

Abstract
Gap junctions serve as intercellular conduits that allow the exchange of small molecular weight molecules (up to 1 kDa) including ions, metabolic precursors and second messengers. Microglia are capable of recognizing peptidoglycan (PGN) derived from the outer cell wall of Staphylococcus aureus, a prevalent CNS pathogen, and respond with the robust elaboration of numerous pro-inflammatory mediators. Based on recent reports demonstrating the ability of tumor necrosis factor-α and interferon-γ to induce gap junction coupling in macrophages and microglia, it is possible that pro-inflammatory mediators released from PGN-activated microglia are capable of inducing microglial gap junction communication. In this study, we examined the effects of S. aureus-derived PGN on Cx43, the major connexin in microglial gap junction channels, and functional gap junction communication using single-cell microinjections of Lucifer yellow (LY). Exposure of primary mouse microglia to PGN led to a significant increase in Cx43 mRNA and protein expression. LY microinjection studies revealed that PGN-treated microglia were functionally coupled via gap junctions, the specificity of which was confirmed by the reversal of activation-induced dye coupling by the gap junction blocker 18-α-glycyrrhetinic acid. In contrast to PGN-activated microglia, unstimulated cells consistently failed to exhibit LY dye coupling. These results indicate that PGN stimulation can induce the formation of a functional microglial syncytium, suggesting that these cells may be capable of influencing neuroinflammatory responses in the context of CNS bacterial infections through gap junction intercellular communication.
Keywords: central nervous system, connexin 43, gap junction, microglia, neuroinflammation, peptidoglycan
Gap junctions are intercellular membrane channels that provide direct cytoplasmic continuity between adjacent cells (Saez et al. 2003). They are composed of hydrophilic transmembrane pores directly linking the cytoplasm of neighboring cells, where each cell contributes a hemichannel (termed a connexon) formed by the assembly of six connexin (Cx) monomers surrounding a central pore (Saez et al. 2003). Cxs are highly homologous proteins encoded by a multigene family, with more than 19 members currently identified (Willecke et al. 2002; Saez et al. 2003). Gap junction coupling can be regulated at several levels including alterations in Cx transcription, translation, stability, post-translational processing (i.e. phosphorylation), insertion/removal from the cell membrane, and channel gating (i.e. influenced by intracellular pH) (Saez et al. 2003). Gap junctions can be classified on the basis of the cell types that are functionally coupled as well as the Cx isoforms that comprise the channels. Examples of the former include channels connecting cells of the same type (homocellular), communications between different cell types (heterocellular), or channels that connect processes of the same cell (auto-cellular). To add another level of complexity to the system, the biochemical composition of gap junction channels influences their biophysical properties and permeabilities to various small molecular weight molecules (Goldberg et al. 1999, 2002). The extensive intercellular coupling and large number of gap junctions in the CNS suggests a syncytium-like organization of glial compartments (Spray et al. 1999; Rouach et al. 2002a; Theis et al. 2005).
Microglia are the resident mononuclear phagocytes of the CNS parenchyma and play an important role in immune surveillance of CNS tissues (Aloisi 2001; Hanisch 2002). Microglia communicate with one another (homocellular) and with other CNS cells (heterocellular) through specific extracellular signals such as cytokines and adhesion molecules (Aloisi 2001; Kielian 2004a). Under normal conditions, microglia show little expression of Cx43 and are not gap junction coupled; however, in vitro exposure to pro-inflammatory cytokines (Eugenin et al. 2001) or Ca2+ ionophore (Martinez et al. 2002) leads to an induction of Cx43 expression concomitant with the establishment of functional gap junction coupling. That microglia may become coupled by gap junctions in vivo was recently suggested by the finding that Cx43 immunoreactivity co-localized with the macrophage/microglial activation marker CD11b following cortical stab wound injury (Eugenin et al. 2001). Therefore, under the appropriate conditions, the inflammatory milieu and physical association of activated microglia may facilitate gap junction formation; however, the functional implications of activation-dependent microglial coupling remain unclear.
Inflammation is a hallmark of various CNS diseases such as bacterial and viral infections, multiple sclerosis, Alzheimer’s disease and cerebral ischemia. Alterations in Cx expression have been associated with several CNS diseases with an inflammatory component, including cerebral ischemia and Alzheimer’s disease (Nagy et al. 1996; Siushansian et al. 2001; Nakase et al. 2003, 2004). Although there is evidence to suggest that activated microglia can become functionally coupled by gap junctions following exposure to pro-inflammatory cytokines (Eugenin et al. 2001), the effects of prevalent CNS bacterial pathogens, such as Staphylococcus aureus, on microglial coupling are not known. S. aureus represents one of the main etiologic agents of brain abscess in humans (Mathisen and Johnson 1997; Townsend and Scheld 1998) and the effects of bacterial infection on the glial syncytia may have a significant impact on disease severity and outcome. Indeed, brain abscess survivors may exhibit numerous long-term deficits in cognition and motor function as well as seizures (Mathisen and Johnson 1997; Townsend and Scheld 1998), all of which may be influenced, in part, by changes in gap junction communication both within the abscess and surrounding normal brain parenchyma. Microglial activation is a hallmark of brain abscess and we have recently demonstrated in our experimental brain abscess model that the inflammatory milieu within evolving brain abscesses potentiates macrophage/microglial activation (Baldwin and Kielian 2004). In addition, we have shown that exposure of primary microglia to either intact heat-inactivated S. aureus or its cell wall product peptidoglycan (PGN) leads to the elaboration of many of the pro-inflammatory mediators that exert well documented effects on glial homocellular gap junction communication, including nitric oxide, tumor necrosis factor (TNF)-α and interleukin (IL)-1β (Bolanos and Medina 1996; John et al. 1999; Eugenin et al. 2001; Kielian et al. 2002, 2004, 2005). It therefore appears likely that microglia may be capable of forming homocellular syncytia mediated by gap junctions in the context of CNS bacterial infections through the autocrine/paracrine action of pro-inflammatory mediators produced by activated cells. In this study, we demonstrated that S. aureus-derived PGN induces a significant increase in Cx43 expression that coincides with the establishment of functional gap junctions in activated microglia.
Materials and methods
Primary microglial cell culture and reagents
Primary microglia were isolated from neonatal C57BL/6 mouse pups (1–8 days of age; Harlan Laboratories, Indianapolis, IN, USA) as described previously (Kielian et al. 2004). The purity of microglial cultures was evaluated by immunohistochemical staining using antibodies against CD11b (BD Pharmingen, San Diego, CA, USA) and glial fibrillary acidic protein (GFAP) (Dako, Carpenteria, CA, USA), and a more sensitive quantitative real-time RT–PCR (qRT–PCR) approach using Taqman probes specific for CD11b and GFAP to identify microglia and astrocytes respectively, as described below. The purity of primary microglia cultures was routinely greater than 95%.
PGN derived from S. aureus was obtained from Fluka (Sigma, St Louis, MO, USA). Throughout the course of these studies, PGN was added to microglial cultures at a final concentration of 10 μg/mL, a dose previously determined to be optimal for inducing maximal pro-inflammatory cytokine expression in microglia without any evidence of cytotoxicity (Kielian et al. 2004, 2005). Rabbit polyclonal antibody reactive with mouse Cx43 was purchased from Zymed Laboratories (South San Francisco, CA, USA). All reagents and culture media were verified to have endotoxin levels < 0.03 EU/mL as determined by Limulus amebocyte lysate assay (Associates of Cape Cod, Falmouth, MA, USA).
RT–PCR
Total RNA from C57BL/6 primary microglia was isolated using Trizol reagent and treated with Dnase1 (both from Invitrogen, Carlsbad, CA, USA) before use in qRT–PCR studies. Primers for GFAP, CD11b and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and TAMRA TaqMan probes were designed as described previously (Esen et al. 2004). An ABI Assays-on-Demand™ Taqman kit (Applied Biosystems, Foster City, CA, USA) was used to examine Cx43 expression. RT reactions were conducted using an iScript cDNA™ synthesis kit (Bio-Rad, Hercules, CA, USA) according to the manufacturer’s instructions. For real-time PCR, reactions were performed in a total volume of 25 μL using an iCycler™ kit (Bio-Rad) and analyzed using an iCycler IQ™ multicolor real-time PCR detection system (Bio-Rad). The levels of Cx43 expression in primary microglia were calculated after normalizing cycle thresholds against the ‘housekeeping’ gene GAPDH and are presented as the fold induction value (2–Δ ΔCt) relative to unstimulated microglia (mean ± SD).
Protein extraction and western blotting
Protein extracts were prepared and quantitated from primary microglia as described previously (Kielian et al. 2004), and the effects of PGN on microglial Cx43 expression were evaluated by western blot analysis. Briefly, microglial protein extracts (25 μg total protein) were electrophoresed on 10% Tris-HCl Ready Gels (Bio-Rad) and transferred to a polyvinylidene difluoride membrane (Immobilon-P, Millipore, Bedford, MA, USA) using a semidry transfer apparatus (Bio-Rad). Blots were probed using a rabbit anti-Cx43 followed by a donkey anti-rabbit IgG–horseradish peroxidase conjugate (Jackson Immunoresearch, West Grove, PA, USA). Duplicate blots were probed with a rabbit actin polyclonal antibody (Sigma) to verify uniformity in gel loading. Blots were developed using ChemiGlow West substrate (Alpha Innotech, San Leandro, CA, USA) and detected by exposure to X-ray film.
Cx43 immunofluorescence staining
Visualization of Cx43 localization in microglia was performed using an indirect immunofluorescence staining procedure. Briefly, primary microglia were seeded into eight-well chamber slides (2 × 105 cells/well) (LabTek; Nalge Nunc International, Naperville IL, USA) and incubated overnight. The following day, cells were treated with 10 μg/mL PGN for 24 or 48 h (two replicates per time point). At the end of each incubation period, cells were washed extensively with phosphate-buffered saline and immediately fixed in ice-cold methanol. Microglia were incubated with a rabbit Cx43 polyclonal antibody overnight at 4°C in a humidified chamber and, following numerous rinses in phosphate-buffered saline/0.3% Tween-20, incubated with biotinylated goat anti-rabbit IgG (Vector Laboratories, Burlingame, CA, USA) for 30 min at room temperature (25°C). Cx43 immunoreactivity was visualized with a streptavidin–Alexa 568 conjugate, and slides were subsequently mounted in ProLong anti-fade reagent (both from Molecular Probes, Eugene, OR, USA), coverslipped and sealed. Images were acquired with a fixed-stage upright epifluorescence microscope (BX51WI; Olympus USA, Melville, NY, USA) using the MetaMorph image analysis program (Universal Imaging, Downington, PA, USA).
Single-cell microinjections of Lucifer yellow (LY)
Microglia cultures were continuously superfused with carbogen (95% O2 and 5% CO2)-saturated artificial CSF at a rate of 3–4 mL/min. The perfusion solution was prewarmed to 37°C using an inline solution heater regulated by a dual-channel heater (both from Warner Instruments, Hamden, CT, USA). Cells were allowed to equilibrate for 15 min before microinjections. Sharp micropipettes (resistance 100–200 MΩ) were prepared from borosilicate filament tubing (outer diameter 1.5 mm, inner diameter 0.86 mm) using a micropipette puller (P-97 Fleming/Brown; both from Sutter Instruments, Novato, CA, USA). Pipettes were back-filled with the gap junction-permeable dye (LY) CH (molecular weight 453 Da; Sigma Chemicals) (4% w/v LY in 150 mM LiCl) with a 34-G microfill syringe needle (World Precision Instruments, Sarasota, FL, USA). LY was injected intracellularly by applying brief hyperpolarizing pulses generated by a microiontophoresis current generator (World Precision Instruments) through a micropipette that was carefully inserted into the cell guided by a micromanipulator (MP-225; Sutter Instruments). In studies designed to confirm the gap junction-dependent nature of LY spread in PGN-activated microglia, cells were treated with the gap junction blocker 18-α-glycyrrhetinic acid (AGA). Briefly, a minimum of three LY intracellular injections were performed in PGN-treated microglial cultures before AGA treatment to confirm the establishment of gap junction-dependent coupling. Subsequently, the same PGN-treated cultures were continuously perfused for 20 min in artificial CSF supplemented with 25 μM AGA to allow sufficient levels of inhibitor to accumulate within the culture medium where upon gap junction-dependent coupling was assessed in the continued presence of AGA. A minimum of 10 individual cells were microinjected with LY in each experimental group (unstimulated, PGN only, PGN + AGA) with results replicated in six independent experiments. LY-injected cells were visualized using an Olympus BX51WI fixed-stage upright epifluorescence microscope equipped with a 40 × water immersion objective lens (numerical aperture 0.8), a 12-bit intensified monochrome charge coupled device camera (CoolSnap ES; Photometrics, Tucson, AZ, USA), a multiple excitation filter controlled through a filter wheel (Prior Scientific, Rockland, MA, USA) and a quad emission filter (Chroma, Rockingham, VT, USA). Images were acquired and processed using MetaMorph software. An overlay of the bright field and background subtracted LY image (excitation wave length 425 nm, emission wave length 540 nm) was constructed to clearly show LY spread into neighboring cells.
Statistical analysis
Significant differences between experimental groups were determined by the Student’s t-test at the 95% confidence interval using Sigma Stat (SPSS Science, Chicago, IL, USA).
Results
S. aureus-derived PGN induces Cx43 expression in primary microglia
Cx43 expression and functional gap junctions have recently been reported in macrophages and microglia following stimulation with pro-inflammatory mediators (Eugenin et al. 2001, 2003) or Ca2+ ionophore (Martinez et al. 2002). However, the effects of Gram-positive bacterial stimuli, such as S. aureus, on regulating Cx expression in microglia remain unknown. Owing to its prevalence as a brain abscess pathogen, we examined the effects of S. aureus-derived PGN on Cx43 mRNA and protein expression using qRT–PCR and western blotting respectively. As shown in Fig. 1, PGN treatment significantly increased Cx43 mRNA expression in primary microglia with maximal levels observed at 24 h after exposure to PGN, the latest time point examined. To verify that the observed changes in Cx43 mRNA expression extended to increased protein levels, western blots were performed. As shown in Fig. 2, PGN-stimulated microglia exhibited increased Cx43 levels, with maximal expression detected at 48 h. Importantly, qRT–PCR using primers for CD11b and GFAP to assess the purity of microglial cultures revealed that the observed changes in microglial Cx43 expression were not due to contaminating astrocytes (Table 1). Examination of additional Cx isoforms, including Cx26 and Cx30, by qRT–PCR revealed low to undetectable expression in activated cells, suggesting that these forms are not major constituents of the Cx pool in microglia (data not shown).
Fig. 1.
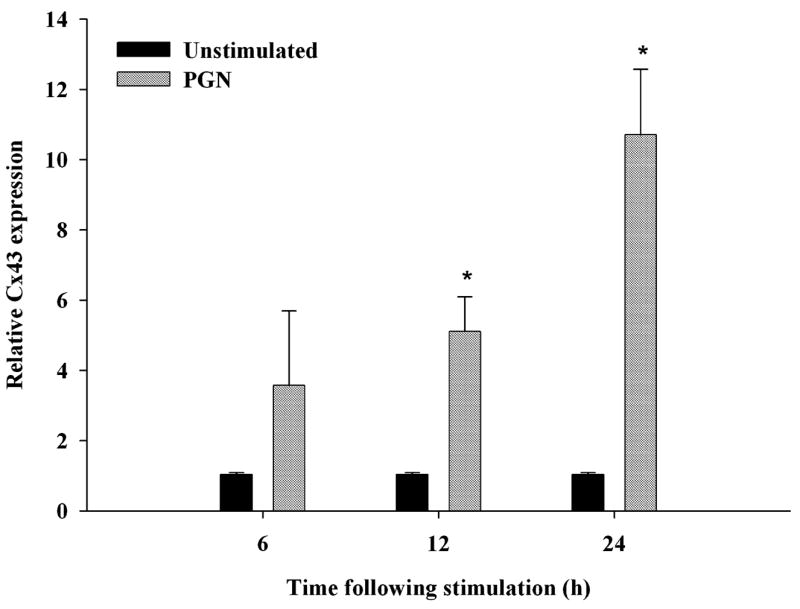
PGN induces Cx43 mRNA expression in microglia. Primary microglia were stimulated with 10 μg/mL PGN for 6, 12 or 24 h, and total RNA was isolated to quantitate changes in Cx43 expression by qRT–PCR. Values are mean ± SD of three independent experiments. *p < 0.05 versus PGN-stimulated microglia (Student’s t-test).
Fig. 2.
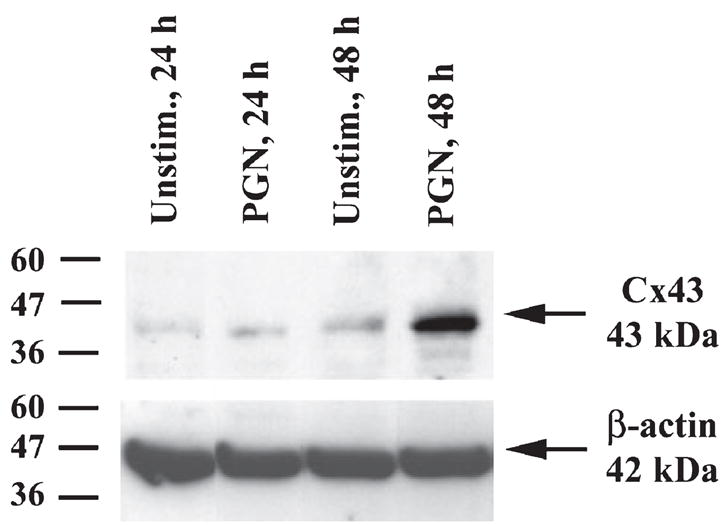
Cx43 protein expression is increased in PGN-stimulated microglia. Protein extracts from microglial whole-cell lysates were prepared at 24 and 48 h following PGN stimulation (10 μg/mL), and Cx43 levels were evaluated by western blot analysis. Duplicate blots were probed with β-actin to control for differences in gel loading. Results are representative of three independent experiments.
Table 1.
Assessment of primary microglial cell culture purity by qRT–PCR
| Treatment | CD11b | GFAP |
|---|---|---|
| Unstimulated | 1.0 | ND |
| PGN, 6 h | 0.6 | ND |
| PGN, 12 h | 0.7 | ND |
| PGN, 24 h | 0.3 | ND |
The level of CD11b expression was calculated after normalizing target signals against GAPDH and is presented as the fold change in mRNA expression relative to unstimulated microglia. Results are representative of three independent experiments. ND, not detected.
To visualize the nature and extent of Cx43 localization in PGN-activated microglia, immunofluorescence staining was performed. Cx43 expression was low or undetectable in unstimulated microglia (Fig. 3). In contrast, Cx43 immunoreactivity was significantly enhanced in PGN-stimulated microglia that were associated with cellular aggregates. The activation-dependent increase in Cx43 was observed as early as 24 h and was maximal at 48 h following activation, the latest time point examined (Fig. 3). Collectively, these findings suggest that PGN is capable of inducing Cx43 expression suggestive of activation-dependent gap junction formation.
Fig. 3.

Cx43 immunoreactivity is enhanced in primary microglia following PGN activation and is associated with cellular aggregates. Primary microglia were stimulated with 10 μg/mL PGN for either 24 or 48 h in eight-chamber tissue culture slides, and Cx43 expression was evaluated by immunofluorescence staining. The red spherical particles depicted in unstimulated cells represent artifactual staining. Results are representative of two independent experiments.
PGN induces functional gap junction communication in primary microglia
Recent evidence suggests that pro-inflammatory mediators are capable of establishing gap junction communication in macrophages and microglia (Eugenin et al. 2001, 2003). However, the effects of Gram-positive bacterial stimulation on microglial gap junction coupling have not yet been examined. To determine whether the observed PGN-dependent increase in microglial Cx43 expression translated into the formation of functional gap junction channels, gap junction communication was evaluated in primary microglia using a single-cell microinjection technique with LY. Gap junction coupling was never detected in unstimulated microglia, as evident from LY containment within the injected cell (Fig. 4a). In contrast, PGN-treated microglia exhibited functional gap junction coupling within 48 h of stimulation, which was demonstrated by the spread of LY into neighboring cells (Fig. 4a). Interestingly, PGN stimulation led to an activation-dependent homotypic aggregation of microglia that coincided with gap junction-coupled cells (Fig. 4a). Importantly, the observed PGN-dependent dye coupling was completely inhibited by the gap junction blocker AGA (Rozental et al. 2000), confirming the activation-dependent induction of gap junction channels in microglia (Figs 4a and b). Quantitation of dye coupling revealed that approximately 50% of PGN-stimulated microglia demonstrated dye transfer to at least one neighboring cell (dye transferred to a mean of three cells), whereas unstimulated and AGA-treated microglia consistently failed to demonstrate any evidence of gap junction-dependent communication (Fig. 4b). Previous studies from our laboratory have established that PGN treatment of primary microglia at the dose used in this study does not induce overt cell death or proliferation as determined by standard 3-[4,5-dimethylthiazol-2-yl]-2,5 diphenyltetrazolium bromide (MTT) cell viability assays (Kielian et al. 2004, 2005), indicating that the observed induction of gap junction coupling is influenced by alterations in cellular activation status. Collectively, these findings indicate that PGN is capable of inducing functional gap junction communication in microglia that may have important implications in regulating glial syncytia and neuronal homeostasis in the context of CNS infectious diseases such as brain abscess.
Fig. 4.

PGN treatment establishes functional gap junction communication in microglia. (a) Primary microglia were stimulated with 10 μg/mL PGN for 48 h, and gap junction communication was examined by single-cell microinjections of the gap junction-permeant dye LY CH. The gap junction-dependent spread of LY in activated microglia was confirmed by incubating PGN-treated cells with the gap junction inhibitor AGA (25 μM) before injections. Microinjected cells are denoted with asterisks. (b) Enumeration of the incidence of gap junction coupling revealed that approximately 50% of primary micro-glia stimulated with 10 μg/mL PGN for 48 h exhibited gap junction-dependent dye transfer to at least one neighboring cell. The horizontal bars representing results for unstimulated and PGN + AGA treatments indicate that microglia in these groups consistently failed to demonstrate any evidence of gap junction-dependent communication. Values are mean ± SD; the data presented are representative of six independent experiments evaluating at least 10 injected cells per treatment group per experiment. **p < 0.001 versus unstimulated microglia (Student’s t-test).
Discussion
The organization of cells into syncytia through the formation of gap junction channels is one mechanism by which local alterations in cell activation can lead to the direct propagation of signals at distant sites within tissues. Recent evidence indicates that macrophages and microglia can become functionally coupled by gap junctions in response to pro-inflammatory stimuli such as cytokines and LPS (Eugenin et al. 2001, 2003). Microglial activation is a hallmark of brain abscess (Baldwin and Kielian 2004; Kielian 2004b) and the pro-inflammatory milieu that ensues subsequent to bacterial infection may modulate gap junction connectivity in situ and thus neuronal homeostasis and viability. Therefore, based on its relevance as a brain abscess pathogen (Mathisen and Johnson 1997; Townsend and Scheld 1998), we examined the ability of S. aureus-derived PGN to induce gap junction communication in primary microglia. Here we demonstrated that functional gap junctions are established in primary microglia following PGN stimulation concomitant with an increase in Cx43 expression. This activation-dependent induction of Cx43 and functional coupling is consistent with a study by Eugenin et al. (2001), in which approximately 55% of microglia were functionally coupled following stimulation with IFN-γ and LPS or TNF-α and IFN-γ, coincident with an increase in Cx43 expression (Eugenin et al. 2001). However, our findings differ from two previous reports of astrocyte–microglia co-cultures in which the authors were unable to demonstrate Cx43 expression in microglia (Rouach et al. 2002b; Faustmann et al. 2003). Two key differences between these previous reports and the present study may account for these discrepancies. The first relates to the absolute numbers of microglia used in the various studies. Both Rouach et al. (2002b) and Faustmann et al. (2003) used astrocyte–microglial co-cultures in which the relative density of microglia was rather sparse. In contrast, our studies utilized nearly confluent monolayers of primary microglia, in which the activation-dependent homotypic adhesion of cells following PGN exposure is thought to provide the initiating signal(s) required for the induction of detectable Cx43 expression and subsequent establishment of gap junction-dependent coupling. As gap junction communication can only occur between cells in extremely close proximity to one another (i.e. 2–3 nm), it appears likely that contact-dependent events may be required to induce Cx43 expression to levels that are easily detectable using standard immunofluorescence or western blotting approaches. This might explain why we observed Cx43 expression in the current study whereas other groups using co-culture paradigms have not. Second, the conditions under which astrocyte–microglia co-cultures were evaluated were substantially different from the PGN treatment used in our experiments. Specifically, Rouach et al. (2002b) and Faustmann et al. (2003) did not expose co-cultures to any exogenous stimuli. This is in marked contrast to the studies reported here, in which primary microglia were treated with the Toll-like receptor 2 agonist PGN, which is a potent inducer of numerous pro-inflammatory mediators in activated microglia (Kielian et al. 2005), factors that are not constitutively produced at high levels in unstimulated cells. Therefore, when comparing these studies, the array and concentration of molecules present within glial-conditioned supernatants is expected to be very different. Collectively, based on our findings it appears that the induction of microglial Cx43 expression is dependent on the presence of a highly activated, inflammatory phenotype. Importantly, the demonstration of microglial coupling in the present study was not due to the effects of contaminating astrocytes as we were unable to detect astrocytes by immunohistochemical staining or a highly sensitive qRT–PCR approach. In addition, we found that PGN treatment of astrocytes inhibits gap junction communication (N. Esen and T. Kielian, unpublished observation), which is diametrically opposed to what is observed in primary microglia. We have termed this differential effect of bacterial stimuli on glial gap junction communication a ‘syncytial switch’ (Kielian and Esen 2004). The extent of gap junction-dependent dye coupling in PGN-activated microglia (i.e. average of three cells) is in close agreement to that observed by Eugenin et al. (2001). However, it is important to consider that the degree of gap junction coupling in activated microglia may be more extensive in vivo when cells are in their native microenvironment and in intimate three-dimensional contact with neighboring cells. Evidence to support this concept is provided by recent studies demonstrating that the level of Cx43 immunoreactivity in activated microglia following a stab wound injury is extensive, although the establishment of functional coupling in situ was not investigated (Eugenin et al. 2001). In addition, the amount of gap junction communication in PGN-activated microglia may be more pronounced if a smaller tracer than LY were injected (for example Alexa Fluor 350, molecular weight 268 Da or Neurobiotin™, 323 Da) as there is evidence to suggest that there is a rank order transfer of molecules via gap junctions that is size and charge dependent (Goldberg et al. 1999, 2002).
In addition to establishing functional gap junction channels, the PGN-dependent increase in microglial Cx43 expression may also lead to changes that are gap junction independent. This possibility is suggested by the finding that Cx43 mRNA and protein expression was significantly increased in response to PGN stimulation, whereas only a modest induction in microglial gap junction-dependent coupling was observed. Indeed, Cxs have been shown to play multiple roles in addition to being an integral component of gap junction channels (Lin et al. 2003; Giepmans 2004; Stout et al. 2004). For example, ATP release facilitated by Cx expression occurs via hemichannels (Cotrina et al. 1998; Stout et al. 2002, 2004). It is intriguing to speculate that the PGN-dependent increase in Cx43 expression leads to ATP release which acts via an autocrine/paracrine loop to stimulate microglial P2X receptors, effectively exacerbating pro-inflammatory mediator production from these cells. Indeed, recent evidence indicates that ATP released from LPS-activated microglia signals through P2X receptors to augment pro-inflammatory cytokine production (Ferrari et al. 1997; Boucsein et al. 2003). With regard to infectious disease, a recent study has suggested that Shigella infection of epithelial cells triggers ATP release through hemichannels, resulting in the activation of purinergic receptors on neighboring cells and bacterial dissemination (Tran Van Nhieu et al. 2003). As we have not yet examined whether ATP is released from PGN-activated microglia, these possibilities remain speculative. Other reported gap junction-independent activities of Cxs include regulation of cell proliferation (Huang et al. 1998; Moorby and Patel 2001; Qin et al. 2002) and resistance to injury via anti-apoptotic actions (Lin et al. 2003). Whether the elevated levels of Cx43 protein observed in PGN-activated microglia contribute to any gap junction-independent phenomena remains an area for future investigation.
Recent studies from our laboratory have demonstrated that PGN stimulation of microglia leads to the elaboration of a wide array of pro-inflammatory mediators and enhanced expression of surface receptors that play a pivotal role in bacterial recognition and antigen presentation (Kielian et al. 2002, 2004, 2005). Based on these activation-dependent effects, changes in Cx43 expression and subsequent microglial gap junction coupling in response to PGN can be envisioned to occur by one of the following mechanisms. First, the direct engagement of microglial pattern recognition receptors (i.e. Toll-like receptor 2) by PGN may induce the immediate activation of signal transduction pathways, such as those involving nuclear factor-κB and mitogen-activated protein kinase (MAPK), leading to direct changes in Cx expression and gap junction coupling. Indeed, recent evidence indicates that MAPK-dependent signal transduction pathways are capable of influencing the phosphorylation state of Cx43 and subsequent gap junctional activity (Warn-Cramer et al. 1996, 1998; Saez et al. 2003). Second, pro-inflammatory mediators produced by PGN-activated microglia, such as TNF-α and IL-1β, may act in an autocrine/paracrine manner to regulate glial gap junction intercellular communication as these cytokines also activate nuclear factor-κB and MAPK pathways upon binding to their cognate receptors (Hanada and Yoshimura 2002). From the results presented here, the kinetics of changes in microglial Cx mRNA and protein expression (i.e. within 12–24 h), together with the delayed gap junction coupling responses observed following PGN exposure (i.e. within 48 h), suggest that these two pathways are probably both involved in regulating glial gap junction communication through signal transduction-dependent mechanisms.
Following exposure to PGN, gap junction-coupled microglia were associated with foci of cellular aggregates. This activation-dependent coupling is thought to result from homotypic interactions facilitated by adhesion molecules whose expression is enhanced upon cell activation, such as intercellular adhesion molecule (ICAM)-1). Indeed, pro-inflammatory stimuli including IFN-γ and LPS have been reported to enhance ICAM-1 gene expression in primary rat microglia (Shrikant et al. 1995; Lee and Benveniste 1999). Furthermore, ICAM-1 facilitates homotypic adhesion (Boyd et al. 1988; Lauener et al. 1990), suggesting that it may promote the formation of gap junction plaques. Collectively, these findings suggest that the establishment of adhesive interactions is a prerequisite for the formation of functional gap junction channels in PGN-activated microglia. Further studies are required to ascertain the relationship between adhesion molecule expression and the induction of gap junction coupling in PGN-stimulated microglia, which may provide insights into factor(s) controlling the nature and extent of microglial/homocellular gap junction communication.
The consequence(s) of the observed PGN-dependent gap junction coupling of microglia are currently unknown, but might be envisioned to affect CNS homeostasis in opposing manners (Kielian and Esen 2004). For example, the induction of functional gap junction communication in activated microglia may serve to transmit local neuroinflammatory signals to maximize bactericidal efficacy. Recent evidence indicates that the activation-dependent coupling of monocytes enhances antigen presentation through the direct transfer of small peptides through gap junction channels (Neijssen et al. 2005). Alternatively, microglial coupling may serve as a mechanism by which host cell viability is reduced through the propagation of damaging factors and/or the continual release of cytotoxic pro-inflammatory mediators. Currently, the biological significance of gap junction communication in PGN-activated microglia remains unknown in the setting of CNS Gram-positive bacterial infections.
In summary, we report that S. aureus-derived PGN induces functional gap junction intercellular communication in primary microglia that coincides with increased Cx43 expression. With regard to the potential impact of these changes in the context of CNS infectious disease, recent studies describing the effects of inflammatory mediators on glial gap junction communication suggest that alterations in the local connections between microglia may lead to far-reaching changes in the function of non-inflamed CNS tissue (Bolanos and Medina 1996; John et al. 1999; Kielian and Esen 2004; Nakase et al. 2004).
Acknowledgments
The authors would like to thank Dr Gerry Dienel for use of equipment during the initial phase of this study and critical review of the manuscript. This work was supported by the Arkansas Biosciences Institute to TK and the National Institute of Neurological Disorders and Stroke-supported Core Facility at University of Arkansas for Medical Sciences (P30 NS047546).
Abbreviations used
- AGA
18-α-glycyrrhetinic acid
- Cx
connexin
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
- GFAP
glial fibrillary acidic protein
- ICAM
intercellular adhesion molecule
- IL
interleukin
- LPS
lipopolysaccharide
- LY
Lucifer yellow
- MAPK
mitogen-activated protein kinase
- MTT
3-[4,5-dimethylthiazol-2-yl]-2,5 diphenyltetrazolium bromide
- PGN
peptidoglycan
- qRT–PCR
quantitative real-time RT–PCR
- TNF
tumor necrosis factor
References
- Aloisi F. Immune function of microglia. Glia. 2001;36:165–179. doi: 10.1002/glia.1106. [DOI] [PubMed] [Google Scholar]
- Baldwin AC, Kielian T. Persistent immune activation associated with a mouse model of Staphylococcus aureus-induced experimental brain abscess. J Neuroimmunol. 2004;151:24–32. doi: 10.1016/j.jneuroim.2004.02.002. [DOI] [PubMed] [Google Scholar]
- Bolanos JP, Medina JM. Induction of nitric oxide synthase inhibits gap junction permeability in cultured rat astrocytes. J Neurochem. 1996;66:2091–2099. doi: 10.1046/j.1471-4159.1996.66052091.x. [DOI] [PubMed] [Google Scholar]
- Boucsein C, Zacharias R, Farber K, Pavlovic S, Hanisch UK, Kettenmann H. Purinergic receptors on microglial cells: functional expression in acute brain slices and modulation of microglial activation in vitro. Eur J Neurosci. 2003;17:2267–2276. doi: 10.1046/j.1460-9568.2003.02663.x. [DOI] [PubMed] [Google Scholar]
- Boyd AW, Wawryk SO, Burns GF, Fecondo JV. Intercellular adhesion molecule 1 (ICAM-1) has a central role in cell-cell contact-mediated immune mechanisms. Proc Natl Acad Sci USA. 1988;85:3095–3099. doi: 10.1073/pnas.85.9.3095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cotrina ML, Lin JH, Alves-Rodrigues A, Liu S, Li J, Azmi-Ghadimi H, Kang J, Naus CC, Nedergaard M. Connexins regulate calcium signaling by controlling ATP release. Proc Natl Acad Sci USA. 1998;95:15 735–15 740. doi: 10.1073/pnas.95.26.15735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esen N, Tanga FY, DeLeo JA, Kielian T. Toll-like receptor 2 (TLR2) mediates astrocyte activation in response to the Gram-positive bacterium Staphylococcus aureus. J Neurochem. 2004;88:746–758. doi: 10.1046/j.1471-4159.2003.02202.x. [DOI] [PubMed] [Google Scholar]
- Eugenin EA, Eckardt D, Theis M, Willecke K, Bennett MV, Saez JC. Microglia at brain stab wounds express connexin 43 and in vitro form functional gap junctions after treatment with interferon-gamma and tumor necrosis factor-alpha. Proc Natl Acad Sci USA. 2001;98:4190–4195. doi: 10.1073/pnas.051634298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eugenin EA, Branes MC, Berman JW, Saez JC. TNF-alpha plus IFN-gamma induce connexin43 expression and formation of gap junctions between human monocytes/macrophages that enhance physiological responses. J Immunol. 2003;170:1320–1328. doi: 10.4049/jimmunol.170.3.1320. [DOI] [PubMed] [Google Scholar]
- Faustmann PM, Haase CG, Romberg S, Hinkerohe D, Szlachta D, Smikalla D, Krause D, Dermietzel R. Microglia activation influences dye coupling and Cx43 expression of the astrocytic network. Glia. 2003;42:101–108. doi: 10.1002/glia.10141. [DOI] [PubMed] [Google Scholar]
- Ferrari D, Chiozzi P, Falzoni S, Hanau S, Di Virgilio F. Purinergic modulation of interleukin-1 beta release from microglial cells stimulated with bacterial endotoxin. J Exp Med. 1997;185:579–582. doi: 10.1084/jem.185.3.579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giepmans BN. Gap junctions and connexin-interacting proteins. Cardiovasc Res. 2004;62:233–245. doi: 10.1016/j.cardiores.2003.12.009. [DOI] [PubMed] [Google Scholar]
- Goldberg GS, Lampe PD, Nicholson BJ. Selective transfer of endogenous metabolites through gap junctions composed of different connexins. Nat Cell Biol. 1999;1:457–459. doi: 10.1038/15693. [DOI] [PubMed] [Google Scholar]
- Goldberg GS, Moreno AP, Lampe PD. Gap junctions between cells expressing connexin 43 or 32 show inverse permselectivity to adenosine and ATP. J Biol Chem. 2002;277:36 725–36 730. doi: 10.1074/jbc.M109797200. [DOI] [PubMed] [Google Scholar]
- Hanada T, Yoshimura A. Regulation of cytokine signaling and inflammation. Cytokine Growth Factor Rev. 2002;13:413–421. doi: 10.1016/s1359-6101(02)00026-6. [DOI] [PubMed] [Google Scholar]
- Hanisch UK. Microglia as a source and target of cytokines. Glia. 2002;40:140–155. doi: 10.1002/glia.10161. [DOI] [PubMed] [Google Scholar]
- Huang RP, Fan Y, Hossain MZ, Peng A, Zeng ZL, Boynton AL. Reversion of the neoplastic phenotype of human glioblastoma cells by connexin 43 (cx43) Cancer Res. 1998;58:5089–5096. [PubMed] [Google Scholar]
- John GR, Scemes E, Suadicani SO, Liu JS, Charles PC, Lee SC, Spray DC, Brosnan CF. IL-1beta differentially regulates calcium wave propagation between primary human fetal astrocytes via pathways involving P2 receptors and gap junction channels. Proc Natl Acad Sci USA. 1999;96:11 613–11 618. doi: 10.1073/pnas.96.20.11613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kielian T. Microglia and chemokines in infectious diseases of the nervous system: views and reviews. Front Biosci. 2004a;9:732–750. doi: 10.2741/1266. [DOI] [PubMed] [Google Scholar]
- Kielian T. Immunopathogenesis of brain abscess. J Neuroinflammation. 2004b;1:16. doi: 10.1186/1742-2094-1-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kielian T, Esen N. Effects of neuroinflammation on glia–glia gap junctional intercellular communication: a perspective. Neurochem Int. 2004;45:429–436. doi: 10.1016/j.neuint.2003.09.010. [DOI] [PubMed] [Google Scholar]
- Kielian T, Mayes P, Kielian M. Characterization of microglial responses to Staphylococcus aureus: effects on cytokine, costimulatory molecule, and Toll-like receptor expression. J Neuroimmunol. 2002;130:86–99. doi: 10.1016/s0165-5728(02)00216-3. [DOI] [PubMed] [Google Scholar]
- Kielian T, McMahon M, Bearden ED, Baldwin AC, Drew PD, Esen N. S. aureus-dependent microglial activation is selectively attenuated by the cyclopentenone prostaglandin 15-deoxy-Delta12,14-prostaglandin J2 (15d-PGJ2) J Neurochem. 2004;90:1163–1172. doi: 10.1111/j.1471-4159.2004.02579.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kielian T, Esen N, Bearden ED. Toll-like receptor 2 (TLR2) is pivotal for recognition of S. aureus peptidoglycan but not intact bacteria by microglia. Glia. 2005;49:567–576. doi: 10.1002/glia.20144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauener RP, Geha RS, Vercelli D. Engagement of the monocyte surface antigen CD14 induces lymphocyte function-associated antigen-1/intercellular adhesion molecule-1-dependent homotypic adhesion. J Immunol. 1990;145:1390–1394. [PubMed] [Google Scholar]
- Lee SJ, Benveniste EN. Adhesion molecule expression and regulation on cells of the central nervous system. J Neuroimmunol. 1999;98:77–88. doi: 10.1016/s0165-5728(99)00084-3. [DOI] [PubMed] [Google Scholar]
- Lin JH, Yang J, Liu S, Takano T, Wang X, Gao Q, Willecke K, Nedergaard M. Connexin mediates gap junction-independent resistance to cellular injury. J Neurosci. 2003;23:430–441. doi: 10.1523/JNEUROSCI.23-02-00430.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez AD, Eugenin EA, Branes MC, Bennett MV, Sae ZJC. Identification of second messengers that induce expression of functional gap junctions in microglia cultured from newborn rats. Brain Res. 2002;943:191–201. doi: 10.1016/s0006-8993(02)02621-5. [DOI] [PubMed] [Google Scholar]
- Mathisen GE, Johnson JP. Brain abscess. Clin Infect Dis. 1997;25:763–779. doi: 10.1086/515541. quiZ. 780–761. [DOI] [PubMed] [Google Scholar]
- Moorby C, Patel M. Dual functions for connexins: Cx43 regulates growth independently of gap junction formation. Exp Cell Res. 2001;271:238–248. doi: 10.1006/excr.2001.5357. [DOI] [PubMed] [Google Scholar]
- Nagy JI, Li W, Hertzberg EL, Marotta CA. Elevated connexin43 immunoreactivity at sites of amyloid plaques in Alzheimer’s disease. Brain Res. 1996;717:173–178. doi: 10.1016/0006-8993(95)01526-4. [DOI] [PubMed] [Google Scholar]
- Nakase T, Fushiki S, Naus CC. Astrocytic gap junctions composed of connexin 43 reduce apoptotic neuronal damage in cerebral ischemia. Stroke. 2003;34:1987–1993. doi: 10.1161/01.STR.0000079814.72027.34. [DOI] [PubMed] [Google Scholar]
- Nakase T, Sohl G, Theis M, Willecke K, Naus CC. Increased apoptosis and inflammation after focal brain ischemia in mice lacking connexin43 in astrocytes. Am J Pathol. 2004;164:2067–2075. doi: 10.1016/S0002-9440(10)63765-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neijssen J, Herberts C, Drijfhout JW, Reits E, Janssen L, Neefjes J. Cross-presentation by intercellular peptide transfer through gap junctions. Nature. 2005;434:83–88. doi: 10.1038/nature03290. [DOI] [PubMed] [Google Scholar]
- Qin H, Shao Q, Curtis H, Galipeau J, Belliveau DJ, Wang T, Alaoui-Jamali MA, Laird DW. Retroviral delivery of connexin genes to human breast tumor cells inhibits in vivo tumor growth by a mechanism that is independent of significant gap junctional intercellular communication. J Biol Chem. 2002;277:29 132–29 138. doi: 10.1074/jbc.M200797200. [DOI] [PubMed] [Google Scholar]
- Rouach N, Avignone E, Meme W, Koulakoff A, Venance L, Blomstrand F, Giaume C. Gap junctions and connexin expression in the normal and pathological central nervous system. Biol Cell. 2002a;94:457–475. doi: 10.1016/s0248-4900(02)00016-3. [DOI] [PubMed] [Google Scholar]
- Rouach N, Calvo CF, Glowinski J, Giaume C. Brain macrophages inhibit gap junctional communication and downregulate connexin 43 expression in cultured astrocytes. Eur J Neurosci. 2002b;15:403–407. doi: 10.1046/j.0953-816x.2001.01868.x. [DOI] [PubMed] [Google Scholar]
- Rozental R, Srinivas M, Spray DC. How to close a gap junction channel: efficacies and potencies of uncoupling agents. In: Bruzzone RAGC, editor. Methods in Molecular Biology. Vol. 154. Humana Press; Totowa: 2000. pp. 447–476. [DOI] [PubMed] [Google Scholar]
- Saez JC, Berthoud VM, Branes MC, Martinez AD, Beyer EC. Plasma membrane channels formed by connexins: their regulation and functions. Physiol Rev. 2003;83:1359–1400. doi: 10.1152/physrev.00007.2003. [DOI] [PubMed] [Google Scholar]
- Shrikant P, Weber E, Jilling T, Benveniste EN. Intercellular adhesion molecule-1 gene expression by glial cells. Differential mechanisms of inhibition by IL-10 and IL-6. J Immunol. 1995;155:1489–1501. [PubMed] [Google Scholar]
- Siushansian R, Bechberger JF, Cechetto DF, Hachinski VC, Naus CC. Connexin43 null mutation increases infarct size after stroke. J Comp Neurol. 2001;440:387–394. doi: 10.1002/cne.1392. [DOI] [PubMed] [Google Scholar]
- Spray DC, Duffy HS, Scemes E. Gap junctions in glia: types, roles, and plasticity. In: Tsacopoulos MA, editor. The Functional Roles of Glial Cells in Health and Disease. Plenum Publishers; New York: 1999. pp. 339–359. [PubMed] [Google Scholar]
- Stout CE, Costantin JL, Naus CC, Charles AC. Intercellular calcium signaling in astrocytes via ATP release through connexin hemichannels. J Biol Chem. 2002;277:10 482–10 488. doi: 10.1074/jbc.M109902200. [DOI] [PubMed] [Google Scholar]
- Stout C, Goodenough DA, Paul DL. Connexins: functions without junctions. Curr Opin Cell Biol. 2004;16:507–512. doi: 10.1016/j.ceb.2004.07.014. [DOI] [PubMed] [Google Scholar]
- Theis M, Sohl G, Eiberger J, Willecke K. Emerging complexities in identity and function of glial connexins. Trends Neurosci. 2005;28:188–195. doi: 10.1016/j.tins.2005.02.006. [DOI] [PubMed] [Google Scholar]
- Townsend GC, Scheld WM. Infections of the central nervous system. Adv Intern Med. 1998;43:403–447. [PubMed] [Google Scholar]
- Tran Van Nhieu G, Clair C, Bruzzone R, Mesnil M, Sansonetti P, Combettes L. Connexin-dependent inter-cellular communication increases invasion and dissemination of Shigella in epithelial cells. Nat Cell Biol. 2003;5:720–726. doi: 10.1038/ncb1021. [DOI] [PubMed] [Google Scholar]
- Warn-Cramer BJ, Lampe PD, Kurata WE, Kanemitsu MY, Loo LW, Eckhart W, Lau AF. Characterization of the mitogen-activated protein kinase phosphorylation sites on the connexin-43 gap junction protein. J Biol Chem. 1996;271:3779–3786. doi: 10.1074/jbc.271.7.3779. [DOI] [PubMed] [Google Scholar]
- Warn-Cramer BJ, Cottrell GT, Burt JM, Lau AF. Regulation of connexin-43 gap junctional intercellular communication by mitogen-activated protein kinase. J Biol Chem. 1998;273:9188–9196. doi: 10.1074/jbc.273.15.9188. [DOI] [PubMed] [Google Scholar]
- Willecke K, Eiberger J, Degen J, Eckardt D, Romualdi A, Guldenagel M, Deutsch U, Sohl G. Structural and functional diversity of connexin genes in the mouse and human genome. Biol Chem. 2002;383:725–737. doi: 10.1515/BC.2002.076. [DOI] [PubMed] [Google Scholar]