

Abstract
Genetic variation among individual humans occurs on many different scales, ranging from gross alterations in the human karyotype to single nucleotide changes. Here we explore variation on an intermediate scale—particularly insertions, deletions and inversions affecting from a few thousand to a few million base pairs. We employed a clone-based method to interrogate this intermediate structural variation in eight individuals of diverse geographic ancestry. Our analysis provides a comprehensive overview of the normal pattern of structural variation present in these genomes, refining the location of 1,695 structural variants. We find that 50% were seen in more than one individual and that nearly half lay outside regions of the genome previously described as structurally variant. We discover 525 new insertion sequences that are not present in the human reference genome and show that many of these are variable in copy number between individuals. Complete sequencing of 261 structural variants reveals considerable locus complexity and provides insights into the different mutational processes that have shaped the human genome. These data provide the first high-resolution sequence map of human structural variation—a standard for genotyping platforms and a prelude to future individual genome sequencing projects.
Human genetic structural variation, including large (more than 1 kilobase pair (kbp)) insertions, deletions and inversions of DNA, is common1–9. These differences are thought to encompass more polymorphic base pairs than single nucleotide differences5,6,9,10. The importance of structural variation to human health and common genetic disease has become increasingly apparent11–14. However, only a small fraction of copy-number variant (CNV) base pairs have been determined at the sequence level15. Most genome-wide approaches for detecting CNVs are indirect, depending on signal intensity differences to predict regions of variation. They therefore provide limited positional information and cannot detect balanced events such as inversions. Because the human genome reference assembly is now viewed as a patchwork of structurally variant sequence1,2, it is expected that sequencing projects of other individuals would reveal previously uncharacterized human euchromatic sequence, in a similar manner to comparisons between the Celera and International Human Genome Project assemblies16–18. We implemented an approach to construct clone-based maps of eight human genomes with the aim of systematically cloning and sequencing structural variants more than 8 kbp in length. We present a validated, structural variation map of these eight human genomes of Asian, European and African ancestry, identify 525 regions of previously uncharacterized ‘novel sequence’, and provide sequence resolution of 261 selected regions of structural variation in the human genome.
Fine-scale map of human genome structural variation
We selected eight individuals as part of the first phase of the Human Genome Structural Variation Project19 (Supplementary Information). This included four individuals of Yoruba Nigerian ethnicity and four individuals of non-African ethnicity20 (Table 1 and Supplementary Information). For each individual we constructed a whole genomic library of about 1 million clones by using a fosmid subcloning strategy21. Each library was arrayed and both ends of each clone insert were sequenced to generate a pair of high-quality end sequences (termed an end-sequence pair (ESP)22). The overall approach generated a physical clone map for each individual human genome, flagging regions discrepant by size or orientation on the basis of the placement of end sequences against the reference assembly (Supplementary Fig. 1)3,19. Across all eight libraries, we mapped 6.1 million clones to distinct locations against the reference sequence (Supplementary Fig. 2; http://hgsv.washington.edu). Of these, 76,767 were discordant by length and/or orientation (Supplementary Fig. 3 and Supplementary Table 1), indicating potential sites of structural variation. About 0.4% (23,742) of the ESPs mapped with only one end to the reference assembly despite the presence of high-quality sequence at the other end (termed one-end anchored (OEA) clones; Supplementary Table 2 and Supplementary Information).
Table 1.
Validated sites of structural variation detected by fosmid end sequence pairs
| Library Information | Nucleotide Variation | Structural Variation | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Library | Population | Coriell ID | Number of Reads | Q30 Bases | Total SNPs | Total INDELs | Mean in silico insert size (kb) | StdDev (kb) | Size Threshold (kb) | Deletions | Insertions | Inversions |
| G248 | NA15510 | 39.89 | 2.75 | 8.25 | 105 | 108 | 48 | |||||
| ABC7 | Yoruba | NA18517 | 2,076,237 | 841,505,234 | 705,558 | 125,307 | 37.59 | 3.88 | 11.64 | 86 | 42 | 59 |
| ABC8 | Yoruba | NA18507 | 3,331,676 | 1,549,030,580 | 1,241,616 | 222,740 | 36.7 | 3.85 | 11.55 | 156 | 101 | 67 |
| ABC9 | Japan | NA18956 | 2,076,828 | 1,032,726,952 | 610,897 | 114,203 | 39.51 | 2.26 | 6.78 | 163 | 186 | 79 |
| ABC10 | Yoruba | NA19240 | 2,118,546 | 1,022,392,331 | 736,451 | 130,646 | 41 | 1.84 | 5.52 | 252 | 297 | 88 |
| ABC11 | China | NA18555 | 1,966,644 | 939,700,332 | 590,518 | 102,650 | 40.03 | 1.77 | 5.31 | 286 | 246 | 67 |
| ABC12 | CEPH | NA12878 | 2,168,656 | 1,005,800,422 | 610,619 | 102,524 | 39.75 | 1.4 | 4.2 | 274 | 258 | 77 |
| ABC13 | Yoruba | NA19129 | 2,053,392 | 1,083,273,138 | 591,310 | 101,565 | 39.29 | 1.77 | 5.31 | 246 | 265 | 83 |
| ABC14 |
CEPH |
NA12156 |
2,021,844 |
1,086,415,113 |
714,848 |
140,498 |
39.44 |
1.72 |
5.16 |
275 |
265 |
98 |
|
Total |
|
|
17,813,823 |
8,560,844,102 |
5,801,817 |
1,040,133 |
|
|
|
1843 |
1768 |
666 |
| Non Redundant Total | 4,044,538 | 795,989 | 747 | 724 | 224 | |||||||
Number of reads: number of sequencing reads generated from each library
Q30 Bases: Number of sequenced bases with a quality score of at least Q30 (99.9% accuracy)
SNPs: single nucleotide variants with respect to the reference genome, no information regarding frequency.
INDELs: Insertion/deletion variants (>1 bp <100 bp in size)
Mean in silico insert size: mean size of clones based on mapping of paired reads
StdDev: Standard deviation of in silico clone insert sizes based on end-sequence pair mapping
Size Threshold: 3 standard deviation size threshold used for detecting variant sites (see Supplemental Methods)
*1,068 SNP and 284 INDELs on the Y chromosome were identified from the single male sample (NA18507)
We undertook three main approaches to validate sites of copy-number variation. First, we selected 3,371 discordant fosmids corresponding to sites supported by two or more overlapping fosmids from the same individual whose apparent insert size deviated from the library mean insert size. These corresponded to 2,990 non-overlapping sites that are supported by multiple independent clones3. Using four multiple complete restriction enzyme digests (MCD analysis), we compared the predicted and expected insert sizes, confirming 1,182 non-redundant sites of copy-number variation (Supplementary Tables 3 and 4). As a secondary validation method, we designed two high-density customized oligonucleotide microarrays targeting a subset of insertion and deletion regions (Supplementary Fig. 4). This analysis recovered an additional 194 regions that had a copy-number difference but were not validated by MCD analysis. Combined with other experimental methods, we validated a total of 1,471 sites of copy-number variation (Fig. 1, Table 1, Supplementary Tables 3 and 4, and Supplementary Information). To assess the heritability of our events, we further intersected validated deletions with single nucleotide polymorphism (SNP) genotyping data (Illumina Human1M BeadChip) collected for 125 HapMap DNAs of African, European and Asian individuals, which included 28 parent–child trios. Although only a subset of the deletion events (n = 130) could be reliably genotyped because of a lack of informative probes (Supplementary Fig. 5 and Supplementary Table 5), the allele frequencies ranged from rare (1%) to common (more than 50%), were generally consistent with Hardy–Weinberg equilibrium, and more than 98% of parent–child transmissions were consistent with mendelian patterns of inheritance (Supplementary Information).
Figure 1. Map of structural variation in the human genome.

The location of 724 insertions (blue), 747 deletions (red) and 224 inversions (green) that have been experimentally validated are mapped onto the human genome (build35). Sites are arranged according to individuals in rows above each chromosome, in order of the nine individual genomic libraries (G248 (first row), then ABC7–ABC14); the Coriell IDs are listed in Table 1. All sites have been validated by array CGH, MCD analysis, or sequencing in at least one reference individual. The location of 525 novel sequence loci are depicted as arrows below each chromosome. Those mapping to gaps (black) are distinguished from those mapping to regions not associated with gaps (orange). The Y chromosome is not shown because samples were primarily from females.
Inversions proved more difficult to validate in a high-throughput manner because the events are balanced and because breakpoints are prone to map in the largest and most complex regions of segmental duplications23–25. We validated 217 inversions by detailed fingerprint analysis and/or sequence analysis. In addition, we validated seven larger ESP-detected inversions by interphase and metaphase fluorescence in situ hybridization (Supplementary Fig. 6, and Supplementary Tables 6 and 7). This included two previously described events: a roughly 5-million-base pair (Mbp) inversion on 8p23.1 and a roughly 1-Mbp inversion on 17q21.3. We detected five novel large inversions, including a 1.2-Mbp inversion on 15q24, a 2.1-Mbp inversion on 15q13, and a 1.7-Mbp inversion on 17q12. Three of these regions correspond to sites of recurrent microdeletion associated with human disease, providing further support for a link between common inversion polymorphisms and genomic disorders26,27. Overall, we found a twofold enrichment for inversions mapping to clustered regions of the X chromosome (Fig. 1 and Supplementary Table 7), consistent with theoretical predictions of increased inversion content based on unusual inverted repeat structures28. These data provide one of the first high-quality inversion maps of the human genome.
In total, we validated and refined the location of 1,695 sites of structural variation across nine diploid human genomes (eight fosmid libraries plus the original genome examined by the fosmid ESP approach (G248)) (Fig. 1, Table 1 and Supplementary Fig. 7). This included 747 deletions, 724 insertions and 224 inversions. A large fraction of the insertion/deletion events (40%) are novel when compared with previous published reports of CNVs. This is particularly unexpected, considering that at least 25% of the human genome now shows some evidence of copy-number variation (The Database of Genomic Variants1, hg17.v2). Many of the events (856, or 50%) were identified in multiple libraries and probably represent common polymorphisms (more than 5% frequency) (Fig. 2); 261 (15%) of the sites were observed in five or more individuals, indicating that the current reference human genome sequence organization may actually represent a minor allele. At 34 loci, all nine individuals were inconsistent with the build35 assembly, identifying the reference allele as rare or as a potential sequence misassembly.
Figure 2. Frequency distribution.
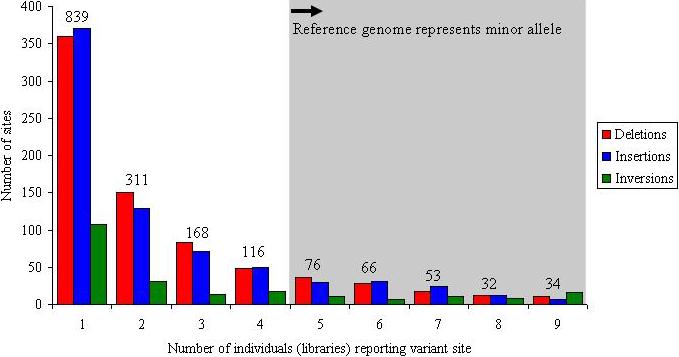
Plot showing the number of times that a particular structural variant was detected on the basis of ESP analysis for nine fosmid libraries (eight HapMap, plus G248): 15% (261 of 1,695) of the sites seem to represent a more common sequence configuration (major allele) with respect to the human reference genome; 49% (839 of 1,695) of the validated sites are observed once, suggesting that saturation has not been achieved. The numbers above the columns report the total number of events for each frequency class.
Using the refined set of CNVs, we compared CNV predictions within eight of the same samples analysed in ref. 5 (Supplementary Information). When we compared the predicted size of intersected sites on the same eight samples, we found that the bacterial artificial chromosome (BAC) array comparative genomic hybridization (CGH) CNVs were substantially (tenfold) larger and showed no correlation with the ESP estimated size (Supplementary Fig. 8). In contrast, we found extremely strong concordance between the sizes estimated from the ESP map and the annotations generated by our targeted high-density array CGH experiments (Supplementary Fig. 8b) and independent predictions on the same eight individuals analysed using the Affymetrix 6.0 platform (Supplementary Information and Supplementary Fig. 8c). We conclude that the BAC array CGH experiments performed in ref. 5 had, in some cases, exquisite sensitivity to detect much smaller events (about 10 kbp) than previously expected. However, our analysis indicates that the current amount of the reference genome sequence represented as CNV in these eight genomes has been overestimated.
Novel human euchromatic sequences
To identify potentially novel euchromatic sequences not present within the reference genome, we first identified clusters of clones in which one end sequence mapped to the human reference assembly but the other end sequence did not, termed OEA clusters (Fig. 3a and Supplementary Information). Pooling results from the first seven genomic libraries, we identified 21,556 OEA clones. Next, we assembled the sequence corresponding to all non-anchored ends by using the TIGR assembler29. This procedure generated 1,736 sequence contigs (n = 4,996 OEA clones) of which 48% (820) had no matches to previously published human sequence assemblies (minimum 100 base pairs (bp) with more than 98% sequence identity). By combining these sequence contigs with the positions of the OEA clusters we identified the map location of 525 regions of novel sequence insertion.
Figure 3. Discovery of novel human sequences that are CNV.

a, Clusters of clones where one end is mapped to the genome (build35) but the other does not map are shown schematically on the basis of their orientation (blue and yellow lines). Three categories are distinguished: clones mapping around a site already spanned by a discordant fosmid ESP (spanned), regions where no discordant clones are identified (unspanned), and clones mapping adjacent to sequence gaps (gap). b, Array CGH experiment based on an oligonucleotide microarray designed to a sequence assembly of these novel sequences (525 distinct loci). Of the spanned and unspanned loci, 45% show copy-number variation (gains, orange; losses, blue) in comparison with a reference sample (NA15510). Each data point represents the average log2 intensity values for all of the probes from a single contig. Within each of the three categories, contigs are ordered on the basis of their chromosomal anchored positions. The bottom row represents the results of one of three self-versus-self hybridizations with sample NA15510. c, A novel insertion of 130 kbp on chromosome 6 identified by OEA fosmid clones (blue and gold arrows) and confirmed by optical mapping of DNA from the GM15510 cell line. Optical images of SwaI-restricted DNA are aligned to the reference (build35) genome. This large insertion maps intergenically to a region rich in conserved sequence elements and is confirmed in all eight libraries. This region does not correspond to a known gap in the human genome and does not appear CNV in our eight samples. d, Validation of a CNV region by fluorescence in situ hybridization. Hemizygous signals are detected by fluorescence in situ hybridization on metaphase chromosomes (with OEA clone ABC7_42397600_G7 as probe), corresponding to samples where no signal intensity difference was observed with respect to the reference by array CGH.
We distinguished three categories of novel insertion (Fig. 3a and Supplementary Fig. 9): 214 of the novel insertion loci intersected with regions identified as insertions with the paired-end sequence approach (see above); 139 putative ‘insertions’ flanked sequence assembly gaps30; and another 172 new sites did not correspond to known gaps or spanned insertions within the human genome. Among these we identified at least 11 regions where we estimate that the insertions are too large (more than 40 kbp in length) to be physically spanned by fosmid ESPs. Examination of these loci in a whole-genome restriction map constructed by optical mapping (Supplementary Information) on one of the same individuals confirms that the majority (8 of 11) correspond to insertions as large as 130 kbp in length (Fig. 3c and Supplementary Table 8).
To assess copy-number variation of these unannotated human sequences, we designed an oligonucleotide microarray specifically for these 525 loci and assessed copy-number status by array CGH (Supplementary Information) among the eight genomes tested (Fig. 3b). Novel sequences not associated with gaps showed the most extensive variation in copy number. For example, we found that 49% of novel sequences associated with fosmid ESPs (spanned insertions) showed evidence of copy-number variation. We note that sequence contigs mapping to the same novel locus (Fig. 3d) often showed the same pattern of copy-number variation. Such regions cannot be genotyped by existing commercial platforms that depend on sequence in the reference genome. The presence of a mapped clone ensures that these regions can be sequenced in their entirety (see below) and incorporated as part of future CNV and SNP genotyping platforms.
Sequence resolution
The acquisition of high-quality, finished sequence corresponding to the breakpoints of a rearrangement is the ultimate form of validation31. We selected 405 clones with predicted structurally variant haplotypes by ESP analysis for full-length sequencing (Supplementary Fig. 10 and Supplementary Table 9), generating 16 Mbp of alternative haplotype sequence. Sequence validation confirmed 230 insertion/deletion loci (median 8.1 kbp, mean 15.3 kbp) and 35 inversions (up to 2 Mbp in size). Validation for 63 sequenced clones could not be conclusively resolved, despite the fact that both fingerprint and ESP analysis confirmed 80% of these ‘ambiguous’ clones as being structurally variant with respect to the reference genome. Detailed sequence analyses revealed that most of these contained large, multi-copy tandem repeat sequences, which confound breakpoint identification and complicate the final sequence assembly of the insert. Including these ambiguous clones, we estimate that 84% (341 of 405) of the clones contained structural variants. The vast majority of the clones that failed to confirm at the sequence level represented putative insertion events (Supplementary Information) as a result of a slight subcloning preference for ‘short insert’ clones.
High-quality finished sequence at the breakpoints allowed us to assess the potential molecular mechanisms underlying larger structural variation events in the human genome (Table 2). Non-allelic homologous recombination between repeated sequences accounts for 47% (124 of 261) of events assigned a mechanism. Recombination between segmental duplications is more common than L1 or Alu-mediated events. Of the inversions, 67% show evidence of large blocks of sequence homology at the breakpoints, with the remainder mediated by shorter common repeat sequences. An additional ten events (4%) involved the expansion or contraction of a variable number of tandem repeats. Retrotransposition accounted for 15% (40 of 261) of events, although this is likely to represent a lower bound given that the detection thresholds exceeded the length of an L1 insertion (6 kbp) for several of the libraries (Table 1). Analysis of structurally variant sequences found a slight enrichment of repetitive DNA for both insertion (58.5%) and deletion (60.8%) events, with 28% of events having a repeat content greater than 90%. Such events are not resolvable with array-based techniques and will probably require directed, PCR-based assays for genotyping.
Table 2.
| Event Type | Total Events | NAHR | NHEJ | VNTR | Retrotransposition |
|---|---|---|---|---|---|
| Insertion | 98 | 41 | 29 | 8 | 20 |
| Deletion | 129 | 49 | 58 | 2 | 20 |
| Inversion |
34 |
34 |
0 |
0 |
0 |
| Total | 261 | 124 | 87 | 10 | 40 |
The mechanism of origin for 261 events were classified as being non-allelic homologous recombination (NAHR), non-homologous end-joining (NHEJ), variable number of tandem repeats (VNTR), or retrotransposition
We compared RefSeq gene annotation between the structurally variant haplotypes and found that 107 distinct gene structures were altered (Supplementary Table 10). Of these genes, 87% belong to members of a gene family, suggesting potential functional redundancy. We specifically examined insertion sequences and found homology for 60 spliced expressed sequence tags and 15 RefSeq gene annotations. Most of these putative gene structures corresponded to duplicated copies of genes or portions of genes (NAIP, BIRCA1, NBPF11, DNM1 and LPA) and/or had homology to genes predicted in either chimpanzee or macaque (ANKRD20A and LOC713531). There are three examples of insertions restricted to coding exons (EPPK1, BAHCC1 and MUC6)—events predicted to alter the composition and structure of the encoded transcripts and proteins. In the case of MUC6 and LPA, these protein length polymorphisms have been associated with H. pylori infection32 and risk of coronary heart disease33, respectively.
We sequenced multiple alleles for the SIRPB1 locus and found evidence for recurrent deletion events on different haplotypes. Sequencing confirmed two distinct deletion alleles having different breakpoints (Fig. 4) embedded within segmental duplications. Both deletion alleles seem to be common and only one of these two results in the loss of an exon, raising the possibility that the two events have different functional consequences despite their extensive overlap. The two different alleles cannot be reliably distinguished by array CGH genotyping because of the presence of duplicated sequences at the boundary and uncertainty in the reference sample genotype (Supplementary Fig. 4). Although we have only begun to survey the sequence organization of a small fraction of our sites, a preliminary analysis of the SNP content of sequenced sites suggests that about 24% of the variants predicted in multiple individuals may be found on different haplotype backgrounds.
Figure 4. Sequence resolution of human structural variation.

Two different deletions within the SIRPB1 gene (exons, red) provide evidence for an independently recurrent deletion event. Both structural variants are probably mediated by non-allelic homologous recombination between segmental duplications (blue bars, arching lines) in direct orientation. Deletion alleles from four different individuals (G248 and ABC8−10) are depicted; deletion 2 (del2, minimal region chromosome 20: 1509210−1542041) eliminates exon 2, whereas deletion 1 (del1, minimal region chromosome 20: 1502353−1533914) does not. Repeat content and orientation are depicted as coloured arrows (green, long interspersed transposable element; purple, short interspersed transposable element; orange, transposon). Predicted and annotated segmental duplications are depicted as indicated.
Other forms of genetic variation
One of the ancillary benefits of sequence-based detection of structural variation is the identification and characterization of other forms of human genetic variation. Because each library represents about 0.3-fold sequence coverage per individual, the ESP pipeline generated about three genomic equivalents (8.5 × 109 bases) of high-quality sequence data from the eight individuals. We mined the existing 13 million end sequences34 and identified 4.0 million non-redundant single nucleotide variants and 796,273 smaller insertion/deletion events (more than 1 bp to less than 100 bp in length); 28% (1.29 million) of the single nucleotide variants and 75% (597,790) of the insertion/deletion variants (indels) were novel when compared with dbSNP (build125). Of the eight HapMap individuals selected in this project, five are common to the ENCODE resequencing project35. We therefore compared our SNP and indel predictions against those ten regions resequenced in the same individuals as a measure of SNP/indel accuracy. On the basis of 1,988 SNP and 120 indel genotypes, we estimated false positive rates of 3.5% (SNPs) and 10.0% (indels).
As expected, the Yoruba African samples showed 15.3% more single nucleotide genetic diversity than non-African samples on the autosomes. The X chromosome shows greater genetic diversity (40%) between African and non-African samples when compared with the autosomes. Because this is one of the first random surveys of sequence data from an ethnically diverse collection of individuals, we also assessed single nucleotide density within 100-kbp windows across the entire genome, identifying regions significantly enriched or depleted in single nucleotide variants (Supplementary Information). After masking sites of segmental duplication, we identified 15 large regions of excess nucleotide variation, ranging in size from 500 kbp to 3 Mbp. These include known sites of increased sequence diversity36,37, for instance HLA and 8p23, as well as several previously undescribed regions such as two large (more than 10 Mbp) regions on each arm of chromosome 16 (Fig. 5). The interval on 8p23 also showed the highest concentration of structural variants validated by our ESP approach (22 distinct variants). The molecular basis for this regional enrichment of genetic diversity across human genomes is unknown, but our preliminary data suggest that structural and single nucleotide variation may correlate.
Figure 5. Regions of enriched SNP density.

Regions of increased single nucleotide variant density identified in eight individuals are shown (ABC7–ABC14 samples ordered from bottom to top). Heterozygosity was calculated in 100-kbp windows, and those windows having a heterozygosity 2 s.d. above the mean are plotted for four chromosomes. Regions of increased heterozygosity, over 1 Mb of genome sequence, are highlighted by red bars.
Discussion
We present a high-resolution integrated map of genetic variation for eight human genomes. We refine the location of 1,695 sites of structural variation (more than about 6 kbp in length), identify 525 regions of novel sequence that harbour highly polymorphic CNVs, and provide single-base-pair sequence resolution for 261 regions of structural variation. These events are placed within the context of 4 million SNPs and 796,273 small indels (1−100 bp in size).
Our detailed analysis of eight human genomes provides significant biological and technological insights into human genetic variation. First, we have discovered and mapped a large number of novel sequences not represented in the human reference genome and show that more than 40% are CNV. These sequences range in size from a few kilobase pairs up to 130 kbp and are randomly distributed, located both within genic and intergenic regions. Although the sequences represent only a fraction of the euchromatin (less than 0.1%), these results strongly argue that the human genome sequence is still incomplete. The role of such sequences in disease association cannot be determined without de novo sequencing of additional genomes and the design of new platforms to genotype these variants specifically on the basis of these ‘new’ sequences.
Second, our refined map of structural variation predicts that the current database of copy-number variation is inflated, which is consistent with previous studies38. An analysis of the same samples with customized high-resolution microarrays and two independent commercial platforms shows an excellent correspondence between ESP-predicted size and commercial SNP platforms (Affymetrix 6.0 arrays and Illumina Human1M BeadChips). The net effect is that there are fewer CNV base pairs per haplotype; consequently, fewer genes and exons are affected. This is an important consideration in view of the fact that CNV maps and databases based largely on BAC-based array CGH are being used to exclude disease-causing variation26,27,39. A comparison of the same eight individuals with the highest-density SNP commercial platforms reveals that more than 50% of the structural variants that we have detected cannot be adequately genotyped, although we note that many more events can be detected than is possible with the fosmid ESP approach. These data argue for the need for customized CNV genotyping platforms based on sequence-validated sites of structural variation.
Third, our sequence analyses suggest that non-allelic homologous recombination is the predominant mechanism (48%; Supplementary Information) altering the larger structural variation landscape of the human genome. This is consistent with several reports confirming that copy-number variation is enriched fourfold to tenfold for regions of segmental duplication3,4,5. However, these findings are in contrast with a recent analysis of two individuals with next-generation ESP sequencing technology40, which reported that events mediated by non-allelic homologous recombination were relatively rare. One possibility for this discrepancy is that shorter reads with lower sequence quality offered by next-generation sequencing technologies may have less power to map within duplication and repeat-rich regions of the genome, thereby missing a large fraction of variation.
The establishment of a clone-based framework for each of these eight genomes provides an important resource for future studies of genetic variation. The clones provide the ability to recover and integrate all forms of genetic variation, ranging from SNPs to larger structural variants within specific haplotypes. The existing end-sequenced clone map permits novel insert sequences anchored within the genome to be mapped and sequenced completely, generating complete alternative human haplotypes. It also permits sequence-based validation of CNV events predicted with other methods and facilitates the targeted resequencing of any genomic region of interest. These full insert sequences will be important in the identification of haplotype-specific ‘tag’ SNPs that may be used to genotype more complex structural variants indirectly and to assess more fully the spectrum of human genetic variation. Thus, these eight genomes can serve as an important benchmark as new genomes become routinely sequenced with next-generation technologies.
METHODS SUMMARY
Library construction and ESP analysis
Fosmid libraries (pCC2Fos vector) were constructed21 from human genomic DNA samples (Coriell Cell Repositories) corresponding to eight HapMap individuals (Table 1). We sequenced about 1 million clones (900 Mbp) for each genome in the form of high-quality ESPs (Supplementary Table 11) and deposited sequences into the NIH trace repository (http://www.ncbi.nlm.nih.gov/Traces). All ESPs were mapped to the human genome assembly (build35) with a previously described algorithm3. Map information, including ESP alignments and corresponding clone IDs of discordant and concordant clones, are available in an interactive browser format and database (http://hgsv.washington.edu).
Validation
Fosmid clones discordant by size (n = 3,371 fosmid clones) were subjected to fingerprint analysis using four multiple complete restriction enzyme digests (MCD analysis) to confirm insert size and eliminate rearranged clones41,42. Two high-density customized oligonucleotide microarrays (Agilent and NimbleGen) were designed to confirm sites of deletion and insertion (GEO accessions GSE10008 and GSE10037). We developed a new, expectation maximization-based clustering approach to genotype deletions with the use of data from the Illumina Human1M BeadChip collected for 125 HapMap DNA samples (Supplementary Information). We found that more than 98% of the children's genotypes were consistent with mendelian transmission on the basis of an analysis of 28 parent–child trios.
Fosmid insert sequencing
We completely sequenced the inserts of 405 fosmid clones from six genomic libraries (210 from G248, 31 from ABC7, 39 from ABC8, 21 from ABC9, 98 from ABC10, and 6 from ABC12) with previously described methods. All sequences have been deposited in GenBank (Supplementary Table 9).
SNP/indel analysis
We identified single nucleotide variants using the ssahaSNP software tool34 and indels (1−100 bp in size) using ssahaSNP in combination with cross_match (http://www.phrap.org; Supplementary Information). SNP and indel variants have been deposited in dbSNP (release 129).
Acknowledgements
We thank the staff from the University of Washington Genome Center and the Washington University Genome Sequencing Center for technical assistance. J.M.K. is supported by a National Science Foundation Graduate Research Fellowship. G.M.C. is supported by a Merck, Jane Coffin Childs Memorial Fund Postdoctoral Fellowship. This work was supported by National Institutes of Health grants HG004120 to E.E.E., D.A.N. and M.V.O., and 3 U54 HG002043 to M.V.O. E.E.E. is an Investigator of the Howard Hughes Medical Institute.
Footnotes
Supplementary Information is linked to the online version of the paper at www.nature.com/nature.
Author Contributions
J.M.K., G.M.C., M.V.O, D.A.N, and E.E.E. contributed to the writing of this paper. The study was coordinated by L.B., M.V.O, R.K., D.R.S., J.M.K. and E.E.E.;
A.B., D.R.S., D.Sa., E.G., H.M.E., K.M., N.T., R.D., W.F.D., and W.T. performed library construction and end sequencing.
E.H., H.S.H., K.A.P., M.V.O., R.K., R.K.W., T.G., and W.G. performed clone insert validation and sequencing.
C.A., D.A.N., E.T., J.D.S., J.S., L.C., M.D., M.M., M.W., T.L.N. and Z.C. provided technical and analytical support.
D.A.P., D.A.A., J.M.Ko. and S.A.M. contributed variation data.
G.M.C., J.M.K., L.B., N.A.Y., N.S., and P.T. designed and analyzed array CGH experiments.
G.M.C., and T.Z. performed the genotype analysis.
F.A. performed FISH experiments.
B.T. and D.S. performed optical mapping experiments.
E.E.E., J.M.K., and L.C. analyzed sequenced clones.
J.C.M. and N.H. identified SNPs and indels.
Supplementary Material
References
- 1.Iafrate AJ, et al. Detection of large-scale variation in the human genome. Nature Genet. 2004;36:949–951. doi: 10.1038/ng1416. [DOI] [PubMed] [Google Scholar]
- 2.Sebat J, et al. Large-scale copy number polymorphism in the human genome. Science. 2004;305:525–528. doi: 10.1126/science.1098918. [DOI] [PubMed] [Google Scholar]
- 3.Tuzun E, et al. Fine-scale structural variation of the human genome. Nature Genet. 2005;37:727–732. doi: 10.1038/ng1562. [DOI] [PubMed] [Google Scholar]
- 4.Sharp AJ, et al. Segmental duplications and copy-number variation in the human genome. Am. J. Hum. Genet. 2005;77:78–88. doi: 10.1086/431652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Redon R, et al. Global variation in copy number in the human genome. Nature. 2006;444:444–454. doi: 10.1038/nature05329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wong KK, et al. A comprehensive analysis of common copy-number variations in the human genome. Am. J. Hum. Genet. 2007;80:91–104. doi: 10.1086/510560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Conrad DF, Andrews TD, Carter NP, Hurles ME, Pritchard JK. A high-resolution survey of deletion polymorphisms in the human genome. Nature Genet. 2006;38:75–81. doi: 10.1038/ng1697. [DOI] [PubMed] [Google Scholar]
- 8.McCarroll SA, et al. Common deletion polymorphisms in the human genome. Nature Genet. 2006;38:86–92. doi: 10.1038/ng1696. [DOI] [PubMed] [Google Scholar]
- 9.Hinds DA, Kloek AP, Jen M, Chen X, Frazer KA. Common deletions and SNPs are in linkage disequilibrium in the human genome. Nature Genet. 2006;38:82–85. doi: 10.1038/ng1695. [DOI] [PubMed] [Google Scholar]
- 10.Cheng Z, et al. A genome-wide comparison of recent chimpanzee and human segmental duplications. Nature. 2005;437:88–93. doi: 10.1038/nature04000. [DOI] [PubMed] [Google Scholar]
- 11.Aitman TJ, et al. Copy number polymorphism in Fcgr3 predisposes to glomerulonephritis in rats and humans. Nature. 2006;439:851–855. doi: 10.1038/nature04489. [DOI] [PubMed] [Google Scholar]
- 12.Gonzalez E, et al. The influence of CCL3L1 gene-containing segmental duplications on HIV-1/AIDS susceptibility. Science. 2005;307:1434–1440. doi: 10.1126/science.1101160. [DOI] [PubMed] [Google Scholar]
- 13.Fellermann K, et al. A chromosome 8 gene-cluster polymorphism with low human β-defensin 2 gene copy number predisposes to Crohn disease of the colon. Am. J. Hum. Genet. 2006;79:439–448. doi: 10.1086/505915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hollox EJ, et al. Psoriasis is associated with increased β-defensin genomic copy number. Nature Genet. 2007;40:23–25. doi: 10.1038/ng.2007.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cooper GM, Nickerson DA, Eichler EE. Mutational and selective effects on copy-number variants in the human genome. Nature Genet. 2007;39:S22–S29. doi: 10.1038/ng2054. [DOI] [PubMed] [Google Scholar]
- 16.Istrail S, et al. Whole-genome shotgun assembly and comparison of human genome assemblies. Proc. Natl Acad. Sci. USA. 2004;101:1916–1921. doi: 10.1073/pnas.0307971100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Khaja R, et al. Genome assembly comparison identifies structural variants in the human genome. Nature Genet. 2006;38:1413–1418. doi: 10.1038/ng1921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Levy S, et al. The diploid genome sequence of an individual human. PLoS Biol. 2007;5:e254. doi: 10.1371/journal.pbio.0050254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Eichler EE, et al. Completing the map of human genetic variation. Nature. 2007;447:161–165. doi: 10.1038/447161a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.The International HapMap Consortium A haplotype map of the human genome. Nature. 2005;437:1299–1320. doi: 10.1038/nature04226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Donahue W, Ebling HM. Fosmid libraries for genomic structural variation detection. Curr. Protocols Hum. Genet. 2007;5:20.1–20.18. doi: 10.1002/0471142905.hg0520s54. [DOI] [PubMed] [Google Scholar]
- 22.Volik S, et al. End-sequence profiling: sequence-based analysis of aberrant genomes. Proc. Natl Acad. Sci. USA. 2003;100:7696–7701. doi: 10.1073/pnas.1232418100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Small K, Iber J, Warren S. Emerin deletion revals a common X-chromosome inversion mediated by inverted repeats. Nature Genet. 1997;16:96–99. doi: 10.1038/ng0597-96. [DOI] [PubMed] [Google Scholar]
- 24.Giglio S, et al. Heterozygous submicroscopic inversions involving olfactory receptor-gene clusters mediate the recurrent t(4;8)(p16;p23) translocation. Am. J. Hum. Genet. 2002;71:276–285. [Google Scholar]
- 25.Stefansson H, et al. A common inversion under selection in Europeans. Nature Genet. 2005;37:129–137. doi: 10.1038/ng1508. [DOI] [PubMed] [Google Scholar]
- 26.Sharp AJ, et al. Discovery of previously unidentified genomic disorders from the duplication architecture of the human genome. Nature Genet. 2006;38:1038–1042. doi: 10.1038/ng1862. </jrn>. [DOI] [PubMed] [Google Scholar]
- 27.Sharp AJ, et al. Characterization of a recurrent 15q24 microdeletion syndrome. Hum. Mol. Genet. 2007;16:567–572. doi: 10.1093/hmg/ddm016. [DOI] [PubMed] [Google Scholar]
- 28.Warburton PE, Giordano J, Cheung F, Gelfand Y, Benson G. Inverted repeat structure of the human genome: the X-chromosome contains a preponderance of large, highly homologous inverted repeats that contain testes genes. Genome Res. 2004;14:1861–1869. doi: 10.1101/gr.2542904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sutton GG, White O, Adams MD, Kerlavage A. TIGR Assembler: a new tool for assembling large shotgun sequencing projects. Genome Sci. Technol. 1995;1:9–19. [Google Scholar]
- 30.Bovee D, et al. Closing gaps in the human genome with fosmid resources generated from multiple individuals. Nature Genet. 2008;40:96–101. doi: 10.1038/ng.2007.34. [DOI] [PubMed] [Google Scholar]
- 31.Scherer SW, et al. Challenges and standards in integrating surveys of structural variation. Nature Genet. 2007;39:S7–S15. doi: 10.1038/ng2093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nguyen TV, et al. Short mucin 6 alleles are associated with H. pylori infection. World J. Gastroenterol. 2006;12:6021–6025. doi: 10.3748/wjg.v12.i37.6021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lackner C, Cohen JC, Hobbs HH. Molecular definition of the extreme size polymorphism in apolipoprotein(a). Hum. Mol. Genet. 1993;2:933–940. doi: 10.1093/hmg/2.7.933. [DOI] [PubMed] [Google Scholar]
- 34.Ning Z, Cox AJ, Mullikin JC. SSAHA: a fast search method for large DNA databases. Genome Res. 2001;11:1725–1729. doi: 10.1101/gr.194201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.ENCODE Project Consortium Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature. 2007;447:799–816. doi: 10.1038/nature05874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sachidanandam R, et al. A map of human genome sequence variation containing 1.42 million single nucleotide polymorphisms. Nature. 2001;409:928–933. doi: 10.1038/35057149. [DOI] [PubMed] [Google Scholar]
- 37.Nusbaum C, et al. DNA sequence and analysis of human chromosome 8. Nature. 2006;439:331–335. doi: 10.1038/nature04406. [DOI] [PubMed] [Google Scholar]
- 38.de Smith AJ, et al. Array CGH analysis of copy number variation identifies 1284 new genes variant in healthy white males: implications for association studies of complex diseases. Hum. Mol. Genet. 2007;16:2783–2794. doi: 10.1093/hmg/ddm208. [DOI] [PubMed] [Google Scholar]
- 39.Sebat J, et al. Strong association of de novo copy number mutations with autism. Science. 2007;316:445–449. doi: 10.1126/science.1138659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Korbel JO, et al. Paired-end mapping reveals extensive structural variation in the human genome. Science. 2007;318:420–426. doi: 10.1126/science.1149504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gillett W, et al. Assembly of high-resolution restriction maps based on multiple complete digests of a redundant set of overlapping clones. Genomics. 1996;33:389–408. doi: 10.1006/geno.1996.0215. [DOI] [PubMed] [Google Scholar]
- 42.Wong GK, Yu J, Thayer EC, Olson MV. Multiple-complete-digest restriction fragment mapping: generating sequence-ready maps for large-scale DNA sequencing. Proc. Natl Acad. Sci. USA. 1997;94:5225–5230. doi: 10.1073/pnas.94.10.5225. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.