

Abstract
Mammalian inflammatory signaling, for which NF-κB is a principal transcription factor, is an exquisite example of how cellular signaling pathways can be regulated to produce different yet specific responses to different inflammatory insults. Mathematical models, tightly linked to experiment, have been instrumental in unraveling the forms of regulation in NF-κB signaling and their underlying molecular mechanisms. Our initial model of the IκB–NF-κB signaling module highlighted the role of negative feedback in the control of NF-κB temporal dynamics and gene expression. Subsequent studies sparked by this work have helped to characterize additional feedback loops, the input–output behavior of the module, crosstalk between multiple NF-κB-activating pathways, and NF-κB oscillations. We anticipate that computational techniques will enable further progress in the NF-κB field, and the signal transduction field in general, and we discuss potential upcoming developments.
Keywords: NF-kB, signal transduction, systems biology
Introduction
The transcription factor NF-κB is a central inflammatory mediator, as it is essential for the majority of gene induction events in response to inflammatory cytokines as well as pathogen-derived substances. In unstimulated cells, NF-κB is bound to IκB proteins which hold it latent in the cytoplasm. Cellular stimulation with inflammatory agents results in IKK-mediated phosphorylation of IκB proteins, their ubiquitination, and proteasome-mediated proteolysis, allowing free NF-κB to accumulate in the nucleus and bind the cognate κB elements in target gene promoters (Box 1; reviewed in Hayden and Ghosh, 2008). Regulation of NF-κB is important for the physiology of inflammation and immune activation, and misregulation of NF-κB activity has been identified as a major culprit of chronic inflammatory diseases and cancer. As such understanding NF-κB regulation has been a major focus of biochemical, mouse genetic, and human disease studies since its discovery more than 20 years ago (Sen and Baltimore, 1986).
Primer on TNFα signaling to NF-κB.
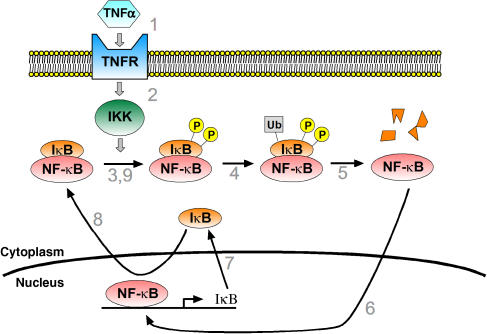
Upon binding of TNFα (1), TNF receptor (TNFR) is activated, leading to activation of the IκB kinase (IKK) (2). IKK dually phosphorylates inhibitor of NF-κB (IκB) (3), which in a basal state holds NF-κB latent in the cytoplasm. Phosphorylated IκB is targeted for ubiquitination (4) and subsequently proteosome-mediated degradation (5). NF-κB, no longer bound to IκB, enters the nucleus (6) where it may modulate gene transcription. The genes for IκB are among the genes that are upregulated by NF-κB (7). Newly synthesized IκBenters the nucleus, binds to NF-κB, and promotes its export to the cytoplasm (8), thereby forming a negative feedback loop that terminates the response. New IκB–NF-κB complexes may enter the feedback loop, beginning with phosphorylation by IKK, if TNF stimulation persists (9). There are three typical isoforms of IκB: IκBα, IκBβ, and IκBɛ. As discussed in the main text, expression of IκBα is robustly induced by NF-κB and was a focus of initial modeling studies of the pathway, whereas NF-κB-induced expression of IκBβ and IκBɛ was a topic of later investigations.
Major components of many signaling pathways that activate NF-κB have been mapped, and this information is often summarized in pathway diagrams (e.g. Box 1). However, the dynamics of molecular level regulation are insufficiently captured by the static representation inherent in such diagrams. Mathematical models, on the other hand, can quantitatively describe how changes in signaling occur in space and time, enabling exploration of signaling pathways in silico (Box 2). The resulting insights can provide a theoretical framework and generate testable predictions for subsequent experimental studies. Experimental results likewise inform the development and refinement of mathematical models with predictive power. In this way, our understanding of cell signaling processes can be progressively advanced (Kearns and Hoffmann, 2008).
Primer on modeling NF-κB pathways using differential equations.
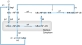
The core of our original model of NF-κB signaling is depicted below as a set of linked biochemical reactions. The diagram omits reactions (e.g. dissociation, reactions involving IκBβ and IκBɛ) that are present in the full model but are not essential to oscillatory behavior. Complexes are denoted by ‘:' and generic sources and sinks for synthesis and degradation are denoted by ‘∅.' Rate parameters are shown above their respective reactions, named according to the convention of the original model (Hoffmann et al, 2002). The input into the model is a step increase in IKK, which is a surrogate for TNFα stimulation. This allows the first reaction, IKK binding to IκBα–NF-κB complex (a7), to proceed. The steps of phosphorylation, ubiquitination, and proteosomal degradation of IκBα within this complex are lumped into a single reaction whose products are free IKK and free NF-κB (r4). NF-κB enters the nucleus, denoted by the suffix ‘n' (k1). This leads to synthesis of IκB mRNA transcript, denoted by the suffix ‘t' (tr2). The half-life of the transcript is determined by tr3. Translation leads to synthesis of new IκBα (tr1), whose half-life is determined by deg1. IκBα can enter (tp1) and leave (tp2) the nucleus, and in the nucleus, IκBα is also denoted with the suffix ‘n.' Nuclear IκBα and NF-κB associate (a4), and together are exported to the cytoplasm (k2). In all, these steps form a negative feedback loop (also described in Box 1), whose overall sequence is shown by the blue arrow. Mass action kinetics are used to convert these biochemical reactions into a system of ordinary differential equations. For example, the equation for the time rate of change of cytoplasmic IκBα–NF-κB complex is given by
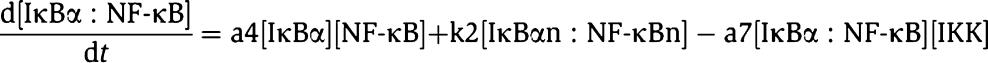
where the terms show increases in the amount of complex due to association of IκBα and NF-κB (a4) and export of nuclear complex (k2), and decreases in the amount of complex due to association with IKK (a7). Equations are written in this way for each chemical species. In the full version of the original model, similar reactions govern the behavior of IκBβ and IκBɛ, resulting in additional differential equations. In this model formulation, the parameters are biochemical rates of association, dissociation, catalysis, transport, synthesis, and degradation. Thus, their values may be quantitatively measured or constrained by biochemical experiments. The procedure we used is summarized in the main text. Finally, to run the model, the initial concentrations of each species must be specified. (Running the model means to numerically solve the differential equations, e.g. with Mathematica's NDSolve function, to determine time courses of the concentrations of each species.) We initialized the model with a biologically plausible total level of NF-κB (0.1 μM) with all other concentrations set to zero. The basal state of the cell (non-stimulated) is simulated by running the model starting from this initial state until it reaches steady state. At steady state, NF-κB is found in the cytoplasm and nucleus, as well as free or complexed with IκB, but is predominantly found complexed in the cytoplasm in accordance with experimental observations. Following a step increase in IKK, the model can be further run to simulate the effects of TNF stimulation.
Here, we review how mathematical modeling has impacted our understanding of signaling through NF-κB pathways. First, we summarize our original mathematical model, which is the predecessor of many models used to study the regulation of NF-κB dynamics (Table I). Then, we describe how mathematical and computational models have been instrumental in increasing our understanding of the control of NF-κB signaling. We also discuss the emerging areas of research in which mathematical models may shed light.
Table 1.
Comparison of published NF-κB models
| Model | Predecessor | Feedback | Major changes from predecessor |
|---|---|---|---|
| The original mathematical model of NF-κB signaling | |||
| Hoffmann et al (2002) | Carlotti et al (2000) | Inducible IκBα Constitutive IκBβ, IκBɛ | • Responsive to IKK stimulus • IκBα negative feedback loop |
| Direct descendants of the original model | |||
| Covert et al (2005) | Hoffmann et al (2002) | Inducible IκBα Constitutive IκBβ, IκBɛ | • LPS stimulus modeled as two additive signals offset in time • Transcription and translation rates were re-fit |
| O'Dea et al (2007) | Hoffmann et al (2002) | Inducible IκBα Constitutive IκBβ, IκBɛ | • IκB degradation rates were updated based on experimental measurements |
| Cheong et al (2006) | Hoffmann et al (2002) | Inducible IκBα, Constitutive IκBβ, IκBɛ | • IKK time-course generator was added • Transcription, translation, and degradation rates were re-fit • Nuclear–cytoplasmic volume ratio was added |
| Kearns et al (2006) | O'Dea et al (2007) | Inducible IκBα Delayed inducible IκBβ, IκBɛ | • IκBβ and IκBɛ are inducible with a 45 min delay • IκB degradation rates were altered to fit new data |
| Werner et al (2005) | Kearns et al (2006) | Inducible IκBα Delayed inducible IκBβ, IκBɛ | • Cubic transcription rate • LPS modeled by using its IKK time course as an input |
| Moss et al (2008) | Identical to the model described in Werner et al (2005) | ||
| O'Dea et al (2008) | Werner et al (2005) | Inducible IκBα Delayed inducible IκBβ, IκBɛ | • Some rate parameters were modified to model the effect of UV-induced NF-κB activity |
| Mathes et al (2008) | Werner et al (2005) | Inducible IκBα Delayed inducible IκBβ, IκBɛ | • Some rate parameters were modified to model the effect of IκBα mutants on NF-κB signaling |
| Basak et al (2007) | Werner et al (2005) | Inducible IκBα, p100 Delayed inducible IκBβ, IκBɛ | • Introduction of the IκB species p100 • LPS or TNF induces IKK2-mediated IκB degradation • LTβ induces IKK1-mediated p100 degradation |
| Analysis of the original model by MR White and colleagues | |||
| Nelson et al (2004) | Identical to the model described in Hoffmann et al (2002) | ||
| Ihekwaba et al (2004) | Identical to the model described in Hoffmann et al (2002) | ||
| Ihekwaba et al (2005) | Identical to the model described in Hoffmann et al (2002) | ||
| Ihekwaba et al (2007) | Hoffmann et al (2002) | Inducible IκBα Constitutive IκBβ, IκBɛ | • Identical to predecessor except some IKK-related parameters changed to match measurements based on experiments where cells were stimulated with IL-1 |
| NF-κB models by M Kimmel and colleagues | |||
| Lipniacki et al (2004) | Hoffmann et al (2002) | Inducible IκBα Inducible A20 | • IκBβ and IκBɛ were removed from predecessor and A20 negative feedback loop was added • New assumptions about IKK activation and deactivation • Nuclear–cytoplasmic volume ratio was added • Transcription and translation rates were re-fit |
| Lipniacki et al (2006) | Lipniacki et al (2004) | Inducible IκBα Inducible A20 | • Stochastic translation and transcription • Some parameters were re-fit |
| Lipniacki et al (2007) | Lipniacki et al (2006) | Inducible IκBα Inducible A20 | • Introduction of TNF receptor and IKK kinase • Stochastic TNF receptor activation and IκBα/A20 transcription |
| Fujarewicz et al (2007) | Lipniacki et al (2004) | Inducible IκBα Inducible A20 | • Equations identical to predecessor but parameters were re-fit |
| Joo et al (2007) | Identical to the model described in Lipniacki et al (2004) | ||
| Other descendants of the original model | |||
| Sung and Simon (2004) | Hoffmann et al (2002) | Inducible IκBα | • IκBβ and IκBɛ are removed from predecessor • NF-κB induction of IκBα has an explicit transcriptional time delay • Some parameters were re-fit |
| Hayot and Jayaprakash (2006) | Hoffmann et al (2002) | Inducible IκBα | • IκBβ and IκBɛ are removed, and IκBα has linear transcription rate • Whole model is stochastic |
| Krishna et al (2006) | Hoffmann et al (2002) | Inducible IκBα | • Reduces predecessor to a three-component system with five dimensionless parameters |
| Park et al (2006) | Hoffmann et al (2002) | Inducible IκBα Constitutive IκBβ, IκBɛ | • Explicit TNF receptor to IKK pathway • IKK activity was affected by factors X and Y representing effects of HBV infection |
| Other NF-κB models | |||
| Cho et al (2003) | None | No inducible factors | • Tree-like signaling pathway structure with no feedback loops • TNFα leads either to apoptosis (FADD) or proliferation (NF-κB) |
| Monk (2003) | None | Inducible IκBα | • Proposes NF-κB oscillations derive from time delay of IκBα transcription |
| Janes et al (2005) | None | • Partial least-squares regression on a large compendium of cytokine signaling data | |
| Janes et al (2006) | Identical to the model described in Janes et al (2005) | ||
| Piotrowska et al (2006) | None | No inducible factors | • Two-component system with five dimensionless parameters • Negative correlation between IκBα and NF-κB is directly assumed • Proliferation rate is a function of NF-κB |
| Pogson et al (2006) | None | No inducible factors | • Agent-based stochastic simulation • Incorporates events from receptor activation to NF-κB nuclear import |
| Rangamani and Sirovich (2007) | None | Inducible IκBα Inducible IAP | • TNFα leads either to apoptosis (caspase) or survival (NF-κB) • IκBβ and IκBɛ are not present • Parameters were taken from a variety of sources |
The original mathematical model of the IκB–NF-κB signaling module
NF-κB activation involves stimulus-induced degradation of its inhibitor IκB, which allows for its translocation to the nucleus. The earliest attempt to capture the dynamics of these events in mathematical equations was aimed at understanding how NF-κB translocation and IκB association/dissociation rate constants keep the majority of NF-κB in an inactive state in resting cells (Carlotti et al, 2000). However, this work did not result in a model that allowed for computational simulations of the full NF-κB activation and attenuation process.
Our interest was to understand the differential functions, if any, of the three IκB isoforms (IκBα, IκBβ, and IκBɛ) that modulate inflammatory activation of NF-κB. Biochemical studies had shown that all three sequester p65–p50, the predominant NF-κB dimer, are degraded in response to stimulation with tumor necrosis factor alpha (TNFα) (Ghosh et al, 1998). Nevertheless, mice deficient in any one of these three IκB proteins have distinct phenotypes, indicating that the IκBs have different and non-overlapping functions (Beg et al, 1995; Klement et al, 1996; Memet et al, 1999; Mizgerd et al, 2002). As time-course data, derived from electrophoretic mobility shift assays (EMSAs), indicated that the three IκB proteins had differential dynamic control, we set out to construct a mathematical model of NF-κB signaling to study the specific roles of each IκB isoform in regulating the temporal control of NF-κB (Hoffmann et al, 2002).
We defined the scope of the model to be that of the IκB–NF-κB signaling module, in which the IKK activity as an input to the model determines the NF-κB activity over time. The model consisted of a system of differential equations based on mass action kinetics of the association/dissociation, synthesis/degradation, and translocation of IKK, IκB, and NF-κB species. Of the 34 independent model parameters, about one-third were derived from the extensive biochemical literature on NF-κB, especially for the parameters of the Michaelis–Menten reactions of IKK-mediated IκB phosphorylation. A further third, especially those parameters relating to species half-lives, transport rates, and IκB–NF-κB affinities, was constrained by published time-course data. We used a genetic approach to reduce the complexity of the signaling module to obtain the data used to fit the remaining parameters (primarily mRNA and protein synthesis). By mouse reverse genetics, we obtained cells deficient in any two of the three IκB isoforms, thereby enabling us to parameter fit three reduced models each containing only one IκB isoform that were then combined into a wild-type cell model.
Exploration of the model with computational simulations resulted in two major insights. First, it described how differential functions of the IκB isoforms could give rise to strikingly different NF-κB dynamics in genetically reduced cells. The role of IκBα, whose expression is induced by NF-κB, was to provide negative feedback. This was aptly demonstrated by pronounced oscillations in NF-κB activity in cells lacking the other isoforms (Figure 1A). The role of IκBβ and IκBɛ was to dampen these oscillations. When all three isoforms were present, the NF-κB response was biphasic, with an initial NF-κB activity rising and falling within ∼1 h, followed by a late activation phase characterized by a steady intermediate level of activity (Figure 1B). Second, we explored the ‘temporal dose–response' characteristics of the NF-κB signaling module by simulating the NF-κB response duration for different stimulus durations. The model predicted that the module would generate the initial phase of 60 min of NF-κB activity even with much shorter stimuli, while only for longer lasting stimuli (>1 h) did the responses have durations proportional to the input duration. This prediction was confirmed by using EMSA on wild-type cells. Moreover, we found experimentally that the initial phase of NF-κB activity is sufficient to drive the expression of a subset of inflammatory genes, while others require longer lasting NF-κB activity. Hence, the functions of IκBα, IκBβ, and IκBɛ combine to allow the signaling module to distinguish between short and longer lasting stimuli. A subsequent study of gene expression in single cells also found that some target genes require longer lasting TNFα stimulation than others (Nelson et al, 2004).
Figure 1.

Schematic of NF-κB dynamics in response to persistent TNFα. (A) Oscillatory time course of NF-κB in response to TNFα in cells whose only classical IκB is IκBα (see also BioModels database http://www.ebi.ac.uk/biomodels, accession ID BIOMD0000000139). (B) Characteristic biphasic time course of NF-κB signaling in response to TNFα in various wild-type cells. NF-κB activity peaks around 30 min, drops to basal levels around 1 h, and rises to an intermediate level thereafter (see also BioModels accession ID BIOMD0000000140).
Two of the more significant advances provided by our study were that temporal dynamics of NF-κB help control the expression of inflammatory genes, and that mathematical modeling could be extremely useful in understanding the molecular mechanisms that regulate NF-κB dynamics. This spurred a number of subsequent modeling studies designed to further understand the regulation of NF-κB dynamics, which we review below. Some of these studies were primarily theoretical in nature and pointed to interesting potential dynamical properties of NF-κB signaling, whereas in others, modeling was tightly integrated with experiment leading to a plethora of unexpected insights into the mechanisms that control NF-κB dynamics.
Mechanisms that control NF-κB dynamics revealed by mathematical models
In this section, we highlight how mathematical and computational models have been applied with impressive success to direct or illuminate experimental studies to characterize additional feedback loops involving NF-κB, IKK dynamics, crosstalk between inflammatory and non-inflammatory inducers of NF-κB activity, and NF-κB oscillations.
Multiple feedback loops
The original mathematical model of the IκB–NF-κB signaling module revealed that NF-κB-induced expression of IκBα provides negative feedback and that this feedback is a major determinant of NF-κB temporal dynamics. Subsequent studies, integrating experimental analysis and computational models, have shown that additional feedback mechanisms also control NF-κB activity. One such loop involves IκBɛ, which like IκBα, is expressed after TNFα stimulation in an NF-κB-dependent manner (Tian et al, 2005). Unlike IκBα, however, IκBɛ transcription is delayed by about 45 min relative to the onset of nuclear NF-κB activity, as revealed by cells deficient in IκBα and IκBβ (Kearns et al, 2006). Intuitively, delayed IκBɛ induction might provide oscillatory feedback in antiphase with IκBα feedback, which combine to provide steady overall levels of IκB with concomitant steady NF-κB activity. A computational model derived from the original model encapsulating this idea predicted that the duration of NF-κB activity in response to a transient (45 min) TNFα stimulation would be prolonged in cells deficient in both IκBα and IκBɛ, compared to cells deficient in only one of these isoforms, or to wild-type cells. This prediction, confirmed by EMSA, indicated that IκBɛ is capable of providing post-induction repression of NF-κB. Likewise, the expression of inflammatory genes is prolonged in the iκBα−/−iκBɛ−/− cells compared to iκBα−/− and wild-type cells, providing functional evidence for the importance of IκBɛ in terminating the inflammatory response (Kearns et al, 2006). Overall, the negative feedbacks provided by IκBα and IκBɛ appear to work in tandem to ensure rapid post-induction repression of NF-κB, while suppressing sustained oscillations, thus solving a classic shortcoming of simple linear control systems (Coughanowr, 1991).
In addition to intracellular feedback due to IκBα and IκBɛ, extracellular feedback might arise through autocrine signaling. A prime example of this phenomenon relative to the NF-κB pathway was found while exploring cell responses to lipopolysaccharide (LPS). LPS is a component of bacterial cell walls that serves as an important signal of infection activating two intracellular pathways that branch at the receptor level, respectively dependent on MyD88 and Trif. The NF-κB activity in response to persistent LPS is normally steady over time, but it is oscillatory when either the Trif- or MyD88-dependent pathway is isolated by knockout of MyD88 and Trif, respectively (Covert et al, 2005). Reminiscent of IκBα and IκBɛ, the Trif- and MyD88-dependent oscillations are out of phase. Computational modeling based on the original model indicated that the reason that the oscillations are out of phase is that the Trif- and MyD88-dependent pathways have similar activation kinetics, but the Trif-dependent pathway is activated 30 min after the MyD88-dependent pathway. A search for the biochemical mechanism underlying this delay uncovered an autocrine signaling loop. Specifically, the MyD88-dependent pathway was found to lead to fast, direct activation of NF-κB, whereas the Trif-dependent pathway resulted in slow indirect NF-κB activation via TNFα production, secretion, and subsequent autocrine signaling (Figure 2). Interestingly, the same autocrine mechanism ensures that NF-κB activity is steady not only in response to persistent LPS but also to transient LPS stimulation as well (Werner et al, 2005).
Figure 2.

Feedback loops in NF-κB signaling. IKK may be activated by the TNFα signaling pathway as well as the MyD88-dependent arm of the LPS signaling pathway. IKK leads to NF-κB activity, which is regulated by a negative feedback loop involving IκB (described in detail in Box 1), as depicted in the lower center. TNFα-induced NF-κB activity also leads to A20 expression, and subsequent decrease in IKK activation. Also, the Trif-dependent arm of the LPS-signaling pathway activates the transcription factor interferon regulatory factor-3 (IRF3), leading to TNFα expression and subsequent autocrine signaling. Thus, A20 and TNF form feedback loops that regulate NF-κB activity.
These discoveries suggest that mathematical modeling will be useful in understanding many other potential feedbacks involved in the regulation of NF-κB. For example, the expression of the third inhibitor isoform, IκBβ, is weakly upregulated by TNFα (Kearns et al, 2006) and, although the removal of this inhibitor does not unmask oscillations, some more subtle signaling defects are likely to be present. The NF-κB subunit RelB (Bren et al, 2001), the p50 subunit precursor p105 (Ten et al, 1992), and the p52 subunit precursor p100 (Lombardi et al, 1995), are all potentially expressed in response to TNFα, which could result in a change in NF-κB dimer composition that could in turn affect all other transcriptionally mediated feedback loops. Likewise, NF-κB may be subject to a wide variety of extracellular feedback mechanisms, especially through autocrine signaling. NF-κB target genes include the cytokines TNFα (Collart et al, 1990; Shakhov et al, 1990), many interleukins (Pahl, 1999), and lymphotoxin-β (LTβ) (Kuprash et al, 1996), all of which are direct activators of NF-κB. Well-defined computational models should prove useful in unraveling such complex feedback-rich signaling systems, addressing among other questions the roles of individual feedbacks and the need for all the feedbacks to be in place.
Control of NF-κB dynamics through IKK
We defined the input of our original mathematical model of the IκB–NF-κB signaling module to be IKK, rather than TNFα or other extracellular ligands. This raised the question of how IKK dynamics control the downstream NF-κB dynamics and how IKK activity itself is regulated.
It is apparent that IKK dynamics are important in controlling the timing of NF-κB activity. Experimentally, we found that the initial phase of NF-κB activity invariantly lasted 60 min in response to different concentrations of TNFα (Cheong et al, 2006), paralleling the response to different durations of exposure to TNFα (Hoffmann et al, 2002). We found that the original pathway model failed to reproduce this behavior, despite an exhaustive attempt to refit the parameter values. This suggested that the model was incomplete, and perhaps omitted an important biochemical interaction needed to explain the observed dynamics. We surmised that IKK, whose regulation was not represented in detail in the original model, played an important role in determining the NF-κB dynamics. By examining the model's responses to various IKK time courses, we found that the fixed duration of the initial phase of NF-κB activity could be explained if the IKK activity was sharply attenuated. Specifically, the model predicted that at any TNFα dose, the IKK activity rises quickly upon exposure to TNFα, peaks after 5–10 min, and drops to a low but positive level after another 10–20 min. Experiments utilizing IKK assays validated this prediction (Cheong et al, 2006), indicating that the specific IKK dynamics are essential for maintaining a normal biphasic NF-κB response. Importantly, this study showed how incongruities between models and experiments can be exploited to further understand the signaling system of interest.
More generally, the NF-κB dynamics are sensitive to the timing and duration of the IKK activity. As discussed above, both in model and experiment, a peaked IKK profile, i.e. one that rises quickly then falls quickly, generates a transient NF-κB response of fixed duration. In contrast, an IKK profile that plateaus, i.e. rises slowly to a sustained level, results in a delayed rise to a sustained level of NF-κB activity. Importantly, these different IKK dynamics help enable stimulus-specific responses (Figure 3). For example, the peaked IKK profile results from transient TNFα stimulation, whereas the sustained IKK activity can result from transient LPS stimulation. Furthermore, these different IKK profiles, which in turn result in the different NF-κB dynamics, allow for some genes to be specifically expressed in response to LPS and others to be specifically expressed in response to TNFα, even though the expression of these genes are all regulated by NF-κB (Werner et al, 2005).
Figure 3.

Schematic of stimulus-specific NF-κB responses. Both TNFα and LPS activate NF-κB through IKK, yet the NF-κB responses to each are different. In response to a 45-min pulse of TNFα, NF-κB activity rises quickly then terminates after approximately 60 min (bottom right). In contrast, in response to a 45-min pulse of LPS, NF-κB activity rises slowly over 2 h (bottom left). The NF-κB response correlates with the IKK activity profile, which is highly peaked in response to TNFα (upper right) but sustained in response to LPS (upper left). This illustrates how IKK helps to mediate stimulus-specific NF-κB responses.
One important determinant of the IKK dynamics is A20, which inhibits IKK activation by modifying the ubiquitination pattern of a subunit of the TNF receptor complex (Wertz et al, 2004). The expression of A20 itself is induced by TNFα in an NF-κB-dependent manner (Figure 2). This fact led to the development of a version of the original model that suggested that A20-mediated negative feedback is sufficient to produce the sharply peaked IKK activity profile resulting from persistent TNFα stimulation (Lipniacki et al, 2004). However, initial experiments could not verify this prediction (Cheong et al, 2006) and the mechanism that leads to a rapid attenuation of TNFα-induced IKK activity remains an open question (Delhase et al, 1999; Cheong et al, 2006; Schomer-Miller et al, 2006). Nonetheless, A20 is clearly important in inhibiting late IKK activity and is required for the drop in NF-κB activity that separates the early and late phases in response to TNFα (Lee et al, 2000; Werner et al, 2005). Computational models thus point to the gap of current knowledge about A20 and IKK regulation in general as a barrier to further understanding of NF-κB dynamics, and modeling work in this area should prove fruitful for additional studies integrating models and experiments.
Crosstalk between the IκB–NF-κB module and other pathways
NF-κB is activated by numerous inflammatory stimuli, such as TNFα and LPS as discussed above, and also by many non-inflammatory stimuli (Hayden and Ghosh, 2004). One such stimulus is LTβ, a cytokine implicated in the normal development of lymph nodes. Unlike classical inflammatory stimuli, LTβ-mediated activation of NF-κB does not occur through the degradation of NF-κB-bound IκB (Beinke and Ley, 2004). Rather, it occurs through degradation of the inhibitory domain of NF-κB-bound p100, an NF-κB protein precursor that, as a homodimeric complex, has IκB-like function. Furthermore, p100 is an NF-κB target gene whose expression can be stimulated by TNFα, leading to potential crosstalk between the TNFα and LTβ pathways. Specifically, an expanded version of the original model that included the classical IκBs and p100 predicted that exposing cells to TNFα leads to a greater percentage of NF-κB molecules bound to p100 instead of to the classical IκBs, thereby priming the cells to subsequent LTβ exposure. Indeed, experimentally, LTβ-induced NF-κB activity can be increased ∼3-fold in TNF-primed versus naïve cells, with concomitant increases in expression of NF-κB-responsive genes (Basak et al, 2007).
Another non-inflammatory activator of NF-κB is ultraviolet (UV) irradiation. One of the effects of UV irradiation is bulk arrest of translation in a dose-dependent manner, which inhibits basal and induced synthesis of IκB. We recently showed, that although NF-κB is liberated when free IκB and NF-κB-bound IκB are gradually turned over, the NF-κB signaling module is actually remarkably robust to such metabolic perturbations (O'Dea et al, 2008). However, UV can dramatically amplify the response to simultaneous inflammatory stimulation. This synergy has implications for how inflammation can enhance the effects of cancer-associated stresses (O'Dea et al, 2008).
NF-κB can also be activated indirectly by signaling pathways that do not principally involve NF-κB. For example, TNFα, through activation of IKK and NF-κB, can induce the secretion of transforming growth factor-alpha (TGFα), leading to autocrine stimulation of the epidermal growth factor receptor. Taken together, TNFα and TGFα induce production of interleukin-1, providing an autocrine signal that can bring about a second episode of IKK and NF-κB activity (Janes et al, 2006). This helps to explain why, for example, IKK activity can be better predicted computationally from the combination of growth factor and inflammatory signaling data versus inflammatory signaling data alone (Janes et al, 2005, 2006).
NF-κB oscillations
Our analyses of the IκB–NF-κB signaling module concluded that oscillations in NF-κB activity, primarily driven by negative feedback through IκBα, underlie biphasic NF-κB dynamics. These oscillations are largely hidden in wild-type cells by the effects of IκBβ and IκBɛ (Hoffmann et al, 2002), and oscillations do not seem to alter gene expression programs when compared to the biphasic response (Barken et al, 2005), raising doubts about the functional significance of oscillations. Nonetheless, the apparent mathematical and biochemical complexity underlying the existence and particular shape of these oscillations intrinsically begs the question of how to generate and control them. These questions have so far been primarily addressed through computational analysis.
Negative feedback is a common way to achieve oscillatory behavior. Indeed, a simple negative feedback system comprised of two components that interact linearly is sufficient to generate oscillations (Hoffmann et al, 2002), but it is important to note that this abstraction is fundamentally different from the IκB–NF-κB module. Linear systems do not require persistent stimulation to exhibit undamped oscillations whereas the module does. Also, the mathematical dependency between individual parameters and oscillation frequency (e.g. monotonic relationship versus existence of an optimum) does not translate even qualitatively from the linear system to the module (R Cheong and A Levchenko, unpublished observations). Thus, components in the module and their nonlinear interactions play important roles in controlling and shaping NF-κB oscillations. Different aspects of NF-κB oscillations, such as the timing and amplitude of peaks and troughs, are sensitive to different parameters in the original model, as measured by sensitivity coefficients (an analog of metabolic control coefficients). Some parameters are predicted to be broadly important for nearly all aspects of oscillations, and they all relate to reactions involving IκBα (Ihekwaba et al, 2004; Joo et al, 2007). These parameters cooperate in a complex, nonlinear way to modulate oscillations (Ihekwaba et al, 2005), and overall, their effects on the timing and amplitude of the initial peak can be rationalized based on their contribution to total IκB levels and the speed of the feedback loop (Cheong et al, 2006; Mathes et al, 2008; Moss et al, 2008). Interestingly, the highly sensitive parameters correlate well with a minimal subset of reactions from the original model that sustain oscillations (Box 2). Additionally, a condensed model involving only NF-κB, IκBα, and IκBα mRNA still oscillates (Krishna et al, 2006), and in principle, a model with only NF-κB and IκBα with transcriptional delay can as well (Monk, 2003). Taken together, these theoretical perspectives indicate that the IκBα portion of the module is indeed the strongest generator of oscillations.
Interest in oscillations was further spurred by observations in which the NF-κB activity spiked repetitively (‘spiky oscillations') in cells overexpressing fluorescent protein-tagged NF-κB or IκBα, with the timing and frequency of spikes varying from cell to cell (Nelson et al, 2004). This is distinctly different than the biphasic dynamics observed in the population average (Hoffmann et al, 2002), and reconciling the two has become an important goal of mathematical analysis. Our statistical analysis of NF-κB activity measured by immunocytochemistry in single wild-type cells indicates that biphasic population dynamics is easily distinguished from an ensemble of individually oscillating cells, regardless of the mechanism underlying spiky oscillations (Barken et al, 2005). The intuitive conclusion, also supported by computational analysis, was that overexpression of NF-κB or IκBα components alters the oscillatory potential of the module. Others have attempted to attribute spiky oscillations and their variations from cell to cell to fluctuations in the rates of the chemical reactions comprising the pathway. Full stochastic simulation of a module in which the only IκB species is IκBα indicates that intrinsic biochemical randomness results in minimal deviation from the deterministic NF-κB response unless transcription and translation rates have been badly estimated (Hayot and Jayaprakash, 2006). Rather, fluctuations in extrinsic factors, such as the number of molecules of active IKK or NF-κB, need to be invoked to reconcile single live cell and average responses. However, these conclusions are at odds with simulations of other IκBα-only models (Lipniacki et al, 2006, 2007), in which only a few biochemical reactions need to be stochastic to generate distributions of responses similar to those obtained in live cells. Differences in parameter values or the inclusion of an A20 feedback loop in the latter models (Lipniacki et al, 2004) may explain these differing conclusions. In any case, at minimum, accurate measurements of IκB transcription and translation rates are needed to test the role of stochasticity in individualized cell responses.
Emerging developments in mathematical modeling of NF-κB signaling
As seen above, computationally oriented studies have led to numerous and varied insights into the molecular mechanisms that regulate NF-κB dynamics and inflammatory gene expression, and will surely continue to do so in the future. In this section, we highlight other aspects of NF-κB biology for which mathematical modeling is likely to play an important role.
Information encoding and decoding
Secretion of NF-κB-activating cytokines like TNFα is one way in which one cell can communicate to another and alter its behavior. One general question is what information is conveyed by secreted signals, how this information is encoded by the signaling cell, and how it is interpreted by the receiving cell. The unique temporal dynamics of NF-κB responses to TNFα provides a model system to address the principles underlying cell–cell communication.
For TNFα, it is possible to use changes in its concentration over time to transmit information about the distance between the signaling and receiving cells. Specifically, in a local infection, a macrophage will secrete a brief pulse of TNFα in a self-limited manner. Because of the effect of diffusion, nearby cells experience temporal patterns of changes in TNFα concentration that depend on the separation distance: the concentration experienced by a cell drops exponentially and while the duration of exposure to the cytokine increases modestly with distance. Experimentally, we observed that NF-κB is able to respond to amounts of TNFα that vary over several orders of magnitude, including very small ones. A model incorporating these observations, therefore, predicted that cells in a wide region around a local infection would mount an inflammatory defense. Moreover, because the amplitude of NF-κB activity scales according to the logarithm of TNFα concentration, the model also predicts that NF-κB responses drop roughly linearly with distance. Thus, cells near the infection would mount a vigorous inflammatory defense, whereas cells further away would have a tempered response, suggesting that the TNFα–NF-κB pathway is optimized so that cells respond in a way commensurate with their distance from danger (Cheong et al, 2006).
We anticipate that mathematical and computational models tightly coupled to experimental analysis will be indispensable in further understanding the information processing characteristics of NF-κB pathways. Because modeling to date has been very successful in demonstrating how dynamic IKK signals are transformed into dynamic NF-κB signals by the IκB–NF-κB module (Werner et al, 2005; Cheong et al, 2006), we especially look forward to progress in understanding events upstream of IKK or downstream of NF-κB. For example, multiple cytokine signaling pathways converge on IKK, but how each transmits information through IKK is poorly understood, as is how multiple cytokines convey information simultaneously through the same module. On the downstream end, different NF-κB-responsive genes are expressed after different durations of NF-κB activity (Hoffmann et al, 2002; Barken et al, 2005), but the basis of these differential responses is unknown. Combining pathway models with mathematical analysis of promoters and enhancers is likely to shed light on this issue (Krishna et al, 2006).
Rational drug targeting
NF-κB is involved in numerous physiologic responses, such as inflammation and apoptosis, and is implicated in myriad diseases like arthritis, autoimmune and inflammatory disorders, and cancer (Kumar et al, 2004). As such, numerous anti-inflammatory compounds are under development to target NF-κB (Karin et al, 2004), and mathematical models are beginning to be used to understand how these potential drugs affect NF-κB signaling.
One initial study in this direction examined the effect of three drug classes—inhibitors of IKK, the proteosome, and nuclear import machinery—on NF-κB oscillations in response to TNFα (Sung and Simon, 2004). The effect of each class was simulated by altering the appropriate kinetic rate parameters in a simplified version of the original model containing only one IκB-like species. In this way, NF-κB oscillations were predicted to be disrupted with high doses of IKK or proteosome inhibitors, or low doses of nuclear import inhibitors. Similarly, another study predicted that an IKK inhibitor dampens the NF-κB response to interleukin-1 (Ihekwaba et al, 2007). These types of simulations could potentially be used to further understand drug specificity or the effect of multiple drugs applied simultaneously.
In addition, drug-targeting studies may benefit from extending this idea further, that is, by performing a ‘computational drug screen.' Each kinetic rate parameter in the IκB–NF-κB module represents a potential target for modulation by a drug, so we are studying how sensitive the biphasic NF-κB response to TNFα is to alterations in each parameter. For example, we find that the initial transient phase but not the late sustained phase of NF-κB activity is robust to variations in the values of the parameters that control the half-life of IκBα (D Barken et al. in preparation). This suggests that even drugs that target reactions within the central NF-κB signaling module may in fact have selective effects, for example, by inhibiting prolonged inflammation without completely abrogating acute responses. This surprising possibility would be difficult to foresee by qualitative reasoning alone, but quantitative predictions provided by modeling are crucial in rationally identifying rate-limiting reactions for specific phases of the NF-κB temporal profile. We anticipate that similar methods will prove useful in rational selection of drug targets to mediate highly specific therapeutic effects.
Other trends in applications of NF-κB models
Paralleling the many physiological roles of NF-κB, models of NF-κB signaling are beginning to be applied in a variety of contexts. For example, TNFα-induced NF-κB dynamics were measured in liver cells infected with or without hepatitis B virus. A model of TNFα signaling to NF-κB suggests that an unknown IKK upregulating factor can reconcile subtle changes in NF-κB dynamics due to infection (Park et al, 2006). Another model examined the role of NF-κB in neural stem cells and predicts that the level of NF-κB activity correlates with the rate of cell proliferation (Piotrowska et al, 2006). Tests of predictions from these and related models are likely to be useful in elucidating the role of NF-κB dynamics in physiological and pathophysiological contexts.
Another trend that we anticipate continuing is merging of the NF-κB signaling models with models of other pathways. Initial steps have been in the areas of modeling LPS-induced TNFα signaling (Covert et al, 2005; Werner et al, 2005), TNF–EGF–insulin crosstalk and autocrine signaling (Janes et al, 2005, 2006), and crosstalk between TNF-induced caspases and NF-κB-induced antiapoptotic factors (Rangamani and Sirovich, 2007). The interest in linking NF-κB models to other pathway models is likely to grow, and since TNFα induces JNK activity, and because interleukins, T-cell receptors, B-cell receptors, and other stimuli activate NF-κB (Hayden and Ghosh, 2004), we can expect expansion into these areas. Elements of some existing models may prove useful in this regard (Schoeberl et al, 2002).
As a corollary to this trend, we expect existing NF-κB models to merge with each other. Most of the published NF-κB models described above are ‘backwards compatible,' in the sense that they recapitulate the essential dynamic properties of their predecessors while demonstrating some new dynamic properties. However, the descendant models are not necessarily compatible with each other. To address this issue, we have, for example, developed a ‘consensus model' that recapitulates a multitude of combined experiments (Hoffmann et al, 2002; Werner et al, 2005; Cheong et al, 2006; Kearns et al, 2006; O'Dea et al, 2007) and is thus increasingly predictive (R Cheong and A Levchenko, in preparation). Advanced parameter fitting techniques are likely to emerge as important tools in developing highly comprehensive consensus models (Fujarewicz et al, 2007).
Finally, an increasing use of the core IκB–NF-κB model is in illustrating new modeling environments and techniques. For example, the NF-κB pathway has been used to illustrate a new technique to graphically represent models in a way that is easy to interpret and yet is mathematically precise (Cho et al, 2003). Another application used NF-κB to exhibit an agent-based stochastic modeling method (Pogson et al, 2006). NF-κB models have also been used to illustrate efficient ways to investigate parameter sensitivity (Fujarewicz et al, 2007; Joo et al, 2007). Models of the IκB–NF-κB signaling module are attractive in these settings because they are usually moderate in size (a few dozen parameters and equations) yet display complex behavior, much of which can be rationalized through careful analysis of these models. As modeling becomes more accessible to non-specialists, we anticipate model analysis and applications will grow rapidly. In fact, one can already interactively explore the NF-κB model online through the Sigmoid project (http://www.sigmoid.org/) (Cheng et al, 2005). The growth and dissemination of such new tools can only contribute to NF-κB modeling efforts.
Conclusion
Signal transduction pathways are dedicated sets of chemical reactions responsible for detection, processing, and delivery of the information about changes in the cell environment to the ‘decision centers' of a cell. Unlike wires and antennas used in human-built devices designed for information transfer, cells are limited to using chemistry as the basis for the sophisticated and robust passing of signals within complex and convoluted intracellular spaces. The underlying complexity may thus be foreign to our anthropomorphic attempts to confer the ideas of wires, transistors, and resistors to sophisticated liquidity of biological processes. Nevertheless, as much as the behavior of electrical circuits can be captured by mathematical equations, so too can the intricacies of signal transduction be understood through computational techniques.
A myriad of soluble signaling molecules, coupled to each other through feedback loops and pathway crosstalk, impinge upon NF-κB. The regulation and dynamics of the resulting signaling network are rich and complex and their underlying mechanisms are not immediately transparent. Mathematical modeling has cut through the haze by helping to summarize experimental observations and develop a deep and coherent understanding of the NF-κB signaling. Such computational approaches are essential for continued advancements in the field of signal transduction, as exemplified by the profound qualitative and quantitative insights obtained thus far for NF-κB signaling.
References
- Barken D, Wang CJ, Kearns J, Cheong R, Hoffmann A, Levchenko A (2005) Comment on ‘oscillations in NF-kappaB signaling control the dynamics of gene expression'. Science 308: 52; author reply 52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basak S, Kim H, Kearns JD, Tergaonkar V, O'Dea E, Werner SL, Benedict CA, Ware CF, Ghosh G, Verma IM, Hoffmann A (2007) A fourth IkappaB protein within the NF-kappaB signaling module. Cell 128: 369–381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beg AA, Sha WC, Bronson RT, Baltimore D (1995) Constitutive NF-kappa B activation, enhanced granulopoiesis, and neonatal lethality in I kappa B alpha-deficient mice. Genes Dev 9: 2736–2746 [DOI] [PubMed] [Google Scholar]
- Beinke S, Ley SC (2004) Functions of NF-kappaB1 and NF-kappaB2 in immune cell biology. Biochem J 382: 393–409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bren GD, Solan NJ, Miyoshi H, Pennington KN, Pobst LJ, Paya CV (2001) Transcription of the RelB gene is regulated by NF-kappaB. Oncogene 20: 7722–7733 [DOI] [PubMed] [Google Scholar]
- Carlotti F, Dower SK, Qwarnstrom EE (2000) Dynamic shuttling of nuclear factor kappa B between the nucleus and cytoplasm as a consequence of inhibitor dissociation. J Biol Chem 275: 41028–41034 [DOI] [PubMed] [Google Scholar]
- Cheng J, Scharenbroich L, Baldi P, Mjolsness E (2005) Sigmoid: a software infrastructure for pathway bioinformatics and systems biology. IEEE Intell Syst 20: 68–75 [Google Scholar]
- Cheong R, Bergmann A, Werner SL, Regal J, Hoffmann A, Levchenko A (2006) Transient IkappaB kinase activity mediates temporal NF-kappaB dynamics in response to a wide range of tumor necrosis factor-alpha doses. J Biol Chem 281: 2945–2950 [DOI] [PubMed] [Google Scholar]
- Cho KH, Shin SY, Lee HW, Wolkenhauer O (2003) Investigations into the analysis and modeling of the TNF alpha-mediated NF-kappa B-signaling pathway. Genome Res 13: 2413–2422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collart MA, Baeuerle P, Vassalli P (1990) Regulation of tumor necrosis factor alpha transcription in macrophages: involvement of four kappa B-like motifs and of constitutive and inducible forms of NF-kappa B. Mol Cell Biol 10: 1498–1506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coughanowr DR (1991) Process Systems Analysis and Control, 2nd edn. Boston: McGraw-Hill, pp 90–103 [Google Scholar]
- Covert MW, Leung TH, Gaston JE, Baltimore D (2005) Achieving stability of lipopolysaccharide-induced NF-kappaB activation. Science 309: 1854–1857 [DOI] [PubMed] [Google Scholar]
- Delhase M, Hayakawa M, Chen Y, Karin M (1999) Positive and negative regulation of IkappaB kinase activity through IKKbeta subunit phosphorylation. Science 284: 309–313 [DOI] [PubMed] [Google Scholar]
- Fujarewicz K, Kimmel M, Lipniacki T, Swierniak A (2007) Adjoint systems for models of cell signaling pathways and their application to parameter fitting. IEEE/ACM Trans Comput Biol Bioinform 4: 322–335 [DOI] [PubMed] [Google Scholar]
- Ghosh S, May MJ, Kopp EB (1998) NF-kappa B and Rel proteins: evolutionarily conserved mediators of immune responses. Annu Rev Immunol 16: 225–260 [DOI] [PubMed] [Google Scholar]
- Hayden MS, Ghosh S (2004) Signaling to NF-kappaB. Genes Dev 18: 2195–2224 [DOI] [PubMed] [Google Scholar]
- Hayden MS, Ghosh S (2008) Shared principles in NF-kappaB signaling. Cell 132: 344–362 [DOI] [PubMed] [Google Scholar]
- Hayot F, Jayaprakash C (2006) NF-kappaB oscillations and cell-to-cell variability. J Theor Biol 240: 583–591 [DOI] [PubMed] [Google Scholar]
- Hoffmann A, Levchenko A, Scott ML, Baltimore D (2002) The IkappaB–NF-kappaB signaling module: temporal control and selective gene activation. Science 298: 1241–1245 [DOI] [PubMed] [Google Scholar]
- Ihekwaba AE, Broomhead DS, Grimley R, Benson N, White MR, Kell DB (2005) Synergistic control of oscillations in the NF-kappaB signalling pathway. Syst Biol (Stevenage) 152: 153–160 [DOI] [PubMed] [Google Scholar]
- Ihekwaba AE, Broomhead DS, Grimley RL, Benson N, Kell DB (2004) Sensitivity analysis of parameters controlling oscillatory signalling in the NF-kappaB pathway: the roles of IKK and IkappaBalpha. Syst Biol (Stevenage) 1: 93–103 [DOI] [PubMed] [Google Scholar]
- Ihekwaba AE, Wilkinson SJ, Waithe D, Broomhead DS, Li P, Grimley RL, Benson N (2007) Bridging the gap between in silico and cell-based analysis of the nuclear factor-kappaB signaling pathway by in vitro studies of IKK2. FEBS J 274: 1678–1690 [DOI] [PubMed] [Google Scholar]
- Janes KA, Albeck JG, Gaudet S, Sorger PK, Lauffenburger DA, Yaffe MB (2005) A systems model of signaling identifies a molecular basis set for cytokine-induced apoptosis. Science 310: 1646–1653 [DOI] [PubMed] [Google Scholar]
- Janes KA, Gaudet S, Albeck JG, Nielsen UB, Lauffenburger DA, Sorger PK (2006) The response of human epithelial cells to TNF involves an inducible autocrine cascade. Cell 124: 1225–1239 [DOI] [PubMed] [Google Scholar]
- Joo J, Plimpton S, Martin S, Swiler L, Faulon JL (2007) Sensitivity analysis of a computational model of the IKK NF-{kappa}B I{kappa}B{alpha} A20 signal transduction network. Ann NY Acad Sci 1115: 221–239 [DOI] [PubMed] [Google Scholar]
- Karin M, Yamamoto Y, Wang QM (2004) The IKK NF-kappa B system: a treasure trove for drug development. Nat Rev Drug Discov 3: 17–26 [DOI] [PubMed] [Google Scholar]
- Kearns JD, Basak S, Werner SL, Huang CS, Hoffmann A (2006) IkappaBepsilon provides negative feedback to control NF-kappaB oscillations, signaling dynamics, and inflammatory gene expression. J Cell Biol 173: 659–664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kearns JD, Hoffmann A (2008) A biochemist's perspective on computational modeling: lessons from NF-kappaB. J Biol Chem (in press) [Google Scholar]
- Klement JF, Rice NR, Car BD, Abbondanzo SJ, Powers GD, Bhatt PH, Chen CH, Rosen CA, Stewart CL (1996) IkappaBalpha deficiency results in a sustained NF-kappaB response and severe widespread dermatitis in mice. Mol Cell Biol 16: 2341–2349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishna S, Jensen MH, Sneppen K (2006) Minimal model of spiky oscillations in NF-kappaB signaling. Proc Natl Acad Sci USA 103: 10840–10845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A, Takada Y, Boriek AM, Aggarwal BB (2004) Nuclear factor-kappaB: its role in health and disease. J Mol Med 82: 434–448 [DOI] [PubMed] [Google Scholar]
- Kuprash DV, Osipovich OA, Pokholok DK, Alimzhanov MB, Biragyn A, Turetskaya RL, Nedospasov SA (1996) Functional analysis of the lymphotoxin-beta promoter. Sequence requirements for PMA activation. J Immunol 156: 2465–2472 [PubMed] [Google Scholar]
- Lee EG, Boone DL, Chai S, Libby SL, Chien M, Lodolce JP, Ma A (2000) Failure to regulate TNF-induced NF-kappaB and cell death responses in A20-deficient mice. Science 289: 2350–2354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipniacki T, Paszek P, Brasier AR, Luxon B, Kimmel M (2004) Mathematical model of NF-kappaB regulatory module. J Theor Biol 228: 195–215 [DOI] [PubMed] [Google Scholar]
- Lipniacki T, Paszek P, Brasier AR, Luxon BA, Kimmel M (2006) Stochastic regulation in early immune response. Biophys J 90: 725–742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipniacki T, Puszynski K, Paszek P, Brasier AR, Kimmel M (2007) Single TNFalpha trimers mediating NF-kappaB activation: stochastic robustness of NF-kappaB signaling. BMC Bioinformatics 8: 376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lombardi L, Ciana P, Cappellini C, Trecca D, Guerrini L, Migliazza A, Maiolo AT, Neri A (1995) Structural and functional characterization of the promoter regions of the NFKB2 gene. Nucleic Acids Res 23: 2328–2336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathes E, O'Dea EL, Hoffmann A, Ghosh G (2008) NF-kappaB denotes IkappaB degradation modes. EMBO J (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Memet S, Laouini D, Epinat JC, Whiteside ST, Goudeau B, Philpott D, Kayal S, Sansonetti PJ, Berche P, Kanellopoulos J, Israel A (1999) IkappaBepsilon-deficient mice: reduction of one T cell precursor subspecies and enhanced Ig isotype switching and cytokine synthesis. J Immunol 163: 5994–6005 [PubMed] [Google Scholar]
- Mizgerd JP, Scott ML, Spieker MR, Doerschuk CM (2002) Functions of IkappaB proteins in inflammatory responses to Escherichia coli LPS in mouse lungs. Am J Respir Cell Mol Biol 27: 575–582 [DOI] [PubMed] [Google Scholar]
- Monk NA (2003) Oscillatory expression of Hes1, p53, and NF-kappaB driven by transcriptional time delays. Curr Biol 13: 1409–1413 [DOI] [PubMed] [Google Scholar]
- Moss BL, Gross S, Gammon ST, Vinjamoori A, Piwnica-Worms D (2008) Identification of a ligand-induced transient refractory period in nuclear factor-{kappa}B signaling. J Biol Chem 283: 8687–8698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson DE, Ihekwaba AE, Elliott M, Johnson JR, Gibney CA, Foreman BE, Nelson G, See V, Horton CA, Spiller DG, Edwards SW, McDowell HP, Unitt JF, Sullivan E, Grimley R, Benson N, Broomhead D, Kell DB, White MR (2004) Oscillations in NF-kappaB signaling control the dynamics of gene expression. Science 306: 704–708 [DOI] [PubMed] [Google Scholar]
- O'Dea EL, Barken D, Peralta RQ, Tran KT, Werner SL, Kearns JD, Levchenko A, Hoffmann A (2007) A homeostatic model of IkappaB metabolism to control constitutive NF-kappaB activity. Mol Syst Biol 3: 111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Dea EL, Kearns JD, Hoffmann A (2008) UV as an amplifier rather than inducer of NF-kappaB activity. Mol Cell (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pahl HL (1999) Activators and target genes of Rel/NF-kappaB transcription factors. Oncogene 18: 6853–6866 [DOI] [PubMed] [Google Scholar]
- Park SG, Lee T, Kang HY, Park K, Cho KH, Jung G (2006) The influence of the signal dynamics of activated form of IKK on NF-kappaB and anti-apoptotic gene expressions: a systems biology approach. FEBS Lett 580: 822–830 [DOI] [PubMed] [Google Scholar]
- Piotrowska MJ, Widera D, Kaltschmidt B, an der Heiden U, Kaltschmidt C (2006) Mathematical model for NF-kappaB-driven proliferation of adult neural stem cells. Cell Prolif 39: 441–455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pogson M, Smallwood R, Qwarnstrom E, Holcombe M (2006) Formal agent-based modelling of intracellular chemical interactions. Biosystems 85: 37–45 [DOI] [PubMed] [Google Scholar]
- Rangamani P, Sirovich L (2007) Survival and apoptotic pathways initiated by TNF-alpha: modeling and predictions. Biotechnol Bioeng 97: 1216–1229 [DOI] [PubMed] [Google Scholar]
- Schoeberl B, Eichler-Jonsson C, Gilles ED, Muller G (2002) Computational modeling of the dynamics of the MAP kinase cascade activated by surface and internalized EGF receptors. Nat Biotechnol 20: 370–375 [DOI] [PubMed] [Google Scholar]
- Schomer-Miller B, Higashimoto T, Lee YK, Zandi E (2006) Regulation of IkappaB kinase (IKK) complex by IKKgamma-dependent phosphorylation of the T-loop and C terminus of IKKbeta. J Biol Chem 281: 15268–15276 [DOI] [PubMed] [Google Scholar]
- Sen R, Baltimore D (1986) Multiple nuclear factors interact with the immunoglobulin enhancer sequences. Cell 46: 705–716 [DOI] [PubMed] [Google Scholar]
- Shakhov AN, Collart MA, Vassalli P, Nedospasov SA, Jongeneel CV (1990) Kappa B-type enhancers are involved in lipopolysaccharide-mediated transcriptional activation of the tumor necrosis factor alpha gene in primary macrophages. J Exp Med 171: 35–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sung MH, Simon R (2004) In silico simulation of inhibitor drug effects on nuclear factor-kappaB pathway dynamics. Mol Pharmacol 66: 70–75 [DOI] [PubMed] [Google Scholar]
- Ten RM, Paya CV, Israel N, Le Bail O, Mattei MG, Virelizier JL, Kourilsky P, Israel A (1992) The characterization of the promoter of the gene encoding the p50 subunit of NF-kappa B indicates that it participates in its own regulation. EMBO J 11: 195–203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian B, Nowak DE, Jamaluddin M, Wang S, Brasier AR (2005) Identification of direct genomic targets downstream of the nuclear factor-kappaB transcription factor mediating tumor necrosis factor signaling. J Biol Chem 280: 17435–17448 [DOI] [PubMed] [Google Scholar]
- Werner SL, Barken D, Hoffmann A (2005) Stimulus specificity of gene expression programs determined by temporal control of IKK activity. Science 309: 1857–1861 [DOI] [PubMed] [Google Scholar]
- Wertz IE, O'Rourke KM, Zhou H, Eby M, Aravind L, Seshagiri S, Wu P, Wiesmann C, Baker R, Boone DL, Ma A, Koonin EV, Dixit VM (2004) De-ubiquitination and ubiquitin ligase domains of A20 downregulate NF-kappaB signalling. Nature 430: 694–699 [DOI] [PubMed] [Google Scholar]