

Summary
Epigenetic alterations of DNA play key roles in determining gene structure and expression. Methylation of the 5-position of cytosine is thought to be the most common modification of the genome in mammals. Studies have generally shown that hypermethylation in gene regulatory regions is associated with inactivation and reduced transcription and that alteration in established methylation patterns during development can affect embryonic viability. Changes in methylation have also been associated with aging and cellular senescence as well as tumorogenesis. DNA methyltransferase 1 (DNMT1) is thought to play an important role in maintaining already established methylation patterns during DNA replication and catalyzes the transfer of a methyl moiety from S-adenosyl-l-methionine (SAM) to the 5-position of cytosines in the CpG dinucleotide. Several studies illustrate changes in activity and transcription of DNMT1 during aging and here we show a comprehensive method of detection of DNMT1 mRNA transcription from senescing cells in culture.
Keywords: DNA methylation, senescence, CpG dinucleotides, tumorigensis, SAM
1. Introduction
DNA methylation of the 5-position of cytosine is considered to be the most common modification of the genome in mammals, and methylation in regulatory regions of genes has been shown to be inversely related to gene expression (1). Methylation patterns of cytosines are associated with many cellular processes, including chromatin structuring (2), development (3), carcinogenesis (4), and aging (5,6). DNA methyltransferase (Dnmt) catalyzes the transfer of a methyl moiety from S-adenoslyl-l-methionine (SAM) to the 5-postion of cytosines in the CpG dinucleotide (7,8). Several Dnmts have been identified in somatic tissues of vertebrates. Dnmt1 is the most abundant methyltransferase in mammalian cells and is primarily associated with maintaining established methylation patterns in newly synthesized DNA (3).
Methylation patterns have been shown to be important in normal embryonic development. For example, monoclonal antibodies against Dnmt1 halt cell division in early-stage Xenopus embryos. Morphological data suggested that cells were arrested in interphase, and this was supported by biochemical analysis that indicated cells had not entered M-phase (9). Dnmt1−/− and Dnmt3b−/− mutations in mice result in embryonic death while mice with mutations Dnmt1,3a−/− die at approx 4 wk of age (10,11). A study investigating DNMT activity in human systems showed that overall maintenance methylation decreased during cellular senescence of WI-38 fibroblasts and that combined de novo methylation initially decreased but later increased as these cells aged (12). Thus many recent studies have illustrated the important role of Dnmts in embryonic development and aging. However, much remains to be elucidated and research aimed at Dnmt activities both in vivo and in vitro is essential to understanding the mechanisms of important biological processes such as aging.
2. Materials
2.1. RNA Extraction and Quantification
Items 1–4 are included in the Qiagen RNeasy Mini Kit (cat. no. 74104).
RNeasy mini spin column.
Buffer RLT.
Buffer RPE.
Buffer RW1.
QIAshredder Qiagen (cat. no. 79656).
β-mercaptoethanol.
Phosphate-buffered saline (PBS).
Ethanol (both 70% and 100%).
Spectrophotometer Cuvet Silica (quartz) (Sigma, St. Louis, MO; cat. no. C-5178).
SmartSpec™ Plus spectrophotometer (Bio-Rad, Hercules, CA).
2.2. Synthesis of cDNA and Sequence-Specific PCR
Items 1–4 are included in the SuperScript™ First-Strand Synthesis System for reverse-transcription (RT)-PCR kit (Invitrogen, Carlsbad, CA; cat. no. 11904-018).
Control RNA.
50 ng/μL random Hexamer mix.
10 mM dNTP mix.
Diethyl pyrocarbonate (DEPC)-treated water.
Thin-walled 0.2 mL PCR tubes.
Gene-specific primers.
PCR Master Mix (Promega, Madison, WI; cat. no. M750B).
Nuclease-free water.
2.3. Gel Electrophoresis
Gel electrophoresis apparatus (Bio-Rad, Hercules, CA).
Gel electrophoresis-grade agarose.
1X TAE buffer.
100 bp DNA molecular marker (Promega, Madison, WI; cat. no. G210A).
6X Loading Dye (Promega, Madison, WI; cat. no. G190A).
10 mg/mL ethidium bromide solution.
Ultraviolet (UV) transilluminator.
3. Methods
3.1. Cell Extract Preparation
The following protocol pertains to adherent aging cell cultures, for cells grown in suspension (see Note 1).
Aspirate media from experimental and control cells.
Wash bottom of flasks with a predetermined amount of 1X PBS warmed to 37°C (see Note 2).
Add a predetermined amount of trypsin-EDTA directly to the bottom of the flasks and incubate at 37°C for 5–6 min (see Note 2).
Collect detached cells by washing the bottom of the flasks with media warmed to 37°C and add the media/trypsin/cell mix to a 15-mL conical tube for centrifugation (see Note 2).
Pellet cells by centrifugation and resuspend pellets in PBS. Count cells using a method determined by the experimenter (see Note 3).
Place 1–3 × 106 cells in a 1.5-mL tube and pellet by centrifugation. Remove all PBS from tubes. Pellets may be frozen at −80°C for later use.
3.2. Cell Lysis and Homogenization
Prepare cell lysis buffer solution by adding 10 μL of β-mercaptoethanol to 1 mL of buffer RLT.
Disrupt pellet obtained as described under Subheading 3.1. by flicking the tube and pipet 350 μL of freshly prepared cell lysis buffer solution to cell pellets obtained as described under Subheading 3.1. It is important to achieve a substantial amount of disruption to the pellet in order to accomplish complete lysis.
To homogenize, pipet lysate into a QIAshredder placed in a 2-mL collection tube and spin for 2 min at maximum speed in a microcentrifuge.
3.3. RNA Extraction
Because of the instability of RNA, it is necessary to perform the following protocol quickly and without too much pause between steps. It is also not recommended to extract RNA from more than six to seven samples at one time.
Add 350 μL of 70% ethanol to homogenized lysate and mix thoroughly by pipetting. Pipet the mixture up and down at least 10 times to mix. Apply the entire sample (700 μL) to an RNeasy mini spin column and centrifuge at 8000g for 30 s.
Discard the liquid in the collection tube only and add 700 μL of buffer RW1 to the column placed in the same collection tube and centrifuge at 8000g for 30 s.
Transfer the RNeasy column to a new collection tube and discard the previously used tube containing flow through from step 3.
Prepare buffer RPE by adding 4 volumes of 100% ethanol to 1 volume of RPE concentrate. Add 500 μL of the prepared RPE buffer to the RNeasy column and centrifuge at 8000g for 30 s.
Discard the liquid in the collection tube and pipet another 500 μL of the prepared RPE into the RNeasy column. Centrifuge at 8000g for 2 min.
Place RNeasy column in a new collection tube and centrifuge for 1 min at maximum speed (see Note 4).
Place RNeasy column in a 1.5-mL collection tube and elute RNA from column by adding 30 μL of RNase-free water. Let column stand for 1 min and centrifuge at 8000g for 1 min.
3.4. RNA Quantification
Be sure to clean the silica cuvet with 1 wash of 70% ethanol followed by two to three washes of sterile water.
Add 495 μL of sterile water to the cuvet. Blank the spectrophotometer. Pipet 5 μl of extracted RNA obtained under Subheading 3.3. Mix by gentle inversion.
Repeat step 2 for each sample to be quantified.
Spectrophotometer concentration reading is micrograms per milliliter. Convert reading to micrograms per microliter.
3.5. First-Strand cDNA Synthesis
For each RNA sample obtained, prepare a mixture containing reagents in Table 1 (n is the amount of sample required to achieve desired concentration of RNA needed for conversion to cDNA).
Incubate each sample at 65°C for 5 min followed by short incubation on ice.
During the incubation in step 2, prepare enough reaction master mix for all samples, including control, as described in Table 2.
Pipet 9 μL of the reaction master mix into each tube containing mixture from step 1, mix briefly by vortexing, and collect samples by brief centrifugation.
Incubate at 25°C for 2 min.
Add 1 μL of SuperScript II reverse transcriptase to each tube and mix gently and collect by brief centrifugation.
Incubate at 25°C for 10 min.
Transfer tubes to 42°C for 50 min.
To terminate the reactions, incubate at 70°C for 15 min and chill on ice.
Collect the samples by brief centrifugation and add 1 μL of RNase H to each tube. Incubate at 37°C for 20 min. Samples are now converted to cDNA and are ready for sequence specific PCR.
Table 1.
RNA and Primer Mix
| Reagent | Sample reaction | Positive RT control |
|---|---|---|
| 1–3 μg Sample RNA | n μL | – |
| Control RNA | – | 1 μL |
| Random hexamers | 1.5 μL | 1.5 μL |
| dNTP mix | 1.5 μL | 1.5 μL |
| Water | to 10 μL | to 10 μL |
Table 2.
cDNA Reaction Master Mix
| Reagent | For each reaction |
|---|---|
| 10X reverse-transcription buffer | 2 μL |
| 25 mM MgCL2 | 4 μL |
| 0.1 M dithiothreitol | 2 μL |
| RNaseOUT Ribonuclease Inhibitor | 1 μL |
3.6. DNMT 1 Gene Amplification by PCR
Prepare a master mix containing the reagents listed in Table 3.
For each reaction, add 3 μL of cDNA sample obtained as described under Subheading 3.5. to a 0.2-mL PCR tube containing 27 μL of prepared master mix.
Mix gently and collect by brief centrifugation.
Perform PCR as follows: 94°C for 5 min; 33 cycles of 94°C for 30 s, 56°C for 40 s, 72°C for 40 s followed by 72°C hold for 7 min.
Table 3.
Sequence-Specific PCR
| Reagent | For each reaction |
|---|---|
| 2X PCR master mix (Promega, Madison, WI; cat. no. M750B) | 15 μL |
| Nuclease-free water | 9 μL |
| Forward: 5′-ACCGCTTCTACTTCCTCGAGGCCTA-3′ | 3 μL of forward and reverse primer mix |
| Reverse: 5′-GTTGCAGTCCTCTGTGAACACTGTGG-3′ |
3.7. Gel Electrophoresis
Prepare a 2% agarose gel by mixing 2 g of agarose with 100 mL of TAE buffer.
Load 2 μL of 6X loading dye and 10 μL of sample obtained as described under Subheading 3.6. into each well.
Run gel for approx 30 min at 100 V. Alternatively, gels can be run at 50 V for approx 1 h.
Mix 20 μL of ethidium bromide with approx 100 mL of TAE buffer and incubate gel for 15 min. Make sure to mix the ethidium bromide/TAE solution thoroughly to ensure even distribution before incubating gel.
View results with UV Transilluminator as shown in Fig. 1.
Fig. 1.
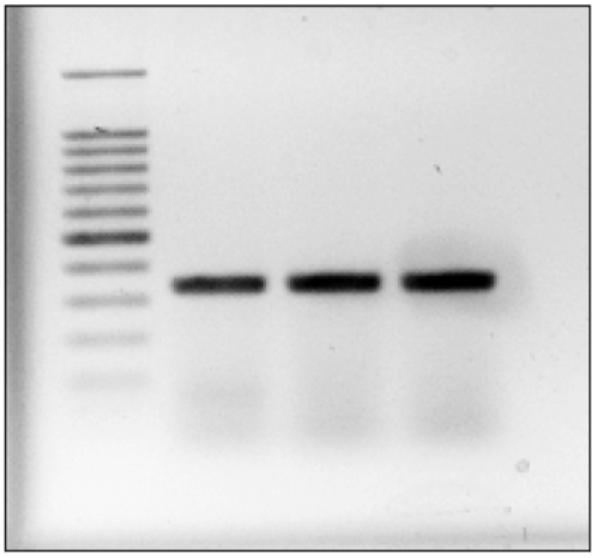
Amplification of the DNMT1 cDNA. Cells were lysed and total mRNA was extracted followed by conversion to cDNA as detailed under Subheadings 3.2., 3.3., and 3.5., respectively. Amplification of cDNA was carried out by PCR and visualized by ethidium bromide staining on a 2% agarose gel.
Footnotes
Free-floating or suspension cells require a slightly different technique for extraction. It is not necessary to detach the cells using trypsin. Cells must be collected and placed in a 15-mL conical tube and then pelleted by centrifugation. Proceed to Subheading 3.1., step 5.
The amount of trypsin added to the plates is determined by the size of the dish used in the experiment. For example, to achieve complete detachment of cells from a 75-mm2 dish, add 3.5 mL of trypsin followed by incubation at 37°C for 5–6 min. It has been shown that weekly trypsinizations do not affect profliferative capacity of the cell culture (13); however, because trypsin is a harsh digestive enzyme, the cells should not be left exposed for longer than necessary. Trypsin is inactivated by the serum component in media and detached cells should be suspended in 5–6 mL of media. The amount of PBS used also varies according to dish size. Use enough to cover the bottom of the dish to effectively wash away media.
Cell counting can be achieved by several different methods. Counting using a hemacytometer is a simple way to determine an approximate cell count. Cells are suspended in PBS as described above and vortexed for approx 1 min to disassociate clumps. Twenty-five microliters of cell suspension is added to a 1.5-mL collection tube and mixed with 75 μL of Trypan Blue dye. Nine microliters of the cell/trypan blue mix is added to the hemacytometer. Cells that are stained dark blue are considered not viable and should not be counted. The use of flow cytometry is also a method of cell counting. Volumetric flow cytometry is used to count absolute cell number in a given volume and provides a more accurate count than counting cells with a hemacytometer.
Subheading 3.1., step 6 is an optional step to eliminate ethanol carryover when eluting your sample RNA. It is highly recommended to perform this additional step since ethanol contamination can result in reduced RNA yields and poor cDNA conversion.
References
- 1.Yeivin A, Razin A. Gene Methylation patterns and expressions. EXS. 1993;64:523–568. doi: 10.1007/978-3-0348-9118-9_24. [DOI] [PubMed] [Google Scholar]
- 2.Adams RL. DNA methylation. The effect of minor bases on DNA-protein interactions. Biochem J. 1990;265:309–320. doi: 10.1042/bj2650309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lei H, Oh Sp., Okano M, et al. De novo DNA cytosine methyltranserase activities in mouse embryonic stem cells. Develompment. 1996;122:3195–3205. doi: 10.1242/dev.122.10.3195. [DOI] [PubMed] [Google Scholar]
- 4.Warnecke PM, Bestor TH. Cytosine methylation and human cancer. Curr Opin Oncol. 2000;12:68–73. doi: 10.1097/00001622-200001000-00012. [DOI] [PubMed] [Google Scholar]
- 5.Cooney CA. Are somatic cells inherently deficient in methylation metabolism? A proposed mechanism for DNA methylation loss, senescence and aging. Growth. 1993;57:261–273. [PubMed] [Google Scholar]
- 6.Tollefsbol TO, Andrews LG. Mechanism for methylation-mediated gene silencing and aging. Med Hypotheses. 1993;41:83–92. doi: 10.1016/0306-9877(93)90040-w. [DOI] [PubMed] [Google Scholar]
- 7.Adams RL, McKay EL, Craig LM, Burdon RH. Mouse DNA methylase: methylation of native DNA. Biochem Biophys Acta. 1979;561:345–357. doi: 10.1016/0005-2787(79)90143-6. [DOI] [PubMed] [Google Scholar]
- 8.Ramsahoye BH, Biniszkiewicz D, Lyko F, Clark V, Bird AP, Jaenisch R. Non-CpG methylation is prevalent in embryonic stem cells and may be mediated by DNA methyltransferase 3a. Proc Natl Acad Sci USA. 2000;97:5237–5242. doi: 10.1073/pnas.97.10.5237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hashimoto H, Suetake I, Tajima S. Monoclonal antibody against DNMT1 arrests the cell division of xenopus early-stage embryos. Exp Cell Res. 2003;286(2):252–262. doi: 10.1016/s0014-4827(03)00060-0. Erratum in: Exp Cell Res289(2), 396. [DOI] [PubMed] [Google Scholar]
- 10.Hansen RS, Wijmenga C, Luo P, et al. The DNMT3B DNA methyltransferase gene is mutated in the ICF immunodeficiency syndrome. Proc Natl Acad Sci USA. 1999;96:14,412–14,417. doi: 10.1073/pnas.96.25.14412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xu GL, Bestor TH, Bourx HD, et al. Chromosome instability and immunodeficiency syndrome caused by mutations in a DNA methyltransferase gene. Nature. 1999;503:178–191. doi: 10.1038/46052. [DOI] [PubMed] [Google Scholar]
- 12.Lopatina N, Haskell JF, Andrews LG, Poole JC, Saldanah S, Tollefsbol TO. Differential maintenance and De novo methylating activity by three DNA methyltransferases in aging and immortalized fibroblasts. J. Cell Biochem. 2002;84:324–334. doi: 10.1002/jcb.10015. [DOI] [PubMed] [Google Scholar]
- 13.Hadley EC, Kruss ED, Cristofalo VJ. Trypsinization frequency and loss of proliferative capacity in WI-38 cells. J. Gerontol. 1979;34:170–176. doi: 10.1093/geronj/34.2.170. [DOI] [PubMed] [Google Scholar]