

SUMMARY
To better understand the signaling properties of oncogenic FGFR3, we performed phospho-proteomics studies to identify potential downstream signaling effectors that are tyrosine phosphorylated in hematopoietic cells expressing constitutively activated leukemogenic FGFR3 mutants. We found that FGFR3 directly tyrosine phosphorylates the serine/threonine kinase p90RSK2 at Y529, which consequently regulates RSK2 activation by facilitating inactive ERK binding to RSK2 that is required for ERK-dependent phosphorylation and activation of RSK2. Moreover, inhibition of RSK2 by siRNA or a specific RSK inhibitor fmk effectively induced apoptosis in FGFR3-expressing human t(4;14)-positive myeloma cells. Our findings suggest that FGFR3 mediates hematopoietic transformation by activating RSK2 in a two-step fashion, promoting both the ERK-RSK2 interaction and subsequent phosphorylation of RSK2 by ERK.
INTRODUCTION
FGFR3 is one of four receptor-tyrosine kinases that respond to fibroblast growth factor (FGF), and negatively regulates bone formation in mammals (Colvin et al., 1996; Deng et al., 1996). FGFR3 is composed of an extracellular ligand-binding domain, a transmembrane domain, and a split cytoplasmic tyrosine kinase domain. FGFR3 is activated by oligomerization induced by ligand binding, and the consequent transautophosphorylation at tyrosine residues in the cytoplasmic domain is required for stimulation of the intrinsic catalytic activity and activation of downstream signaling pathways (Hart et al., 2001; Webster and Donoghue, 1996).
Recent studies have suggested that FGFR3 may play a significant role in the pathogenesis and disease progression of some hematopoietic malignancies including multiple myeloma. Multiple myeloma (MM) is a clonal proliferation of terminally differentiated plasma cells and is among the most common hematologic malignancies in patients over the age of 65. Recurrent translocations involving 14q32 into the immunoglobulin heavy (IgH)-chain switch region are frequent in human multiple myeloma cells (Bergsagel et al., 1996; Bergsagel and Kuehl, 2001). The translocations usually result in dysregulated expression of several heterogeneous partners including c-myc, cyclin D1 (Chesi et al., 1996), c-maf (Chesi et al., 1998a), and FGFR3 (Chesi et al., 1997). The t(4;14) translocation involving FGFR3 has been identified in approximately 15% of multiple myeloma patients and cell lines (Chesi et al., 1997; Chesi et al., 1998b). In some cases, the translocated FGFR3 gene contains an activating mutation K650E that, when present in the germ line, causes thanatophoric dysplasia type II (TDII) (Tavormina et al., 1995). FGFR3 is also involved in the t(4;12)(p16;p13)-associated peripheral T cell lymphoma (PTCL) that progresses into acute myeloid leukemia (AML), which generates the TEL-FGFR3 fusion protein with constitutive tyrosine kinase activity (Yagasaki et al., 2001).
SIGNIFICANCE.
Dysregulated tyrosine kinases play a pathogenic role in diverse forms of hematopoietic malignancies and thus represent potential therapeutic targets. Identification of critical downstream signaling effectors will provide not only the signaling basis of tyrosine kinase-induced hematopoietic transformation but also potential alternative targets in treatment of relevant diseases. Here we report a two-step model that leukemogenic FGFR3 activates RSK2 by both assisting inactive ERK binding via tyrosine phosphorylation of RSK2 at Y529 and activating the MEK/ERK pathway. Targeting RSK2 effectively induced apoptosis in FGFR3-expressing human t(4;14)-positive myeloma cells, suggesting RSK2 is a critical signaling effector in FGFR3-mediated hematopoietic transformation. RSK2 may represent an alternative therapeutic target in the treatment of diverse human malignancies associated with dysregulated FGFR3.
Ectopic expression of FGFR3 has been demonstrated to mediate transformation in hematopoietic cells. In a murine bone marrow transplantation (BMT) model, mice transplanted with bone marrow cells transduced by retroviral vectors carrying wild-type FGFR3 or FGFR3 TDII mutant exclusively develop lethal pro-B or pre-B cell lymphomas, respectively (Li et al., 2001). Moreover, we and others have demonstrated the therapeutic efficacy of small molecule tyrosine kinase inhibitors including PKC412, PD173074, and SU5402, which effectively inhibit FGFR3, in murine hematopoietic Ba/F3 cells; FGFR3-expressing t(4;14)-positive primary MM cell lines including KMS11, KMS18, and OPM-2; as well as in BMT and xenograft murine models (Chen et al., 2005a; Grand et al., 2004; Paterson et al., 2004; Trudel et al., 2004).
In humans, activating mutations of FGFR3 do not occur concurrently in the same myeloma cells with activating mutations of K-ras and N-ras, which are present in ~40% of multiple myeloma patients. Thus, FGFR3 may share the signaling pathways with ras-activating mutations such as the Ras/Raf/MEK/MAPK pathway and play a similar role in multiple myeloma progression (Chesi et al., 2001). We have reported that both leukemogenic FGFR3 TDII and TEL-FGFR3 activate the MEK/ERK pathway (Chen et al., 2005b). The Ser/Thr kinase RSK2 is a substrate of ERK and belongs to a family containing four members, RSK1–RSK4. RSK family members share structural and functional similarities, and contain two distinct kinase domains, both of which are catalytically functional (reviewed in Blenis, 1993; Frodin and Gammeltoft, 1999). The C-terminal kinase domain (CTD) is responsible for autophosphorylation at Ser386 (numbering based on the murine RSK2 amino acid sequence) that is critical in RSK activation, whereas the N-terminal kinase domain (NTD) is believed to phosphorylate exogenous substrates of RSK (Fisher and Blenis, 1996) (Figure 1A). RSK plays an active role in antiapoptosis signaling by phosphorylating BAD (Shimamura et al., 2000), C/EBPβ (Buck et al., 2001), and death-associated protein (DAP) kinase (Anjum et al., 2005) to protect cells from apoptosis. RSK has also been implicated in cell cycle regulation and has been found to phosphorylate and inhibit Myt1, a p34cdc2 inhibitory kinase in Xenopus extracts (Palmer et al., 1998).
Figure 1. p90RSK2 Is Tyrosine Phosphorylated in FGFR3-Expressing Hematopoietic Ba/F3 Cells.

(A) Schematic diagram of p90RSK2 shows domain structure and residues phosphorylated during RSK2 activation by ERK and PDK1 (upper panel). N/CTD, N/C-terminal kinase domain. S386 is autophosphorylated by activated CTD. The two phosphorylated tyrosine residues (Y488 and Y529) identified in the proteomics studies are indicated. Lower panel shows MS spectra of phospho-Tyr peptide fragments of RSK2 with xCorr = 4.1592 for peptide containing Y488 and xCorr = 4.8611 for peptide containing Y529.
(B) TEL-FGFR3 requires its tyrosine kinase activity to induce tyrosine phosphorylation of RSK2 in Ba/F3 cells. Ba/F3 cells stably expressing TEL-FGFR3 were cultured in media with serum and IL-3 withdrawal in the presence or absence of PKC412 for 4 hr prior to harvest. Ba/F3 cells treated with IL-3 withdrawal were included as controls.
(C) RSK2 is tyrosine phosphorylated in Ba/F3 cells expressing TEL-FGFR3 or FGFR3 TDII but not in cells expressing the kinase-defective mutants TEL-FGFR3 K508R or FGFR3 TDII FF4F.
The precise mechanism of RSK activation remains elusive. The current model suggests that ERK-dependent activation of RSK contains a series of sequential events. First, inactive ERK binds to the C terminus of RSK in quiescent cells, and that this interaction is an absolute requirement for activation of RSK (Gavin and Nebreda, 1999; Roux et al., 2003; Smith et al., 1999). Second, when a stimulating signal such as mitogen comes, ERK is activated and phosphorylates RSK at Thr577 (murine RSK2 numbering) in the activation loop of the CTD domain and Ser369 and Thr365 in the linker region between the two kinase domains, leading to activation of the RSK CTD domain. Third, activation of CTD domain results in autophosphorylation of Ser386 in the linker region, which provides a docking site for 3-phosphoinositide-dependent protein kinase 1 (PDK1) (Frodin et al., 2000). PDK1 in turn phosphorylates Ser227 in the activation loop of the NTD domain, allowing RSK to phosphorylate its downstream targets (Jensen et al., 1999). Last, activated NTD autophosphorylates Ser749 at the C-terminal domain of RSK, which results in dissociation of active ERK from RSK (Roux et al., 2003).
Here we present a two-step model that, in addition to FGFR3-mediated activation of the MEK/ERK pathway, FGFR3 tyrosine phosphorylates RSK2 at Y529, which regulates the activation of the serine/threonine kinase RSK2 by allowing inactive ERK to bind RSK2 in the initial step. Moreover, inhibition of RSK2 by specific siRNA or a highly specific small molecule RSK inhibitor fmk (Cohen et al., 2005) induced significant apoptosis in human t(4;14)-positive, FGFR3-expressing myeloma cells, suggesting RSK2 is a critical effector in FGFR3 mediated transformation.
RESULTS
RSK2 Is Specifically Tyrosine Phosphorylated in FGFR3-Expressing Hematopoietic Ba/F3 Cells
To better understand the signaling properties of leukemogenic FGFR3, we performed a mass spectrometry-based proteomics study to identify the profile of tyrosine phosphorylated proteins in murine Ba/F3 cells stably expressing the TEL-FGFR3 fusion. Ba/F3 cells require IL-3 for cell survival and proliferation, and constitutively activated TEL-FGFR3 confers IL-3-independent proliferation to Ba/F3 cells (Chen et al., 2005b). We identified a large spectrum of proteins that are tyrosine phosphorylated in Ba/F3 cells stably expressing TEL-FGFR3 compared to control cells in the absence of IL-3, many of which are important for cell growth control and tumorigenesis including previously reported STAT5 and PLCγ (data not shown). Among these proteins, we identified p90 ribosomal S6 kinase 2 (RSK2) as a potential FGFR3 downstream effector due to its critical role in cell proliferation and survival. The upper panel of Figure 1A shows a schematic illustration of p90RSK2 and the serine/threonine residues that are phosphorylated by ERK and PDK1 for activation. RSK2 was identified to be specifically tyrosine-phosphorylated at Y488 and Y529 due to expression of the constitutively activated TEL-FGFR3 in the proteomics studies (Figure 1A, lower panel). Both Y488 and Y529 are located outside of the activation loop in the CTD domain of RSK2.
We next confirmed the tyrosine-phosphorylation of RSK2 in hematopoietic cells expressing FGFR3. Control Ba/F3 cells and cells stably expressing TEL-FGFR3 were cultured in the presence or absence of the FGFR3 inhibitor PKC412 (Chen et al., 2005a). Immunoblotting results showed that RSK2 was tyrosine phosphorylated in Ba/F3 cells expressing TEL-FGFR3, whereas PKC412 treatment abolished tyrosine phosphorylation of RSK2 by inhibiting TEL-FGFR3 (Figure 1B). Moreover, tyrosine-phosphorylation of RSK2 was abolished in cells stably expressing a kinase-dead K508R mutant form of TEL-FGFR3 (Chen et al., 2005b) (Figure 1C, left).
Tyrosine-phosphorylated RSK2 was also detected in Ba/F3 cells stably expressing leukemogenic full-length FGFR3 TDII mutant with the activating mutation K650E, which was further activated in the presence of ligand acidic FGF (aFGF) (Chen et al., 2005b) (Figure 1C, right). In contrast, cells stably expressing the kinase defective mutant FGFR3 TDII FF4F (Y647/Y648) (Chen et al., 2005b) were unable to induce tyrosine phosphorylation of RSK2. Together, these data suggest that expression of leukemogenic, constitutively activated FGFR3 variants results in tyrosine phosphorylation of RSK2, which requires FGFR3 kinase activity.
RSK2 Is Activated in Hematopoietic Cells Expressing Leukemogenic FGFR3 Variants
We next tested whether expression of constitutively activated FGFR3 variants results in activation of RSK2. As shown in Figure 2A, endogenous RSK2 was highly activated as assessed by phosphorylation at Ser386 in control Ba/F3 cells in the presence of IL-3 compared to cells subjected to IL-3 withdrawal. RSK2 was also activated in Ba/ F3 cells expressing ligand-independent TEL-FGFR3, as well as in cells expressing FGFR3 TDII where Ser386 phosphorylation was enhanced in the presence of ligand aFGF (Figure 2A). In addition, PKC412 treatment or kinase-defective mutation K508R significantly attenuated the TEL-FGFR3-induced phosphorylation of RSK2 at Ser386 (Figure 2B).
Figure 2. Constitutively Activated FGFR3 Variants Activate RSK2 in Hematopoietic Cells.
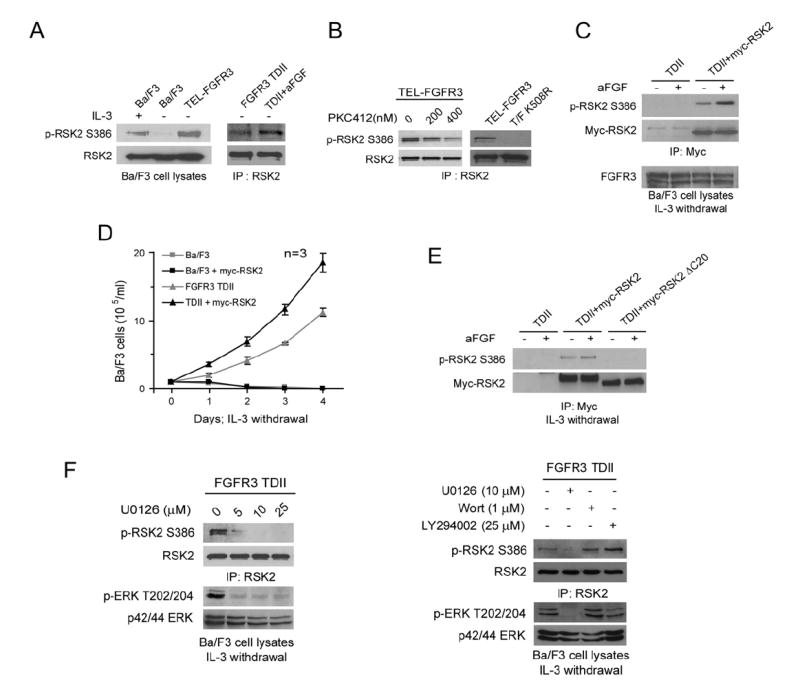
(A) Left panel shows activation of endogenous RSK2 as assessed by phosphorylation at Ser386 in cells expressing TEL-FGFR3. Ba/F3 cells in the presence or absence of IL-3 were included as controls. Right panel shows that RSK2 is further activated in Ba/F3 cells stably expressing FGFR3 TDII in the presence of aFGF ligand.
(B) Tyrosine kinase activity is required for TEL-FGFR3-dependent activation of RSK2. Left: Ba/F3 cells stably expressing TEL-FGFR3 were cultured in the presence of increasing concentrations of PKC412. Right: Cells expressing the kinase-defective mutant TEL-FGFR3 (T/F) K508R were included as a negative control.
(C) Expression of myc-RSK2 enhanced FGFR3 TDII-dependent activation of RSK2. Myc-tagged RSK2 was stably transduced into Ba/F3 cells expressing FGFR3 TDII. Phosphorylation level of myc-RSK2 at Ser386 was assessed by western blotting.
(D) Expression of myc-RSK2 enhanced FGFR3 TDII conferred IL-3-independent proliferation of Ba/F3 cells. Cells were cultured in absence of IL-3 and counted daily; control Ba/F3 cells were included. The data are presented as means ± SD (n = 3).
(E) FGFR3 activates RSK2 through the MEK/ERK pathway. Control RSK2 ΔC20 mutant harbors a deletion of 20 amino acids at the C terminus that are required for ERK binding.
(F) Treatment of MEK inhibitor U0126 abolishes FGFR3-dependent RSK2 activation. Ba/F3 cells stably expressing FGFR3 TDII were treated with serum and IL-3 withdrawal and aFGF stimulation for 4 hr followed by 90 min treatment of increasing concentrations of U0126 (left) or 10 μM U0126 with PI3K inhibitors wortmannin and LY294002 as negative controls (right). Western blotting was performed to detect phosphorylation and expression levels of RSK2 and ERK.
Moreover, in cells stably expressing both FGFR3 TDII and myc-tagged RSK2, the exogenous myc-RSK2 was highly phosphorylated and activated. TDII-dependent activation of myc-RSK2 was further enhanced in the presence of aFGF (Figure 2C). In addition, stable expression of myc-tagged RSK2 enhanced FGFR3 TDII-induced IL-3 independent proliferation of Ba/F3 cells. As shown in Figure 2D, in the absence of IL-3, control Ba/F3 cells or cells stably expressing myc-RSK2 alone underwent apoptotic cell death, suggesting that overexpression of myc-RSK2 is not oncogenic in the absence of active FGFR3 signaling. In contrast, stable expression of TDII conferred IL-3-independence to Ba/F3 cell lines, which was enhanced by coexpression of myc-RSK2 as assessed by proliferative rate (Figure 2D). Together, these data indicate that RSK2 may function as a downstream signaling effector and contribute to FGFR3-mediated hematopoietic transformation.
FGFR3 Activates RSK2 through the MEK/ERK Pathway
We next tested whether ERK is required for RSK2 activation by FGFR3. Compared with myc-RSK2, a myc-RSK2 mutant, ΔC20, which has a deletion of the 20 amino acids at the C-terminus required for ERK binding (Gavin and Nebreda, 1999), was not phosphorylated at Ser386 in the presence of FGFR3 TDII even following aFGF stimulation (Figure 2E). This observation suggests that FGFR3-dependent activation of RSK2 requires ERK binding. This is in consonance with the data that in Ba/F3 cells stably expressing FGFR3 TDII, treatment of MEK1 inhibitor U0126, but not the PI3K inhibitors wortmannin and LY294002, effectively inhibited phosphorylation at RSK2 Ser386 (Figure 2F) as well as RSK2 kinase activity in an in vitro kinase assay (see Figure S1 in the Supplemental Data available with this article online).
FGFR3-Dependent Tyrosine Phosphorylation at Y529 Is Required for Phosphorylation and Activation of RSK2 by ERK
To further elucidate the role of tyrosine phosphorylation induced by FGFR3 in RSK2 activation, we characterized a group of RSK2 mutants with single or double Y→F substitutions at Y488 and Y529. Retroviral vectors encoding distinct myc-tagged RSK2 mutants with a puromycin resistance gene were stably transduced into Ba/F3 cells that already stably expressed FGFR3 TDII. Myc-RSK2 proteins were immunoprecipitated and assayed for specific phosphorylation at the three ERK-dependent phosphorylation sites including Thr365, Ser369, and Thr577 as well as at Ser386 as the index of RSK2 activation. As shown in Figure 3A, myc-RSK2 and Y488F mutant were highly phosphorylated at all of the aforementioned residues induced by FGFR3 TDII in the presence of ligand aFGF, whereas phosphorylation at these residues was completely abolished in the control myc-RSK2 ΔC20 mutant that does not bind ERK. In contrast, myc-RSK2 Y529F and double mutant Y488/529F (2F) demonstrated decreased phosphorylation levels of Thr365, Ser369, Thr577, and Ser386, suggesting substitution of Y529 attenuates ERK-dependent phosphorylation and activation of RSK2 induced by FGFR3 TDII (Figure 3A).
Figure 3. FGFR3-Dependent Tyrosine Phosphorylation at Y529 Is Required for Phosphorylation and Activation of RSK2 by ERK.
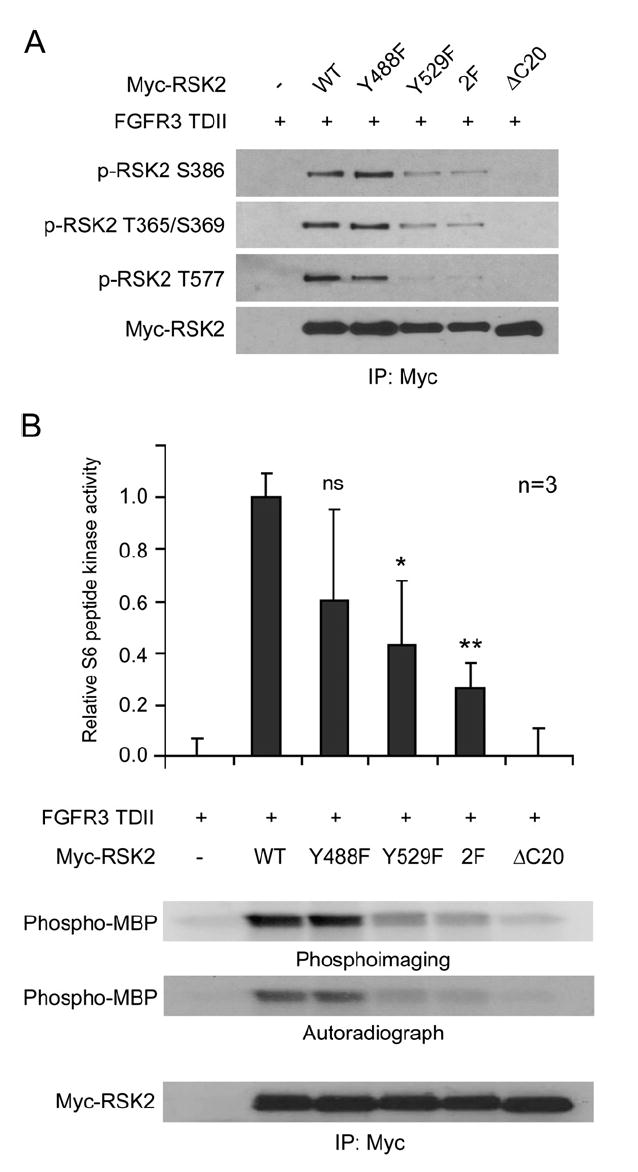
(A) Substitution of Y529 on RSK2 attenuates ERK-dependent phosphorylation and activation of RSK2.
(B) Y529F decreases RSK2 kinase activity in the presence of FGFR3 TDII. Ba/F3 cells stably expressing FGFR3 TDII and distinct myc-tagged RSK2 variants were cultured in the presence of aFGF with withdrawal of IL-3 and serum for 4 hr. Immunocomplexes of RSK2 variants were isolated and incubated with equal amount of S6-peptide (top) or MBP (middle) as exogenous substrates, as well as [γ-32P] ATP. The phosphorylation of S6 peptide was normalized to readings from reactions using RSK2 immunocomplexes from cells stably expressing FGFR3 TDII alone (0.0) and cells coexpressing TDII and myc-RSK2 wild-type (1.0). The data were presented as mean ± SD; the p values were determined by Student’s t test; ns = not significant; 2F = Y488/529F. Bottom: Parallel immunoprecipitation and western blotting results confirmed equal amounts of myc-RSK2 proteins in each kinase reaction.
We also tested the kinase activity of these RSK2 mutants in in vitro kinase assays. The myc-RSK2 variants were immunoprecipitated from cell lysates of the respective Ba/F3 cell lines stably coexpressing FGFR3 TDII. The immunocomplexes were incubated with a specific exogenous substrate S6 peptide in the presence of [γ-32P] ATP. Both myc-RSK2 Y529F and 2F mutants incorporated significantly less 32P into S6 peptide than did the myc-RSK2 wild-type, whereas the negative control myc-RSK2 ΔC20 mutant completely lost the ability to phosphorylate S6 peptide (Figure 3B, top), correlating with the data for ERK-dependent phosphorylation. In contrast, mutation at Y488 did not significantly affect the RSK2 kinase activity. Similar results were obtained by using MBP (myelin basic protein) as a nonspecific exogenous substrate (Vaidyanathan and Ramos, 2003) (Figure 3B, middle). Immunoblotting showed that equal amounts of myc-RSK2 immunoprecipitates were applied in each sample (Figure 3B, bottom).
Substitution of Y529 Attenuates Inactive ERK Binding to RSK2
Inactive ERK interacts with RSK2 in quiescent cells, which is prior to and required for ERK-dependent phosphorylation and activation of RSK2 (Roux et al., 2003). We next tested whether FGFR3-induced phosphorylation at Y529 may regulate RSK2/ERK interaction. The Ba/F3 cell lines stably expressing FGFR3 TDII and the respective Myc-RSK2 variants were treated with the MEK1 inhibitor U0126, since active ERK readily dissociates from RSK2 (Roux et al., 2003). As shown in Figure 4A, the immunoprecipitation results showed that substitution of Y529 in myc-RSK2 decreased ERK association compared to the myc-RSK2 wild-type and Y488F mutant, whereas the negative control myc-RSK2 ΔC20 mutant completely lost the ability to interact with inactive ERK. No phospho-ERK was detected in the immunocomplexes of each RSK2 variant by western blot (data not shown) suggesting that the coimmunoprecipitated ERK was inactive upon U0126 treatment. These data suggest that tyrosine phosphorylation by FGFR3 regulates RSK2 activation by facilitating inactive ERK binding.
Figure 4. FGFR3 Directly Phosphorylates RSK2 at Y529 to Facilitate Inactive ERK Binding to RSK2.

(A) Y529F mutation attenuates inactive ERK binding to RSK2. Ba/F3 cells stably expressing FGFR3 TDII and distinct myc-tagged RSK2 variants were cultured in the absence of serum for 4 hr in the presence of U0126 (10 μM) prior to coimmunoprecipitation.
(B) Global structure of RSK2 proteins is not altered by point mutations, which was determined by similar partial protease digestion patterns of (His)6-RSK2 WT and variants. Recombinant proteins (2.5 μg) were incubated with 0.5 unit of chymotrypsin for 30 min at 37°C.
(C) RSK2 Y529 is specifically phosphorylated in cells expressing FGFR3, detected by an antibody specifically recognizes phospho-Y529 of RSK2.
(D) FGFR3 directly phosphorylates RSK2 at Y529. Purified recombinant RSK2 (rRSK2) C-terminal kinase domain (CTD) and CTD-Y529F proteins were incubated with recombinant FGFR3 kinase domain (rFGFR3) that is constitutively activated. Phosphorylation at Y529 in rRSK2 CTD was detected by specific antibody pRSK2 (Y529).
(E) RSK2 Y529 is specifically phosphorylated by FGFR3, which consequently facilitates inactive ERK binding. GST-tagged RSK2 wild-type or Y529F mutant were pulled down by beads from transfected 293T cell lysates and treated with protein tyrosine phosphatase (PTP), followed by treatment of activated rFGFR3. The beads were then incubated with U0126-treated 293T cell lysates. Reconstituted Y529 phosphorylation and inactive ERK in the complex of bead-bound GST-RSK2 were detected by immunoblotting.
(F) Y529F mutation does not intrinsically affect the kinase activation of RSK2. Purified rRSK2 CTD variants were incubated with or without activated recombinant ERK in an in vitro kinase assay. The phosphorylation of the specific CTD-tide peptide substrate in each reaction was normalized to readings from reaction of rRSK2 CTD WT with activated ERK (1.0) (mean ± SD).
We also evaluated the structural properties of the mutant proteins compared to RSK2 wild-type by analysis of the relative stability of proteins to limited proteolytic digestion with chymotrypsin (Zhang et al., 1997). Purified recombinant His-tagged RSK2 WT and mutants including Y488F, Y529F, and Y488/529F were incubated with chymotrypsin, and the resultant digestion patterns of the mutant proteins were similar compared to RSK2 WT (Figure 4B) suggesting that the global structure of each mutant protein was not altered and decreased kinase activation or inactive ERK-binding of RSK2 Y529F mutant is not due to structural alterations.
FGFR3 Directly Phosphorylates RSK2 at Y529 and Consequently Facilitates Inactive ERK Binding
To determine whether FGFR3-dependent RSK2 Y529 phosphorylation physiologically occurs in cells, we generated an antibody that specifically recognizes phospho-Y529 of RSK2. By using this antibody, we observed that RSK2 WT and Y488F mutant, but not Y529F mutant, were specifically tyrosine phosphorylated at Y529 in FGFR3 TDII-expressing 293T cells (Figure 4C, left) and Ba/F3 cells (Figure 4C, middle). Phosphorylation at Y529 was also abolished in TDII or TEL-FGFR3 stable cells treated with PKC412 that inhibits FGFR3 (Figure 4C, right).
We then determined whether FGFR3 phosphorylates RSK2 at Y529 directly or indirectly by activating other tyrosine kinases. In an in vitro kinase assay, purified recombinant RSK2 C-terminal kinase domain (CTD) proteins were incubated with recombinant FGFR3 kinase domain that is constitutively activated (Invitrogen). As shown in Figure 4D, wild-type RSK2 CTD domain was highly tyrosine phosphorylated at Y529 by FGFR3, whereas Y529 phosphorylation was abolished in the RSK2 CTD Y529F mutant. Using a pantyrosine phosphorylation antibody pY99, we observed comparable tyrosine phosphorylation levels in both RSK2 CTD WT and Y529F mutant (Figure 4D), suggesting that FGFR3 directly phosphorylates RSK2 at multiple sites, including Y529.
Next we tested whether the tyrosine phosphorylation of RSK2 at Y529 by FGFR3 precedes the inactive ERK binding to RSK2. We performed a GST pull-down assay in which the bead-bound, GST-tagged RSK2 WT or Y529F mutant proteins were dephosphorylated by protein tyrosine phosphatase (PTP) first to remove tyrosine phosphorylation, then treated with recombinant active FGFR3 followed by incubation with U0126-treated 293T cell lysates. As shown in Figure 4E, Y529 phosphorylation was reconstituted in GST-RSK2 WT along with increased inactive ERK binding, upon FGFR3 treatment. In contrast, no ERK binding to Y529F mutant was enhanced despite a minimal phosphorylation of RSK2 Y529 mutant detected by the specific phospho-Y529 antibody, which may be due to the residual nonspecific background of the antibody.
Although the Y529F mutation does not seem to alter the RSK2 global structure (Figure 4B), this mutation may intrinsically affect the kinase properties of RSK2. To test this possibility, we performed an in vitro kinase assay. Recombinant RSK2 CTD variants were incubated with or without activated recombinant ERK in the presence of a specific RSK2 CTD peptide substrate (Cohen et al., 2005). Previous studies have shown that in a similar in vitro kinase assay, purified active ERK is able to phosphorylate and activate an immunoprecipitated RSK2 (1–729) truncation mutant that cannot bind to ERK (Smith et al., 1999), which suggests that probably under this condition, the docking site mediated interaction is less critical for ERK to phosphorylate RSK2. As shown in Figure 4F, all of RSK2 CTD variants demonstrated comparable kinase activity in the presence of active ERK. These data suggest that Y529F mutation does not intrinsically alter the kinase activation of RSK2, and the mutant proteins fold properly.
Targeting RSK2 by a Specific Small Molecule RSK Inhibitor fmk Attenuates FGFR3-Induced Cytokine-Independent Growth in Ba/F3 Cells
Next we tested whether RSK2 is a critical signaling effector in FGFR3-mediated transformation signaling. We evaluated a specific RSK inhibitor, fmk, which is a fluoromethylketone molecule that was designed to specifically exploit two selectivity filters of RSK. Fmk potently inactivates the CTD auto-kinase activity of RSK1 and RSK2 with high specificity in mammalian cells (Figure 5A; Cohen et al., 2005). As shown in Figure 5B, fmk effectively inhibits FGFR3 TDII and TEL-FGFR3-induced IL-3-independent growth of Ba/F3 cells in a dose-responsive manner through attenuation of RSK2 activation as assessed by Ser386 phosphorylation (Figure 5C) but not inhibition of phosphorylation and activation of FGFR3 TDII or ERK (data not shown). Cells expressing FGFR3 TDII in the absence of ligand aFGF were more sensitive to fmk treatment compared to TDII stable cells cultured in the presence of aFGF (Figure 5B). This difference may be due to the possibility that constitutively activated FGFR3 TDII could be further activated by ligand, which consequently results in enhanced RSK2 activation (Figure 2A). The inhibition of RSK2 by fmk is not attributable to nonspecific cytotoxicity due to the lack of inhibition of control Ba/F3 cells by fmk in the presence of IL-3 (Figure 5B).
Figure 5. RSK Inhibitor fmk Inhibits Cytokine-Independent Proliferation of Ba/F3 Cells Conferred by FGFR3.
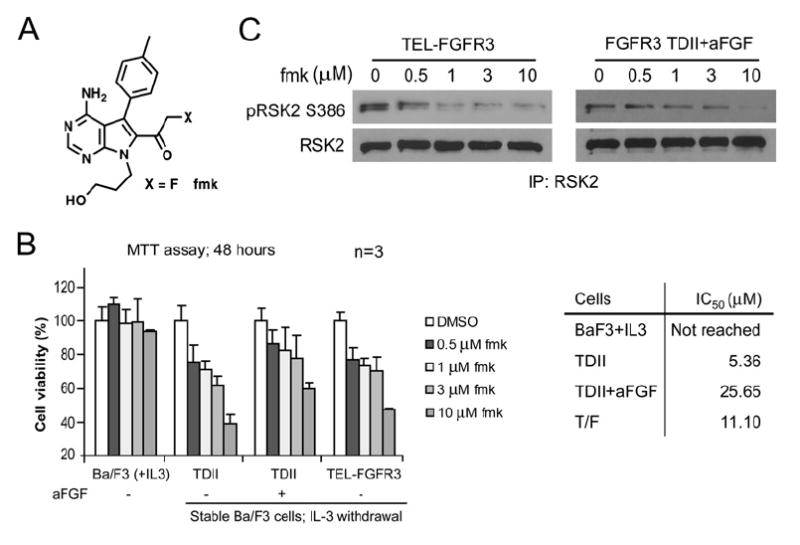
(A) Schematic representation of the specific RSK inhibitor, fmk (Cohen et al., 2005).
(B) Dose response analysis of Ba/F3 cells stably expressing FGFR3 TDII or TEL-FGFR3 to fmk. The relative cell viability was normalized to the viability of cells in the absence of fmk (mean ± SD). Right panel shows the cellular IC50 (μM) of distinct cell lines.
(C) Inhibitory effects of fmk on phosphorylation of RSK2 in Ba/F3 cells stably expressing TEL-FGFR3 or FGFR3 TDII.
RSK2 Is Specifically Phosphorylated at Y529 in FGFR3-Expressing, t(4;14)-Positive Human Myeloma Cell Lines
We also evaluated the role of RSK2 in human t(4;14)-positive, FGFR3-expressing multiple myeloma. RSK2 was highly phosphorylated at Y529 and activated as assessed by phosphorylation levels of Ser386 in four different t(4;14)-positive human myeloma cell lines (HMCLs). KMS11 cells express FGFR3 harboring a single activating mutation Y373C in the transmembrane domain. OPM1 cells overexpress the FGFR3 TDII mutant. H929 express FGFR3 wild-type, whereas LP1 cells express FGFR3 with a polymorphism of F384L in the transmembrane domain (Chesi et al., 2001; Golla et al., 1997; Ronchetti et al., 2001). In contrast, RSK2 was not phosphorylated at Y529 in three t(4;14)-negative HMCLs that do not express FGFR3 (Figure 6A). Moreover, RSK2 was phosphorylated at Ser386 and activated in RPMI8226 cells but not in ANBL6 and U266 cells (Figure 6A). This difference may be due to the active ras K12 mutation harbored by RPMI8226 cells (Chesi et al., 2001). Although U266 cells harbor a BRAF V599E mutation (Ng et al., 2003), RSK2 was not detected as Ser386 phosphorylated and activated (Figure 6A).
Figure 6. Targeting RSK2 by Specific siRNA Induces Apoptosis in FGFR3-Expressing, t(4;14)-Positive Human Myeloma Cell Lines.
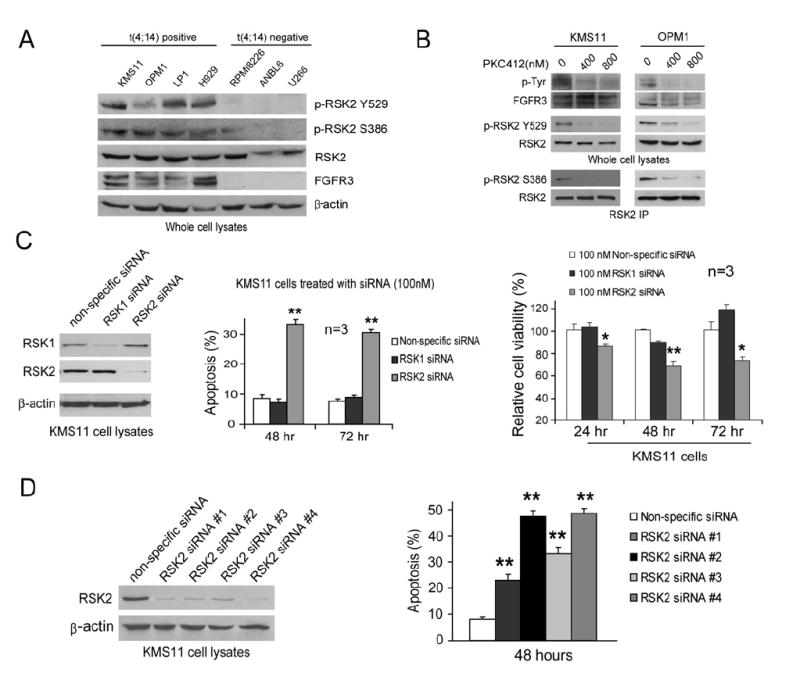
(A) RSK2 is specifically tyrosine-phosphorylated at Y529 and activated in FGFR3-expressing, t(4;14)-positive human myeloma cell lines.
(B) Inhibition of FGFR3 by PKC412 abolishes Y529 phosphorylation as well as activation of RSK2 in t(4;14)-positive KMS11 and OPM1 cells.
(C) Targeted downregulation of RSK2 but not RSK1 induces apoptotic cell death in KMS11 cells. Left: Transfection of siRNA targeting RSK1 or RSK2 specifically decreases RSK1 or RSK2 protein expression in KMS11 cells, respectively. β-actin was detected as a loading control. Middle: Induction of apoptosis by siRNA targeting RSK2 but not RSK1 in KMS11 cells. Cells were transfected with distinct siRNA for 48 hr and 72 hr prior to annexin V staining and flow cytometry analysis. The apoptotic population was characterized as annexin V positive cells. Right: KMS11 cells were transfected with distinct siRNAs for indicated periods. The relative cell viability was normalized to the viability of cells transfected by a nonspecific siRNA. The data were presented as mean ± SD (n = 3).
(D) All of the 4 individual siRNA in the RSK2 siRNA pool in Figure 6C significantly downregulated RSK2 protein expression and induced apoptosis in KMS11 cells (mean ± SD).
In consonance with these data, treatment of PKC412 that inhibits FGFR3 significantly attenuated FGFR3-dependent Y529 phosphorylation as well as activation of RSK2 in both t(4;14)-positive KMS11 and OPM1 cells (Figure 6B).
Targeted Downregulation of RSK2 but Not RSK1 Induces Apoptosis in FGFR3-Expressing Human Myeloma Cells
We next utilized pools of siRNA specifically targeting RSK1 or RSK2 and a nonspecific siRNA as a negative control to test whether targeted downregulation of RSK2 could induce apoptosis in FGFR3-expressing HMCLs. Both RSK1 and RSK2 siRNAs were highly specific in decreasing their respective target protein expression in KMS11 cells (Figure 6C, left). Transfection of RSK2 specific siRNA induced significant apoptosis in KMS11 cells as assessed by increased annexin V positivity (Figure 6C, middle) as well as decreased cell viability (Figure 6C, right) compared to cells transfected with nonspecific siRNA. In contrast, RSK1 siRNA failed to induce apoptotic cell death in KMS11 cells (Figure 6C). Similar results were obtained in another t(4;14)-positive HMCL, OPM1 (data not shown).
In order to examine potential off-target effects of individual siRNAs in the RSK2 siRNA pool, we tested the effects of four siRNAs targeting RSK2 with separate targeting sequences (Figure 6D). All of the four tested siRNAs effectively downregulated the protein expression of RSK2 and induced comparable apoptosis in KMS11 cells as observed using the pool of siRNA targeting RSK2 (Figure 6C). These data together suggest that RSK2 but not RSK1 may play a critical role in FGFR3-mediated transformation signaling.
fmk Induces Apoptosis in FGFR3-Expressing, t(4;14)-Positive HMCLs as Well as Primary Myeloma Cells
We observed that the specific RSK inhibitor fmk inhibited RSK2 activation as assessed by decreased phosphorylation levels of Ser386 in human t(4;14)-positive, FGFR3-expressing myeloma cell lines KMS11 and OPM1 (Figure 7A). Fmk treatment induced significant apoptosis in KMS11 cells in a dose-dependent manner, as assessed by increasing annexin V positivity and emergence of cleaved PARP (Figure 7B). KMS11 cells treated with the FGFR3 inhibitor PKC412 (Chen et al., 2005a) were included as a positive control. Fmk also induced significant apoptosis in other t(4;14)-positive, FGFR3-expressing HMCLs including OPM1, LP1, and KMS18 cells expressing FGFR3 with a single activating mutation G384D in the transmembrane domain (Ronchetti et al., 2001) (Figure 7C). KMS18 cells were less sensitive to fmk treatment compared to the other t(4;14)-positive HMCLs, which may be due to the relatively low RSK2 Ser386 phosphorylation and activation levels (data not shown) resulting from the combinatorial effects of multiple oncogenic mutations in different cell lines (discussed below). RPMI8226 cells that harbor an activating ras K12 mutation that leads to RSK2 activation (Figure 6A) also responded to fmk (Figure 7C). In contrast, HMCLs that do not express FGFR3 or harbor ras mutations including ANBL-6 (IL-6 dependent) and U266 (harboring BRAF V599E mutation) were resistant to fmk treatment (Figure 7C), presumably due to the lack of RSK2 activation in both cell lines (Figure 6A).
Figure 7. Targeting RSK2 by fmk Induces Significant Apoptosis in FGFR3-Expressing, t(4;14)-Positive Human Myeloma Cell Lines and Primary Human Myeloma Cells.
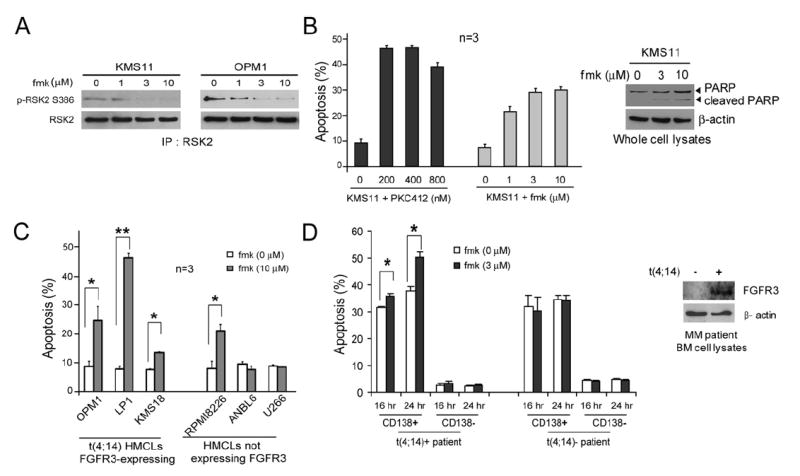
(A) fmk treatment inhibits RSK2 activation in t(4;14)-positive KMS11 and OPM1 cells in a dose-dependent manner.
(B) fmk effectively induces apoptosis in t(4;14)-positive KMS11 cells in a dose-dependent manner. Cells were treated with increasing concentrations of FGFR3 inhibitor PKC412 or fmk for 6 hr prior to FACS analysis. The apoptotic population was characterized as the fraction of annexin V positive cells of total treated cells (left; mean ± SD). Cleaved PARP was detected by western blotting (right).
(C) fmk induces significant apoptosis in human t(4;14)-positive OPM1, LP1, and KMS18 myeloma cells (mean ± SD).
(D) fmk induces significant apoptotic cell death in primary t(4;14)-positive multiple myeloma cells. Freshly isolated bone marrow mononuclear cells (BMNCs) from t(4;14)-positive and negative patients were incubated in the absence or presence of fmk for 16 and 24 hr. Cells were stained with annexin V-FITC and analyzed by flow cytometry. Myeloma cells were identified by CD138 labeling. The apoptotic population was characterized as the fraction of annexin V positive cells of total CD138 positive or negative cells (mean ± SD).
Moreover, we observed that fmk induced significant apoptosis in primary CD138-positive, FGFR3-expressing myeloma cells from a t(4;14)-positive multiple myeloma patient, but not in the control CD138-negative cells from the same patient, nor primary samples from a t(4;14)-negative patient as a control (Figure 7D). These data provide “proof of principle” that not only suggest the therapeutic potential of targeting RSK2 by fmk in t(4;14)-positive, FGFR3-expressing multiple myeloma, but also demonstrate that fmk has minimal nonspecific cytotoxicity in human myeloma cells.
DISCUSSION
Our data support a two-step model by which FGFR3 activates RSK2 and mediates transformation signals in hematopoietic cells. The first step involves tyrosine phosphorylation at Y529 of RSK2 by FGFR3, which facilitates binding of the inactive form of ERK to RSK2 in the initial step of ERK-dependent RSK2 activation (Figure 8, Step 1). This binding, which is required for phosphorylation and activation of RSK2 by ERK, in turn promotes the second step where ERK is activated via the Ras/Raf/ MEK/MAPK pathway downstream of FGFR3, leading to ERK-mediated phosphorylation and activation of RSK2 (Figure 8, Step 2). Thus, FGFR3 plays a dual role in the activation of RSK2 by both assisting inactive ERK binding to RSK2 and activating ERK to phosphorylate and activate RSK2. Moreover, inhibition of RSK2 by specific siRNA or small molecule inhibitor fmk effectively induces apoptosis in human t(4;14)-positive myeloma cells that express FGFR3, which demonstrates the importance of the RSK2 pathway in FGFR3-related myeloma. These studies therefore demonstrate that RSK2 is a critical signaling effector of FGFR3 and may represent a potential therapeutic target in hematologic malignancies associated with dysregulated FGFR3.
Figure 8. Proposed Two-Step Model that FGFR3 Activates RSK2 to Mediate Hematopoietic Transformation Involving Tyrosine Phosphorylation at Y529 in RSK2.
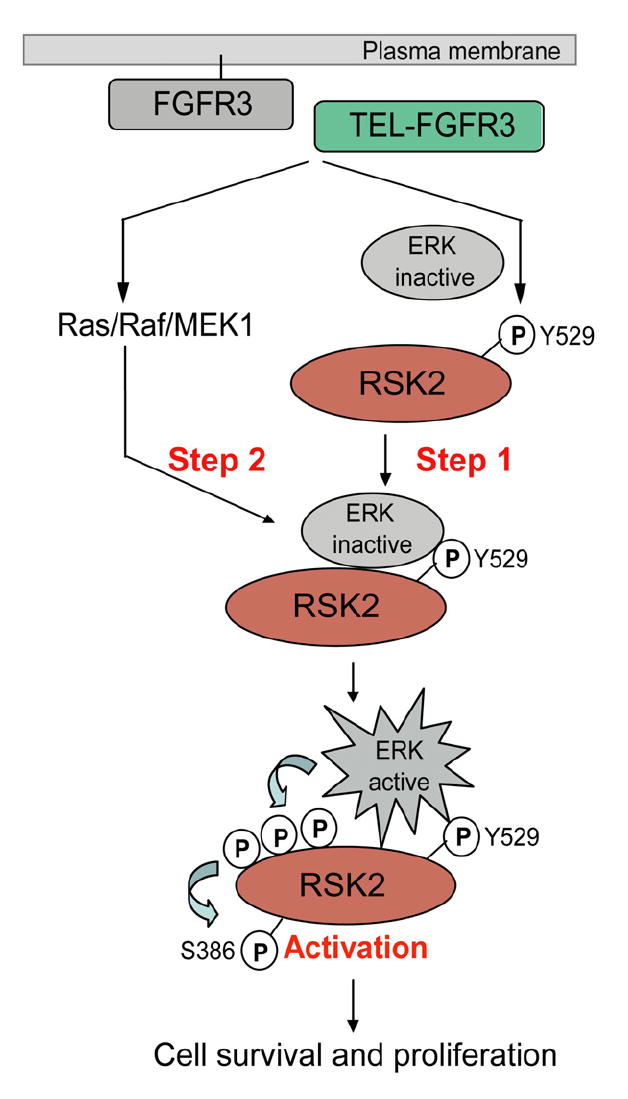
FGFR3 phosphorylates RSK2 at Y529 to facilitate recruitment of the inactive form of ERK to RSK2 (Step 1), which subsequently promotes the phosphorylation and activation of RSK2 by ERK when activated by FGFR3 (Step 2).
Tyrosine phosphorylation at Y529 may provide an additional docking site to promote the binding of inactive ERK to the C terminus of RSK2. Future detailed structural studies would illuminate this process. FGFR3 might not be the only upstream tyrosine kinase that phosphorylates RSK2 at Y529 as we have observed that upon treatment of EGF, RSK2 is tyrosine phosphorylated at Y529 and activated in 293T cells that do not express FGFR3 (S.K., S.D., T.-L.G., A.G., S.L., H.J.K., R.D.P., and J.C., unpublished data). This suggests that phosphorylation at Y529 might be a general requirement for RSK2 activation through the ERK/MAPK pathway. Further studies to identify the alternative upstream tyrosine kinase(s) of RSK2 as well as the role of phospho-tyrosine residues besides Y529 in the activation and function of RSK2 are warranted.
Although Y529 is highly homologous in both RSK1 and RSK2, RSK1 was not detected to be tyrosine phosphorylated in our proteomics studies (data not shown). This is in consonance with our observations that FGFR3 specifically activates RSK2 but fails to significantly activate RSK1 in Ba/F3 cells (data not shown) as well as the lack of apoptosis induced following siRNA knockdown of RSK1 in FGFR3-expressing human myeloma cells (Figure 6C). Thus, FGFR3 may specifically signal through RSK2 to mediate transformation signaling.
Fmk as a highly specific RSK inhibitor induces significant apoptosis in primary CD138-positive myeloma cells from FGFR3-expressing, t(4;14)-positive multiple myeloma patient with minimal nonspecific cytotoxicity (Figure 7D). Interestingly, the sensitivity to fmk is different among t(4;14)-positive HMCLs. Fmk induces significant apoptosis in KMS11, OPM1, and LP1 cells, whereas KMS18 cells are relatively resistant (Figures 7B and 7C). This difference suggests that there may be other oncogenic abnormalities that are not responsive to fmk treatment in KMS18 myeloma cells in addition to FGFR3. For example, in addition to activation of FGFR3, the t(4;14)(p16;q32) also results in creation of a chimeric fusion transcript between IGH and MMSET (Multiple Myeloma SET domain) (Chesi et al., 1998b). Larger numbers of cell lines will need to be evaluated to determine efficacy of fmk in HMCLs that overexpress FGFR3.
Fmk as a first generation RSK inhibitor shows promising but so far limited effectiveness in treatment of FGFR3-expressing myeloma cells, compared to the FGFR3 inhibitor PKC412 (Figure 7B). Fmk was designed to specifically target the CTD auto-kinase domain of RSK1, 2, and 4; however, it cannot completely abrogate the phosphorylation of Ser386 of RSK2 (Figure 5C;Cohen et al., 2007;Cohen et al., 2005). Cohen et al. recently reported that a fmk derivative, fmk-pa, inhibits RSK Ser386 phosphorylation at saturating concentrations following stimulation of phorbol ester, but has no effect on RSK activation by lipopolysaccharide (Cohen et al., 2007). These findings together suggest that RSK CTD-dependent autophosphorylation at Ser386 is context dependent, and alternative kinases may exist and bypass the CTD requirement and phosphorylate Ser386 in the RSK2 hydrophobic motif, which therefore limits the therapeutic effects of fmk. Indeed, PDK1 can phosphorylate RSK at Ser386 in vitro, and Ser386 is also within the identified consensus phosphorylation motif of RSK NTD domain (Richards et al., 2001). On the other hand, the RSK2 NTD transkinase domain is responsible for phosphorylation of RSK2 substrates such as histone H3 and BAD. Thus, targeting RSK CTD and NTD should have different physiological effects, which warrants future studies to test other potent RSK inhibitors that inhibit RSK NTD, such as BI-D1870 (Sapkota et al., 2007), or compounds target both kinase domains of RSK2. Such inhibitors may have enhanced therapeutic efficacy to inhibit FGFR3-mediated transformation signaling.
Fmk is also able to induce significant apoptotic cell death in the t(4;14)-negative human myeloma cell line RPMI8226 that harbors an active RAS K12 mutation (Figure 7C), suggesting a wider therapeutic implication of targeting RSK in treatment of both FGFR3-positive and -negative multiple myeloma. Activating mutations of FGFR3 have been identified in human bladder and cervical carcinomas (Cappellen et al., 1999). Thus, our findings may also have therapeutic implications with regard to solid tumors associated with dysregulation of FGFR3.
EXPERIMENTAL PROCEDURES
Proteomics Studies
Phosphopeptides were prepared using PhosphoScan Kit (Cell Signaling Technology, Inc.). In brief, 2 to 3×108 Ba/F3 cells (~20–40 mg total protein) and cells that stably express TEL-FGFR3 fusion were treated with IL-3- and serum-withdrawal for 4 hr prior to preparation of cell lysates as described (Rush et al., 2005). Protein extracts from whole cell lysates were trypsin digested. Tyrosine-phosphorylated peptides were enriched by Immunoaffinity Purification (IAP) using phosphotyrosine antibody P-Tyr-100 and analyzed by liquid chromatography coupled with mass spectrometry. Tandem mass spectra were collected in a data-dependent manner with an LTQ ion trap mass spectrometer (ThermoFinnigan).
Reagents
RSK specific inhibitor fmk was described previously (Cohen et al., 2005). SiRNA was ordered from Dharmacon, Chicago, IL. CTD-tide (RRQLFRGFSFVAK) was synthesized by American Peptide Company, Sunnyvale, CA. Murine RSK2 in pKH3-RSK2 (generously provided by Dr. Morten Frodin at Glostrup Hospital, Denmark) was myc-tagged by PCR and subcloned into pMSCV-puro derived Gateway destination vectors as described (Chen et al., 2005b). Mutations Y488F and/or Y529F were introduced into RSK2 by using QuikChange-XL site-directed mutagenesis kit (Stratagene, La Jolla, CA). The pET28b-His6-RSK2 CTD (aa 415–740) was described previously (Cohen et al., 2005). pET28b-(His)6-RSK2 variants were generated for bacterial protein purification. RSK2 variants were subcloned into pDEST27 (Invitrogen, Carlsbad, CA) for GST-tagged RSK2 expression in mammalian cells.
Cell Culture
Ba/F3 cells were cultured in RPMI 1640 medium in presence of 10% fetal bovine serum (FBS) and 1.0 ng/ml interleukin-3 (IL-3) (R & D Systems, Minneapolis, MN). HMCLs were cultured in RPMI 1640 medium with 10% FBS. 293T cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) with 10% FBS.
Retroviral Infections, Ba/F3 Cells IL-3 Independent Proliferation Assay, and Apoptosis Assay
RSK2 expressing Ba/F3 cell lines were generated by retroviral transduction as described (Chen et al., 2005b) by using Ba/F3 cells stably expressing FGFR3 TDII (Chen et al., 2005b) with pMSCV-puro plasmids encoding myc-tagged RSK2 variants, followed by antibiotic selection. For cell viability assays, 1 × 105 Ba/F3 cells stably expressing FGFR3 were cultured in 24-well plates with media containing increasing concentrations of fmk, acidic FGF (10nM; R&D system, Minneapolis, MN), and heparin (30 μg/ml; Sigma, St. Louis, MO) in the absence of IL-3. The relative cell viability at each experimental time point was determined by using the Celltiter96AQueous One solution proliferation kit (Promega, Madison, WI). For apoptosis assays, 1 × 106 human myeloma cells were treated with fmk or PKC412 for 6 hr prior to being stained with annexin V-FITC (BD PharMingen, San Diego, CA) and analysis by FACS for apoptotic populations. The primary patient samples were analyzed as previously described (David et al., 2005). Briefly, bone marrow mononuclear cells (BMNCs) were ficolled from bone marrow samples from multiple myeloma patients. 1 × 106/ml cells were cultured in 12-well plate and incubated with 0 or 3 μM of fmk for 16 and 24 hr. The cells were stained with annexin V-FITC and CD138-PE as the recommendations of the manufacturers, followed by FACS analysis. All clinical samples were obtained with informed consent with approval by the Emory University Institutional Review Board.
Antibodies
Phospho-Tyr antibody pY99 and antibodies against RSK1, RSK2, and FGFR3 were from Santa Cruz Biotechnology, Santa Cruz, CA; antibodies against myc, p44/42 ERK, phospho-p44/42 ERK (Thr202/ Tyr204), phospho-RSK (Ser380), phospho-RSK (Thr359/Ser363), phospho-RSK (Thr573), and PARP were from Cell Signaling Technology (CST), Danvers, MA; phospho-Tyr antibody clone 4G10 was from Upstate, Lake Placid, NY; and antibodies against GST and β-actin were from Sigma, St. Louis, MO. Specific antibody against phospho-RSK2 (Tyr529) was generated by CST.
Purification of Recombinant RSK2 Proteins and Limited Proteolytic Digestion
(His)6-tagged RSK2 proteins were purified by sonication of high expressing BL21(DE3)pLysS cells obtained from 250 ml of culture with IPTG-induction for 4 hr. Cellular lysates were resolved by centrifugation and loaded onto a Ni-NTA column in 20 mM imidazole. After a step of 2× washing, the protein was eluted with 250 mM imidazole. Proteins were desalted on a PD-10 column and the purification efficiency was examined by silver staining and western blotting. The limited proteolytic digestion was performed using chymotrypsin (Zhang et al., 1997).
In Vitro Kinase Assays
The S6 peptide kinase assay was carried out according to the manufacturer’s protocol (Upstate Biotechnology) using RSK2 immunoprecipitates. To determine the ability of FGFR3 to phosphorylate RSK2, 500 ng of purified recombinant RSK2 variants were incubated with 500 ng of recombinant active FGFR3 (Invitrogen, Carlsbad, CA) in 10 mM HEPES (pH 7.5), 150 mM NaCl, 1 mM DTT, 0.01% Trixton-X-100, 10 mM MnCl2, and 200 μM ATP for 30 min at 30°C. Phosphorylation of Y529 RSK2 was detected by specific phospho-antibody. To determine kinase activity of RSK2 CTD variants, purified recombinant RSK2 CTD proteins (500 nM) were incubated with 500 nM of active ERK (Invitrogen, Carlsbad, CA) in 20 mM HEPES [pH 8.0], 10 mM MgCl2, 2 mM tris-(2-carboxyethyl)-phosphine (TCEP), and 200 μM ATP for 1 hr at 30°C. Kinase reactions were initiated by the addition of 5 μCi of [γ-32P] ATP and 100 μM peptide substrate (CTD-tide), followed by incubation for 20 min at room temperature. Kinase activity was determined using the standard disk phospho-cellulose assay.
Reconstitution of RSK2 Tyrosine Phosphorylation by FGFR3
GST-tagged RSK2 constructs were transfected into 293T cells using Lipofectamine 2000 (Invitrogen, Carlsbad, CA). Twenty-four hours posttransfection, cells were lysed, and GST-RSK2 variants were pulled down by Glutathione Sepharose 4B beads (Amersham Bioscience, Piscataway, NJ), followed by treatment of 50U of YOP phosphatase (New England Biolab, Beverly, MA) at 30°C for 1 hr in 1 mg/ml BSA and 1 × YOP reaction buffer [50 mM Tris (pH7.0), 100 mM NaCl, 2 mM Na2EDTA, 5 mM DTT]. The beads were then washed with PBS, followed by FGFR3 kinase reaction at 30°C for 30 min as described above. The treated beads were washed with PBS, followed by incubation with 293T cell lysates pretreated with 10 μM U0126 for 90 min, prior to SDS-PAGE and western blotting to detect association of inactive ERK.
Supplemental Data
The Supplemental Data include one supplemental figure and can be found with this article online at http://www.cancercell.org/cgi/content/full/12/3/201/DC1/.
Acknowledgments
We gratefully acknowledge the critical reading of the manuscript by Drs. Benjamin Lee and Brian Huntly. We thank Claire Torre for the generous help with primary patient sample analysis. Drs. Ruan Hong and Qingyuan Ge (Cell Signaling Technology, Inc.) kindly helped us with generation of phospho-RSK2 (Y529) antibody. This work was supported in part by NIH grant CA120272 (J.C.), the Leukemia and Lymphoma Society (J.C.), and the Multiple Myeloma Research Foundation (J.C. and S.L.). J.C. is a Georgia Cancer Coalition Distinguished Cancer Scholar. T.-L.G., A.G., and R.D.P. are employed by Cell Signaling Technology, Inc., whose product was studied in the present work. D.F. is employed by Novartis Pharma AG.
References
- Anjum R, Roux PP, Ballif BA, Gygi SP, Blenis J. The tumor suppressor DAP kinase is a target of RSK-mediated survival signaling. Curr Biol. 2005;15:1762–1767. doi: 10.1016/j.cub.2005.08.050. [DOI] [PubMed] [Google Scholar]
- Bergsagel PL, Chesi M, Nardini E, Brents LA, Kirby SL, Kuehl WM. Promiscuous translocations into immunoglobulin heavy chain switch regions in multiple myeloma. Proc Natl Acad Sci USA. 1996;93:13931–13936. doi: 10.1073/pnas.93.24.13931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergsagel PL, Kuehl WM. Chromosome translocations in multiple myeloma. Oncogene. 2001;20:5611–5622. doi: 10.1038/sj.onc.1204641. [DOI] [PubMed] [Google Scholar]
- Blenis J. Signal transduction via the MAP kinases: Proceed at your own RSK. Proc Natl Acad Sci USA. 1993;90:5889–5892. doi: 10.1073/pnas.90.13.5889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buck M, Poli V, Hunter T, Chojkier M. C/EBPbeta phosphorylation by RSK creates a functional XEXD caspase inhibitory box critical for cell survival. Mol Cell. 2001;8:807–816. doi: 10.1016/s1097-2765(01)00374-4. [DOI] [PubMed] [Google Scholar]
- Cappellen D, De Oliveira C, Ricol D, de Medina S, Bourdin J, Sastre-Garau X, Chopin D, Thiery JP, Radvanyi F. Frequent activating mutations of FGFR3 in human bladder and cervix carcinomas. Nat Genet. 1999;23:18–20. doi: 10.1038/12615. [DOI] [PubMed] [Google Scholar]
- Chen J, Lee BH, Williams IR, Kutok JL, Mitsiades CS, Duclos N, Cohen S, Adelsperger J, Okabe R, Coburn A, et al. FGFR3 as a therapeutic target of the small molecule inhibitor PKC412 in hematopoietic malignancies. Oncogene. 2005a;24:8259–8267. doi: 10.1038/sj.onc.1208989. [DOI] [PubMed] [Google Scholar]
- Chen J, Williams IR, Lee BH, Duclos N, Huntly BJ, Donoghue DJ, Gilliland DG. Constitutively activated FGFR3 mutants signal through PLCgamma-dependent and -independent pathways for hematopoietic transformation. Blood. 2005b;106:328–337. doi: 10.1182/blood-2004-09-3686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chesi M, Bergsagel PL, Brents LA, Smith CM, Gerhard DS, Kuehl WM. Dysregulation of cyclin D1 by translocation into an IgH gamma switch region in two multiple myeloma cell lines. Blood. 1996;88:674–681. [PubMed] [Google Scholar]
- Chesi M, Bergsagel PL, Shonukan OO, Martelli ML, Brents LA, Chen T, Schrock E, Ried T, Kuehl WM. Frequent dysregulation of the c-maf proto-oncogene at 16q23 by translocation to an Ig locus in multiple myeloma. Blood. 1998a;91:4457–4463. [PubMed] [Google Scholar]
- Chesi M, Brents LA, Ely SA, Bais C, Robbiani DF, Mesri EA, Kuehl WM, Bergsagel PL. Activated fibroblast growth factor receptor 3 is an oncogene that contributes to tumor progression in multiple myeloma. Blood. 2001;97:729–736. doi: 10.1182/blood.v97.3.729. [DOI] [PubMed] [Google Scholar]
- Chesi M, Nardini E, Brents LA, Schrock E, Ried T, Kuehl WM, Bergsagel PL. Frequent translocation t(4;14)(p16.3;q32.3) in multiple myeloma is associated with increased expression and activating mutations of fibroblast growth factor receptor 3. Nat Genet. 1997;16:260–264. doi: 10.1038/ng0797-260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chesi M, Nardini E, Lim RS, Smith KD, Kuehl WM, Bergsagel PL. The t(4;14) translocation in myeloma dysregulates both FGFR3 and a novel gene, MMSET, resulting in IgH/MMSET hybrid transcripts. Blood. 1998b;92:3025–3034. [PubMed] [Google Scholar]
- Cohen MS, Hadjivassiliou H, Taunton J. A clickable inhibitor reveals context-dependent autoactivation of p90 RSK. Nat Chem Biol. 2007;3:156–160. doi: 10.1038/nchembio859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen MS, Zhang C, Shokat KM, Taunton J. Structural bioinformatics-based design of selective, irreversible kinase inhibitors. Science. 2005;308:1318–1321. doi: 10.1126/science1108367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colvin JS, Bohne BA, Harding GW, McEwen DG, Ornitz DM. Skeletal overgrowth and deafness in mice lacking fibroblast growth factor receptor 3. Nat Genet. 1996;12:390–397. doi: 10.1038/ng0496-390. [DOI] [PubMed] [Google Scholar]
- David E, Sun SY, Waller EK, Chen J, Khuri FR, Lonial S. The combination of the farnesyl transferase inhibitor lonafarnib and the proteasome inhibitor bortezomib induces synergistic apoptosis in human myeloma cells that is associated with down-regulation of p-AKT. Blood. 2005;106:4322–4329. doi: 10.1182/blood-2005-06-2584. [DOI] [PubMed] [Google Scholar]
- Deng C, Wynshaw-Boris A, Zhou F, Kuo A, Leder P. Fibroblast growth factor receptor 3 is a negative regulator of bone growth. Cell. 1996;84:911–921. doi: 10.1016/s0092-8674(00)81069-7. [DOI] [PubMed] [Google Scholar]
- Fisher TL, Blenis J. Evidence for two catalytically active kinase domains in pp90rsk. Mol Cell Biol. 1996;16:1212–1219. doi: 10.1128/mcb.16.3.1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frodin M, Gammeltoft S. Role and regulation of 90 kDa ribosomal S6 kinase (RSK) in signal transduction. Mol Cell Endocrinol. 1999;151:65–77. doi: 10.1016/s0303-7207(99)00061-1. [DOI] [PubMed] [Google Scholar]
- Frodin M, Jensen CJ, Merienne K, Gammeltoft S. A phosphoserine-regulated docking site in the protein kinase RSK2 that recruits and activates PDK1. EMBO J. 2000;19:2924–2934. doi: 10.1093/emboj/19.12.2924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavin AC, Nebreda AR. A MAP kinase docking site is required for phosphorylation and activation of p90(rsk)/MAPKAP kinase-1. Curr Biol. 1999;9:281–284. doi: 10.1016/s0960-9822(99)80120-1. [DOI] [PubMed] [Google Scholar]
- Golla A, Lichmer P, von Gernet S, Winterpacht A, Fairley J, Murken J, Schuffenhauer S. Phenotypic expression of the fibroblast growth factor receptor 3 (FGFR3) mutation P250R in a large craniosynostosis family. J Med Genet. 1997;34:683–684. doi: 10.1136/jmg.34.8.683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grand EK, Chase AJ, Heath C, Rahemtulla A, Cross NC. Targeting FGFR3 in multiple myeloma: Inhibition of t(4;14)-positive cells by SU5402 and PD173074. Leukemia. 2004;18:962–966. doi: 10.1038/sj.leu.2403347. [DOI] [PubMed] [Google Scholar]
- Hart KC, Robertson SC, Donoghue DJ. Identification of tyrosine residues in constitutively activated fibroblast growth factor receptor 3 involved in mitogenesis, Stat activation, and phosphatidylinositol 3-kinase activation. Mol Biol Cell. 2001;12:931–942. doi: 10.1091/mbc.12.4.931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen CJ, Buch MB, Krag TO, Hemmings BA, Gammeltoft S, Frodin M. 90-kDa ribosomal S6 kinase is phosphorylated and activated by 3-phosphoinositide-dependent protein kinase-1. J Biol Chem. 1999;274:27168–27176. doi: 10.1074/jbc.274.38.27168. [DOI] [PubMed] [Google Scholar]
- Li Z, Zhu YX, Plowright EE, Bergsagel PL, Chesi M, Patterson B, Hawley TS, Hawley RG, Stewart AK. The myeloma-associated oncogene fibroblast growth factor receptor 3 is transforming in hematopoietic cells. Blood. 2001;97:2413–2419. doi: 10.1182/blood.v97.8.2413. [DOI] [PubMed] [Google Scholar]
- Ng MH, Lau KM, Wong WS, To KW, Cheng SH, Tsang KS, Chan NP, Kho BC, Lo KW, Tong JH, et al. Alterations of RAS signalling in Chinese multiple myeloma patients: Absent BRAF and rare RAS mutations, but frequent inactivation of RASSF1A by transcriptional silencing or expression of a non-functional variant transcript. Br J Haematol. 2003;123:637–645. doi: 10.1046/j.1365-2141.2003.04664.x. [DOI] [PubMed] [Google Scholar]
- Palmer A, Gavin AC, Nebreda AR. A link between MAP kinase and p34(cdc2)/cyclin B during oocyte maturation: p90(rsk) phosphorylates and inactivates the p34(cdc2) inhibitory kinase Myt1. EMBO J. 1998;17:5037–5047. doi: 10.1093/emboj/17.17.5037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paterson JL, Li Z, Wen XY, Masih-Khan E, Chang H, Pollett JB, Trudel S, Stewart AK. Preclinical studies of fibroblast growth factor receptor 3 as a therapeutic target in multiple myeloma. Br J Haematol. 2004;124:595–603. doi: 10.1111/j.1365-2141.2004.04814.x. [DOI] [PubMed] [Google Scholar]
- Richards SA, Dreisbach VC, Murphy LO, Blenis J. Characterization of regulatory events associated with membrane targeting of p90 ribosomal S6 kinase 1. Mol Cell Biol. 2001;21:7470–7480. doi: 10.1128/MCB.21.21.7470-7480.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronchetti D, Greco A, Compasso S, Colombo G, Dell’Era P, Otsuki T, Lombardi L, Neri A. Deregulated FGFR3 mutants in multiple myeloma cell lines with t(4;14): Comparative analysis of Y373C, K650E and the novel G384D mutations. Oncogene. 2001;20:3553–3562. doi: 10.1038/sj.onc.1204465. [DOI] [PubMed] [Google Scholar]
- Roux PP, Richards SA, Blenis J. Phosphorylation of p90 ribosomal S6 kinase (RSK) regulates extracellular signal-regulated kinase docking and RSK activity. Mol Cell Biol. 2003;23:4796–4804. doi: 10.1128/MCB.23.14.4796-4804.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rush J, Moritz A, Lee KA, Guo A, Goss VL, Spek EJ, Zhang H, Zha XM, Polakiewicz RD, Comb MJ. Immunoaffinity profiling of tyrosine phosphorylation in cancer cells. Nat Biotechnol. 2005;23:94–101. doi: 10.1038/nbt1046. [DOI] [PubMed] [Google Scholar]
- Sapkota GP, Cummings L, Newell FS, Armstrong C, Bain J, Frodin M, Grauert M, Hoffmann M, Schnapp G, Steegmaier M, et al. BI-D1870 is a specific inhibitor of the p90 RSK (ribosomal S6 kinase) isoforms in vitro and in vivo. Biochem J. 2007;401:29–38. doi: 10.1042/BJ20061088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimamura A, Ballif BA, Richards SA, Blenis J. Rsk1 mediates a MEK-MAP kinase cell survival signal. Curr Biol. 2000;10:127–135. doi: 10.1016/s0960-9822(00)00310-9. [DOI] [PubMed] [Google Scholar]
- Smith JA, Poteet-Smith CE, Malarkey K, Sturgill TW. Identification of an extracellular signal-regulated kinase (ERK) docking site in ribosomal S6 kinase, a sequence critical for activation by ERK in vivo. J Biol Chem. 1999;274:2893–2898. doi: 10.1074/jbc.274.5.2893. [DOI] [PubMed] [Google Scholar]
- Tavormina PL, Shiang R, Thompson LM, Zhu YZ, Wilkin DJ, Lachman RS, Wilcox WR, Rimoin DL, Cohn DH, Wasmuth JJ. Thanatophoric dysplasia (types I and II) caused by distinct mutations in fibroblast growth factor receptor 3. Nat Genet. 1995;9:321–328. doi: 10.1038/ng0395-321. [DOI] [PubMed] [Google Scholar]
- Trudel S, Ely S, Farooqi Y, Affer M, Robbiani DF, Chesi M, Bergsagel PL. Inhibition of fibroblast growth factor receptor 3 induces differentiation and apoptosis in t(4;14) myeloma. Blood. 2004;103:3521–3528. doi: 10.1182/blood-2003-10-3650. [DOI] [PubMed] [Google Scholar]
- Vaidyanathan H, Ramos JW. RSK2 activity is regulated by its interaction with PEA-15. J Biol Chem. 2003;278:32367–32372. doi: 10.1074/jbc.M303988200. [DOI] [PubMed] [Google Scholar]
- Webster MK, Donoghue DJ. Constitutive activation of fibroblast growth factor receptor 3 by the transmembrane domain point mutation found in achondroplasia. EMBO J. 1996;15:520–527. [PMC free article] [PubMed] [Google Scholar]
- Yagasaki F, Wakao D, Yokoyama Y, Uchida Y, Murohashi I, Kayano H, Taniwaki M, Matsuda A, Bessho M. Fusion of ETV6 to fibroblast growth factor receptor 3 in peripheral T-cell lymphoma with a t(4;12)(p16;p13) chromosomal translocation. Cancer Res. 2001;61:8371–8374. [PubMed] [Google Scholar]
- Zhang L, Wang H, Liu D, Liddington R, Fu H. Raf-1 kinase and exoenzyme S interact with 14–3-3zeta through a common site involving lysine 49. J Biol Chem. 1997;272:13717–13724. doi: 10.1074/jbc.272.21.13717. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The Supplemental Data include one supplemental figure and can be found with this article online at http://www.cancercell.org/cgi/content/full/12/3/201/DC1/.