

Abstract
As a normal consequence of aging in men, testosterone levels significantly decline in both serum and brain. Age-related testosterone depletion results in increased risk of dysfunction and disease in androgen-responsive tissues, including brain. Recent evidence indicates that one deleterious effect of age-related testosterone loss in men is increased risk for Alzheimer's disease (AD). We discuss recent findings from our laboratory and others that identify androgen actions implicated in protecting the brain against neurodegenerative diseases and begin to define androgen cell signaling pathways that underlie these protective effects. Specifically, we focus on the roles of androgens as (1) endogenous negative regulators of β-amyloid accumulation, a key event in AD pathogenesis, and (2) neuroprotective factors that utilize rapid non-genomic signaling to inhibit neuronal apoptosis. Continued elucidation of cell signaling pathways that contribute to protective actions of androgens should facilitate the development of targeted therapeutic strategies to combat AD and other age-related neurodegenerative diseases.
Keywords: androgen, testosterone, Alzheimer's disease, neuroprotection, β-amyloid, cell signaling
Introduction
Findings from clinical and basic science studies indicate that testosterone and its androgen metabolites have a wide range of beneficial actions in the central nervous system (CNS). Androgen actions not only influence the development of the CNS, but also help to maintain its proper function in adulthood. However, as a normal consequence of aging in men, both circulating (Feldman et al., 2002; Gray et al., 1991) and brain (Rosario et al., 2004) levels of testosterone exhibit gradual but eventually robust and functionally significant depletion. This age-related androgen loss has senescent effects in androgen-responsive tissues throughout the body, as demonstrated by both impaired function and increased vulnerability to disease (Morley, 2001). As an androgen-responsive tissue, the brain is also thought to suffer deleterious consequences of age-related androgen depletion. Neural manifestations of androgen deficiency in aging males include disturbances in mood, cognition, and libido (Gooren and Kruijver, 2002; Morley, 2001; Swerdloff and Wang, 2003). Recent evidence suggests that androgen depletion in men also increases the risk of developing age-related neurodegenerative disorders including Alzheimer's disease (AD) (Rosario and Pike, 2007). How androgen depletion contributes to CNS dysfunction is not clear, but likely includes the diminished activation of androgen signaling pathways that affect behavior, neuron viability, and regulation of specific pathologies. Further, the cellular and molecular mechanisms that underlie androgen mediated cell signaling pathways remain incompletely defined.
In this review, we discuss recent work from our group and others that begins to identify and characterize androgen signaling pathways that are relevant to protective actions against neurodegenerative diseases. In particular, recent findings from our laboratory indicate that androgens not only regulate β-amyloid (Aβ) accumulation – perhaps the key event in AD pathogenesis – but also modulate vulnerability of neurons to toxic insults. Because cellular effects of sex steroid hormones are often mediated by a number of independent and interactive mechanisms, it is likely that protective actions of androgens involve several different signaling pathways. We will focus primarily on two recently identified androgen signaling pathways that are dependent upon activation of intracellular androgen receptors (AR): a classic genomic pathway involved in regulation of Aβ and a rapid non-genomic pathway that contributes to protection against neuronal apoptosis.
Age-related androgen depletion and Alzheimer's disease
In the past few years, converging observations from several groups have identified increased risk of AD as one of several neural consequences of age-related testosterone depletion in men. In one of the initial studies, Hogervorst and colleagues (2001) reported significantly reduced serum levels of testosterone from men with AD in comparison to age-matched, non-demented men. In subsequent studies by this group, both total (Hogervorst et al., 2004; Hogervorst et al., 2003; Lehmann et al., 2004) and free (Hogervorst et al., 2002) testosterone levels were found to be significantly lower in men without AD irrespective of potentially confounding such indices as age, education, body mass index, smoking, alcohol use, diabetes, and hormone therapy. Similar findings of low testosterone in men with AD have been reported in several (Almeida et al., 2004; Moffat et al., 2004; Paoletti et al., 2004; Rasmuson et al., 2002; Watanabe et al., 2004) but not all (Pennanen et al., 2004) studies. Interestingly, low testosterone has also been linked to several other neurodegenerative diseases, including Parkinson's disease (Okun et al., 2004), vascular dementia (Watanabe et al., 2004), amyotropic lateral sclerosis (Militello et al., 2002), and Huntington's disease (Markianos et al., 2005).
Despite demonstrating a relationship between low testosterone and the diagnosis of AD in men, the previous studies did not determine whether testosterone loss was a comorbid characteristic of AD or, more importantly, a contributing factor to disease pathogenesis. Circulating levels of testosterone are known to decrease as a consequence of several disorders, including diabetes, kidney disease, and liver disease (Albaaj et al., 2006; Barrett-Connor et al., 1990; Elewaut ei al., 1979; Gray et al., 1991). To investigate the potential role of testosterone in contributing to development of AD, we investigated the relationship between brain levels of testosterone and amounts of AD neuropathology (Rosario et al., 2004). Brain samples of midfrontal gyrus that had been rapidly collected and frozen from male cases with brief postmortem delays (average 4.6 hours) were used to extract, purify, and analyze tissue levels of testosterone and estradiol. We found that in neuropathologically normal men, brain levels of testosterone exhibited a robust age-related decline between 50−100 years of age that appeared to reach nadir at approximately 80 years of age (Rosario et al., 2004). We found no age-related change in brain levels of estradiol in these men. These data are generally consistent with the literature on age-related changes in circulating levels of testosterone and estradiol in men except that the extent of testosterone depletion appears to be more severe in brain.
Next, we compared brain levels of sex steroids in men with and without AD neuropathology, as defined by Braak staging criteria. We observed an approximately 50% decrease in brain testosterone in men with AD aged 60−80 years in comparison to age-matched men lacking any evidence of AD or other neuropathology, an effect that was statistically significant by analysis of covariance with age as the covariable (Rosario et al., 2004). To address the issue of whether depletion of brain testosterone is a contributing factor to AD pathogenesis or merely a disease consequence, we also examined a third group of men that were determined to have mild neuropathological changes (Braak stage 2−3) consistent with very early stages of AD. Importantly, brain levels of testosterone were significantly depleted in men with mild neuropathological changes to the same degree as men with severe AD neuropathology, indicating that testosterone loss likely precedes development of AD. We observed no relationship between brain levels of estradiol and the presence or absence of AD neuropathology. Consistent with our findings are longitudinal data that demonstrate circulating levels of testosterone are lower in men with AD up to 10 years prior to clinical diagnosis of dementia (Moffat et al., 2004). Together, these findings suggest that testosterone depletion occurs prior to development of AD and thus may act as a contributing factor to AD pathogenesis.
In subsequent work, we have found that the relationship between low testosterone and increased risk for AD does not appear to apply to women. Recently, we have completed a more extensive analysis of the relationships between age, AD neuropathological status, and brain levels of sex steroid hormones in both men and women. Interestingly, we found that women with AD exhibit lower brain levels of estradiol, an effect we did not observe in men (ERR and CJP, unpublished observations). However, in contrast to our observations in men (Rosario et al., 2004), we observed neither an age-related change in brain levels of testosterone in neuropathologically normal women nor a significant difference in brain testosterone between women with and without AD neuropathology (ERR and CJP, unpublished observations). These results are consistent with observations of circulating total testosterone, which was found to be decreased in men but not women with AD (Hogervorst et al., 2001). Together, these results suggest sex-specific relationships between age-related changes in sex steroid hormones and vulnerability to AD and perhaps other neurodegenerative diseases. Interestingly, these relationships may also be affected by apolipoprotein E ε4 status, which is associated with higher salivary T levels in men but lower salivary T levels in women (Berteau-Levy et al., 2007). One important area that remains to be clearly elucidated is how testosterone depletion in aging men contributes to AD pathogenesis.
Androgens regulate β-amyloid accumulation
Perhaps the key event in initiating and driving AD pathogenesis is the accumulation of β-amyloid protein (Aβ) (Hardy and Selkoe, 2002). In the past several years, findings from our research group and others indicate that androgens act as endogenous negative regulators of Aβ accumulation. Consequently, the age-related depletion of testosterone likely diminishes the ability of the brain to adequately regulate Aβ, resulting in increased Aβ accumulation and development of AD. The mechanisms by which androgens regulate Aβ accumulation have yet to be fully determined but apparently involve at least two pathways.
One mechanism by which androgens may influence Aβ is via aromatization to estradiol and activation of estrogen pathways. Previous studies have shown that estrogen can reduce levels of soluble Aβ (Greenfield et al., 2002; Levin-Allerhand et al., 2002; Manthey et al., 2001; Petanceska et al., 2000; Xu et al., 1998; Zheng et al., 2002) by a mechanism that involves regulating the processing and or trafficking of amyloid precursor protein (APP) (Greenfield et al., 2002; Jaffe et al., 1994), the parental protein of Aβ. APP is proteolytically cleaved at the amino-and carboxyl-termini of Aβ by β-secretase and γ-secretase, respectively, to generate the 40−42 amino acid Aβ peptide (Sinha and Lieberburg, 1999). However, APP is alternatively processed within the Aβ sequence by α-secretase, which prevents formation of full-length Aβ. This nonamyloidogenic processing of APP results in secretion of soluble APP-α(sAPPα), which can be used as an index of APP metabolism and, indirectly, Aβ production.
In the first study to investigate the potential regulatory actions of androgens on Aβ, Gouras and colleagues (2000) (Gouras et al., 2000) evaluated the effect of testosterone on APP metabolism and Aβ production in cultured cortical neurons and a neuroblastoma cell line. They found that, in parallel to the actions of estrogen, prolonged treatment of cultures with testosterone resulted in elevated levels of sAPPα and reduced levels of Aβ (Gouras et al., 2000). Their data clearly demonstrated that testosterone can regulate Aβ levels, but did not indicate whether the mechanism involves AR-dependent pathways or aromatization to estradiol and activation of established estrogen pathways. Because aromatase is present in neurons and can effectively metabolize testosterone into estradiol (Melcangi et al., 1992; Poletti et al., 1997), an estrogen pathway is certainly reasonable. In fact, a subsequent study by Goodenough and colleagues (2000) indicated aromatization to estradiol underlies testosterone regulation of APP and, presumably, Aβ. Like Gouras et al. (2000), these researchers found that testosterone increased sAPPα levels, indicating non-amyloidogenic processing of APP (Goodenough et al., 2000). However, the testosterone-mediated increase in sAPPα was blocked by aromatase inhibition, suggesting that testosterone regulation of Aβ occurs at least in part through estrogen pathways. Perhaps consistent with these cell culture findings are observations from men with prostate cancer in which treatment with anti-androgen therapy reduced circulating levels of testosterone and estradiol and increased Aβ (Almeida et al., 2004; Gandy et al., 2001). Although experimental evidence demonstrates that testosterone can regulate Aβ by an estrogen-mediated mechanism, the significance of this pathway to AD risk in men is questioned by our findings that AD in men is associated with brain levels of testosterone but not estradiol (Rosario et al., 2004) and that soluble Aβ levels in male rats are reduced by DHT but not estradiol (Ramsden et al., 2003a).
A second mechanism by which androgens regulate Aβ accumulation involves estrogen-independent androgen signaling pathways. To begin distinguishing between androgen versus estrogen actions of testosterone in regulation of Aβ accumulation, we used utilized the testosterone metabolite dihydrotestosterone (DHT) in a series of rodent studies. DHT is a potent AR ligand that is produced from testosterone by the enzyme 5α-reducatse. Importantly, DHT is not aromatized to estradiol, although it can be metabolized to form 5α-androstan-3β, 17β-diol which has some agonist effects on estrogen receptor β (Lund et al., 2006). In our first study of this issue, we evaluated how brain levels of soluble Aβ in adult male rats were affected by gonadectomy (GDX) and subsequent treatment with vehicle, DHT, or estradiol. If androgens negatively regulate Aβ accumulation, we predicted that GDX-induced androgen depletion would increase Aβ levels in brain. In agreement with this hypothesis, analysis of soluble Aβ in whole brain by a sensitive and specific ELISA several weeks after GDX showed a significant increase in Aβ in GDX versus sham GDX male rats (Ramsden et al., 2003a). Further, this GDX-induced increase in Aβ was prevented in male rats treated with subcutaneous, slow-release hormone pellets containing DHT. Notably, GDX male rats treated with estradiol pellets did not show any reduction in Aβ (Ramsden et al., 2003a). Our finding that estradiol treatment did not reduce Aβ in GDX male rats is in contrast to observations that estradiol can reduce Aβ in GDX female rodents (Carroll et al., 2007; Petanceska et al., 2000; Levin-Allerhand et al., 2002; Zheng et al., 2002). Similarly, DHT but not estradiol increases hippocampal spine density in male rats (Leranth et al., 2003) although estradiol positively regulates hippocampal spine density in female rats (Woolley and McEwen, 1993). We have observed parallel sex differences in the neuroprotective effects of DHT and estradiol in male and female rats (MR and CJP, unpublished observations). These data suggest that some neural actions of sex steroid hormones are sex-dependent, with estradiol showing significant effects in females but not males. Together, these findings suggest that brain levels of Aβ in males are regulated by androgens and that the underlying mechanism involves, at least in part, androgen signaling pathways that are independent of estrogen.
If androgens are in fact endogenous regulators of Aβ, one might expect that changes in androgen levels more subtle than GDX-induced depletion may affect Aβ levels. In considering this possibility, we examined whether inherent differences in circulating DHT levels in gonadally intact, adult male rats predicted brain levels of soluble Aβ. We observed that male rats with relatively high DHT levels showed lower levels of the two primary forms of Aβ protein, Aβ1−40 and Aβ1−42, than male rats with relatively low DHT levels (Fig. 1). This finding predicts that the even the gradual age-related loss of testosterone associated with normal male aging may promote Aβ accumulation, a possibility supported by recent observations in our lab. First, we have found that in men lacking AD but characterized by mild neuropathological changes, brain levels of testosterone are inversely correlated with brain levels of soluble Aβ (ERR and CJP, unpublished observations). Further, in male brown Norway rats, we have found that age-related decreases in testosterone and DHT are associated with increased brain levels of Aβ (ERR and CJP, unpublished observations). One interesting aspect of the data in aging rats is that the observed changes in androgens and Aβ occurred prior to significant increases in levels of luteinizing hormone, a variable linked to testosterone loss that some have argued also contributes to regulation of Aβ (Casadesus et al., 2005).
Figure 1.

Low serum DHT levels are associated with high brain levels of soluble Aβ. Gonadally intact, adult (age 4 mo) male Sprague-Dawley rats (N=7) were assayed for both serum levels of DHT (determined by radioimmunoassay) and brain levels of soluble Aβ1−40 (A) and Aβ1−42 (B) variants of Aβ peptide (determined by sandwich-capture ELISA from diethylamine-extracted homogenates of hemi-brains) using previously described methods (Ramsden et al., 2003a).
Considering the testosterone and Aβ literature, one obvious and clinically important prediction is that low testosterone may promote the development of AD neuropathology. To investigate this issue, we evaluated how androgen status affects the development of AD-like neuropathology in the 3xTg-AD triple transgenic mouse model of AD (Oddo et al., 2003). We observed that depletion of endogenous androgens by GDX in male 3xTg-AD mice at age 3 mo resulted in a significant increase in accumulation of Aβ in subiculum, hippocampus CA1, and amygdala at age 6 mo (Rosario et al., 2006). This GDX-induced increase in Aβ was associated with a significant worsening in performance in spontaneous alternation behavior, a hippocampal dependent task of working memory. However, continuous treatment with DHT beginning at the time of GDX prevented the increase in Aβ accumulation and worsening in behavioral performance (Rosario et al., 2006). These data confirm in another paradigm that androgen pathways regulate Aβ accumulation and predict a protective role against AD.
Although research findings have established that androgens can regulate Aβ accumulation through estrogen-independent, androgen pathways, the signaling mechanism(s) underlying this action remain to be elucidated. Recent work from our laboratory has begun to define one androgen signaling pathway involved in regulation of Aβ. Levels of Aβ reflect the balance between Aβ production from APP and Aβ clearance. In brain, Aβ clearance is mediated in large part by the Aβ-catabolizing enzyme neprilysin (Iwata et al., 2005). Recently, the neprilysin gene was reported to contain several androgen response elements (ARE), sites at which activated AR may interact with and affect transcription (Shen et al., 2000; Zheng et al., 2006). Thus, we reasoned that androgens may regulate Aβ accumulation by a classic genomic mechanism involving increased expression of neprilysin, which in turn would promote Aβ degradation and thereby decrease Aβ levels. Conversely, androgen depletion would be predicted to reduce neprilysin expression and consequently limit Aβ clearance and promote Aβ accumulation. Consistent with this proposed mechanism, we have found that neprilysin levels in adult male rat brain are reduced by GDX, an effect that is prevented by DHT treatment (unpublished observations, MY and CJP). Further, we observed that DHT increases neprilysin in cultured neurons by an AR-dependent mechanism and that the ability of DHT to reduce Aβ levels is blocked by pharmacological inhibition of neprilysin (unpublished observations, MY and CJP). Together, these data begin to characterize a classic genomic pathway of AR action that contributes to androgen regulation of Aβ accumulation (Fig. 3).
Figure 3.
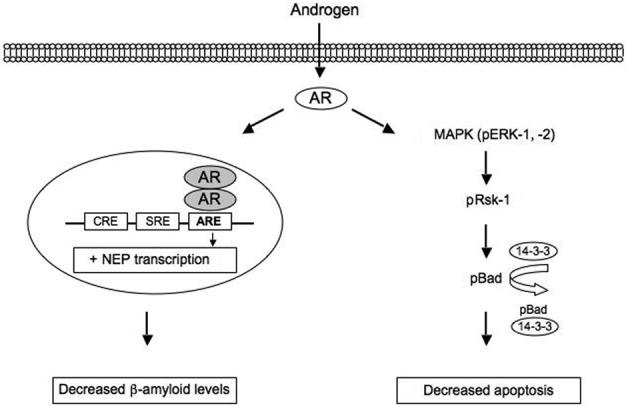
Recent studies in our laboratory indicated two AR-dependent protective pathways. First, androgens decrease levels of the AD-related protein Aβ by a classic genomic mechanism involving activated AR interaction with ARE sites on the neprilysin gene, which results in increased expression of this Aβ-catabolizing enzyme. Second, we find that androgens reduce neuronal apoptosis by an AR-dependent, non-genomic signaling cascade involving activation of MAPK/ERK, followed by activating phosphorylation (p) of Rsk, and inactivating phosphorylation of the pro-apoptotic protein Bad. Both pathways appear to involve activation of intracellular rather than membrane-associated AR.
Androgens regulate neuron viability
In addition to reducing Aβ accumulation, another androgen action that may contribute to its protective role against AD and other age-related neurodegenerative diseases is promotion of neuron viability. During neural development, androgens regulate neuron viability through both androgen and estrogen pathways in several sexually dimorphic brain regions (Lund et al., 2000; Nuñez et al., 2000; Sengelaub et al., 1989; Yang et al., 2004). Androgens can also protect motoneurons in developmental and adult models of injury (Tetzlaff et al., 2006).
More applicable to this review is whether androgens influence neuron viability in brain areas vulnerable to neurodegeneration in AD and related disorders. To begin addressing this issue, we investigated whether androgens function as endogenous neuroprotective agents for the AR-abundant hippocampus, a brain region severely affected by AD. We found that four weeks following GDX, androgen depleted male rats exhibited significantly more neuron loss in the pyramidal layer of hippocampus CA2/3 and in the hilus of dentate gyrus in response to systemic administration of the excitotoxin kainate than did sham GDX male rats (Ramsden et al., 2003b). However, continuous DHT treatment initiated two weeks after GDX protected against the increase in neuron loss associated with androgen depletion (Ramsden et al., 2003b). These data suggest that androgen loss increases the vulnerability of the adult brain to toxic insult and that androgen treatment can protect against this diminished neural resistance. It appears likely that several mechanisms may contribute to androgen neuroprotection.
One mechanism of androgen protection involves seizure inhibition. In seizure-related lesion paradigms similar to our kainate model, Frye and colleagues found that acute androgen treatment reduced hippocampal damage (Frye and McCormick, 2000; Frye and Reed, 1998). Their data suggests a protective mechanism that involves inhibition of seizure activity by the DHT metabolite 5α-androstane-3α, 17β-diol (3α-diol) (Rhodes and Frye, 2004; Rhodes et al., 2004). Attenuation of seizure severity will reduce subsequent neuronal injury in these models. 3β-diol appears to attenuate seizures by acting at the GABAA receptor to increase chloride conductance and thereby suppressing excitatory signaling. However, this mechanism does not appear to contribute to androgen neuroprotection observed in our paradigms. First, DHT treatment did not reduce kainate-induced seizures in our model (Ramsden et al., 2003b), presumably because of densensitization to anxiolytic androgen effects with the prolonged hormone treatment used in our model. In addition, unlike testosterone and DHT, 3α-diol does induce direct neuroprotection in our cell culture paradigms (Pike, 2001), discussed below.
Another general mechanism of androgen neuroprotection involves testosterone aromatization to estrogen. Similar to our observations in kainate-treated rats, Azcoitia et al. (2001) found that GDX of adult male rodents caused increased vulnerability to excitotoxic insult. They also found that lesion-induced neuron loss in GDX animals was reduced by acute treatment with testosterone but not with DHT. In addition, they observed that estradiol treatment in this model was neuroprotective and that aromatase inhibition blocked testosterone protection (Azcoitia et al., 2001). These data clearly indicate a role of estrogen in contributing to at least some aspects of androgen neuroprotection. However, as with the 3α-diol pathway, the role of estrogen pathways does not fully explain androgen neuroprotection. Suggesting an androgen mechanism of neuroprotection, treatment of GDX male rats with estradiol in our kainate model failed to protect against neuron loss (MR and CJP, unpublished observations). It is unclear why our observations differ from those of Azcoitia and colleagues (2001), but two potentially important differences between the studies are brain region and hormone treatment regimen. We analyzed neuron viability in the CA2/CA3 region of hippocampus following extended (two-week) hormone treatment, whereas Azcoitia et al. (2001) examined hilus of the dentate gyrus after acute hormone treatment. Thus, there appear to be specific parameters that determine which pathway of androgen action contributes most strongly to neuroprotection.
Additional evidence of androgen neuroprotection mediated by androgen pathways distinct from 3-αdiol actions and independent of estrogen pathways comes from several cell culture studies. In cultured neurons, androgens can protect against a variety of insults, including serum deprivation (Brooks et al., 1998; Hammond et al., 2001), oxidative stress (Ahlbom et al., 2001), and Aβ (Nguyen et al., 2005; Park et al., 2007; Pike, 2001; Zhang et al., 2004). In our studies, we have found that testosterone provides partial protection against Aβ toxicity (Fig. 2), by activating a rapid, non-genomic pathway (Pike, 2001) that involves MAPK signaling (Nguyen et al., 2005). This testosterone neuroprotection is mimicked by DHT but not 3α-diol and is not attenuated by antagonists to estrogen receptors (Pike, 2001). Similarly, Hammond et al. (2001) observed neuroprotection with the non-aromatizable androgen mibolerone and found that the aromatase inhibition did not block testosterone neuroprotection. In a recent study by Park et al., (2007) androgen and estrogen were found to have mechanistic differences in neuroprotection against Aβ. Definitive evidence for an AR-dependent mechanism of androgen neuroprotection came from our observation that DHT was protective against Aβ in PC12 cells stably transfected with AR but not in PC12 cells stably transfected with empty vector. Also indicating an AR-dependent mechanism was the finding that androgen neuroprotection was blocked by the anti-androgen flutamide (Ahlbom et al., 2001; Hammond et al., 2001; Zhang et al., 2004). Thus, androgens can protect neurons by several mechanisms, including AR-dependent pathways that have yet to be defined.
Figure 2.

Testosterone partially protects neurons from Aβ toxicity. Cultured rat hippocampal neurons were pretreated for 1 h with 0 nm (A, C) or 10 nM testosterone (B, D) then exposed for 24 h to 0 μM (A, B) or 25 μM Aβ1−40 (C, D). Images show fluorescent staining of live cells (green) labeled with calcein acetoxymethyl ester and dead cells (red) labeled with ethidium homodimer, as previously described (Pike, 2001). The amount of Aβ-induced cell loss is less in cells treated with testosterone (D) than those treated with vehicle (C).
Potential mechanisms of androgen neuroprotection
One general mechanism that may contribute to AR-dependent androgen neuroprotection is the activation of classic genomic pathways that increase or decrease expression of genes associated with cell survival. First, as noted above, androgen neuroprotection in several culture paradigms is blocked by the anti-androgen flutamide, which was developed to inhibit classic ARE-mediated gene transcription and thus would antagonize AR-dependent genomic mechanisms of androgen neuroprotection. Uncertain are the proteins affected by genomic androgen pathways that participate in neuroprotection. Proposed candidates include members of the heat shock protein family, which are stress-induced proteins that can contribute to cellular protection (Kelly and Yenari, 2002). Zhang et al. (2004) found that androgen neuroprotection was associated with increased expression of heat shock protein 70 (Hsp70). They also found that treatment with recombinant Hsp70 mimicked the protective effect of testosterone in their paradigm. Another possibility is that androgens increase expression of various proteins with antioxidant functions (Ahlbom et al., 2001; Chisu et al., 2006). For example, Ahlbom et al. (2001) found that testosterone protection of cerebellular granule cells against hydrogen peroxide was characterized by a two-fold increase in catalase activity. Definitive demonstration of an androgen-regulated protein with a requisite role in androgen neuroprotection remains an unfulfilled but promising area of investigation.
To begin defining pathways of AR-dependent neuroprotection in our culture paradigms, one of our strategies has been to find mechanistic similarities and differences in the insults that androgens do and do not protect against. In a recently completed study, we compared the ability of testosterone and DHT to protect against a panel of different toxic insults that we empirically determined induced either an apoptotic or non-apoptotic mode of neuronal death. Specifically, we found that Aβ, staurosporine, and Apoptosis Activator II caused apoptosis as defined by the presence of pyknotic nuclei (Kim et al., 2000; Weil et al., 1996; Wiegele et al., 1998) and attenuation of cell death by pharmacological inhibitors of caspase activation and protein synthesis inhibitors (Colussi and Kumar, 1999; Sperandio et al., 2000; Tsujimoto, 1998). In contrast, we found that hydrogen peroxide, iron, calcium ionophore A23187, and 3-nitroproprionate induced non-apoptotic cell death according to our criteria (TVN and CJP, unpublished observations). Upon comparing the ability of androgens to provide neuroprotection against these insults, we found that testosterone and DHT significantly reduced cell death induced by all of the apoptotic insults but by none of the non-apoptotic insults (TVN and CJP, unpublished observations). These new findings suggest that AR-dependent neuroprotection observed in our culture systems involves a mechanism that specifically inhibits pathways of apoptosis. Further, because our initial data suggested a rapidly acting mechanism of androgen neuroprotection (Pike, 2001), our next step has been to focus on rapid cell signaling pathways that antagonize apoptosis.
Non-genomic MAPK signaling contributes to androgen neuroprotection
Although relatively little is known about androgen activation of signal transduction pathways in neurons, androgens are known to activate several signaling events in non-neuronal cells. For example, androgens can rapidly act via messenger systems such as cyclic adenosine 3', 5'-monophosphate in heart (Rubin et al., 1999), inositol 1, 4, 5-trisphosphate and 1, 2-diacylglycerol in osteoblasts (Lieberherr and Grosse, 1994), phospholipase C in macrophages (Benten et al., 1999), and calcium in T cells and macrophages (Wunderlich et al., 2002), largely through membrane-associated AR. In addition, androgens can activate signal transduction pathways that have relevance to cell viability, both in non-neuronal and neuronal cells. For example, androgens can activate phosphatidylinositol-3 kinase(PI3K)/Akt signaling by AR-dependent mechanisms (Baron et al., 2004; Gatson et al., 2007; Sun et al., 2003). Lin et al. (1999) found that the viability of human prostate LNCaP cells depends on PI3K/Akt signaling, as these cells undergo apoptosis following treatment with specific inhibitors of PI3K/Akt. However, DHT can interrupt apoptosis and increase cell survival (Lin et al., 1999), possibly by restoring PI3K/Akt signaling. Likewise, pharmacological inhibition of PI3K induces apoptosis in prostate cancer cells, which DHT and R1881 block (Kimura et al., 2001). These findings indicate a relationship between androgens and the PI3K/Akt cell survival pathway.
A second signaling pathway relevant to apoptosis that can be activated by androgens is the mitogen-activated protein kinase/extracellular signal-regulated protein kinase (MAPK/ERK) cascade. This cascade consists of a series of sequentially activated kinases with numerous downstream targets relevant to regulation of cell viability (Grewal et al., 1999). Androgen-induced activation of MAPK signaling has been shown to positively regulate cell survival against apoptosis in non-neuronal cells induced by hypoxia (Han and Holtzman, 2000), growth factor/serum withdrawal (Hetman et al., 1999), hydrogen peroxide (Ikeyama et al., 2002), and chemotherapy (Pan et al., 2002). For example, DHT increases levels of phosphorylated/active ERK-1 and ERK-2 protein in prostate cancer cells (Peterziel et al., 1999). Not only do androgens activate MAPK/ERK signaling, but activation may lead to inhibition of apoptosis in non-neuronal cells. Kousteni et al. (2001) found that DHT activates Src/Shc/ERK signaling in mouse osteoblasts and osteocytic cell lines, resulting in attenuation of apoptosis. The MAPK/ERK inhibitor PD98059 and U0126, as well as the Src inhibitor PP1 blocked the androgen activation of Src/Shc/ERK signaling and ensuing protection. Furthermore, the AR antagonist flutamide inhibited androgen activation of MAPK/ERK (Kousteni et al., 2001), suggesting the involvement of AR in androgen-induced rapid signaling and protection against apoptosis.
Recently, we investigated the possibilities that androgens may activate MAPK/ERK signaling in neurons and that MAPK/ERK pathways may contribute to androgen neuroprotection. First, we found that testosterone and DHT rapidly and transiently induced activation of MAPK/ERK in cultured hippocampal neurons as evidenced by phosphorylation of ERK-1 and ERK-2 (Nguyen et al., 2005). Importantly, we also observed that androgen neuroprotection in this culture system requires activation of a MAPK/ERK signaling pathway, as pharmacological inhibition of MAPK/ERK signaling by MEK inhibitors blocked both androgen-induced ERK phosphorylation and androgen neuroprotection. In our PC12 cell paradigm, we observed parallel findings of androgen activation of a neuroprotective MAPK/ERK signaling in cell lines stably transfected with AR but not in either wild type or empty vector-transfected cell lines (Nguyen et al., 2005). Perhaps in contrast to our findings, testosterone did not increase ERK phosphorylation in cerebellar granule cells (Wong et al., 2003). Because levels of AR in cerebellum are significantly lower than that observed in hippocampus (Simerly et al., 1990), these contrary observations may reflect regional differences in AR expression. Thus, our findings indicate that one mechanism of androgen neuroprotection is characterized by AR-dependent, rapid, and transient activation of a MAPK/ERK pathway.
Both membrane-associated and intracellular AR may contribute to androgen regulation of MAPK/ERK signaling. In our studies, membrane-impermeable testosterone-BSA conjugates failed to activate MAPK/ERK signaling in both hippocampal neuron and AR-transfected PC12 cell cultures (Nguyen et al., 2005), suggesting that neuroprotective androgen signaling via MAPK/ERK does not involve membrane-associated AR. Similarly, Gatson and colleagues found that androgens activate MAPK as well as PI3K signaling in C6 glial cell lines, presumably though intracellular AR (Gatson et al., 2006). They also observed that DHT was able to protect cultured astrocytes from cell death (Gatson and Singh, 2007). Upon evaluating the effects of DHT-BSA conjugates in their cultures, they found an opposite effect, namely that DHT-BSA suppressed MAPK and PI3K signaling (Gatson et al., 2006) and increased cell death (Gatson et al., 2007). One possibility is that, at least in glial cells, androgens can exert opposing effects on cell signaling pathways and cell viability depending upon whether they act preferentially on membrane or intracellular receptors.
Because MAPK/ERK signaling has many downstream targets potentially relevant to apoptosis, there may be more than one MAPK/ERK pathway contributing to androgen neuroprotection. One downstream effector of MAPK/ERK signaling that our data indicates is relevant to androgen neuroprotection is MAPK-activated protein kinase, also known as p90 ribosomal S6 kinase (Rsk), which is an established regulator of cell viability (Bonni et al., 1999; Zhu et al., 2002). MAPK activates Rsk proteins by phosphorylation (Frodin and Gammeltoft, 1999; Richards et al., 1999). MAPK/ERK activation of Rsk-1 can result in Rsk-mediated phosphorylation and consequent inactivation of Bad, a pro-apoptotic member of the Bcl-2 family (Shimamura et al., 2000; Tan et al., 2000). Bad is thought to alter mitochondrial integrity by interacting with anti-apoptotic proteins Bcl-2 and Bcl-xL, interrupting with their dimerization (Chao and Korsmeyer, 1998; Lutz, 2000). Bad may also dislocate pro-apoptotic Bax, normally bound by Bcl-2 or Bcl-xL, thus freeing and permitting Bax to move to the mitochondria (Cheng et al., 2001). Phosphorylation of Bad at Ser112, however, inactivates it by facilitating interaction with chaperone protein 14−3−3 (Datta et al., 2000). In agreement with this established pathway, we found that MAPK/ERK activation by androgens was followed by activation of Rsk-1, and then phosphorylation of Bad at Ser112. Interruption of this pathway by either blocking ERK or Rsk phosphorylation prevented downstream phosphorylation of Bad and blocked androgen neuroprotection (Nguyen et al., 2005).
There are many MAPK/ERK-Rsk downstream targets in addition to Bad phosphorylation that may affect neuron survival. Relevant effectors of Rsk (Frodin and Gammeltoft, 1999) include c-Fos (Chen et al., 1993), estrogen receptor (Joel et al., 1998), NFκB/IκBα(Schouten et al., 1997), Elk-1 (Jin et al., 2002), and glycogen synthase kinase-3 (Eldar-Finkelman et al., 1995). Downstream targets of MAPK/ERK-Rsk signaling are not necessarily independent, suggesting the possibility of multiple, perhaps converging protective pathways. For example, in a cerebellar granule cell paradigm of neuronal apoptosis, Rsk-induced inactivation of Bad was found to be protective whereas inhibition of CREB activation, another target of MAPK/ERK-Rsk signaling, increased apoptosis (Bonni et al., 1999). Thus, it is possible that MAPK/ERK-Rsk signaling in androgen neuroprotection might regulate neuron viability by two general pathways: i) rapid, non-genomic mechanisms of neuroprotection involving post-translational, phosphorylation-dependent changes in protein activity such as we found with Bad, and ii) late, indirect genomic mechanisms involving activation of transcription factors such as CREB.
Androgen activation of CREB signaling
Based in part on its association with MAPK/ERK signaling, another important signaling molecule potentially relevant to androgen neuroprotective actions is CREB. In addition to being a downstream effector of MAPK/ERK signaling (Bonni et al., 1999; Dolmetsch et al., 2001; Frodin and Gammeltoft, 1999; Morikawa et al., 2004; Vanhoutte et al., 1999; Voulalas et al., 2005), CREB activity is regulated by several other signaling pathways including PI3K/Akt (Perkinton et al., 1999), protein kinase A (Impey et al., 1998; Vitolo et al., 2002), Ca2+/calmodulin-dependent protein kinase IV (Deisseroth et al., 1998; Redmond et al., 2002), and protein kinase C (PKC) (Roberson et al., 1999; Zhao and Brinton, 2003). In neurons, CREB activation is known to regulate a variety of neurotrophic and neuroprotective effects (Crino et al., 1998; Finkbeiner, 2000; Kelly et al., 2000). It is not known whether androgens activate CREB in neurons, although androgens have been shown to activate CREB signaling in non-neural cells (Fix et al., 2004; Unni et al., 2004; Walker, 2003). In fact, androgen-induced, AR-dependent MAPK/ERK-CREB signaling in prostate cancer cells attenuates apoptosis (Kousteni et al., 2003).
To begin investigating the role of CREB signaling in protective actions of androgens, we determined whether androgens activate CREB in primary hippocampal neuron cultures. We found that androgens rapidly increased CREB phosphorylation and that this androgen-induced CREB phosphorylation was dependent upon intracellular AR activation (TVN and CJP, unpublished observations). Somewhat unexpectedly, pharmacological inhibition of the upstream CREB signaling pathways MAPK/ERK, PI3K/Akt, PKA, or CaMKIV did not block the androgen-induced CREB phosphorylation. However, both pharmacological inhibition and depletion of PKC blocked CREB phosphorylation (TVN and CJP, unpublished observations), suggesting a novel AR-dependent, PKC-dependent CREB signaling pathway in neurons. Thus far, the significance of this pathway in terms of androgen protective effects has yet to be determined. While androgen-induced CREB phosphorylation has been observed in non-neuronal cell types (Fix et al., 2004; Unni et al., 2004; Walker, 2003), our finding that androgen activation of CREB is PKC-dependent has not been reported to the best of our knowledge. Continued investigation of this signaling pathway promises to provide important new insight into the mechanisms underlying many neural actions of androgens.
Antiandrogens and AR-dependent neuroprotection
Recently, our group made a set of unexpected findings about the actions of antiandrogens that has provided new insight into mechanisms of AR-dependent androgen signaling that contribute to neuroprotection. Most research groups have reported that the antiandrogen flutamide inhibited androgen neuroprotection in cell culture (Ahlbom et al., 2001; Hammond et al., 2001; Zhang et al., 2004), a finding interpreted as evidence of an AR-dependent mechanism. However, in our initial study of androgen neuroprotection, we found that flutamide mimicked rather than blocked testosterone neuroprotection (Pike, 2001). What do our observations indicate about the role of AR in neuroprotection? Can AR mediate androgen neuroprotection if an AR antagonist is as protective as testosterone and DHT?
The answer to the antiandrogen neuroprotection conundrum may reside in the growing knowledge that, although antiandrogens competitively bind to AR and typically antagonize classic AR actions (Kemppainen et al., 1992; Kemppainen and Wilson, 1996), antiandrogens do not always function as pure AR antagonists. There are two general types of antiandrogens, steroidal and non-steroidal (Gaillard-Moguilewsky, 1991; Singh et al., 2000). Steroidal antiandrogens such as cyproterone acetate can exhibit partial agonist properties with AR and other members of the steroid receptor family (Berrevoets et al., 2002). However, flutamide and other non-steroidal antiandrogens are generally considered pure antiandrogens that lack AR agonist activity (Berrevoets et al., 2002; Kemppainen et al., 1992). Nonetheless, accumulating data suggest that both steroidal and non-steroidal antiandrogens can at times lack antagonist actions and or behave as partial AR agonists in non-neuronal cells (Kemppainen and Wilson, 1996; Lu et al., 1999; Wong et al., 1995). For example, in some experimental paradigms, antiandrogens unsuccessfully blocked AR-dependent, androgen-induced cell signaling (Baron et al., 2004; Benten et al., 1999; Estrada et al., 2003; Fix et al., 2004; Lieberherr and Grosse, 1994) and even mimicked such signaling (Evangelou et al., 2000; Peterziel et al., 1999; Zhu et al., 1999).
To further investigate the neuroprotective actions of antiandrogens, we evaluated the abilities of the antiandrogens flutamide and cyproterone acetate both to antagonize classic, ARE-mediated gene transcription and to regulate AR-dependent, androgen-induced neuroprotection. As expected, we found that testosterone and DHT time-dependently increased the expression of 5α-reductase type I, an androgen-responsive gene, in cultured hippocampal neurons and in PC12 cells stably transfected with AR. Further, consistent with their established AR antagonist properties, we found that flutamide and cyproterone acetate exhibited dose-dependent inhibition of the androgen-induced increase in 5α-reductase expression (Nguyen et al., 2007). Thus, both androgens and antiandrogens exhibited their classic AR agonist and antagonist properties, respectively, in our culture paradigms. Interestingly, we found that flutamide and cyproterone acetate failed to block testosterone and DHT protection against Aβ, suggesting perhaps that classic genomic pathways are not involved in the observed androgen neuroprotection. Instead, both antiandrogens protected against Aβ at a level comparable to testosterone and DHT (Nguyen et al., 2007). One interpretation of these data is that antiandrogens are functioning as androgen agonists in activating a non-genomic, cell signaling pathway that contributes to neuroprotection. We also observed that the dose-response curves for the antiandrogen antagonist (ie., inhibition of androgen-induced 5α-reductase expression) and agonist (ie., protection against Aβ) actions were very similar, suggesting that the two actions may be mediated by interaction with AR. To demonstrate that antiandrogen neuroprotection is AR-dependent, we showed that flutamide and cyproterone acetate exert neuroprotection only in AR-containing cells (Nguyen et al., 2007). Finally, we found that, like testosterone and DHT, flutamide and cyproterone acetate protect cells specifically against apoptotic insults, suggesting a shared mechanism of neuroprotection.
Our finding of AR-dependent neuroprotection by antiandrogens suggests that the AR agonist versus antagonist functions of antiandrogens likely vary according to specific types of AR actions. Antiandrogens appear to antagonize classic genomic AR actions across all paradigms. Thus, observations that androgen neuroprotection is attenuated by antiandrogens in specific paradigms suggests an AR-dependent genomic mechanism, but does not necessarily exclude a rapid, AR-dependent non-genomic mechanism. This explanation would fit the findings of Ahlbom et al. (2001) and Zhang et al. (2004) in which flutamide blocked theorized genomic mechanisms of androgen neuroprotection involving increased expression of catalase and Hsp70, respectively. In contrast, antiandrogens may exert AR agonist properties for some nongenomic androgen pathways. In this case, antiandrogen binding to AR may be sufficient to induce AR activation of specific cell signaling cascades, presumably including the neuroprotective MAPK/ERK pathway observed in our culture models. This hypothesis would account for our observations of dual agonist/antagonist actions of antiandrogens in neuroprotection and match our findings of a rapid, non-genomic neuroprotective pathway specific to apoptotic insults. Another example is the finding that flutamide mimicked rather than inhibited DHT-induced increases in hippocampal spine density (MacLusky et al., 2004), an androgen action found to be independent of classic AR-dependent genomic mechanisms (MacLusky et al., 2006). However, antiandrogens are not predicted to be AR agonists for all non-genomic signaling pathways and thus findings that antiandrogens block androgen protection and or fail to mimic androgen protection are expected. For example, Gatson et al. (2007) implicate PI3K signaling in androgen protection of astrocytes, a pathway that is antagonized by flutamide.
The dual agonist/antagonist nature of antiandrogens has potentially important clinical implications. Development of selective AR modulators (SARMs) that activate protective actions in brain (e.g., reduce Aβ accumulation and neuron death, increase spine density) without exerting unwanted agonist effects in prostate holds great promise for the prevention and treatment of dysfunction and disease that is associated with normal, age-related androgen depletion in men. Similar strategies have been employed in the development of selective estrogen receptor modulators or SERMs including tamoxifen and raloxifene, which exhibit tissue-specific estrogen receptor agonist (e.g., bone, uterus) and antagonist (e.g., breast) actions (Musa et al., 2007). Recently, several new SARMs have been developed that exhibit tissue-specific AR agonist activity (Kearbey et al., 2007; Ostrowski et al., 2007; Wu et al., 2006), although their actions in brain have yet to be reported.
Conclusions
In this review, we have discussed evidence that androgens have numerous beneficial actions in brain that not only promote normal neural function, but also can protect the brain from age-related neurodegenerative diseases. However, as a consequence of normal male aging, both circulating and brain levels of androgens significantly decline, which is predicted to reduce protective androgen actions in brain. In fact, recent work from several groups has clearly demonstrated that age-related testosterone loss is a risk factor for development of AD in men. Because low testosterone appears to precede AD, loss of androgen and presumably diminished androgen signaling may directly contribute to AD pathogenesis. Understanding how androgens mediate their protective effects will provide valuable insight into both normal neural functioning and vulnerability to androgen-related diseases such as AD.
We discuss two candidate androgen actions that may be particularly important to AD, regulation of Aβ accumulation and neuronal vulnerability to apoptosis (Fig. 3). First, data from several paradigms clearly demonstrate that androgens act as endogenous negative regulators of Aβ accumulation. Although the mechanism(s) underlying this androgen action remain incompletely defined, recent evidence from our laboratory shows that androgens reduce Aβ levels by activating a classic genomic, AR-dependent pathway resulting in increased expression of the Aβ-catabolizing enzyme neprilysin (Fig. 3). Second, findings from several research groups have demonstrated that androgens regulate neuron viability across the lifespan. Androgen neuroprotection appears to be mediated by several different mechanisms, including a rapid, non-genomic MAPK signaling pathway that is AR-dependent and specifically inhibits apoptosis (Fig. 3). Interestingly, we find that both steroidal and non-steroidal antiandrogens also activate AR-dependent neuroprotection, reinforcing the feasibility of SARM development that can offer neural-specific protection presumably without adversely affecting tissues such as prostate. We posit that the continued identification and mechanistic elucidation of androgen pathways that modulate disease vulnerability hold significant promise for the design of therapeutic strategies to combat androgen-related health problems in the aging population.
Acknowledgment
This work was supported by NIA grant AG23739 (CJP). ERR was supported by NS52143.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ahlbom E, et al. Testosterone protects cerebellar granule cells from oxidative stress-induced cell death through a receptor mediated mechanism. Brain Res. 2001;892:255–62. doi: 10.1016/s0006-8993(00)03155-3. [DOI] [PubMed] [Google Scholar]
- Albaaj F, et al. Prevalence of hypogonadism in male patients with renal failure. Postgrad Med J. 2006;82:693–696. doi: 10.1136/pgmj.2006.045963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almeida OP, et al. One year follow-up study of the association between chemical castration, sex hormones, beta-amyloid, memory and depression in men. Psychoneuroendocrinology. 2004;29:1071–1081. doi: 10.1016/j.psyneuen.2003.11.002. [DOI] [PubMed] [Google Scholar]
- Azcoitia I, et al. Brain aromatase is neuroprotective. J Neurobiol. 2001;47:318–29. doi: 10.1002/neu.1038. [DOI] [PubMed] [Google Scholar]
- Baron S, et al. Androgen receptor mediates non-genomic activation of phosphatidylinositol 3-OH kinase in androgen-sensitive epithelial cells. J Biol Chem. 2004;279:14579–14586. doi: 10.1074/jbc.M306143200. [DOI] [PubMed] [Google Scholar]
- Barrett-Connor E, et al. Endogenous sex hormone levels in older adult men with diabetes mellitus. Am J Epidemiol. 1990;132:895–901. doi: 10.1093/oxfordjournals.aje.a115732. [DOI] [PubMed] [Google Scholar]
- Benten WP, et al. Functional testosterone receptors in plasma membranes of T cells. Faseb J. 1999;13:123–33. doi: 10.1096/fasebj.13.1.123. [DOI] [PubMed] [Google Scholar]
- Berrevoets CA, et al. Antiandrogens: selective androgen receptor modulators. Mol Cell Endocrinol. 2002;198:97–103. doi: 10.1016/s0303-7207(02)00373-8. [DOI] [PubMed] [Google Scholar]
- Berteau-Pavy F, et al. Effects of sex and APOE e4 on object recognition and spatial navigation in the elderly. Neuroscience. 2007;147:6–17. doi: 10.1016/j.neuroscience.2007.03.005. [DOI] [PubMed] [Google Scholar]
- Bonni A, et al. Cell survival promoted by the Ras-MAPK signaling pathway by transcription-dependent and -independent mechanisms. Science. 1999;286:1358–62. doi: 10.1126/science.286.5443.1358. [DOI] [PubMed] [Google Scholar]
- Brooks BP, et al. A cell culture model for androgen effects in motor neurons. J Neurochem. 1998;70:1054–1060. doi: 10.1046/j.1471-4159.1998.70031054.x. [DOI] [PubMed] [Google Scholar]
- Carroll JC, et al. Progesterone and estrogen regulate Alzheimer-like neuropathology in female 3xTg-AD mice. J Neurosci. 2007 doi: 10.1523/JNEUROSCI.2718-07.2007. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casadesus G, et al. Evidence for the role of gonadotropin hormones in the development of Alzheimer disease. Cell Mol Life Sci. 2005;62:293–8. doi: 10.1007/s00018-004-4384-0. [DOI] [PubMed] [Google Scholar]
- Chao DT, Korsmeyer SJ. BCL-2 family: regulators of cell death. Ann Rev Immunology. 1998;16:395–419. doi: 10.1146/annurev.immunol.16.1.395. [DOI] [PubMed] [Google Scholar]
- Chen RH, et al. Phosphorylation of the c-Fos transrepression domain by mitogen-activated protein kinase and 90-kDa ribosomal S6 kinase. Proc Natl Acad Sci USA. 1993;90:10952–6. doi: 10.1073/pnas.90.23.10952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng EH, et al. BCL-2, BCL-X(L) sequester BH3 domain-only molecules preventing BAX- and BAK-mediated mitochondrial apoptosis. Mol Cell. 2001;8:705–11. doi: 10.1016/s1097-2765(01)00320-3. [DOI] [PubMed] [Google Scholar]
- Chisu V, et al. Effects of testosterone on differentiation and oxidative stress resistance in C1300 neuroblastoma cells. Neuro Endocrinol Lett. 2006;27:807–12. [PubMed] [Google Scholar]
- Colussi PA, Kumar S. Targeted disruption of caspase genes in mice: what they tell us about the functions of individual caspases in apoptosis. Immunol Cell Biol. 1999;77:58–63. doi: 10.1046/j.1440-1711.1999.00788.x. [DOI] [PubMed] [Google Scholar]
- Crino P, et al. Presence and phosphorylation of transcription factors in developing dendrites. Proc Natl Acad Sci USA. 1998;95:2313–2318. doi: 10.1073/pnas.95.5.2313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datta SR, et al. 14−3−3 proteins and survival kinases cooperate to inactivate BAD by BH3 domain phosphorylation. Mol Cell. 2000;6:41–51. [PubMed] [Google Scholar]
- Deisseroth K, et al. Translocation of calmodulin to the nucleus supports CREB phosphorylation in hippocampal neurons. Nature. 1998;392:198–202. doi: 10.1038/32448. [DOI] [PubMed] [Google Scholar]
- Dolmetsch RE, et al. Signaling to the nucleus by an L-type calcium channel-calmodulin complex through the MAP kinase pathway. Science. 2001;294:333–9. doi: 10.1126/science.1063395. [DOI] [PubMed] [Google Scholar]
- Eldar-Finkelman H, et al. Inactivation of glycogen synthase kinase-3 by epidermal growth factor is mediated by mitogen-activated protein kinase/p90 ribosomal protein S6 kinase signaling pathway in NIH/3T3 cells. J Biol Chem. 1995;270:987–90. doi: 10.1074/jbc.270.3.987. [DOI] [PubMed] [Google Scholar]
- Elewaut A, et al. Testosterone metabolism in normal males and male cirrhotics. Z Gastroenterol. 1979;17:402–405. [PubMed] [Google Scholar]
- Estrada M, et al. Testosterone stimulates intracellular calcium release and mitogen-activated protein kinases via a G protein-coupled receptor in skeletal muscle cells. Endocrinology. 2003;144:3586–97. doi: 10.1210/en.2002-0164. [DOI] [PubMed] [Google Scholar]
- Evangelou A, et al. Down-regulation of transforming growth factor beta receptors by androgen in ovarian cancer cells. Cancer Res. 2000;60:929–935. [PubMed] [Google Scholar]
- Feldman HA, et al. Age trends in the level of serum testosterone and other hormones in middle-aged men: longitudinal results from the Massachusetts male aging study. J Clin Endocrinol Metab. 2002;87:589–598. doi: 10.1210/jcem.87.2.8201. [DOI] [PubMed] [Google Scholar]
- Finkbeiner S. CREB couples neurotrophin signals to survival messages. Neuron. 2000;25:11–14. doi: 10.1016/s0896-6273(00)80866-1. [DOI] [PubMed] [Google Scholar]
- Fix C, et al. Testosterone activates mitogen-activated protein kinase and the cAMP response element binding protein transcription factor in Sertoli cells. Proc Natl Acad Sci USA. 2004;101:10919–10924. doi: 10.1073/pnas.0404278101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frodin M, Gammeltoft S. Role and regulation of 90 kDa ribosomal S6 kinase (RSK) in signal transduction. Mol Cell Endocrinol. 1999;151:65–77. doi: 10.1016/s0303-7207(99)00061-1. [DOI] [PubMed] [Google Scholar]
- Frye CA, McCormick CM. Androgens are neuroprotective in the dentate gyrus of adrenalectomized female rats. Stress. 2000;3:185–94. doi: 10.3109/10253890009001122. [DOI] [PubMed] [Google Scholar]
- Frye CA, Reed TA. Androgenic neurosteroids: anti-seizure effects in an animal model of epilepsy. Psychoneuroendocrinology. 1998;23:385–99. doi: 10.1016/s0306-4530(98)00009-2. [DOI] [PubMed] [Google Scholar]
- Gaillard-Moguilewsky M. Pharmacology of antiandrogens and value of combining androgen suppression with antiandrogen therapy. Urology. 1991;37:5–12. doi: 10.1016/0090-4295(91)80095-o. [DOI] [PubMed] [Google Scholar]
- Gandy S, et al. Chemical andropause and amyloid-beta peptide. JAMA. 2001;285:2195–6. doi: 10.1001/jama.285.17.2195-a. [DOI] [PubMed] [Google Scholar]
- Gatson JW, et al. Dihydrotestosterone differentially modulates the mitogen-activated protein kinase and the phosphoinositide 3-kinase/Akt pathways through the nuclear and novel membrane androgen receptor in C6 cells. Endocrinology. 2006;147:2028–2034. doi: 10.1210/en.2005-1395. [DOI] [PubMed] [Google Scholar]
- Gatson JW, Singh M. Activation of a membrane-associated androgen receptor promotes cell death in primary cortical astrocytes. Endocrinology. 2007;148:2458–64. doi: 10.1210/en.2006-1443. [DOI] [PubMed] [Google Scholar]
- Goodenough S, et al. Testosterone stimulates rapid secretory amyloid precursor protein release from rat hypothalamic cells via the activation of the mitogen-activated protein kinase pathway. Neurosci Lett. 2000;296:49–52. doi: 10.1016/s0304-3940(00)01622-0. [DOI] [PubMed] [Google Scholar]
- Gooren LJ, Kruijver FP. Androgens and male behavior. Mol Cell Endocrinol. 2002;198:31–40. doi: 10.1016/s0303-7207(02)00366-0. [DOI] [PubMed] [Google Scholar]
- Gouras GK, et al. Testosterone reduces neuronal secretion of Alzheimer's beta-amyloid peptides. Proc Natl Acad Sci USA. 2000;97:1202–5. doi: 10.1073/pnas.97.3.1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray A, et al. Age, disease, and changing sex hormone levels in middle-aged men: results of the Massachusetts Male Aging Study. J Clin Endocrinol Metab. 1991;73:1016–1025. doi: 10.1210/jcem-73-5-1016. [DOI] [PubMed] [Google Scholar]
- Greenfield JP, et al. Estrogen lowers Alzheimer beta-amyloid generation by stimulating trans-Golgi network vesicle biogenesis. J Biol Chem. 2002;277:12128–36. doi: 10.1074/jbc.M110009200. [DOI] [PubMed] [Google Scholar]
- Grewal SS, et al. Extracellular-signal-regulated kinase signalling in neurons. Curr Opin Neurobiol. 1999;9:544–53. doi: 10.1016/S0959-4388(99)00010-0. [DOI] [PubMed] [Google Scholar]
- Hammond J, et al. Testosterone-mediated neuroprotection through the androgen receptor in human primary neurons. J Neurochem. 2001;77:1319–26. doi: 10.1046/j.1471-4159.2001.00345.x. [DOI] [PubMed] [Google Scholar]
- Han BH, Holtzman DM. BDNF protects the neonatal brain from hypoxic-ischemic injury in vivo via the ERK pathway. J Neurosci. 2000;20:5775–5781. doi: 10.1523/JNEUROSCI.20-15-05775.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science. 2002;297:353–6. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- Hetman M, et al. Neuroprotection by brain-derived neurotrophic factor is mediated by extracellular signal-regulated kinase and phosphatidylinositol 3-kinase. J Biol Chem. 1999;274:22569–22580. doi: 10.1074/jbc.274.32.22569. [DOI] [PubMed] [Google Scholar]
- Hogervorst E, et al. Low free testosterone is an independent risk factor for Alzheimer's disease. Exp Gerontol. 2004;39:1633–1639. doi: 10.1016/j.exger.2004.06.019. [DOI] [PubMed] [Google Scholar]
- Hogervorst E, et al. Testosterone and gonadotropin levels in men with dementia. Neuro Endocrinol Lett. 2003;24:203–208. [PubMed] [Google Scholar]
- Hogervorst E, et al. Apolipoprotein E varepsilon 4 and testosterone interact in the risk of Alzheimer's disease in men. Intl J Ger Psych. 2002;17:938–40. doi: 10.1002/gps.714. [DOI] [PubMed] [Google Scholar]
- Hogervorst E, et al. Serum total testosterone is lower in men with Alzheimer's disease. Neuroendocrinol Lett. 2001;22:163–8. [PubMed] [Google Scholar]
- Ikeyama S, et al. Loss in oxidative stress tolerance with aging linked to reduced extracellular signal-regulated kinase and Akt kinase activities. FASEB J. 2002;16:114–116. doi: 10.1096/fj.01-0409fje. [DOI] [PubMed] [Google Scholar]
- Impey S, et al. Cross talk between ERK and PKA is required for Ca2+ stimulation of CREB-dependent transcription and ERK nuclear translocation. Neuron. 1998;21:869–883. doi: 10.1016/s0896-6273(00)80602-9. [DOI] [PubMed] [Google Scholar]
- Iwata N, et al. Metabolism of amyloid-beta peptide and Alzheimer's disease. Pharmacol Ther. 2005;108:129–48. doi: 10.1016/j.pharmthera.2005.03.010. [DOI] [PubMed] [Google Scholar]
- Jaffe AB, et al. Estrogen regulates metabolism of Alzheimer amyloid beta precursor protein. J Biol Chem. 1994;269:13065–8. [PubMed] [Google Scholar]
- Jin K, et al. MEK and ERK protect hypoxic cortical neurons via phosphorylation of Bad. J Neurochem. 2002;80:119–25. doi: 10.1046/j.0022-3042.2001.00678.x. [DOI] [PubMed] [Google Scholar]
- Joel PB, et al. pp90rsk1 regulates estrogen receptor-mediated transcription through phosphorylation of Ser-167. Mol Cell Biol. 1998;18:1978–84. doi: 10.1128/mcb.18.4.1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kearbey JD, et al. Selective Androgen Receptor Modulator (SARM) treatment prevents bone loss and reduces body fat in ovariectomized rats. Pharm Res. 2007;24:328–35. doi: 10.1007/s11095-006-9152-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly A, et al. Protein synthesis in entorhinal cortex and long-term potentiation in dentate gyrus. Hippocampus. 2000;10:431–437. doi: 10.1002/1098-1063(2000)10:4<431::AID-HIPO9>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- Kelly S, Yenari MA. Neuroprotection: heat shock proteins. Curr Med Res Opin. 2002;18(Suppl 2):s55–s60. [PubMed] [Google Scholar]
- Kemppainen JA, et al. Androgen receptor phosphorylation, turnover, nuclear transport, and transcriptional activation. Specificity for steroids and antihormones. J Biol Chem. 1992;267:968–74. [PubMed] [Google Scholar]
- Kemppainen JA, Wilson EM. Agonist and antagonist activities of hydroxyflutamide and Casodex relate to androgen receptor stabilization. Urology. 1996;48:157–163. doi: 10.1016/s0090-4295(96)00117-3. [DOI] [PubMed] [Google Scholar]
- Kim DH, et al. Okadaic acid induces cycloheximide and caspase sensitive apoptosis in immature neurons. Mol Cells. 2000;10:83–89. doi: 10.1007/s10059-000-0083-8. [DOI] [PubMed] [Google Scholar]
- Kimura K, et al. Androgen blocks apoptosis of hormone-dependent prostate cancer cells. Cancer Res. 2001;61:5611–5618. [PubMed] [Google Scholar]
- Kousteni S, et al. Nongenotropic, sex-nonspecific signaling through the estrogen or androgen receptors: dissociation from transcriptional activity. Cell. 2001;104:719–30. [PubMed] [Google Scholar]
- Kousteni S, et al. Kinase-mediated regulation of common transcription factors accounts for the bone-protective effects of sex steroids. J Clin Invest. 2003;111:1651–64. doi: 10.1172/JCI17261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehmann DJ, et al. The androgen receptor CAG repeat and serum testosterone in the risk of Alzheimer's disease in men. J Neurol Neurosurg Psychiatry. 2004;75:163–164. [PMC free article] [PubMed] [Google Scholar]
- Leranth C, et al. Gonadal hormones affect spine density in the CA1 hippocampal subfield of male rats. J Neurosci. 2003;23:1588–1592. doi: 10.1523/JNEUROSCI.23-05-01588.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levin-Allerhand JA, et al. 17Alpha-estradiol and 17beta-estradiol treatments are effective in lowering cerebral amyloid-beta levels in AbetaPPSWE transgenic mice. J Alzheimers Dis. 2002;4:449–57. doi: 10.3233/jad-2002-4601. [DOI] [PubMed] [Google Scholar]
- Lieberherr M, Grosse B. Androgens increase intracellular calcium concentration and inositol 1,4,5-trisphosphate and diacylglycerol formation via a pertussis toxin-sensitive G-protein. J Biol Chem. 1994;269:7217–23. [PubMed] [Google Scholar]
- Lin J, et al. The phosphatidylinositol 3′-kinase pathway is a dominant growth factor-activated cell survival pathway in LNCaP human prostate carcinoma cells. Cancer Res. 1999;59:2891–7. [PubMed] [Google Scholar]
- Lu S, et al. Neural androgen receptor regulation: effects of androgen and antiandrogen. J Neurobiol. 1999;41:505–12. doi: 10.1002/(sici)1097-4695(199912)41:4<505::aid-neu6>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- Lund TD, et al. The androgen 5alpha-dihydrotestosterone and its metabolite 5alpha-androstan-3beta, 17beta-diol inhibit the hypothalamo-pituitary-adrenal response to stress by acting through estrogen receptor beta-expressing neurons in the hypothalamus. J Neurosci. 2006;26:1448–56. doi: 10.1523/JNEUROSCI.3777-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lund TD, et al. Pre- or postnatal testosterone and flutamide effects on sexually dimorphic nuclei of the rat hypothalamus. Dev Brain Res. 2000;120:261–6. doi: 10.1016/s0165-3806(00)00013-4. [DOI] [PubMed] [Google Scholar]
- Lutz RJ. Role of the BH3 (Bcl-2 homology 3) domain in the regulation of apoptosis and Bcl-2-related proteins. Biochem Soc Trans. 2000;28:51–6. doi: 10.1042/bst0280051. [DOI] [PubMed] [Google Scholar]
- MacLusky NJ, et al. Androgen effects on hippocampal CA1 spine synapse numbers are retained in Tfm male rats with defective androgen receptors. Endocrinology. 2006;147:2392–2398. doi: 10.1210/en.2005-0673. [DOI] [PubMed] [Google Scholar]
- MacLusky NJ, et al. Effects of dehydroepiandrosterone and flutamide on hippocampal CA1 spine synapse density in male and female rats: implications for the role of androgens in maintenance of hippocampal structure. Endocrinology. 2004;145:4154–4161. doi: 10.1210/en.2004-0477. [DOI] [PubMed] [Google Scholar]
- Manthey D, et al. Estrogen induces a rapid secretion of amyloid beta precursor protein via the mitogen-activated protein kinase pathway. Eur J Biochem. 2001;268:4285–91. doi: 10.1046/j.1432-1327.2001.02346.x. [DOI] [PubMed] [Google Scholar]
- Markianos M, et al. Plasma testosterone in male patients with Huntington's disease: relations to severity of illness and dementia. Ann Neurol. 2005;57:520–525. doi: 10.1002/ana.20428. [DOI] [PubMed] [Google Scholar]
- Melcangi RC, et al. Intracellular signalling systems controlling the 5 alpha-reductase in glial cell cultures. Brain Res. 1992;585:411–5. doi: 10.1016/0006-8993(92)91247-c. [DOI] [PubMed] [Google Scholar]
- Militello A, et al. The serum level of free testosterone is reduced in amyotrophic lateral sclerosis. J Neurol Sci. 2002;195:67–70. doi: 10.1016/s0022-510x(01)00688-8. [DOI] [PubMed] [Google Scholar]
- Moffat SD, et al. Free testosterone and risk for Alzheimer disease in older men. Neurology. 2004;62:188–93. doi: 10.1212/wnl.62.2.188. [DOI] [PubMed] [Google Scholar]
- Morikawa Y, et al. Fasting-induced activation of mitogen-activated protein kinases (ERK/p38) in the mouse hypothalamus. J Neuroendocrinol. 2004;16:105–112. doi: 10.1111/j.0953-8194.2004.01135.x. [DOI] [PubMed] [Google Scholar]
- Morley JE. Androgens and aging. Maturitas. 2001;38:61–71. doi: 10.1016/s0378-5122(00)00192-4. discussion 71−3. [DOI] [PubMed] [Google Scholar]
- Musa MA, et al. Medicinal chemistry and emerging strategies applied to the development of selective estrogen receptor modulators (SERMs). Curr Med Chem. 2007;14:1249–61. doi: 10.2174/092986707780598023. [DOI] [PubMed] [Google Scholar]
- Nguyen TV, et al. Androgens activate mitogen-activated protein kinase signaling: Role in neuroprotection. J Neurochem. 2005;94:1639–1651. doi: 10.1111/j.1471-4159.2005.03318.x. [DOI] [PubMed] [Google Scholar]
- Nguyen TV, et al. Flutamide and cyproterone acetate exert agonist effects: induction of androgen receptor-dependent neuroprotection. Endocrinology. 2007;148:2936–43. doi: 10.1210/en.2006-1469. [DOI] [PubMed] [Google Scholar]
- Nuñez JL, et al. Androgens reduce cell death in the developing rat visual cortex. Dev Brain Res. 2000;125:83–8. doi: 10.1016/s0165-3806(00)00126-7. [DOI] [PubMed] [Google Scholar]
- Oddo S, et al. Triple-transgenic model of Alzheimer's disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron. 2003;39:409–21. doi: 10.1016/s0896-6273(03)00434-3. [DOI] [PubMed] [Google Scholar]
- Okun MS, et al. Plasma testosterone levels in Alzheimer and Parkinson diseases. Neurology. 2004;62:411–413. doi: 10.1212/01.wnl.0000106840.72938.84. [DOI] [PubMed] [Google Scholar]
- Ostrowski J, et al. Pharmacological and x-ray structural characterization of a novel selective androgen receptor modulator: potent hyperanabolic stimulation of skeletal muscle with hypostimulation of prostate in rats. Endocrinology. 2007;148:4–12. doi: 10.1210/en.2006-0843. [DOI] [PubMed] [Google Scholar]
- Pan ZZ, et al. Gamma-synuclein promotes cancer cell survival and inhibits stress- and chemotherapy drug-induced apoptosis by modulating MAPK pathways. J Biol Chem. 2002;277:35050–35060. doi: 10.1074/jbc.M201650200. [DOI] [PubMed] [Google Scholar]
- Paoletti AM, et al. Low androgenization index in elderly women and elderly men with Alzheimer's disease. Neurology. 2004;62:301–303. doi: 10.1212/01.wnl.0000094199.60829.f5. [DOI] [PubMed] [Google Scholar]
- Park SY, et al. Caspase-3- and calpain-mediated tau cleavage are differentially prevented by estrogen and testosterone in beta-amyloid-treated hippocampal neurons. Neuroscience. 2007;144:119–27. doi: 10.1016/j.neuroscience.2006.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pennanen C, et al. Serum testosterone levels in males with Alzheimer's disease. J Neuroendocrinol. 2004;16:95–98. doi: 10.1111/j.0953-8194.2004.01133.x. [DOI] [PubMed] [Google Scholar]
- Perkinton MS, et al. Ca(2+)-permeable AMPA receptors induce phosphorylation of cAMP response element-binding protein through a phosphatidylinositol 3-kinase-dependent stimulation of the mitogen-activated protein kinase signaling cascade in neurons. J Neurosci. 1999;19:5861–5874. doi: 10.1523/JNEUROSCI.19-14-05861.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petanceska SS, et al. Ovariectomy and 17beta-estradiol modulate the levels of Alzheimer's amyloid beta peptides in brain. Neurology. 2000;54:2212–7. doi: 10.1212/wnl.54.12.2212. [DOI] [PubMed] [Google Scholar]
- Peterziel H, et al. Rapid signalling by androgen receptor in prostate cancer cells. Oncogene. 1999;18:6322–9. doi: 10.1038/sj.onc.1203032. [DOI] [PubMed] [Google Scholar]
- Pike CJ. Testosterone attenuates beta-amyloid toxicity in cultured hippocampal neurons. Brain Res. 2001;919:160–5. doi: 10.1016/s0006-8993(01)03024-4. [DOI] [PubMed] [Google Scholar]
- Poletti A, et al. Expression of androgen-activating enzymes in cultured cells of developing rat brain. J Neurochem. 1997;68:1298–303. doi: 10.1046/j.1471-4159.1997.68031298.x. [DOI] [PubMed] [Google Scholar]
- Ramsden M, et al. Androgens modulate beta-amyloid levels in male rat brain. J Neurochem. 2003a;87:1052–5. doi: 10.1046/j.1471-4159.2003.02114.x. [DOI] [PubMed] [Google Scholar]
- Ramsden M, et al. Androgens modulate neuronal vulnerability to kainate lesion. Neuroscience. 2003b;122:573–8. doi: 10.1016/j.neuroscience.2003.08.048. [DOI] [PubMed] [Google Scholar]
- Rasmuson S, et al. Increased levels of adrenocortical and gonadal hormones in mild to moderate Alzheimer's disease. Dement Geriatr Cogn Disord. 2002;13:74–79. doi: 10.1159/000048637. [DOI] [PubMed] [Google Scholar]
- Redmond L, et al. Calcium regulation of dendritic growth via CaM kinase IV and CREB-mediated transcription. Neuron. 2002;34:999–1010. doi: 10.1016/s0896-6273(02)00737-7. [DOI] [PubMed] [Google Scholar]
- Rhodes ME, Frye CA. Androgens in the hippocampus can alter, and be altered by, ictal activity. Pharmacol Biochem Behav. 2004;78:483–93. doi: 10.1016/j.pbb.2004.04.020. [DOI] [PubMed] [Google Scholar]
- Rhodes ME, et al. Gonadal, adrenal, and neuroactive steroids' role in ictal activity. Brain Res. 2004;1000:8–18. doi: 10.1016/j.brainres.2003.12.023. [DOI] [PubMed] [Google Scholar]
- Richards SA, et al. Ribosomal S6 kinase 1 (RSK1) activation requires signals dependent on and independent of the MAP kinase ERK. Current Biology : Cb. 1999;9:810–20. doi: 10.1016/s0960-9822(99)80364-9. [DOI] [PubMed] [Google Scholar]
- Roberson ED, et al. The mitogen-activated protein kinase cascade couples PKA and PKC to cAMP response element binding protein phosphorylation in area CA1 of hippocampus. J Neurosci. 1999;19:4337–4348. doi: 10.1523/JNEUROSCI.19-11-04337.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosario ER, et al. Androgens regulate the development of neuropathology in a triple transgenic mouse model of Alzheimer's disease. J Neurosci. 2006;26:13384–13389. doi: 10.1523/JNEUROSCI.2514-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosario ER, et al. Age-related testosterone depletion and the development of Alzheimer disease. JAMA. 2004;292:1431–1432. doi: 10.1001/jama.292.12.1431-b. [DOI] [PubMed] [Google Scholar]
- Rosario ER, Pike CJ. Androgen regulation of β-amyloid protein and the risk of Alzheimer's disease. Brain Res. Rev. 2007 doi: 10.1016/j.brainresrev.2007.04.012. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubin JM, et al. Positive inotropism induced by androgens in isolated left atrium of rat: evidence for a cAMP-dependent transcriptional mechanism. Life Sci. 1999;65:1035–45. doi: 10.1016/s0024-3205(99)00334-3. [DOI] [PubMed] [Google Scholar]
- Schouten GJ, et al. IkappaB alpha is a target for the mitogen-activated 90 kDa ribosomal S6 kinase. Embo J. 1997;16:3133–44. doi: 10.1093/emboj/16.11.3133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sengelaub DR, et al. Hormonal control of neuron number in sexually dimorphic spinal nuclei of the rat: III. Differential effects of the androgen dihydrotestosterone. J Comp Neurol. 1989;280:637–44. doi: 10.1002/cne.902800413. [DOI] [PubMed] [Google Scholar]
- Shen R, et al. Identification and characterization of two androgen response regions in the human neutral endopeptidase gene. Mol Cell Endocrinol. 2000;170:131–42. doi: 10.1016/s0303-7207(00)00326-9. [DOI] [PubMed] [Google Scholar]
- Shimamura A, et al. Rsk1 mediates a MEK-MAP kinase cell survival signal. Current Biology : Cb. 2000;10:127–35. doi: 10.1016/s0960-9822(00)00310-9. [DOI] [PubMed] [Google Scholar]
- Simerly RB, et al. Distribution of androgen and estrogen receptor mRNA-containing cells in the rat brain: an in situ hybridization study. J Comp Neurol. 1990;294:76–95. doi: 10.1002/cne.902940107. [DOI] [PubMed] [Google Scholar]
- Singh SM, et al. Androgen receptor antagonists (antiandrogens): structure-activity relationships. Curr Med Chem. 2000;7:211–247. doi: 10.2174/0929867003375371. [DOI] [PubMed] [Google Scholar]
- Sinha S, Lieberburg I. Cellular mechanisms of beta-amyloid production and secretion. Proc Natl Acad Sci USA. 1999;96:11049–53. doi: 10.1073/pnas.96.20.11049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sperandio S, et al. An alternative, nonapoptotic form of programmed cell death. Proc Natl Acad Sci USA. 2000;97:14376–14381. doi: 10.1073/pnas.97.26.14376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun M, et al. Activation of phosphatidylinositol 3-kinase/Akt pathway by androgen through interaction of p85alpha, androgen receptor, and Src. J Biol Chem. 2003;278:42992–3000. doi: 10.1074/jbc.M306295200. [DOI] [PubMed] [Google Scholar]
- Swerdloff RS, Wang C. Three-year follow-up of androgen treatment in hypogonadal men: preliminary report with testosterone gel. Aging Male. 2003;6:207–211. [PubMed] [Google Scholar]
- Tan Y, et al. BAD Ser-155 phosphorylation regulates BAD/Bcl-XL interaction and cell survival. J Biol Chem. 2000;275:25865–9. doi: 10.1074/jbc.M004199200. [DOI] [PubMed] [Google Scholar]
- Tetzlaff JE, et al. Motoneuron injury and repair: New perspectives on gonadal steroids as neurotherapeutics. J Mol Neurosci. 2006;28:53–64. doi: 10.1385/jmn:28:1:53. [DOI] [PubMed] [Google Scholar]
- Tsujimoto Y. Role of Bcl-2 family proteins in apoptosis: apoptosomes or mitochondria? Genes Cells. 1998;3:697–707. doi: 10.1046/j.1365-2443.1998.00223.x. [DOI] [PubMed] [Google Scholar]
- Unni E, et al. Changes in androgen receptor nongenotropic signaling correlate with transition of LNCaP cells to androgen independence. Cancer Res. 2004;64:7156–7168. doi: 10.1158/0008-5472.CAN-04-1121. [DOI] [PubMed] [Google Scholar]
- Vanhoutte P, et al. Glutamate induces phosphorylation of Elk-1 and CREB, along with c-fos activation, via an extracellular signal-regulated kinase-dependent pathway in brain slices. Mol Cell Biol. 1999;19:136–46. doi: 10.1128/mcb.19.1.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitolo OV, et al. Amyloid beta -peptide inhibition of the PKA/CREB pathway and long-term potentiation: reversibility by drugs that enhance cAMP signaling. Proc Natl Acad Sci USA. 2002;99:13217–13221. doi: 10.1073/pnas.172504199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voulalas PJ, et al. Metabotropic glutamate receptors and dopamine receptors cooperate to enhance extracellular signal-regulated kinase phosphorylation in striatal neurons. J Neurosci. 2005;25:3763–3773. doi: 10.1523/JNEUROSCI.4574-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker WH. Nongenomic actions of androgen in Sertoli cells. Curr Top Dev Biol. 2003;56:25–53. doi: 10.1016/s0070-2153(03)01006-8. [DOI] [PubMed] [Google Scholar]
- Watanabe T, et al. Small dense low-density lipoprotein and carotid atherosclerosis in relation to vascular dementia. Metabolism. 2004;53:476–482. doi: 10.1016/j.metabol.2003.11.020. [DOI] [PubMed] [Google Scholar]
- Weil M, et al. Constitutive expression of the machinery for programmed cell death. J Cell Biol. 1996;133:1053–1059. doi: 10.1083/jcb.133.5.1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiegele G, et al. Apoptosis and necrosis during ischaemia in renal tubular cells (LLCPK1 and MDCK). Nephrol Dial Transplant. 1998;13:1158–1167. doi: 10.1093/ndt/13.5.1158. [DOI] [PubMed] [Google Scholar]
- Wong C, et al. Androgen receptor antagonist versus agonist activities of the fungicide vinclozolin relative to hydroxyflutamide. J Biol Chem. 1995;270:19998–20003. doi: 10.1074/jbc.270.34.19998. [DOI] [PubMed] [Google Scholar]
- Wong JK, et al. Estrogens and ICI182,780 (Faslodex) modulate mitosis and cell death in immature cerebellar neurons via rapid activation of p44/p42 mitogen-activated protein kinase. J Neurosci. 2003;23:4984–95. doi: 10.1523/JNEUROSCI.23-12-04984.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolley CS, McEwen BS. Roles of estradiol and progesterone in regulation of hippocampal dendritic spine density during the estrous cycle of the rat. J Comp Neurol. 1993;336:293–306. doi: 10.1002/cne.903360210. [DOI] [PubMed] [Google Scholar]
- Wu D, et al. Pharmacokinetics and metabolism of a selective androgen receptor modulator in rats: implication of molecular properties and intensive metabolic profile to investigate ideal pharmacokinetic characteristics of a propanamide in preclinical study. Drug Metab Dispos. 2006;34:483–94. doi: 10.1124/dmd.105.006643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wunderlich F, et al. Testosterone signaling in T cells and macrophages. Steroids. 2002;67:535–8. doi: 10.1016/s0039-128x(01)00175-1. [DOI] [PubMed] [Google Scholar]
- Xu H, et al. Estrogen reduces neuronal generation of Alzheimer beta-amyloid peptides. Nature Med. 1998;4:447–51. doi: 10.1038/nm0498-447. [DOI] [PubMed] [Google Scholar]
- Yang SL, et al. Perinatal androgenization prevents age-related neuron loss in the sexually dimorphic nucleus of the preoptic area in female rats. Dev Neurosci. 2004;26:54–60. doi: 10.1159/000080712. [DOI] [PubMed] [Google Scholar]
- Zhang Y, et al. Estrogen and androgen protection of human neurons against intracellular amyloid beta1–42 toxicity through heat shock protein 70. J Neurosci. 2004;24:5315–21. doi: 10.1523/JNEUROSCI.0913-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao L, Brinton RD. Vasopressin-induced cytoplasmic and nuclear calcium signaling in embryonic cortical astrocytes: dynamics of calcium and calcium-dependent kinase translocation. J Neurosci. 2003;23:4228–4239. doi: 10.1523/JNEUROSCI.23-10-04228.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng H, et al. Modulation of A(beta) peptides by estrogen in mouse models. J Neurochem. 2002;80:191–6. doi: 10.1046/j.0022-3042.2001.00690.x. [DOI] [PubMed] [Google Scholar]
- Zheng R, et al. Multiple androgen response elements cooperate in androgen regulated activity of the type 1 neutral endopeptidase promoter. Mol Cell Endocrinol. 2006;259:10–21. doi: 10.1016/j.mce.2006.07.004. [DOI] [PubMed] [Google Scholar]
- Zhu X, et al. Androgen stimulates mitogen-activated protein kinase in human breast cancer cells. Mol Cell Endocrinol. 1999;152:199–206. doi: 10.1016/s0303-7207(99)00031-3. [DOI] [PubMed] [Google Scholar]
- Zhu Y, et al. Transforming growth factor-beta 1 increases bad phosphorylation and protects neurons against damage. J Neurosci. 2002;22:3898–909. doi: 10.1523/JNEUROSCI.22-10-03898.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]