Mb (primers CTCF1 to CTCF4, Supplementary Table 3); while fosmid WI2-2802K11 (5’ of CTCF) was only positive on the der(16) and two primer pairs between 66.202 and 66.203 Mb (primers CTCF5 and CTCF6) gave products only with the der(11). This agrees with the oligonucleotide array mapping, which places the breakpoints at about 66.197 and 66.142 Mb, with around 50 kb of sequence present on both products, i.e. duplicated between the two products of translocation. Similarly, the balanced breakpoint on 8q22 in HCC1187, and its translocation partner breakpoint on 1p, were mapped first by FISH with BAC or fosmid probes, then by PCR on sorted chromosomes (result not shown). Finally, PCR with pairs of primers, one on each chromosome, gave a product showing fusion of the genome between 101059276 bp on 8q in the RGS22 gene and 84234152 bp on 1p in the TTL7 gene (Supplementary Table 4). This would fuse RGS22 and TTL7 in opposite directions of transcription so no fusion transcript is predicted.
Overall about one-third of all breaks were confirmed using at least two techniques.
An apparently novel feature of the balanced breakpoints was that some overlapped, i.e. a small amount of sequence was present on both products of the translocation, resulting in net duplication. In three cases from two cell lines, where oligonucleotide hybridisation was performed on both products of the translocation, there was a clear overlap: the complex t(11;16) that breaks at CTCF in HCC1187 has net duplication of about 50 kb of CTCF and its upstream region, supported by FISH and PCR data; and in HCC1806 the balanced 7p15.2 breaks at TAX1BP1 overlap by about 80 kb, and the balanced 6p21.1 breaks at FOXP4 overlap by about 50 kb. Low-resolution results suggested that there would be other examples, notably in the case of the reciprocal translocation t(3;16) in HCC1806, where around 1Mb of chromosome 16 appears duplicated by 1Mb array analysis.
Genes at balanced breakpoints
Many of the breakpoints of the balanced translocations proved to be within or adjacent to genes, several of which are plausible candidates for translocation targets (see Discussion) (Table 2). For example, in HCC1806, the breakpoint at 40 Mb on 22q of the reciprocal translocation t(12;22)(q13.2;q13.2) proved to be within the EP300/p300 gene (Figure 3f). The location of the break was confirmed by FISH with BACS: on the der(12), RP11-12M9 was negative but RP11-422A16 was positive, while on the der(22), both were positive.
To show that at least some of the translocations resulted in fusion transcripts, two examples were investigated further.
RIF1-PKD1L1: a classic gene fusion
Mapping of the reciprocal translocation between chromosomes 2 and 7 in HCC1806 suggested that they would result in fusion between the RIF1 and PKD1L1 genes (Supplementary Table 2), so we investigated expression of the two genes. The two products of the translocation are the der(2)t(13;11;2;7), sorted in chromosome fraction ‘peak P’, and the der(7)t(2;7), sorted in ‘peak G’ (Figure 4a and Table 1).
Figure 4.
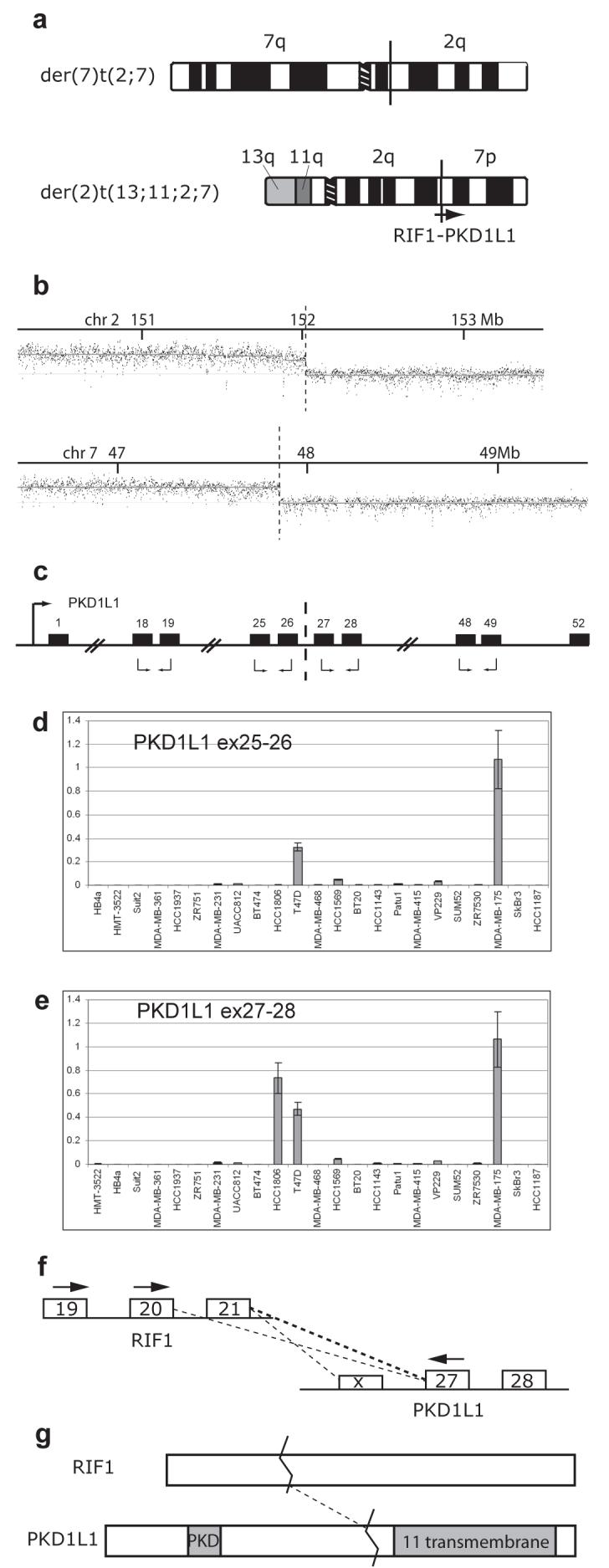
Translocation of PKD1L1 results in fusion with RIF1 and upregulation of expression of the PKD1L1 3′ end. (a) Schematic representation of the products of the reciprocal translocation between chromosomes 2 and 7 in HCC1806. The approximate location of the RIF1 and PKD1L1 genes is shown on the der(2) where fusion occurs. The junction on the der(7) has not been mapped to the gene level on chromosome 2. (b) Hybridisation of der(2)t(13;11;2;7) to a custom Nimblegen oligonucleotide array covering specified regions on chromosomes 2 and 7. Breakpoints are indicated with a broken line. (c) Schematic representation of PKD1L1 (not to scale). Relevant exons are shown as numbered black boxes, the promoter is indicated with a black arrow and the breakpoint in HCC1806 is indicated with a broken line. Primer pairs used in quantitative RT-PCR are shown with black arrows below the relevant exons. (d and e) mRNA expression levels for PKD1L1 exons 25-26 (d) and exons 27-28 (e) in a panel of 21 breast cancer cell lines, and two ‘normal’ breast lines, HB4a (immortalised normal lumenal cells), and HMT3522 (breast epithelium from fibrocystic disease). Expression levels (y axis, mean of three replicates with standard error) are shown relative to the highest expressing cell line, MDA-MB-175, using GAPDH expression as a reference. Results for exons 18-19 were similar to (d) and for exons 48-49 similar to (e) (result not shown). (f) exons of RIF1 and PKD1L1 that flank the translocation junction. PCR primers used to amplify the cDNA junction are indicated with black arrows. The dotted lines show the junctions detected by these PCRs. PKD1L1 exon X is an undocumented alternative exon present in one of the junction PCR products. (g) RIF1 and PKD1L1 proteins. Both are large, of the order of 2,500 amino acids. The fusion consists of the first third of RIF1 joined to the transmembrane domains of PKD1L1.
Hybridisation of the der(2)t(13;11;2;7) to an oligonucleotide array showed that chromosome breakpoints were at 152.017 Mb on chromosome 2, in between exons 21 and 22 of RIF1 (RefSeq NM_018151.3), and at 47.869 Mb on chromosome 7 between exons 26 and 27 of PKD1L1 (Refseq NM_138295.2) (Figure 4b). The breakpoints on the der(7) product were reciprocal at the resolution of the BAC arrays available, i.e. 1Mb for chromosome 2 and tiling-path resolution for chromosome 7(47.80-47.91 Mb).
RIF1 is reported to be expressed widely (Silverman et al, 2004). Quantitative RT-PCR, using primer pairs in adjacent exons (Figure 4c), showed that PKD1L1 mRNA is expressed at low or negligible levels in most breast cancer cell lines, two ‘normal’ breast cell lines (Figure 4d and e), and commercial normal breast mRNA samples. Similar very low expression was seen for exons proximal to the breakpoint in HCC1806, but exons distal to the breakpoint were dramatically upregulated to 200 times the level of expression of the 5′ end. This is as expected if the translocation creates a fusion transcript that is expressed from the RIF1 promoter. Incidentally, expression was also high in cell lines T-47D and MDA-MB-175 for both proximal and distal exon pairs tested (Figure 4d and 4e).
The predicted RIF1-PKD1L1 fusion transcript was detected by RT-PCR using primers in exon 20 of RIF1 (exon 20 was used to permit concurrent detection of RIF1 expression with an exon 21 primer) and exon 27 of PKD1L1 (Figure 4f). A major band of about 350 bp was visible on agarose gel electrophoresis, but cloning of the product gave two clone insert sizes, 342 and 242 bp. Sequencing showed they were derived from two alternatively spliced fusion transcripts: the expected fusion of RIF1 exon 21 to PKD1L1 exon 27, and direct fusion of exon 20 to exon 27, skipping RIF1 exon 21. Quantitative PCR was then used to determine that the relative abundance of the two transcripts was around 6:1 respectively. The primers and junction sequences are in Supplementary Tables 3 and 4. The fusion transcripts were verified independently by PCR using a primer in exon 19 of RIF1 to prevent accidental reamplification of PCR product, and a third junction was cloned splicing from exon 19 of RIF1 to a cryptic exon upstream of PKD1L1 exon 27. To show the presence of the complete gene fusion, longer RIF1-PKD1L1 cDNA fragments were cloned, from exon 1 of RIF1 to exon 28 of PKD1L1 and from exon 19 of RIF1 to exon 57 of PKD1L1 (Supplementary Figure 6; primers are given in Supplementary Table 3).
TAX1BP1-AHCY fusion
In HCC1806, there was a balanced 7p15.2 break, one side in a der(7) (Peak L, Table 1) apparently joined to 8q23.3, the other in two der(20)s (Peaks i and M), joined to a piece of chromosome 20 extending from 20p to 20q11.22 (Supplementary Figure 5). FISH with BACs showed the 7p15.2 break to be joined to 20q (result not shown). On the der(20) peak M, the chromosome 7 break is at 27.752-27.7543 Mb, in the first intron of TAX1BP1 (Figure 3e), while the chromosome 20 break is slightly 5′ of the first exon of AHCY, S-adenosylhomocysteine hydrolase (Supplementary Figure 5; Supplementary Table 2). This would be expected to fuse the first exon of TAX1BP1 to the first splice acceptor in AHCY, presumably the second exon of AHCY. The breakpoint on the der(7) peak L is at 27.67 Mb, so that about 80 kb is common to both products of the rearrangement.
The predicted TAX1BP1-AHCY fusion transcript was detected by RT-PCR, fusing exon 1 of TAX1BP1 and exon 2 of AHCY (Supplementary Figure 5c and d). The primers and sequence across the junction are given in Supplementary Tables 3 and 4.
Discussion
Array Painting
Our results illustrate the power of array painting to map the complex chromosome translocations in cancer cell lines. In principle, array CGH should be able to identify many of the breakpoints as step changes in copy number, but array painting proved superior in three respects: many breakpoints were balanced and therefore invisible to array CGH; the much higher signal-to-noise ratio gave much clearer breakpoint identification than corresponding array CGH; and where a cell line had several breakpoints on the same chromosome, array painting assigned them to particular chromosomes. We were able to resolve most chromosome species in these cell lines, and where two or more chromosomes sorted together, the SKY karyotype enabled us to identify which they were. The remaining limitations of array painting are that for complex translocations of multiple fragments, the order and orientation of fragments might not be clear; inversions are not detected; and cell lines are needed—although breakpoints found can subsequently be screened for in paraffin sections of tumours by FISH (e.g. Huang et al, 2004).
Many translocations were reciprocal or partly balanced
Overall, about one-quarter (26%) of the breakpoints were balanced, to 1 Mb resolution (our definition of balanced is discussed above), and almost half (46%) of the abnormal chromosomes contained at least one breakpoint that was balanced (Figure 2). This contrasts with the standard view that the overwhelming majority of translocations in epithelial cancers are unbalanced (see references in next paragraph). The reason for the discrepancy is mainly that such translocations are difficult to identify in these complex karyotypes, and we are not aware of any previous estimates of translocations balanced for one chromosome—likely examples can be found, for example, in our reverse chromosome painting (Morris et al, 1997).
We also found six reciprocal translocations plus two additional almost-reciprocal translocations in the three cell lines, again more than expected. There were at most five candidate examples in the SKY karyotypes: not only did all these prove to be reciprocal, but there were three additional examples that had been missed, either because they were disguised by previous or subsequent rearrangements, or because one chromosome fragment was too small to be classified by SKY analysis. However, our chosen lines are not representative—they had been selected because the SKY analysis showed one or two probable reciprocal translocations. SKY analysis had suggested that carcinoma cell lines have on average around 0.4 reciprocal translocations per line (Davidson et al, 2000; Abdel-Rahman et al, 2001; Sirivatanauksorn et al, 2001; Roschke et al, 2003; Grigorova et al, 2005). We now expect this to be a substantial underestimate.
Some of the breaks balanced to 1Mb resolution showed small overlaps between the breakpoints when mapped to high resolution, i.e. net duplication of sequence at the breakpoint. We believe this is a novel observation, in contrast to net loss at breakpoints which is already recognised (e.g. Sinclair et al, 2000; Gribble et al, 2005). Such duplication would usually be missed in conventional FISH analysis.
Many balanced breaks were in genes of interest
For each of the 19 pairs of balanced breakpoints that were not at centromeres, at least one side of the break was mapped to higher resolution. This showed that at least 13 were in genes (Table 2), and at least 8 of these genes (EP300/p300, PIP5K2A, FOXP4, CTCF, TAX1BP1, RGS22, PKD1L1, RIF1) were clearly candidate targets of oncogenic rearrangement— by fusion, promoter insertion or simply inactivation—supporting the view that some of these translocations target specific genes.
Among the broken genes, we were able to demonstrate two gene fusions: TAX1BP1 was fused to AHCY and RIF1 was fused to PKD1L1. In other cases, the regions that appeared to be joined to them were either not in annotated genes, were in genes in the opposite orientation, or have not yet been mapped to high resolution. Some of these translocations may lead to gene inactivation rather than gene fusion.
Fusion genes: RIF1-PKD1L1
The reciprocal translocation between chromosomes 2 and 7 in HCC1806 resembled many classic chromosome translocations, producing a fusion transcript expressed at a much higher level than the normal copy of the gene (PKD1L1) that forms the 3′ end of the fusion gene (Figure 4).
However, the consequences appear more complex, as two alternatively spliced fusions were detected by PCR: the expected RIF1 exon 21 to PKD1L1 exon 27 fusion and in addition a less abundant direct RIF1 exon 20 to PKD1L1 exon 27 fusion. The former is predicted to result in an out-of-frame fusion protein with two stop codons shortly after the junction; while the latter should be in frame. These predictions are unlikely to be in error as exon 20 of RIF1 has stop codons in the other reading frames. Thus the more abundant mRNA encodes a truncated RIF1, while a minor mRNA should encode a fusion protein of a large fragment of RIF1 to the multiple transmembrane segments of PKD1L1 (Figure 4g). There is evidence that both RIF1 and PKD1L1 might be relevant to breast cancer, so both these protein products may be functional.
RIF1 is a component of the ATM-mediated response to DNA damage signalling, and inhibition of RIF1 results in radiosensitivity and a defect in the intra-S-phase checkpoint (Silverman et al, 2004). (It was originally characterised in yeast as Rap1-Interacting Factor1, associated with telomeres, but in mammals its role appears different). In the mutation screen of Sjoblom et al (2006), one breast cancer cell line had two non-conservative point mutations in RIF1. Together with the present truncation this suggests that modification of RIF1 may play a role in breast cancer, perhaps in genetic instability.
In the case of PKD1L1 (polycystic kidney disease 1 like-1), a close homologue, PKDREJ, is point mutated in breast cancers (Sjoblom et al 2006). The PKD1 family (PKD1, PKD1L1 to 3, PKDREJ) are 11-transmembrane signalling molecules that heterodimerise with transient receptor potential (TRP) signalling molecules of the PKD2 family and mediate sensing of fluid flow, taste, and pH (Delmas, 2005; Huang et al, 2006). Mutations in PKD1 and PKD2 are responsible for polycystic kidney disease, which may be a non-malignant overgrowth syndrome in which PKD1 and 2 behave like tumour suppressor genes. PKD1 also interacts with the mTOR pathway (Shillingford et al, 2006). Little is known about PKD1L1, though it is reported to be expressed in whole mammary gland (Yuasa et al, 2002). The translocation preserves the transmembrane segments of PKD1L1 and so one possibility is that it creates a dominant negative, inhibiting normal activity of a heterodimer of a PKD1 and a PKD2 family member.
Fusion genes: TAX1BP1-AHCY
A fusion transcript TAX1BP1-AHCY was demonstrated in HCC1806. TAX1BP1 (Tax binding protein) is widely expressed, is a target for HIV Tax protein, interacts with the NF-kappaBeta signalling pathway, and is a coactivator of nuclear receptors (Chin et al). AHCY (S-adenosyl homocysteine hydrolase; EC3.3.1.1) is thought to control methylation by regulating the intracellular concentration of adenosylhomocysteine (UniProt database). The fusion transcript fuses the first exon of TAX1BP1 to the second exon of AHCY, but it is not clear whether this would be translated: exon 1 of TAX1BP1 has an ATG but is thought to be non-coding; translation of AHCY is thought to begin in the first exon and, while there are four in-frame methionines in Exon 2, none are in a Kozak consensus sequence. As in other cases, the translocation had a small duplication at the junction which appeared to leave an intact TAXB1P1 gene on the other chromosome translocation product. The presence of two different der(20)s incorporating this fusion (peaks M and i) suggests that this rearrangement may have been a relatively early event.
Other genes at balanced breaks
EP300/p300, on chromosome 22, is a target of occasional point mutation in breast and other epithelial cancers, which appear to inactivate the gene (Gayther et al, 2000), and is a target of gene fusion to MLL and MOZ in chromosome translocations described in acute myeloid leukaemias (Ida et al, 1997; Chaffanet et al, 2000). It is a histone acetyltransferase and transcription co-activator and a target of Adenovirus E1a. The breakpoint in EP300 in HCC1806 was close to exon 22 (of 31 exons; Refseq NM_001429), retaining part or all of the histone acetyltransferase domain (Ogryzko et al, 1996) and the two zinc finger domains at the 3′ end. The two AML breakpoints are respectively in introns 1 and 14. However, the attached breakpoint on chromosome 12 is within the human ANKRD52/FLJ34236 gene, which is transcribed in the opposite direction, so unless there is additional rearrangement the effect may simply be inactivation of p300. The other product of the translocation was not mapped to high resolution.
PIP5K2A is a phosphatidyl-inositol 5-kinase on 10p12.2, disrupted at the breakpoint of a reciprocal translocation to 13q21.31 in HCC1806. We were unable to identify any genes at the 13q21.1 breakpoint, so the gene might simply be inactivated but as the oligonucleotide array suggests duplication of sequences flanking the 10p12.2 breakpoint, there is likely to be additional rearrangement.
FOXP4 is a member of the forkhead box transcription factor family. Members of the related FOXO subfamily are fused by translocation in acute lymphoblastic leukaemias and alveolar rhabdomyosarcomas (Carlsson & Mahlapuu, 2002). A single FOXP4 somatic mutation was identified in a breast tumour in the mutation screen of Sjoblom et al (2006), in a motif that is highly conserved in vertebrates.
CTCF (CCCTC-binding factor) mediates control of gene expression by DNA methylation, by binding to unmethylated, but not methylated, CCCTC sites. Its best-known role is in mediating imprinting and chromatin conformation at the classic imprinted locus IGF2-H19 (Murrell et al, 2004), which is a target of recurrent retrovirus integration in MMTV-induced mouse mammary tumours (Theodorou et al, 2007). Again, the breaks in this gene were not exactly balanced as judged by oligonucleotide array hybridisation, implying partial duplication of about 60 kb of the gene. On the chromosome retaining the 5′ end the break was mapped to intron 2, upstream of the coding sequence. On the other, the break was 11 kb upstream of the 5′ end and the corresponding break on the partner chromosome 11 was within the first intron of AMPD3, which is in the opposite orientation, so while CTCF appears not to be fused its promoter activity might be altered.
RGS22 is regulator of G protein signalling 22. It may be significant that RGS17 is fused by translocation in MCF7 (Hahn et al, 2004).
Although, so far, we have only observed breaks in these genes in individual cell lines, we have previously shown that tumour sections can be screened for breaks by FISH, so that the prevalence of breaks in a given gene can be assessed in tumour tissue (Huang et al, 2004).
Genes at the breakpoints of unbalanced translocations
Some unbalanced breakpoints that were not joined to balanced breaks were mapped with sufficient precision to show that they were in genes of interest. For example, the 6p21.1 breakpoint in the der(1)t(1;6) of HCC1187, is within the TRERF1 gene, retaining the 3′ end of the gene. TRERF1 is a transcription co-activator that has been identified as a gene giving tamoxifen resistance in vitro (Dorssers & Veldscholte, 1997; Genbank AM404182). Similarly the 8q23.3 breakpoint in the der(7)t(8;7;17) of HCC1806 is within the TRPS1 gene, shown to be overexpressed in breast cancer (Radvanyi et al, 2005) and the 7q23.2 breakpoint, also in the der(7)t(8;7;17), is within the BCAS3 gene, which is fused in the breast cancer cell line MCF7 (Barlund et al, 2002). In addition, NRG3, a homologue of NRG1, is both translocated and amplified in the der(10)t(13;10;19) of HCC1187.
Translocations as a component of mutation in cancer
Our observations suggest that translocations are an important part of the mutation load of the common epithelial cancers, consistent with an overall picture of mutations in cancers being very numerous, diverse and variable. This is clear both from mutation screens (Sjoblom et al, 2006) and surveys of chromosome translocations (Mitelman et al, 2005). Although a few mutations are highly recurrent (APC, p53, 9;22 translocation) the vast majority occur in less than 20% of cases of a given tumour or leukaemia. The high number of genes we have found potentially affected by translocation should not be surprising: if a typical breast cancer has 20 genes point-mutated (Sjoblom et al, 2006) why should other changes not affect as many genes? We also see a suggestion that point mutation and translocation may target the same gene (EP300 and perhaps RIF1) or the same pathway (translocation of PKD1L1, and point mutation of its relative PKDREJ).
We chose to focus on balanced rearrangements because a priori they have a higher probablility of targeting genes at their breakpoints (rather than, for example, leading to loss of heterozygosity of a region). However, some unbalanced breakpoints will probably also target genes and our limited data supports this (see above). The known DOC4-NRG1 fusion gene of MDA-MB-175, for example, is at an unbalanced translocation (Wang et al, 1999; Liu et al, 1999).
The neglect of chromosome translocations in the common epithelial cancers can be ascribed to various factors (Mitelman et al, 2004). Classical cytogenetic analysis of epithelial tumours is exceptionally difficult and so there has been a lack of detailed data. There was also an expectation that important chromosome translocations would be highly recurrent, which is not generally true (Mitelman et al, 2004), and translocations in carcinomas were thought to be mostly unbalanced and thus could be ascribed to tumour suppressor loss.
Conclusion
Our data establishes that array painting is a very effective way to map substantial numbers of translocation breakpoints and supports the emerging view that chromosome rearrangements that fuse, activate or otherwise alter genes at their breakpoints may play an important role in common epithelial cancers, as they do in leukaemias.
Materials and Methods
Breast cancer cell lines of the HCC series (Gazdar et al., 1998) were obtained from ATCC, and grown in RPMI1640 medium. ZR-75-30 (Engel et al., 1978) and HMT3522 (Briand et al, 1987) were from Prof. M. J. O’Hare (LICR/UCL Breast Cancer Laboratory, University College Medical School, London, UK and grown in 50:50 DMEM: F12 medium. Other cell lines were as in Davidson et al (2000) or Alsop et al (2006).
FISH on metaphases using chromosome paints (kindly supplied by Professor M. Ferguson-Smith, Department of Veterinary Medicine, University of Cambridge), BAC probes, and fosmids was performed essentially as described (Alsop et al, 2006; Pole et al, 2006). Fosmids were from the WI2 library distributed by the Wellcome Trust Sanger Institute. Whole-genome array CGH was as described (Pole et al, 2006). BAC arrays were prepared as described by Fiegler et al (2003a [DOP and 1Mb set]). The 1Mb array used the 1Mb clone set of Fiegler et al (2003a [DOP and 1Mb set]). The 8p tiling-path clone set used the 8p clones of the Vancouver tiling path clone set (Krzywinski et al, 2004) combined with clones from our 8p12 array (Huang et al, 2004) and supplementary clones to reduce the maximum gap between clones—average inter midpoint spacing is 135kb (SLC, unpublished). The other tiling-path arrays are described elsewhere (Seng et al, 2005; Ichimura et al, 2006; and KI & VPC, manuscripts in preparation). All tiling-path arrays incorporated reference BACs spaced at either 5 or 10Mb over the autosomal genome.
Flow sorting of chromosomes was by standard methods (Ng & Carter, 2006). Between 16-24 hours after subculture, cells were treated with 0.1 μg/ml colcemid for 6-24 hours. Mitotic cells from adherent cell cultures were harvested by mitotic shake-off and treated with hypotonic solution and polyamine buffer as described previously (Ng & Carter, 2006). Chromosomes were stained overnight with Hoechst 33258 (5 μg/ml final) and chromomycin A3 (40 μg/ml final) at 4°C. The stained chromosome suspension was treated with 10 mM trisodium citrate and 25 mM sodium sulphite for 1 hour before being analysed and flow sorted on a MoFlo cell sorter (DAKO, Fort Collins, CO). For hybridisation to metaphase chromosomes, aliquots of 300 chromosomes were flow sorted, amplified by degenerate oligonucleotide primed PCR (DOP-PCR), then reamplified with labelled dUTP (Morris et al, 1997). For array hybridisation, 5000 chromosomes were flow sorted.
For array hybridisation, aliqots of 5000 chromosomes were precipitated and redissolved in 1μl TE; amplified with phi29 polymerase using the GenomiPhi kit (GE Healthcare, Buckinghamshire, UK); and amplified DNA cleaned up using MicroSpin G-50 columns (GE Healthcare, Buckinghamshire, UK). Hybridisation of this DNA to BAC arrays was as described for array-CGH (Pole et al, 2006). 300 ng amplified DNA was labelled (Cy3 for test and Cy5 for amplified pooled normal female DNA as reference) using random primers and exo- Klenow fragment in 150 μl (BioPrime Labeling Kit, Invitrogen) overnight at 37°C, and purified by spin column chromatography using MicroSpin G-50 columns. Test and reference DNA were precipitated with 90 μg human C0t-1 DNA and 400 μg yeast tRNA, dissolved in 30μl hybridisation buffer, denatured at 70°C for 10 min, preincubated at 37°C for 1 hour, then hybridised to arrays at 37°C for 24 hours in a humid hybridisation chamber (containing 2X SSC, 20% (v/v) formamide).
Arrays were washed in 1X PBS/0.05% (v/v) Tween 20 (10 min, room temperature), in 50% (v/v) formamide/0.5X SSC (30 min, 42°C), and in 1X PBS/0.05% (v/v) Tween 20 (10 min, room temperature) and dried by spinning. The arrays were scanned on an Axon 4100A scanner (Axon Instruments) and the images analysed using GenePix Pro 4.1 Software (Axon Instruments). Analysis of arrays was as in Pole et al. (2006). Briefly, using Microsoft Excel, the spot intensity after background subtraction was compared to the median intensity of the control Drosophila BAC clones on the arrays and spots were rejected if spot intensity was less than 2X the Drosophila clone median. Test/reference ratios were calculated and normalised to the median ratio for all reference BACs (for 1Mb arrays, all autosomal BACs). Spots were rejected if duplicate spot ratios differed by more than 25%. The mean of the log2 ratios was plotted against chromosome position according to the NCBI Build 36 of the Human Genome Sequence. To score breakpoints, a chromosome piece was counted as present if more than 3 adjacent BACs on the 1Mb array gave a positive hybridisation, i.e. at a level equal to the log2 ratio of other pieces of chromosome present in the same peak. This means that some very small amplifications or pieces will have been missed.
Hybridisation, of the same amplified DNA preparations, to high-resolution custom oligonucletide arrays was purchased as a service from NimbleGen Systems (Madison, WI). Each custom 385,000-oligonucleotide array was designed to cover around 10 breakpoints. For each breakpoint a region of typically 5 Mb spanning the breakpoint region (defined by the 1Mb array) was covered at an average spacing of around 120 bp. Data was averaged over 300 bp or 1 kb intervals and manually inspected.
PCR primers are given in Supplementary Table 3. To map genomic breakpoints by PCR, selected sequences were amplified from GenomiPhi-amplified chromosomes. For Quantitative RT-PCR, RNA prepared using Trizol (Invitrogen) was reverse transcribed using oligo-dT priming and Superscript III (Invitrogen) and quantitated by PCR using the SYBR green system and an ABI PRISM 7900HT Sequence Detection System (both Applied Biosystems). GAPDH was used to normalise expression levels for each cell line.
Supplementary Material
Supplementary information accompanies the paper on the Oncogene website (http://www.nature.com/onc).
Acknowledgements
We thank Mira Grigorova for unpublished SKY data; Elizabeth Batty for help with array analysis; Danita Pearson, Martin McCabe, the Wellcome Trust Sanger Institute Microarray Facility, the CRUK Microarray Facility, Institute of Cancer Research, and the University of Cambridge Department of Pathology Microarray Facility for array production. This work was supported by Cancer Research UK grant number C1023/A4392, Breast Cancer Campaign, Breast Cancer Research Trust, Samantha Dickson Brain Tumour Trust, the Wellcome Trust (NPC, BLN) and studentships from Cambridge Commonwealth Trust and the Sackler Foundation (KAB) and MRC (SLC).
References
- Abdel-Rahman WM, Katsura K, Rens W, Gorman PA, Sheer D, Bicknell D, Bodmer WF, Arends MJ, Wyllie AH, Edwards PA. Spectral karyotyping suggests additional subsets of colorectal cancers characterized by pattern of chromosome rearrangement. Proc Natl Acad Sci U S A. 2001;98:2538–43. doi: 10.1073/pnas.041603298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adeyinka A, Kytola S, Mertens F, Pandis N, Larsson C. Spectral karyotyping and chromosome banding studies of primary breast carcinomas and their lymph node metastases. Int J Mol Med. 2000;5:235–40. doi: 10.3892/ijmm.5.3.235. [DOI] [PubMed] [Google Scholar]
- Alsop AE, Teschendorff AE, Edwards PA. Distribution of breakpoints on chromosome 18 in breast, colorectal, and pancreatic carcinoma cell lines. Cancer Genet Cytogenet. 2006;164:97–109. doi: 10.1016/j.cancergencyto.2005.09.011. [DOI] [PubMed] [Google Scholar]
- Barlund M, Monni O, Weaver JD, Kauraniemi P, Sauter G, Heiskanen M, Kallioniemi OP, Kallioniemi A. Cloning of BCAS3 (17q23) and BCAS4 (20q13) genes that undergo amplification, overexpression, and fusion in breast cancer. Genes Chromosomes Cancer. 2002;35:311–7. doi: 10.1002/gcc.10121. [DOI] [PubMed] [Google Scholar]
- Briand P, Petersen OW, Van Deurs B. A new diploid nontumorigenic human breast epithelial cell line isolated and propagated in chemically defined medium. In Vitro Cell Dev Biol. 1987;23:181–8. doi: 10.1007/BF02623578. [DOI] [PubMed] [Google Scholar]
- Carlsson P, Mahlapuu M. Forkhead transcription factors: key players in development and metabolism. Dev Biol. 2002;250:1–23. doi: 10.1006/dbio.2002.0780. [DOI] [PubMed] [Google Scholar]
- Chaffanet M, Gressin L, Preudhomme C, Soenen-Cornu V, Birnbaum D, P\’ebusque MJ. MOZ is fused to p300 in an acute monocytic leukemia with t(8;22) Genes Chromosomes Cancer. 2000;28:138–44. doi: 10.1002/(sici)1098-2264(200006)28:2<138::aid-gcc2>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- Chin KT, Chun AC, Ching YP, Jeang KT, Jin DY. Human T-cell leukemia virus oncoprotein tax represses nuclear receptor-dependent transcription by targeting coactivator TAX1BP1. Cancer Res. 2007;67:1072–81. doi: 10.1158/0008-5472.CAN-06-3053. [DOI] [PubMed] [Google Scholar]
- Davidson JM, Gorringe KL, Chin SF, Orsetti B, Besret C, Courtay-Cahen C, Roberts I, Theillet C, Caldas C, Edwards PA. Molecular cytogenetic analysis of breast cancer cell lines. Br J Cancer. 2000;83:1309–17. doi: 10.1054/bjoc.2000.1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, Teague J, Woffendin H, Garnett MJ, Bottomley W, Davis N, Dicks E, Ewing R, Floyd Y, Gray K, Hall S, Hawes R, Hughes J, Kosmidou V, Menzies A, Mould C, Parker A, Stevens C, Watt S, Hooper S, Wilson R, Jayatilake H, Gusterson BA, Cooper C, Shipley J, Hargrave D, Pritchard-Jones K, Maitland N, Chenevix-Trench G, Riggins GJ, Bigner DD, Palmieri G, Cossu A, Flanagan A, Nicholson A, Ho JW, Leung SY, Yuen ST, Weber BL, Seigler HF, Darrow TL, Paterson H, Marais R, Marshall CJ, Wooster R, Stratton MR, Futreal PA. Mutations of the BRAF gene in human cancer. Nature. 2002;417:949–54. doi: 10.1038/nature00766. [DOI] [PubMed] [Google Scholar]
- Delmas P. Polycystins: polymodal receptor/ion-channel cellular sensors. Pflugers Arch. 2005;451:264–76. doi: 10.1007/s00424-005-1431-5. [DOI] [PubMed] [Google Scholar]
- Dorssers LC, Veldscholte J. Identification of a novel breast-cancer-anti-estrogen-resistance (BCAR2) locus by cell-fusion-mediated gene transfer in human breast-cancer cells. Int J Cancer. 1997;72:700–5. doi: 10.1002/(sici)1097-0215(19970807)72:4<700::aid-ijc24>3.0.co;2-d. [DOI] [PubMed] [Google Scholar]
- Dutrillaux B. Pathways of chromosome alteration in human epithelial cancers. Adv Cancer Res. 1995;67:59–82. doi: 10.1016/s0065-230x(08)60710-1. [DOI] [PubMed] [Google Scholar]
- Engel LW, Young NA, Tralka TS, Lippman ME, O’Brien SJ, Joyce MJ. Establishment and characterization of three new continuous cell lines derived from human breast carcinomas. Cancer Res. 1978;38:3352–64. [PubMed] [Google Scholar]
- Fiegler H, Carr P, Douglas EJ, Burford DC, Hunt S, Scott CE, Smith J, Vetrie D, Gorman P, Tomlinson IP, Carter NP. DNA microarrays for comparative genomic hybridization based on DOP-PCR amplification of BAC and PAC clones. Genes Chromosomes Cancer. 2003a;36:361–74. doi: 10.1002/gcc.10155. [DOI] [PubMed] [Google Scholar]
- Fiegler H, Gribble SM, Burford DC, Carr P, Prigmore E, Porter KM, Clegg S, Crolla JA, Dennis NR, Jacobs P, Carter NP. Array painting: a method for the rapid analysis of aberrant chromosomes using DNA microarrays. J Med Genet. 2003b;40:664–70. doi: 10.1136/jmg.40.9.664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gayther SA, Batley SJ, Linger L, Bannister A, Thorpe K, Chin SF, Daigo Y, Russell P, Wilson A, Sowter HM, Delhanty JD, Ponder BA, Kouzarides T, Caldas C. Mutations truncating the EP300 acetylase in human cancers. Nat Genet. 2000;24:300–3. doi: 10.1038/73536. [DOI] [PubMed] [Google Scholar]
- Gazdar AF, Kurvari V, Virmani A, Gollahon L, Sakaguchi M, Westerfield M, Kodagoda D, Stasny V, Cunningham HT, Wistuba II, Tomlinson G, Tonk V, Ashfaq R, Leitch AM, Minna JD, Shay JW. Characterization of paired tumor and non-tumor cell lines established from patients with breast cancer. Int J Cancer. 1998;78:766–74. doi: 10.1002/(sici)1097-0215(19981209)78:6<766::aid-ijc15>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- Gribble SM, Kalaitzopoulos D, Burford DC, Prigmore E, Selzer RR, Ng BL, Matthews NS, Porter KM, Curley R, Lindsay SJ, Baptista J, Richmond TA, Carter NP. Ultra-high resolution array painting facilitates breakpoint sequencing. J Med Genet. 2007;44:51–8. doi: 10.1136/jmg.2006.044909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gribble SM, Prigmore E, Burford DC, Porter KM, Ng BL, Douglas EJ, Fiegler H, Carr P, Kalaitzopoulos D, Clegg S, Sandstrom R, Temple IK, Youings SA, Thomas NS, Dennis NR, Jacobs PA, Crolla JA, Carter NP. The complex nature of constitutional de novo apparently balanced translocations in patients presenting with abnormal phenotypes. J Med Genet. 2005;42:8–16. doi: 10.1136/jmg.2004.024141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grigorova M, Lyman RC, Caldas C, Edwards PA. Chromosome abnormalities in 10 lung cancer cell lines of the NCI-H series analyzed with spectral karyotyping. Cancer Genet Cytogenet. 2005;162:1–9. doi: 10.1016/j.cancergencyto.2005.03.007. [DOI] [PubMed] [Google Scholar]
- Hahn Y, Bera TK, Gehlhaus K, Kirsch IR, Pastan IH, Lee B. Finding fusion genes resulting from chromosome rearrangement by analyzing the expressed sequence databases. Proc Natl Acad Sci U S A. 2004;101:13257–61. doi: 10.1073/pnas.0405490101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang AL, Chen X, Hoon MA, Chandrashekar J, Guo W, Tr\“ankner D, Ryba NJ, Zuker CS. The cells and logic for mammalian sour taste detection. Nature. 2006;442:934–8. doi: 10.1038/nature05084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang HE, Chin SF, Ginestier C, Bardou VJ, Ad\’ela\“ide J, Iyer NG, Garcia MJ, Pole JC, Callagy GM, Hewitt SM, Gullick WJ, Jacquemier J, Caldas C, Chaffanet M, Birnbaum D, Edwards PA. A recurrent chromosome breakpoint in breast cancer at the NRG1/neuregulin 1/heregulin gene. Cancer Res. 2004;64:6840–4. doi: 10.1158/0008-5472.CAN-04-1762. [DOI] [PubMed] [Google Scholar]
- Ichimura K, Mungall AJ, Fiegler H, Pearson DM, Dunham I, Carter NP, Collins VP. Small regions of overlapping deletions on 6q26 in human astrocytic tumours identified using chromosome 6 tile path array-CGH. Oncogene. 2006;25:1261–71. doi: 10.1038/sj.onc.1209156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ida K, Kitabayashi I, Taki T, Taniwaki M, Noro K, Yamamoto M, Ohki M, Hayashi Y. Adenoviral E1A-associated protein p300 is involved in acute myeloid leukemia with t(11;22)(q23;q13) Blood. 1997;90:4699–704. [PubMed] [Google Scholar]
- Krzywinski M, Bosdet I, Smailus D, Chiu R, Mathewson C, Wye N, Barber S, Brown-John M, Chan S, Chand S, Cloutier A, Girn N, Lee D, Masson A, Mayo M, Olson T, Pandoh P, Prabhu AL, Schoenmakers E, Tsai M, Albertson D, Lam W, Choy CO, Osoegawa K, Zhao S, de Jong PJ, Schein J, Jones S, Marra MA. A set of BAC clones spanning the human genome. Nucleic Acids Res. 2004;32:3651–60. doi: 10.1093/nar/gkh700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Baker E, Eyre HJ, Sutherland GR, Zhou M. Gamma-heregulin: a fusion gene of DOC-4 and neuregulin-1 derived from a chromosome translocation. Oncogene. 1999;18:7110–4. doi: 10.1038/sj.onc.1203136. [DOI] [PubMed] [Google Scholar]
- McMullan TW, Crolla JA, Gregory SG, Carter NP, Cooper RA, Howell GR, Robinson DO. A candidate gene for congenital bilateral isolated ptosis identified by molecular analysis of a de novo balanced translocation. Hum Genet. 2002;110:244–50. doi: 10.1007/s00439-002-0679-5. [DOI] [PubMed] [Google Scholar]
- Mehra R, Tomlins SA, Shen R, Nadeem O, Wang L, Wei JT, Pienta KJ, Ghosh D, Rubin MA, Chinnaiyan AM, Shah RB. Comprehensive assessment of TMPRSS2 and ETS family gene aberrations in clinically localized prostate cancer. Mod Pathol. 2007;20:538–44. doi: 10.1038/modpathol.3800769. [DOI] [PubMed] [Google Scholar]
- Mitelman F. Recurrent chromosome aberrations in cancer. Mutat Res. 2000;462:247–53. doi: 10.1016/s1383-5742(00)00006-5. [DOI] [PubMed] [Google Scholar]
- Mitelman F, Johansson B, Mertens F. Fusion genes and rearranged genes as a linear function of chromosome aberrations in cancer. Nat Genet. 2004;36:331–4. doi: 10.1038/ng1335. [DOI] [PubMed] [Google Scholar]
- Mitelman F, Mertens F, Johansson B. Prevalence estimates of recurrent balanced cytogenetic aberrations and gene fusions in unselected patients with neoplastic disorders. Genes Chromosomes Cancer. 2005;43:350–66. doi: 10.1002/gcc.20212. [DOI] [PubMed] [Google Scholar]
- Morris JS, Carter NP, Ferguson-Smith MA, Edwards PA. Cytogenetic analysis of three breast carcinoma cell lines using reverse chromosome painting. Genes Chromosomes Cancer. 1997;20:120–39. doi: 10.1002/(sici)1098-2264(199710)20:2<120::aid-gcc3>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- Murrell A, Heeson S, Reik W. Interaction between differentially methylated regions partitions the imprinted genes Igf2 and H19 into parent-specific chromatin loops. Nat Genet. 2004;36:889–93. doi: 10.1038/ng1402. [DOI] [PubMed] [Google Scholar]
- Ng BL, Carter NP. Factors affecting flow karyotype resolution. Cytometry A. 2006;69:1028–36. doi: 10.1002/cyto.a.20330. [DOI] [PubMed] [Google Scholar]
- Ogryzko VV, Schiltz RL, Russanova V, Howard BH, Nakatani Y. The transcriptional coactivators p300 and CBP are histone acetyltransferases. Cell. 1996;87:953–9. doi: 10.1016/s0092-8674(00)82001-2. [DOI] [PubMed] [Google Scholar]
- Persson K, Pandis N, Mertens F, Borg A, Baldetorp B, Killander D, Isola J. Chromosomal aberrations in breast cancer: a comparison between cytogenetics and comparative genomic hybridization. Genes Chromosomes Cancer. 1999;25:115–22. [PubMed] [Google Scholar]
- Pole JC, Courtay-Cahen C, Garcia MJ, Blood KA, Cooke SL, Alsop AE, Tse DM, Caldas C, Edwards PA. High-resolution analysis of chromosome rearrangements on 8p in breast, colon and pancreatic cancer reveals a complex pattern of loss, gain and translocation. Oncogene. 2006;25:5693–706. doi: 10.1038/sj.onc.1209570. [DOI] [PubMed] [Google Scholar]
- Radvanyi L, Singh-Sandhu D, Gallichan S, Lovitt C, Pedyczak A, Mallo G, et al. The gene associated with trichorhinophalangeal syndrome in humans is overexpressed in breast cancer. Proc Natl Acad Sci U S A. 2005;102:11005–10. doi: 10.1073/pnas.0500904102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roschke AV, Tonon G, Gehlhaus KS, McTyre N, Bussey KJ, Lababidi S, Scudiero DA, Weinstein JN, Kirsch IR. Karyotypic complexity of the NCI-60 drug-screening panel. Cancer Res. 2003;63:8634–47. [PubMed] [Google Scholar]
- Rowley JD. The critical role of chromosome translocations in human leukemias. Annu Rev Genet. 1998;32:495–519. doi: 10.1146/annurev.genet.32.1.495. [DOI] [PubMed] [Google Scholar]
- Seng TJ, Ichimura K, Liu L, Tingby O, Pearson DM, Collins VP. Complex chromosome 22 rearrangements in astrocytic tumors identified using microsatellite and chromosome 22 tile path array analysis. Genes Chromosomes Cancer. 2005;43:181–93. doi: 10.1002/gcc.20181. [DOI] [PubMed] [Google Scholar]
- Shillingford JM, Murcia NS, Larson CH, Low SH, Hedgepeth R, Brown N, Flask CA, Novick AC, Goldfarb DA, Kramer-Zucker A, Walz G, Piontek KB, Germino GG, Weimbs T. The mTOR pathway is regulated by polycystin-1, and its inhibition reverses renal cystogenesis in polycystic kidney disease. Proc Natl Acad Sci U S A. 2006;103:5466–71. doi: 10.1073/pnas.0509694103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silverman J, Takai H, Buonomo SB, Eisenhaber F, de Lange T. Human Rif1, ortholog of a yeast telomeric protein, is regulated by ATM and 53BP1 and functions in the S-phase checkpoint. Genes Dev. 2004;18:2108–19. doi: 10.1101/gad.1216004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinclair PB, Nacheva EP, Leversha M, Telford N, Chang J, Reid A, Bench A, Champion K, Huntly B, Green AR. Large deletions at the t(9;22) breakpoint are common and may identify a poor-prognosis subgroup of patients with chronic myeloid leukemia. Blood. 2000;95:738–43. [PubMed] [Google Scholar]
- Sirivatanauksorn V, Sirivatanauksorn Y, Gorman PA, Davidson JM, Sheer D, Moore PS, Scarpa A, Edwards PA, Lemoine NR. Non-random chromosomal rearrangements in pancreatic cancer cell lines identified by spectral karyotyping. Int J Cancer. 2001;91:350–8. doi: 10.1002/1097-0215(200002)9999:9999<::aid-ijc1049>3.3.co;2-3. [DOI] [PubMed] [Google Scholar]
- Sjoblom T, Jones S, Wood LD, Parsons DW, Lin J, Barber T, Mandelker D, Leary RJ, Ptak J, Silliman N, Szabo S, Buckhaults P, Farrell C, Meeh P, Markowitz SD, Willis J, Dawson D, Willson JK, Gazdar AF, Hartigan J, Wu L, Liu C, Parmigiani G, Park BH, Bachman KE, Papadopoulos N, Vogelstein B, Kinzler KW, Velculescu VE. The Consensus Coding Sequences of Human Breast and Colorectal Cancers. Science. 2006;314:268–274. doi: 10.1126/science.1133427. [DOI] [PubMed] [Google Scholar]
- Soda M, Choi YL, Enomoto M, Takada S, Yamashita Y, Ishikawa S, Fujiwara SI, Watanabe H, Kurashina K, Hatanaka H, Bando M, Ohno S, Ishikawa Y, Aburatani H, Niki T, Sohara Y, Sugiyama Y, Mano H. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature. 2007;448 doi: 10.1038/nature05945. epub11July. [DOI] [PubMed] [Google Scholar]
- Theodorou V, Kimm MA, Boer M, Wessels L, Theelen W, Jonkers J, Hilkens J. MMTV insertional mutagenesis identifies genes, gene families and pathways involved in mammary cancer. Nat Genet. 2007;39:759–769. doi: 10.1038/ng2034. [DOI] [PubMed] [Google Scholar]
- Tomlins SA, Rhodes DR, Perner S, Dhanasekaran SM, Mehra R, Sun XW, Varambally S, Cao X, Tchinda J, Kuefer R, Lee C, Montie JE, Shah RB, Pienta KJ, Rubin MA, Chinnaiyan AM. Recurrent fusion of TMPRSS2 and ETS transcription factor genes in prostate cancer. Science. 2005;310:644–8. doi: 10.1126/science.1117679. [DOI] [PubMed] [Google Scholar]
- Vogelstein B, Kinzler KW. Cancer genes and the pathways they control. Nat Med. 2004;10:789–99. doi: 10.1038/nm1087. [DOI] [PubMed] [Google Scholar]
- Volik S, Zhao S, Chin K, Brebner JH, Herndon DR, Tao Q, Kowbel D, Huang G, Lapuk A, Kuo WL, Magrane G, De Jong P, Gray JW, Collins C. End-sequence profiling: sequence-based analysis of aberrant genomes. Proc Natl Acad Sci U S A. 2003;100:7696–701. doi: 10.1073/pnas.1232418100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang XZ, Jolicoeur EM, Conte N, Chaffanet M, Zhang Y, Mozziconacci MJ, Feiner H, Birnbaum D, P\’ebusque MJ, Ron D. gamma-heregulin is the product of a chromosomal translocation fusing the DOC4 and HGL/NRG1 genes in the MDA-MB-175 breast cancer cell line. Oncogene. 1999;18:5718–21. doi: 10.1038/sj.onc.1202950. [DOI] [PubMed] [Google Scholar]
- Yuasa T, Venugopal B, Weremowicz S, Morton CC, Guo L, Zhou J. The sequence, expression, and chromosomal localization of a novel polycystic kidney disease 1-like gene, PKD1L1, in human. Genomics. 2002;79:376–86. doi: 10.1006/geno.2002.6719. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.