

Abstract
Manipulating gene expression in primary neurons has been a goal for many scientists for over 20 years. Vertebrate central nervous system neurons are classically difficult to transfect. Most lipid reagents are inefficient and toxic to the cells, and time-consuming methods such as viral infections are often required to obtain better efficiencies. We have developed an efficient method for the transfection of cerebellar granule neurons and hippocampal neurons with standard plasmid vectors. Using 96-well electroporation plates, square-wave pulses can introduce 96 different plasmids into neurons in a single step. The procedure results in greater than 20% transfection efficiencies and requires only simple solutions of nominal cost. In addition to enabling the rapid optimization of experimental protocols with multiple parameters, this procedure enables the use of high content screening methods to characterize neuronal phenotypes.
INTRODUCTION
The expression of proteins in cells using promoter driven cDNAs is a widely used approach for studying the function of proteins and analyzing molecular networks. Over the past 20 years, a number of approaches have been developed to allow transfection of cDNA containing plasmids into cells, especially cell lines. However, primary cells, such as neurons and T cells, have been resistant to most transfection methods, requiring the use of time-consuming and expensive viral-based methods. Neurons have been particularly challenging to transfect, with low efficiency of transfection up until the past few years (1). Recently, Amaxa (2) has introduced the nucleofection method, which achieves neuronal transfection efficiencies of >20% (3,4). A single cuvette is used in this method to electroporate a million or more neurons at a time. Each electroporation requires the use of an expensive proprietary reagent. For testing multiple genes, millions of neurons have to be harvested, and electroporations have to be performed one at a time. This presents two major problems for testing many genes. First, the repetitive action of electroporating single samples, and then manually placing them correctly in their destination, lends itself to variability and error. Second, the extended time it takes to sequentially transfect each gene leaves the neurons in a toxic environment leading to even lower viabilities and efficiencies.
High content screening (HCS) uses automated acquisition of images of cells in multiwell plates combined with detailed quantitative image analysis to obtain multiple parameters about cell morphology and molecular expression. HCS is ideally suited for screening various kinds of libraries for their effects on cell proliferation, apoptosis, or other cell biological processes. It is ideal for quantitatively studying the morphology of neurons and how various agents alter process growth. The paucity of efficient methods for testing cDNAs in mammalian neurons using HCS has restricted the usefulness of HCS approaches to drug and compound libraries and inhibited genome style studies. To overcome this roadblock, we have sought to develop an inexpensive method that would allow multiple transfections at once— with identical conditions except for the gene to be transfected. This electroporation approach involves a mixture of a minimal set of components, all at reasonable volumes and with small numbers of cells, such that experiments can be performed quickly with multichannel pipets or 96-well liquid handlers.
MATERIALS AND METHODS
Primary Neuron Culture
Postnatal day 8–11 mouse cerebella were prepared as described previously (5). Briefly, cerebella were harvested from ketamine-euthanized mice and minced with a razor blade. The cerebellar pieces were incubated in 0.05% trypsin-EDTA (Invitrogen, Carlsbad, CA, USA) for 20 min at 37° C, with occasional swirling. The trypsin was inactivated by adding horse serum to 10% and diluted with Hank’s balanced salt solution (HBSS; 5.4 mM KCl, 0.44 mM KH2PO4, 131 mM NaCl, 4.2 mM NaHCO3, 0.34 mM Na2HPO4, 10 mM HEPES). The cells were triturated sequentially with large- and small-bore flame-polished glass pipets in the presence of 1 mg/mL DNase I (Sigma, St. Louis, MO, USA). Hoechst dye (Invitrogen) was added during this step. The cells were spun and resuspended in HBSS for counting. Centrifugation steps were all performed at 115× g for up to 7 min. Solutions and cells were kept at room temperature throughout the procedure. Preparations yielded >90% cerebellar granule neurons (CGNs).
Rat hippocampal slices were purchased from BrainBits LLC (Springfield, IL, USA), and dissociated neurons were prepared using a modified version of their protocol. Briefly, half hippocampal slices were incubated for 30 min in 2.5% trypsin and 100 μL 30 mg/mL DNase and then rinsed four to five times with Hibernate-E, no Ca2+, media plus B27 (BrainBits LLC). Cells were triturated using flame-polished small- and large-bore glass pipets. After triturating, chunks were allowed to settle, and then the supernatant was transferred to new tubes, and the cells counted. Volumes were kept small (<2 mL) throughout the process. Preparations commonly yield >95% neurons.
Transfection
Typically, 100,000 neurons were resuspended in 100 μL intracellular buffer (INB) solution (135 mM KCl, 0.2 mM CaCl2, 2 mM MgCl2, 10 mM HEPES, 5 mM EGTA, pH 7.3), which results in about 3 nM free calcium. Cells (90–100 μL) are added to a 96-well, 2-mm gap, electroporation plate (HT-P96-2; Harvard Apparatus/BTX, Holliston, MA, USA), and 1–5 μg plasmid cDNA were also added to each well for a total volume of no more than 120 μL. The plate is sealed with a 3M ScotchPad tape sheet (Qiagen, Valencia, CA, USA). All solutions were removed from the cold 10 min prior to transfection to allow them to come to room temperature. The electroporation plate was placed inside the Model HT-200 plate handler attached to an ECM 830 square wave pulse generator (Harvard Apparatus/BTX). This system was used to deliver pulses to the plate, one column at a time. For CGNs, one pulse was delivered with 340–350 V, 900 μs pulse length. For hippocampal neurons, two pulses were delivered, each with 900 μs pulse lengths and an approximate 2-s interval between. The first pulse was 140 V and the second 340 V.
Once transfection was complete, a fraction of the cells was immediately transferred to 96-well assay ViewPlates™ (Packard Instrument Company, Meriden, CT, USA), containing neurobasal complete (NBC) media (supplemented 1:50 with B27), 2 mM GlutaMAX™ media, 25 mM KCl, in Neurobasal™-A media (all from Invitrogen) preincubated to 37° C and 5% CO2. Ten to twenty thousand cells were used per assay plate well, allowing up to six replicates per transfection condition. After plating, cells were placed in a 37° C incubator maintained at 5% CO2.
Immunohistochemistry
Cells were left 2 days in vitro, then fixed, permeabilized, and stained. Briefly, fixation was performed for 45 min using 180 μL/well of 4% paraformaldehyde, 0.01% glutaraldehyde in 0.1 M phosphate buffer, pH 7.4. Cells were rinsed three times with phosphate-buffered saline (PBS) followed by a blocking solution (BS) of 0.2% gelatin and 0.03% Triton® X-100 in PBS. After blocking for 1 h, 100 μL E7 mouse anti-β-tubulin primary antibody (diluted in BS) were added to each well and incubated overnight. The plate was washed three times with PBS and incubated with Alexa Fluor® 555 goat anti-mouse secondary antibody (Invitrogen) in BS. The fixation and staining procedures were performed using a liquid handling instrument (BioRobot® 3000; Qiagen), but could be done using multichannel pipets. Human L1 (see Figure 4) was stained with a monoclonal mouse anti-human L1 antibody—7B5. It was produced in the laboratory (3) and used 1/500 diluted in BS.
Figure 4. Co-expression of two plasmids.
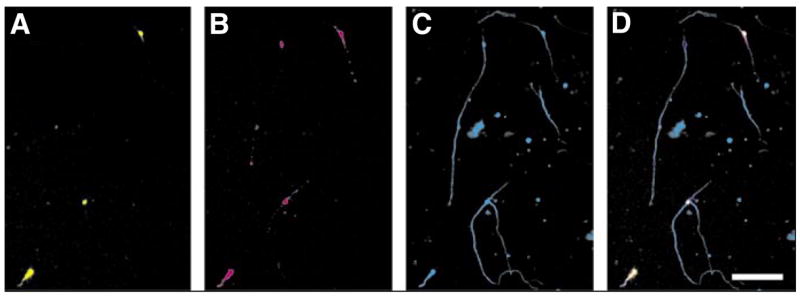
Green fluorescent protein (GFP) and the cell adhesion molecule L1 were cotransfected into cerebellar granule neurons (CGNs). The neurons were kept in culture for 48 h before being fixed and stained. (A) GFP fluorescence, (B) L1 immunofluorescence, and (C) β-tubulin immunofluorescence were used to determine transfection efficiencies. (D) A merge of the three channels in panels A, B, and C. Note that there are three cells expressing GFP and L1, and a fourth cell that is only expressing L1. Scale bar, 100 μm.
Microscopy and Analysis
The fixed and stained assay plate was analyzed by an HCS microscope, the KineticScan® Reader (Cellomics, Pittsburgh, PA, USA). This instrument automatically took five images per well in three channels: (i) nuclei (Hoechst); (ii) neuron cell body and neurites (β-tubulin); and (iii) green fluorescent protein (GFP). A software suite built into the Cellomics package (Extended Neurite Outgrowth v1) processed the images and automatically identified neurons based on a valid nucleus and cell body. The program then quantified a variety of parameters and returned a report about each cell that satisfied predetermined thresholds and criteria. We used information about the number of cells, number of nuclei, length and number of neurites, and average fluorescence intensity in the cell body for further analysis.
Quantification
GFP transfection was determined first by finding the distribution of intensities in the green channel in wells in which GFP plasmids were not added. These intensities represented fluorescence signal that did not result from GFP. A threshold was set at 4 standard deviations above the mean in wells with no GFP, and cells with higher average intensities were considered GFP-expressing. GFP-expressing cells were then counted to determine transfection efficiency and were grouped to obtain average measures for each parameter studied (neurite length, tubulin intensity, cell body area, etc).
RESULTS
Transfection Efficiency
Optimization was first attempted in mouse CGNs, harvested from postnatal day 8–10 wild-type mice. Preparations from mouse cerebellum at this age yield a relatively homogenous population of neurons (6). Figure 1A demonstrates that increasing voltages gave higher transfection efficiency. However, increasing voltages resulted in lower viability (Figure 1B). Varying the number of cells in the 96-well electroporation plate did not significantly change either transfection efficiency (Figure 1C) or viability (data not shown) in CGNs. Of particular importance, it was possible to successfully electroporate very small numbers of neurons, only 25,000 per transfection well (see Supplementary Figure S1, B and C, available online at www.BioTechniques.com). Transfection efficiencies were also reliable across an entire 96-well plate (see Supplementary Figure S1, B and D). In addition, a calcium-free intracellular buffer (7) provided significantly better transfection efficiency than standard extracellular buffers or media (Figure 1D). Using conditions near 340 V, 100,000 cells, and INB buffer, 14 other independent experiments were performed. The transfection efficiency for these was an average of 26.8% with a standard deviation of 8.6% for the transfection of GFP in CGNs (see Supplementary Figure S1A). Hippocampal neurons were transfected with an average efficiency of 17.3% ± 3.2% (n = 4).
Figure 1. Electroporation in cerebellar granule cells.
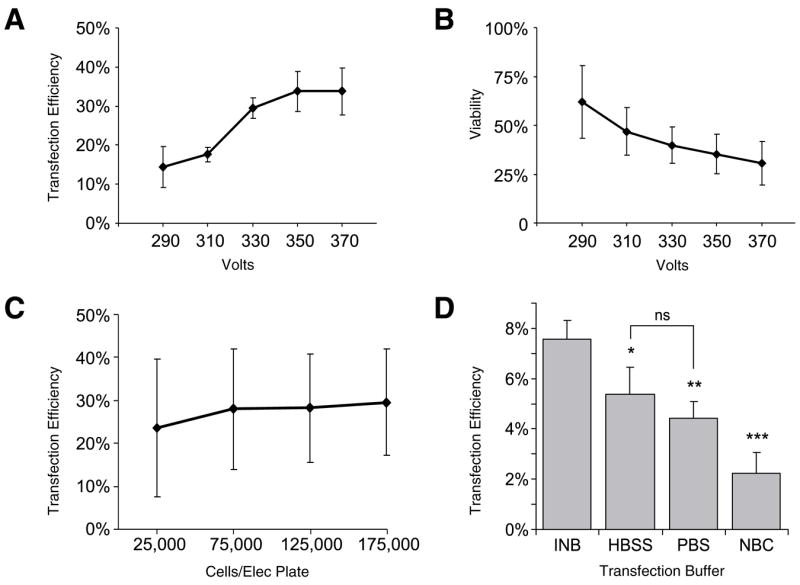
(A) Cerebellar cells are optimally transfected at voltages above 340 V, with an approximately 30% transfection efficiency. (B) The viability of the neurons in each transfection well decreases as the voltage increases, thus the lowest voltage that produces reasonable transfection efficiency is optimal. (C) The number of cells per well in the 96-well electroporation plate makes no significant difference in transfection efficiency for cell numbers examined. Standard deviations are large because data was averaged across the five voltages. (D) An intracellular buffer (INB) gave better transfection efficiency compared with Hank’s balanced salt solution (HBSS), phosphate-buffered saline (PBS), and neurobasal complete (NBC) media. Data points in panels A, B, C, and D are presented as means ± standard deviation. Panels A and B, n = 4; panel C, n = 8; panel D, n = 3. ns, not significantly different.
The amount of plasmid DNA per transfection can affect expression level and transfection efficiency (see Supplementary Figure S2). For the plasmids we tested, 1–5 μg DNA in 100 μL transfection volume resulted in successful transfection.
Expression
The expression levels of the proteins encoded by various transfected plasmids were analyzed using a KineticsScan Reader HCS machine. GFP was expressed in CGNs and in hippocampal neurons (Figure 2). The GFP expression levels ranged from just above background to more than 100 standard deviations from the nontransfected levels (Figure 2E). Expression of GFP in CGNs was seen in some cells at 6 h after transfection. By 10 h, about 6% of all the nuclei (live and dead) had GFP positive cell bodies. In addition to GFP, human L1CAM was transfected with transfection efficiency of 9.9% ± 5.4% (n = 12), and GAP43-YFP was transfected with transfection efficiency of 29.8% ± 9.6% (n = 6).
Figure 2. Expression of green fluorescent protein (GFP) in cerebellar granule neurons (CGNs) and hippocampal neurons.
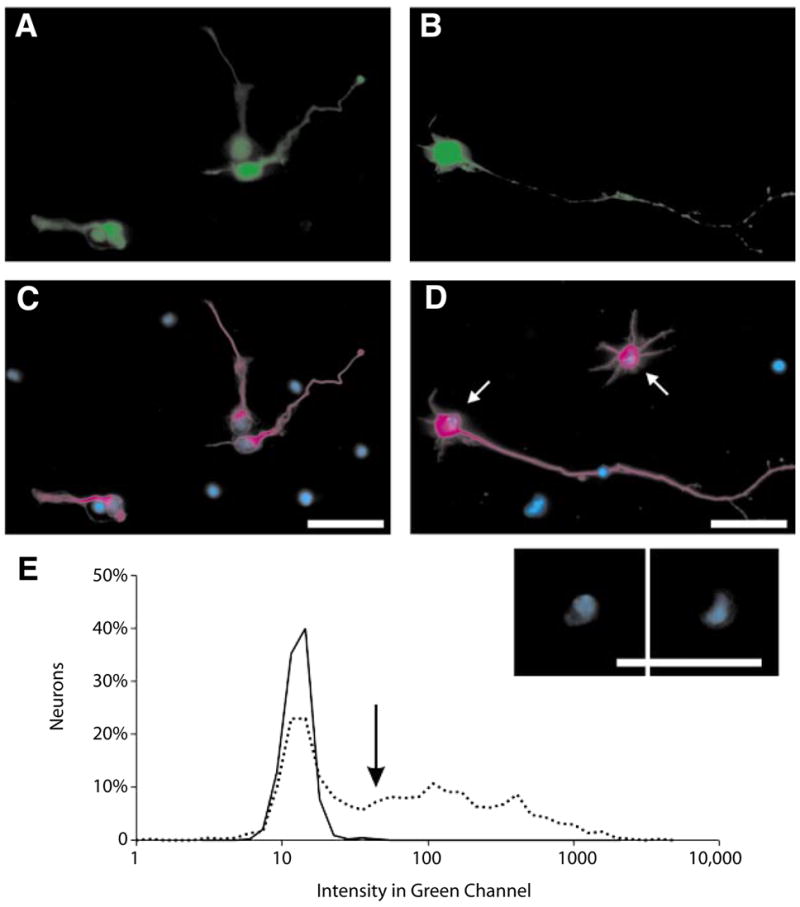
(A and C) Mouse CGNs 24 h after transfection. (B and D) Rat hippocampal neurons 48 h after transfection. (A and B) GFP fluorescence is displayed in green. (C and D) β-Tubulin staining in magenta, and nuclear staining (Hoechst) is in blue. An inset (below panel D) shows nuclear morphology for the neurons indicated by the arrows in panel D. Note that all β-tubulin positive neurons in panel A are expressing GFP. Dead neurons are nearby with distinct nuclear morphology and Hoechst intensity. Scale bar, 25 μm. (E) Fluorescent intensity in the GFP (green) channel for control, nontransfected neurons (solid line), and GFP-transfected neurons (dotted line). Many neurons expressing GFP have intensities 2 orders of magnitude greater than the mean of the control intensities. The threshold for a neuron to be considered GFP positive is indicated by the arrow (see the Materials and Methods section).
Viability
Because transfection often has adverse effects on neuronal viability, we evaluated parameters of cell health and viability in transfected neurons 1 or 2 days after plating (examples of data from entire 96-well plates are provided in Supplementary Figure S1, C and E). An average of 37.0% ± 16.3% of the CGNs originally plated (18,000) remained alive after 2 days (n = 14 experiments). CGNs that were resuspended in INB but not electroporated had an average viability of 67.1% ± 9.7% (n = 4 experiments). CGNs that were electroporated in INB, but with no cDNA had an average viability of 34.9% ± 8.7% (n = 7 experiments). Longer periods of incubation in the transfection buffer (INB) result in lower viability of cells. In our hands, if the time spent in INB is <15 min, the reduction in viability is minimal. Usually a 96-well plate can be set up within this time. Transfected cells were healthy with normal morphology and normal expression of proteins such as β-tubulin (Figure 3A). The nuclei of GFP-expressing cells had the same appearance as wild-type cultured neurons, by Hoechst staining, and were clearly not pyknotic (example Figure 2, C and D, inset). Distributions of cell areas (Figure 3B) and neurite length (Figure 3C) for GFP-expressing cells were similar to those for nonexpressing or nontransfected cells. Thus, the transfected cells were viable, healthy, and good candidates for study. After 6 days, we found that the number of GFP-expressing neurons stayed the same. The total number of neurons in the plate decreased (data not shown). Taking into account percent survival, transfection efficiency, and the optics of 96-well plates, we normally found 50–100 transfected cells to study in the central one-third of the well. These are generally sufficient numbers of neurons for studies in which single cells can be assayed.
Figure 3. Viability and health of transfected cerebellar granule neurons (CGNs).
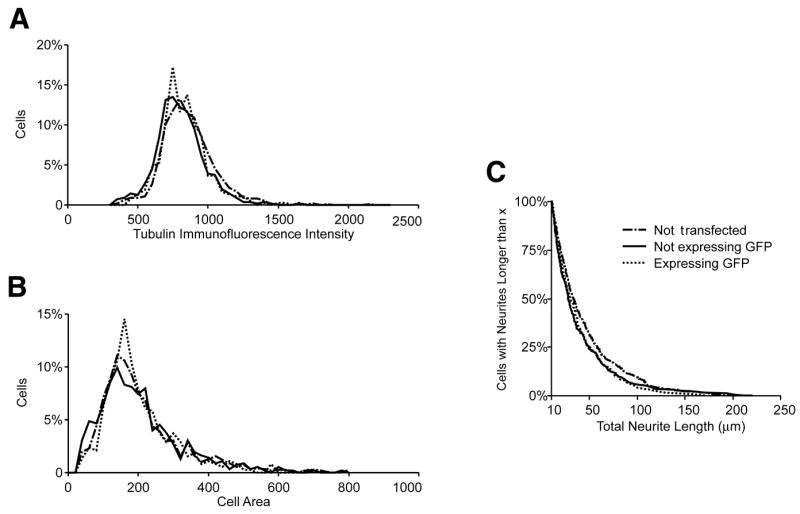
Data are presented as standard histograms for (A) tubulin immunofluorescence and (B) cell body area. (C) Total neurite length profile is presented as a cumulative probability histogram. In all plots, dashed lines represent neurons that were electroporated without plasmid (not transfected, n = 539). Solid lines represent neurons that were electroporated with green fluorescent protein (GFP) cDNA, but were not expressing GFP after 48 h (n = 263). Dotted lines represent GFP-expressing neurons in the same wells (n = 210). Total neurite length was included only for values >10 μm. In the histograms, the y-axis reports percentage of total neurons, normalizing for the different number of cells in the three populations.
Identifying Transfected Cells by Co-Expression
With 20%–40% transfection efficiency, it is important to identify the transfected neurons so they can be analyzed as a distinct population. One method for such identification is cotransfection of a test plasmid with a reporter such as GFP. By optimizing the ratio of the two genes (2:5; GFP reporter:gene of interest), 90% of GFP positive cells were found to co-express a second cDNA of interest (Figure 4). Transfection of two plasmids, such as L1 and GFP did not affect neurite length (P = 0.08), cell area (P = 0.11), or tubulin intensity (P = 0.23) compared with neurons expressing GFP alone (Kruskal-Wallis test, see Supplementary Figure S3). For many applications, such as screening libraries of genes, it is not practical to directly analyze expression of every experimental gene. Thus, by cotransfecting a reporter gene, the experimenter can ensure a high probability of co-expression and use reporter expression as a surrogate for expression of the experimental gene.
DISCUSSION
The introduction of HCS methods into academic and biotech laboratories has enabled the screening of drugs and compounds for effects on cell differentiation, cell signaling pathways, and various pathological conditions such as cancer. The extension of these methods to incorporate genomic approaches has been hindered in part by a lack of efficient and inexpensive ways to transfect primary cells, such as neurons, with hundreds of different DNAs. We have developed a 96-well-based electroporation method that overcomes this hurdle. In addition to cDNA plasmids, vector-based short hairpin RNA (shRNA) could also be used to knockdown gene expression. Thus, libraries of shRNAs that block the expression of genes throughout the genome could be screened with this method. In our hands, knockdown of gene expression with small interfering RNAs (siRNAs) in neurons requires different transfection parameters, which we have not yet optimized.
Buffers
Different minimal buffers were examined for use in electroporation. Extracellular buffers such as HBSS, PBS, L-15, and Neurobasal media were tested. HBSS and PBS worked but still failed to give reasonable transfection efficiencies. Next, an intracellular buffer was tested with a simple formulation based on those used in the electrodes for whole-cell patch clamping of neurons. This buffer turned out to be essential for the success of our system. Due to the large numbers of cDNAs that need to be tested in genome-wide or even smaller screens, it is significant that this methodology use a buffer produced inexpensively with off-the-shelf chemicals. The proprietary reagents often used for transfection are rendered unnecessary—drastically reducing the cost of each transfection and making screens of large sets of genes possible for laboratories with modest budgets.
While we have not investigated the issue systematically, a variety of experiments indicated that the presence of Ca2+ in buffers used to dissociate primary neurons is deleterious to cell survival and that the presence of Ca2+ during electroporation is especially harmful. We make every effort to prevent the introduction of Ca2+ into the various buffers. We now use Ca2+-free Hibernate media to dissociate our neurons.
Electroporation in Neurons
Several laboratories have reported successful transfection and protein expression in neurons after electroporation. Mertz and colleagues (8) used a Bio-Rad Laboratories system to transfect cerebellar neurons in basal media. They used 5 million cells/well and had a transfection efficiency of 10.4% with a viability of 44%. This technique most closely resembles ours but requires the use of a much larger number of cells and is not high-throughput. Teruel et al. (9) used another method in which hippocampal neurons were electroporated after plating. They called this method microporation, and they reported between 1% and 30% transfection efficiency. This system could potentially be modified to use multiple microporation heads and thereby be made compatible with high-throughput applications. Organotypic culture systems are also commonly used as in situ ways to study neurons. Murphy and Messer (10) used BTX paddle electrodes to electroporate slice cultures and reported 26% transfection efficiency.
Amaxa Biosystems (Cologne, Germany) has helped to lead neuroscientists to more reliable and effective transfections with the introduction of their Nucleofector™ technology (2). Recently, Leclere and colleagues (11) used the Nucleofector technology successfully in transfection of retinal ganglion cells and dorsal root ganglia. Using Amaxa system’s proprietary solutions and transfection conditions, Leclere et al. achieved 28% and 20% transfection efficiency, respectively, with these two neuronal populations.
The electroporation methods described above have enabled a variety of studies on neuronal differentiation, synapse formation, and neuronal disease. However, they are not appropriate for projects in which hundreds or thousands of targets need to be screened. A significant problem with earlier methods concerns the numbers of cells needed per transfection. We have optimized the BTX transfection system to work with as few as 25,000 cells per transfection. This is significantly less than the 1 million or more required by most of the other methods listed above. Given that 6 million neurons can be derived from one postnatal mouse cerebellum, there are enough cells to test 240 different cDNAs using the present method. The alternatives mentioned above would require 40 mice for an experiment of similar scale.
Supplementary Material
Acknowledgments
We would like to thank Cristina Vila, Robin Smith, Anthony Oliva, Alexis Tapanes-Castillo, and the rest of the Lemmon/Bixby laboratory, as well as Darcie Moore, Tony DeFazio, Ella Bossy-Wetzel, and Akos Gerencser for their assistance and advice on this project. This work was supported by the Miami Project to Cure Paralysis, the Buoniconti Fund, DOD W81XWH-05-1-0061, and grant no. 2396 from the Paralyzed Veterans of America Research Foundation. W.J.B. is a recipient of a Lois Pope LIFE Scholar award. V.P.L. holds the Walter G. Ross Chair in Developmental Neuroscience at the University of Miami.
Footnotes
COMPETING INTERESTS STATEMENT
The authors declare no competing interests.
To purchase reprints of this article, contact: Reprints@BioTechniques.com
References
- 1.Washbourne P, McAllister AK. Techniques for gene transfer into neurons. Curr Opin Neurobiol. 2002;12:566–573. doi: 10.1016/s0959-4388(02)00365-3. [DOI] [PubMed] [Google Scholar]
- 2.Hamm A, Krott N, Breibach I, Blindt R, Bosserhoff AK. Efficient transfection method for primary cells. Tissue Eng. 2002;8:235–245. doi: 10.1089/107632702753725003. [DOI] [PubMed] [Google Scholar]
- 3.Cheng L, Lemmon V. Pathological missense mutations of neural cell adhesion molecule L1 affect neurite outgrowth and branching on an L1 substrate. Mol Cell Neurosci. 2004;27:522–530. doi: 10.1016/j.mcn.2004.08.005. [DOI] [PubMed] [Google Scholar]
- 4.Dityateva G, Hammond M, Thiel C, Ruonala MO, Delling M, Siebenkotten G, Nix M, Dityatev A. Rapid and efficient electroporation-based gene transfer into primary dissociated neurons. J Neurosci Methods. 2003;130:65–73. doi: 10.1016/s0165-0270(03)00202-4. [DOI] [PubMed] [Google Scholar]
- 5.Beattie CE, Siegel RE. Developmental cues modulate GABAA receptor subunit mRNA expression in cultured cerebellar granule neurons. J Neurosci. 1993;13:1784–1792. doi: 10.1523/JNEUROSCI.13-04-01784.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Keilhauer G, Faissner A, Schachner M. Differential inhibition of neurone-neurone, neurone-astrocyte and astrocyte-astrocyte adhesion by L1, L2 and N-CAM antibodies. Nature. 1985;316:728–730. doi: 10.1038/316728a0. [DOI] [PubMed] [Google Scholar]
- 7.Majoul I, Straub M, Hell SW, Duden R, Soling HD. KDEL-cargo regulates interactions between proteins involved in COPI vesicle traffic: measurements in living cells using FRET. Dev Cell. 2001;1:139–153. doi: 10.1016/s1534-5807(01)00004-1. [DOI] [PubMed] [Google Scholar]
- 8.Mertz KD, Weisheit G, Schilling K, Luers GH. Electroporation of primary neural cultures: a simple method for directed gene transfer in vitro. Histochem Cell Biol. 2002;118:501–506. doi: 10.1007/s00418-002-0473-4. [DOI] [PubMed] [Google Scholar]
- 9.Teruel MN, Blanpied TA, Shen K, Augustine GJ, Meyer T. A versatile microporation technique for the transfection of cultured CNS neurons. J Neurosci Methods. 1999;93:37–48. doi: 10.1016/s0165-0270(99)00112-0. [DOI] [PubMed] [Google Scholar]
- 10.Murphy RC, Messer A. Gene transfer methods for CNS organotypic cultures: a comparison of three nonviral methods. Mol Ther. 2001;3:113–121. doi: 10.1006/mthe.2000.0235. [DOI] [PubMed] [Google Scholar]
- 11.Leclere PG, Panjwani A, Docherty R, Berry M, Pizzey J, Tonge DA. Effective gene delivery to adult neurons by a modified form of electroporation. J Neurosci Methods. 2005;142:137–143. doi: 10.1016/j.jneumeth.2004.08.012. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.