

Abstract
The A-to-B form transition has been examined in three DNA duplexes, d(CGCGAATTCGCG)2, d(CGCGAATTGCGC), and d(CGCAAATTTCGC), using circular dichroism spectroscopy, ultraviolet resonance Raman (UVRR) spectroscopy, and molecular dynamics (MD) simulation. Circular dichroism spectra confirm that these molecules adopt the A form under conditions of reduced water activity. UVRR results, obtained under similar conditions, suggest that the transition involves a series of intermediate forms between A and B. Cooperative and distinct transitions were observed for the bases and the sugars. Independent MD simulations on d(CGCGAATTCGCG)2 show a spontaneous change from the A to B form in aqueous solution and describe a kinetic model that agrees well with UVRR results. Based on these observations, we predict that the mechanism of the transition involves a series of A/B hybrid forms and is sequential in nature, similar to previous crystallographic studies of derivatized duplexes. A simulation in which waters were restrained in the major groove of B DNA shows a rapid, spontaneous change from B to A at reduced water activity. These results indicate that a quasiergodic sampling of the solvent distribution may be a problem in going from B to A at reduced water activity in the course of an MD simulation.
INTRODUCTION
The B form of DNA is well known to be predominant in biological systems. However, conformational changes from B in the direction of A DNA are implicated in protein-DNA and drug-DNA interactions, and there is leading evidence for a functional role for A as well as B forms in genome structure and function (1). The A-to-B transition in DNA has also served as a prototype case for testing out and validating empirical energy functions and force fields used in molecular dynamics simulations on nucleic acids (2–4). A number of recent experimental (5–8) and theoretical (4,9–12) studies have reported on this system. Most studies to date have focused on the preferential stability of A and B DNA as a function of water activity and salt concentrations, whereas the mechanism of the B-to-A transition has received less attention.
Unique new information on this problem can now be obtained from ultraviolet resonance Raman (UVRR) spectroscopy, which monitors the local structures of individual nucleotide bases in a sequence. Molecular dynamics (MD) simulations can provide a detailed computational model for the conformational change, which must of course be experimentally validated. We report herein a combined experimental/theoretical study of the B-to-A transition in selected DNA oligonucleotides based on UVRR spectroscopy and MD computer simulation. The particular objective of this study is to elucidate base-pair sequence and structural effects on the transition mechanism. UVRR spectroscopy has been applied to explicitly examine different base types through the intensity and frequency of their vibrational modes as a function of water activity.
Experiments were carried out on three dodecamer sequences, d(CGCGAATTCGCG), d(CGCGAATTGCGC), and d(CGCAAATTTCGC). Corresponding MD simulations including water and counterions performed on d(CGCGAATTCGCG) with an A-form initial structure and the spontaneous transition of the A- to B-form dodecamer during the initial phase of the MD simulation provides a theoretical model of the transition mechanism (Fig. 1). The essential results of the experimental and theoretical studies are in close accord, and are found to support the idea of a sequence-dependent, sequential mechanism for the transition rather than a concerted, all-or-none mechanism. Detailed analyses of DNA structures and solvation obtained from the MD simulations provide further information leading to an improved understanding of the transition mechanism. In addition, some new insights into sampling issues in obtaining conformational transitions in MD on DNA for mixed solvent systems have been obtained.
FIGURE 1.

Structural representation of the d(CGCGAATTCGCG)2 DNA sequence. (a) Canonical B-form DNA structure. (b) An ensemble of structures derived from a 5-ns-long MD simulation starting from the canonical B-form structure. (c) An ensemble of structures derived from a 5-ns-long MD simulation starting from the canonical A-form DNA structure. (d) Canonical A-form structure. The nucleotides are color coded as follows: red, adenine; blue, thymine; green, guanine; and yellow, cytosine.
BACKGROUND
The structural difference between B and A forms of DNA resides essentially in the sugar puckers (φ) and the helix-base parameters x-displacement (XDP), inclination (INC), and slide (SLD), according to the definition of these parameters (13). In B DNA the nucleotide basepairs are perpendicular to the helix axis (INC = 0) and centered (XDP, SLD = 0), and the sugar pucker is C2′-endo. In the A form, the basepairs are tilted (INC = 20°) such that they are displaced from the helix axis by −4.0 Å, and the sugar pucker is C3′-endo.
DNA is unique in the extent to which its structure depends on solvation. The early fiber diffraction experiments that provided experimental data crucial to the discovery of the double helix also revealed that DNA structure was sensitive to the relative humidity of the sample fibers (14). The B form is stable at relative humidities of 95% and the A form is preferentially stabilized below 74%. Subsequent fiber diffraction by Arnott and co-workers (15) elucidated the molecular structures of the canonical B and A forms of DNA. The preferential stability of B- and A-form DNA in solution was first reported by Ivanov and co-workers (16) based on CD spectroscopy, and was found to depend on water activity in a manner that parallels the results on fibers. Malenkov et al. reported the B-to-A transition to be cooperative (17).
There has been considerable research into the forces involved in the preferential stability of right-handed DNA helices and the A-to-B transition. Solvent accessibility (18), base-stacking interactions (19), the economics of phosphate hydration (20), hydrophobic pressure, enthalpic stabilization of B form by the minor-groove spine of hydration (21,22), and electrostatic effects associated with the explicit organization of mobile counterions in the vicinity of the polyanionic double helix (23–26) have all been invoked as plausible and possible explanations. Based on analysis of MD simulation trajectories, Jayaram et al. (27) proposed that the molecular origins of the conformational preferences of A and B DNA in water and 85% EtOH lie primarily in the differential free-energy contributions from interphosphate repulsion, counterion condensation, and solvation.
Recently, Vargason et al. (8,28) reported a crystallographic map of the conversion of B to A DNA based on a set of 13 crystal structures of d(GGCGCC), with various structural intermediates trapped by methylating or brominating the cytosine bases. This study provided a model for the B-to-A conformational transition in terms of a set of distinct conformational intermediates, each of which is an equilibrium state with respect to the free energy of the particular crystal. The duplex in some instances was indicated to be partly A form and partly B form, depending on local sequence composition, and some of the intermediate structures bear little resemblance to either the starting or ending states. If the mechanism of the transition can be successfully modeled based on these intermediate structures, the conversion of SLD precedes the change in base-pair inclination in going from B to A, and changes in XDP occur continuously. In these results, sugar puckers show a reasonably sharp transition point from B-like C2′-endo to A-like C3′-endo structures (with some intermediate structures exhibiting a mixture of C2′- and C3′-endo-type sugars). In the intermediate structures, an extension and unwinding of the helix occurs along with the change in sugar pucker. It remains to be established whether these results from crystallography carry over from equilibrium states to dynamics and are applicable to the solution as well as to the crystalline state. Experimental data on the dynamics of the B-to-A transition are relatively sparse. A study by Jose and Porshke (29) reports on the dynamics of the A-to-B transition based on stopped-flow, electric-field-jump experiments. They report time constants for the transition in the range of 10 μs, and present evidence for a significant activation-energy barrier in going from B- to A-form DNA.
A number of CD and related studies on DNA have been reported which characterized the sequence dependence of the preferential stability of A and B forms of DNA (20). In general, ApA and ApT steps are relatively rigid and resist transition from B to A, and the B→A transition is viable only for sequences with a sufficient number of GC basepairs (30,31). Similarly, conversion to the A form using Co3+(NH3)6 requires GpG steps and has not been demonstrated with sequences containing a majority of AT basepairs (25,32). As noted above, the B form is favored under conditions of high water activity in solution or high relative humidity in fibers. When the water activity is lowered, or the relative humidity of a DNA fiber is <∼75%, B-form sequences convert spontaneously to A form. Several groups (33,34) have examined the experimental data on the preferential stability as a function of sequence and B/A philicity has been expressed based on dimeric and trimeric base-pair steps. It is important to note that the solution structures of the A- and B-DNA endpoints of the transition are expected to contain some structural differences relative to the ideal canonical A and B forms of DNA inferred from fiber diffraction (35).
In addition to CD, infrared (36–38), and Raman (5,39) spectroscopic methods have been applied to the study of B- and A-form DNA. Raman spectroscopic markers for the B and A forms of DNA are well established (5,39). Certain structural parameters of DNA can be obtained from these methods, particularly the configuration of the sugar rings, the angles of phosphates, and overall base geometries. A disadvantage of these methods, including CD spectroscopy, is that the information on sequence dependence cannot generally be resolved. NMR studies involving 2D nuclear Overhauser effect spectroscopy do not resolve the DNA helical parameters, but the recent addition of reduced dipolar coupling has measurably improved these determinations. However, elucidation of a reliable molecular structure from NMR data remains challenging.
UVRR has introduced some new dimensions to the study of nucleic acid structure (40–42). The resonance enhancement intrinsic to UVRR spectroscopy makes it possible to selectively investigate the behavior of individual types of nucleotide bases and, in the case of propitious sequences, individual bases. As a consequence of this resonance selective enhancement and detection, stacking interactions and H-bonding of relevant functional groups can be attributed to distinct residue types. Thus, UVRR spectroscopy can be a source of more detailed information regarding local structure of DNA as a function of sequence and conformation.
In previous studies, the UVRR method has proven useful in evaluating the structure and stacking of DNA molecules, cation coordination, and H-bonding (43,44). From measurements of the premelting transition in DNA A-tracts, the relative strength of cross-strand 3-centered H-bonds could be determined (45). More recently, UVRR spectroscopy has been applied to the study of protein-DNA interactions, and subtle changes in DNA structure associated with protein binding were readily detected (46–48).
MD computer simulation including explicit consideration of solvent has been applied extensively to model the dynamic structure of DNA oligonucleotides (Fig. 1) (49–54). MD simulations are found to be generally in close accord with NMR-derived solution structures (7,55–59). MD simulations have provided new insights into the solution structures of these molecules, which include essentially straight A-tracts (60), highly flexible purine-pyrimidine steps that can serve as a locus for axis bending (56), and support for the “non-A-tract” model of DNA curvature (49). MD studies of sequence effects on B-form DNA structures has been advanced by recent simulations on all 136 unique tetranucleotide base-pair steps (61,62) and has led also to improvements in nucleic acid force fields (63).
The interconversion from B to A DNA has been investigated via molecular dynamics simulations as described in several recent articles (2,4,11,12,64). Cheatham and Kollman (2) were the first to report MD studies of A- and B-form conformational stability in explicit solvent. Subsequently, Sprous et al. (4) reported MD simulations on d(CGCGAATTCGCG)2 in A and B forms. The motivation for these studies was directed more toward assessing the performance of force fields, with the A-to-B transition as a prototype case. Both Cheatham et al. (2) and Sprous et al. (4) independently observed that in MD based on the AMBER parm94 force field, B-form DNA was stable in water and the A form was stable in a mixed ethanol/water solvent with reduced water activity. The MD model A-form DNA in aqueous solution was observed to convert spontaneously into the B form. However, both studies reported that B-form DNA did not spontaneously convert to the A form in the low-water-activity mixed solvent at 300 K. This was initially addressed as a force-field limitation and a subsequent local alteration of the force field was designed to make this transition more favorable (65). However, the performance of the resulting MD model DNA deteriorated with respect to other structural features. In these studies, the expectation of a B-to-A transition at low water activity was based on generic patterns of environmental effects on DNA conformational stability rather than specific behavior of a DNA sequence and the question of whether in fact this sequence was observed experimentally to form A DNA at low water activity was raised by Pichler et al. based on infrared studies of hydrated films (38). The exact solvent conditions that favored A- or B-form DNA for this sequence were not well delineated. Elucidation of these issues, as well as the mechanism underlying them, motivated the study described here.
Recently, Pastor studied the B-to-A transition in a TATA box sequence using MD based on the CHARMM nucleic acids force field (12) and found evidence in support of a sequential “slide first, roll later” mechanism that is opposed by DNA electrostatics and favored by increasing condensation of sodium ions. The latter phenomenon was noted earlier by Sprous et al. (4) and figured significantly in the free energy calculations of Jayaram et al. (27), with cross correlations between various theoretical studies providing some enhanced validation of the results. Banavali and Roux (64) recently reported a free energy profile for the B-to-A transition of d(CTCGAG) in water with minimal salt based on the CHARMM force field. The free energy difference between canonical A and B forms was found to be at least 2.8 kcal/mol, and the simulation with root mean-square deviation constraints sampled a continuum of hybrid structures with no well defined local minima between the two forms. The details of this calculation raise a number of additional issues, since some base-flipping events and other effects were noted that are presumably artifacts. Also the MD structures of A and B DNA deviate significantly from the idealized canonical forms, which are not appropriate endpoints for a determination of the free energy difference in solution.
In this study, the mechanism of the A-to-B transition is revisited to address the relative importance of base step, solvation, and electrostatics in the transition. The combined methodologies of UVRR spectroscopy and MD simulation provide new insights into the transition, since the UVRR results identify regions of change and stability, and the MD simulations provide a structural basis for interpretation of Raman data. These results particularly point to a sequential mechanism for the transition, in which reorganization of water plays a central role.
METHODS
Oligonucleotides
Experiments were carried out on three dodecamer sequences, d(5′-CGCGAATTCGCG-3′), d(5′-CGCGAATTGCGC-3′), and d(5′-CGCAAATTTCGC-3′), as a function of water activity. The 5′ to 3′ sequences are shown; however, all experiments were performed on duplexes with the appropriate complementary strands. Oligonucleotides were synthesized in 1-μmol quantities (Integrated DNA Technologies, Coralville, IA). The deblocked and desalted oligomers were purified by polyacrylamide gel electrophoresis. Purity was checked by analytical gel electrophoresis. For all spectroscopic experiments, samples were dialyzed against a buffer containing 0.1 M NaCl and 0.67 mM NaPO4, pH 7.0. For the trifluoroethanol (TFE) experiments, only buffer was used, as minimal salt concentrations were chosen to promote oligonucleotide solubility. Duplex samples were prepared by heating to 90°C in a water bath for 5 min, followed by slow cooling to room temperature. Annealed samples were subsequently diluted 10-fold with either TFE or buffer, to produce two 1 × 10−4-M samples (strand), one in buffer (0% TFE) and one in 85% TFE.
CD spectroscopy
CD experiments were performed using a Jasco Spectropolarimeter J-810 with a six-cell Peltier temperature controller. Spectral scans were measured from 300 to 190 nm, with a scan speed of 20 nm/min and an 8-s response time. Three scans were averaged at each molar hydration point.
UVRR spectroscopy
A Q-switched, Nd:YLF pumped Ti:sapphire laser system (Quantronix, East Setauket, New York) was used to generate the excitation wavelengths, by frequency-tripling or quadrupling the output of the Ti:sapphire using barium borate crystals, as previously described (43,45). Samples were contained in a 3 × 3-mm quartz cuvette and continuously stirred for the duration of the experiment. All spectra were collected at 15-min intervals in separate cycles; if any degradation in the sample was observed, that scan was discarded. The 0% and 85% TFE spectra arise from 2 h of averaged data, and all other points result from 1 h of averaged data. Spectra were acquired with a slit width of 170 μm and calibrated against ethanol, acetone, and pentane. Relative spectral shifts are accurate to 0.25 cm−1 and absolute frequencies are accurate to 1 cm−1. Samples were examined at room temperature and were normalized to the TFE band occurring at 1459 cm−1. Curve fitting of the spectra was accomplished with a mixed Lorentzian and Gaussian function, using constant peak frequencies. All data manipulation and analyses were performed using GramsAI (ThermoGalactic, Salem, NH).
Percent relative molar hydration (MH) was calculated as the mole fraction of water at each TFE concentration using the equation  where n refers to the number of moles of water or TFE. Concentrations of DNA and salt were neglected, as they remained constant. Transition midpoints were treated independently and analyzed using a Boltzmann function with Origin v. 6.0. (MicroCal, Northampton, MA):
where n refers to the number of moles of water or TFE. Concentrations of DNA and salt were neglected, as they remained constant. Transition midpoints were treated independently and analyzed using a Boltzmann function with Origin v. 6.0. (MicroCal, Northampton, MA): 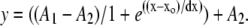
Molecular dynamics simulations
Four simulations on the duplex d(CGCGAATTGCGC) are considered here. Simulations on both the B and A forms of this sequence reported earlier were all redone and extended to much longer trajectories. Two of these involve the simulation of the canonical A-form structure in an ∼85% (v/v) ethanol/water mixture, i.e., ∼30% relative MH, and in water, and the third and fourth are of the canonical B-form structure in water and at 30% MH. Considerable new analysis of solvation was carried out for this project. Details of the calculations are as follows: The AMBER utility NUCGEN was employed to create the canonical A- and B-DNA starting structures based on fiber diffraction data (35). For simulations in the ethanol/water mixture, the united-atom OPLS-ethanol model (66), which is computationally effective and closely approximates the correct density, heat capacity, and heat of vaporization observed for ethanol (67), together with the TIP3P water molecules, was used at a concentration of 30% MH relative to ethanol. The simulation protocol is similar to those reported earlier (4,9). A “biphasic” initial configuration, as described by Cheatham et al. (9), was constructed by adding TIP3P water molecules in the first 6 Å from the DNA surface followed by the requisite number of ethanol molecules to make up the 30% MH ethanol/water solvent mixture. For the simulations in water, the TIP3P (68) water model was employed for the solvent such that the solvent extends up to 13 Å from the surface of the solute in all the directions.
Electroneutrality was established by adding 22 sodium cations (69) to the system. The positions of the ions were randomized such that they were at least 5 Å from the DNA and 5 Å away from each other. All MD simulations were performed in TPN ensemble using AMBER version 7 or 8 and the parm94 version of the Cornell et al. force field (70), and employing the particle-mesh Ewald procedure (71,72) for the treatment of long-range interactions. The MD protocol of minimization, heating, equilibration, and production dynamics was carried out in the following steps. Minimization involved 100 steps of steepest descent followed by 250 steps of conjugate gradient method. Heating from 0 to 300 K was done over 10 ps, followed by equilibration at 300 K for an additional 40 ps with SHAKE constraints (73). During the equilibration phase, a flat well restraint on the C1′-C2′-C3′-C4′ torsion was introduced to maintain the angle between 30° and 40° in the A-form structure, set at 25 kcal/mol/deg2 for the first 30 ps and then reduced to 5 kcal/mol/deg2. The production simulations were pursued without any restraints for simulation times of 10 ns and longer using 2-fs time steps.
The analysis of the composition of a molecular fluid requires an interpretation of the statistical distribution functions in structural and energetic terms. A theoretical approach for this problem was mapped out several years ago for pure liquids by Ben-Naim (74) based on the generalized molecular distribution functions and the closely related quasicomponent distribution function involves developing the distribution of particles with certain well-defined values of a compositional characteristic on the statistical state of the system (75). The basis for a general compositional analysis of the statistical state of molecular fluids must be a unique definition of the local solution environment of each identifiable substructure-atom, function group, or subunit of the solute. The proximity criterion accomplishes this by uniquely identifying each solvent molecule with a well-defined solute entity in each configuration. The proximity analysis partitions the solvent according to the proximal solute atoms and calculates distribution properties for each partition. The solute atoms can be partitioned independently or into functional groups defined by their chemical identity. All the proximity calculations were performed using the MMC program (76). The solvent-accessible surface area calculations on the snapshots in the MD trajectory have been performed using the program SurfRacer 3.0 (77). The atomic radius set reported by Alden and Kim (18) was employed. A spherical probe radius of 1.4 Å corresponding to the regular radius of a water molecule was employed in all the solvent-accessible surface area calculations.
RESULTS
CD spectra
CD spectra for all three duplexes considered in this study were examined to make sure they each exhibited the expected preferential stability of the B and A forms at high and low water activities and were suitable for our study. The CD spectrum of the d(CGCGAATTCGCG)2 duplex is shown as a function of increasing relative percentages of TFE in Fig. 2 a. At relatively low concentrations of TFE (>77% MH), the spectrum exhibits a maximum at 283 nm and a minimum at 252 nm of comparable magnitude. These conservative features above 220 nm are consistent with those previously observed for B-form DNA (78). As the TFE concentration is increased, the maximum at 283 nm decreases in intensity and shifts to 270 nm (Fig. 2, inset). In previous work, spectroscopic features of A-form DNA at low ionic strength included a peak at 270 nm, the shape of which was strongly sequence-dependent (16,31). To ensure that the transition was not associated with the palindromic nature of the d(CGCGAATTCGCG) sequence, similar CD studies were performed with other DNA dodecamers, which are of similar base composition but different sequence. For all duplexes examined, a decrease in ellipticity at 250 nm coupled with a shift in maximum to 270 nm is observed with increasing concentrations of TFE (Fig. S2 in Supplementary Material, Data S1). The magnitude of the ellipticity at 270 nm measured at 41% MH lies in the 10–20 Δɛ (M−1cm−1) range. This is in good accord with previous measurements of the B-to-A conversion in TFE, in which peaks of similar ellipticity were observed at 270 nm and were identified as being diagnostic of the A form (16,31). For all duplexes examined, the spectrum obtained at 41% MH exhibits an increase in ellipticity at 270 nm, a decrease at 250 nm and an increase at 210 nm relative to the B-form spectrum. These spectral features have been previously assigned to A-form DNA (33,78) and indicate that the d(CGCGAATTCGCG)2 duplex has adopted the A form with increasing concentrations of TFE.
FIGURE 2.

(a) CD spectra of d(CGCGAATTCGCG)2 DNA at increasing percentage of molar hydration. Inset shows an expansion of the region between 230 and 300 nm. DNA concentration was 100 μm (strand). (b) Intensity at 252 nm plotted as a function of percent molar hydration.
The CD intensity at 252 nm for d(CGCGAATTCGCG)2 is plotted as a function of molar hydration in Fig. 2 b. The change in intensity occurs over a relatively narrow range of hydration, consistent with previous studies. The results indicate that the global change in conformation from B- to A-form DNA occurs at 65% molar hydration. The d(CGCGAATTGCGC)2 duplex, which is exactly the same base composition as the d(5′-CGCGAATTCGCG-3′) duplex, has a transition midpoint of 68.3%, whereas the d(5′-CGCAAATTTCGC-3′) duplex exhibits a transition midpoint at 63.2% molar hydration (Fig. S2 in Data S1). As expected, the increase in number of AT basepairs in going to d(5′-CGCAAATTTCGC-3′) results in a transition midpoint at a significantly lower level of hydration, that is, the more AT-rich sequence converts to the B form at lower water activity, consistent with conventional wisdom on the relative propensities of the basepairs to form A- and B-form DNA. In summary, the CD studies indicate that A/B interconversion does occur in each of the sequences studied. The average transition midpoint is consistent with the general idea of the preferential stability of B form at high water activity and A form at low water activity and of A/B philicity as a function of sequence.
UVRR spectra: ribosyl conformation
Three different excitation wavelengths were used to gain a molecular picture of the DNA oligonucleotides changing from A to B form in solution (Fig. 3). To selectively monitor the deoxyribose-phosphate backbone, an excitation wavelength of 210 nm was used (43,45). At this excitation wavelength, the peak intensities of ribose modes for dG residues are preferentially enhanced (41,42). The vibrational frequencies of ribose modes are sensitive to conformation, with the C2′-endo configuration associated with a peak at 685 cm−1 and the C3′-endo configuration associated with a peak at 665 cm−1 (5,39). This feature is often used as an indicator of DNA conformation, but the results presented below indicate that ribose conformation does not necessarily reflect an overall A- or B-form structure. In this work, we find that this mode shifts to lower frequency as the relative hydration is decreased (Fig. 3). A transition midpoint of 68.3 ± 2.3% is obtained from a plot of the frequency as a function of relative hydration (Fig. 4); this value is within the range of that determined by CD spectroscopy (see above) (Table 1).
FIGURE 3.

UVRR spectra of the sequence d(CGCGAATTCGCG), excited at 210, 240, and 260 nm. Far left panel shows an expanded view of the region containing modes assigned to the dG C2′-endo and C3′-endo sugar puckers. Spectra are shown at 41%, 70%, and 100% molar hydration. Vibrational modes referenced in the main text are indicated on the 100% molar hydration spectra. Peaks appearing at 1460 and 1283 cm−1 are assigned to residual TFE modes present in the spectra after subtraction. Each spectrum results from 1 h of acquisition.
FIGURE 4.

Plots of changes in (a) sugar pucker vibrational mode frequency, (b) AT mode intensity, (c) GC mode intensity, and (d) dT carbonyl mode frequency as a function of molar hydration, as observed by UVRR for d(CGCGAATTCGCG)2. The left axis scale corresponds to the squares in each plot, and the right axis scale to the stars. Transition midpoints and ΔΔG values for each component are reported in Table 2. Original data are shown in Fig. 3.
TABLE 1.
Relaxation times of some global d(CGCGAATTCGCG)2 basepair-axis parameters and pseudorotation phase angles
| DNA Base | τXDP | τINC | τPHA1 | τPHA2 |
|---|---|---|---|---|
| C1-G24 | 0.128 | 0.128 | 0.021 | 0.558 |
| G2-C23 | 0.145 | 0.065 | 0.091 | 0.423 |
| C3-G22 | 0.159 | 0.048 | 0.094 | 0.021 |
| G4-C21 | 0.178 | 0.078 | 0.192 | 0.035 |
| A5-T20 | 0.152 | 0.064 | 0.256 | 0.151 |
| A6-T19 | 0.171 | 0.071 | 0.005 | 0.025 |
| T7-A18 | 0.153 | 0.061 | 0.008 | 0.011 |
| T8-A17 | 0.216 | 0.066 | 0.031 | 0.043 |
| C9-G16 | 0.182 | 0.054 | 0.030 | 0.034 |
| G10-C15 | 0.070 | 0.063 | 2.582 | 0.074 |
| C11-G14 | 0.111 | 0.063 | 0.027 | 0.024 |
| G12-C13 | 0.739 | 0.351 | 0.008 | 0.007 |
| Average for G and C bases | 0.141 | 0.062 | 0.502 | 0.102 |
| Average for A and T bases | 0.173 | 0.066 | 0.075 | 0.058 |
| Total average | 0.157 | 0.064 | 0.288 | 0.080 |
Parameters were calculated from the first-order exponential decay fit of the x displacement (XDP), inclination (INC), and phase angle of sugar pucker in strands 1 (PHA1) and 2 (PHA2).
To estimate the relative populations of the C2′-endo and C3′-endo configurations, the Raman bands were fit assuming that only these two components were contributing to the observed band. This fitting indicates that even at 41.5% MH, the population of dG ribose modes in the C3′-endo configuration is 94% and not 100% (Fig. 5 a). This reflects a situation wherein the dynamic structure of the duplex in solution involves both B-form and A-form ribose structures in the Boltzmann ensemble. Because UVRR spectroscopy superimposes all dG ribose modes, it is not possible to distinguish which individual ribose moieties have adopted the C3′-endo configuration at 41.5% MH and which ones have remained in the C2′-endo configuration. The sugar conformation of other bases cannot be examined because of spectral interference from TFE bands.
FIGURE 5.

(a) Population of C3′-endo and C2′-endo ribose conformations determined from the fitting of UVRR bands at 665 and 685 cm−1 (Fig. 3). (b) Normalized frequency of sugar puckers for the d(CGCGAATTCGCG)2 sequence calculated over the course of the 60-ns B-DNA simulation in water and the 20-ns A-DNA simulation in 85% water/ethanol.
UVRR spectra: base interactions
An advantage of the UVRR technique is the ability to selectively enhance contributions from the different bases through judicious choice of excitation wavelength. Thus, dG residues are enhanced through an excitation wavelength of 240 nm and dA residues contribute strongly to the spectrum when excited at a wavelength of 260 nm. Contributions from dT bases are also observed using an excitation wavelength of 260 nm. Under these excitation conditions the modes that are observed primarily arise from ring-stretching vibrations. Because of the effect of Raman hypochromism, which is directly related to absorption hypochromism, the intensity of the base modes can be related to base-stacking interactions. For all of the modes examined, the intensity of the modes increases upon adopting the B form (Fig. 3). The increase in intensity is attributed to a reduction in base-stacking interactions because of the greater distance or rise in base steps (3.4 Å for B vs. 2.8 Å for A) and the more pronounced helical twist between base steps (36° for B vs. 33° for A) in B-form DNA. The increased intensity upon adoption of the B form has also been observed in other Raman studies of DNA conformation (5,39,47).
The intensity changes of the 1362 and 1485 cm−1 modes obtained with an excitation wavelength of 240 nm are shown as a function of molar hydration in Fig. 4. Based on a comparison of the relative Raman cross sections (41,42,79), these modes arise primarily from dG stretching motions rather than dA residues because of the 240-nm excitation wavelength. The mode occurring at 1362 cm−1 arises from dG C2=N3-C4-N9 stretching motions, with contributions from N7 ring-stretching vibrations and the 1485-cm−1 mode arises from C2-H and C8-H bending modes, coupled with C8-C9 stretching motions. Since these motions lie primarily in the plane of the ring, the enhancement mechanism is associated with the absorption of the base itself and changes in intensity can be related to changes in hypochromicity associated with stacking interactions. The transition midpoints obtained from the intensity changes as a function of molar hydration yield a value of 85 ± 1.0% (Table 2). Interestingly, this transition midpoint differs significantly from that obtained for the change in ribose conformation and suggests that the base-stacking geometry of the dG residues changes at a higher molar hydration compared to the ribose.
TABLE 2.
UVRR-observed transition midpoints and ΔΔG values for the d(CGCGAATTCGCG) sequence
| Tm (% MH) | ΔΔG (kcal/mol) | |
|---|---|---|
| Sugars | ||
| Δν 685 | 68.35 ± 2.3 | −3.02 |
| Δν 1420 | 72.2 ± 0.7 | −3.05 |
| AT | ||
| ΔI 1332 | 69.2 ± 1.4 | −3.03 |
| ΔI 1575 | 75.3 ± 2.5 | −3.09 |
| GC | ||
| ΔI 1362 | 84.6 ± 1.1 | −3.17 |
| ΔI 1483 | 85.4 ± 1.5 | −3.18 |
| T C4=O | 87.5 ± 6.0 | −3.19 |
Using an excitation wavelength of 260 nm, the intensities of modes at 1332 and 1575 cm−1 can also be monitored as a function of hydration (Fig. 4). These modes, which arise primarily from dA residues at this excitation wavelength, result from imidazole and pyrimidine ring-stretching vibrations, respectively. The intensity change as a function of molar hydration yields transition midpoints of 69% and 75%, respectively. These midpoints are suggestive of a mean transition midpoint for the AT basepair of 72% MH, which differs significantly from that obtained for the ring-stretching modes arising from dG residues (Fig. 4). Thus, these results indicate that dA residues do not change their base-stacking geometries under the same conditions as dG residues and that the dA residues require significantly lower levels of molar hydration to convert to the A form. For both base types, the transition midpoints observed are at higher water activity relative to that observed for the ribose conformation. These findings are in good qualitative agreement with the structure of the d(CCCCGGGG)2 duplex sequence determined by CD and NMR spectroscopic techniques (6), in which the base-stacking properties were found to be A-like and the sugar pucker was observed to be B-like. The NMR-determined structure was interpreted to be an intermediate in the A-to-B transition (6).
UVRR spectra of the A and B forms of DNA also provide some insight into the relative hydration of the grooves through measurement of H-bonding strength. The relative strength of H-bonding interactions is inferred from the carbonyl stretching frequency of relevant functional groups. A decrease in frequency results from a reduction in the force constant of the carbonyl bond and is correlated with increased strength of H-bonding (45,80). The thymine C4=O group points into the major groove and is readily monitored using an excitation wavelength of 210 nm (41,45). Under conditions of increasing concentrations of TFE, the frequency of the C4=O shifts from 1662 to 1653 cm−1 (Fig. 4). The downshift in frequency is indicative of stronger H-bonding in A-form than in B-form DNA. Crystallographic structures have revealed a more ordered water structure in the major groove of A-form DNA, and the decrease in frequency of the C4=O is attributed to stronger H-bonding with locally ordered water molecules. Examination of the MD simulations and canonical structures also indicates that H-bonding geometries of the A and B forms are not substantively different.
In summary, the UVRR results make it possible to monitor the ribose conformations, as well as the base stacking of dG, dA, and dT. An examination of the series of equilibrium states of the system as the relative hydration is increased reveals that the sugar moieties change to C2′-endo followed by A- to B-type changes in base-stacking interactions. There is evidence of stronger H-bonding to solvent in the major groove of A-form DNA than in the B-form. The UVRR results thus support a cooperative but sequential mechanism for the B-to-A transition, as opposed to an all-or-none, simple two-state mechanism.
MD simulations
Since earlier reported MD involved relatively short trajectories, MD simulations on B and A forms of d(CGCGAATTCGCG) were extended to 60 ns and 20 ns, respectively. The overall behavior of the extended simulations was essentially similar to that reported (Fig. 1) (4), and provides a more rigorous basis for detailed analysis of the solvation. The MD simulation most relevant to the mechanism of the B-to-A transition is a simulation on the d(CGCGAATTCGCG) duplex in aqueous solution, beginning with the sequence in the canonical A form. A comparison of the canonical A- and B-form structures with the ensemble of structures generated by the MD simulations is given in Fig. 1. During the initial phase of the MD, there is a rapid interconversion of the sequence from A- to B-form, and a detailed structural analysis provides the MD-predicted mechanism. In going from A to B, the sugar puckers and the helical parameter INC transition to B-form values within 500–750 ps. The basepair displacement XDP converts from A to B values in the 750–1250 ps time range. The A-to-B interconversion is complete by 1500 ps of MD, and the resulting B-form structure is quite similar to that found in the simulation of the sequence in aqueous solution that began with the canonical B form. This finding agrees with experiment, i.e., the B form is the preferred conformation in aqueous solution.
To be more quantitative, we have monitored the relaxation times of these properties in the course of the A-to-B transition observed during the simulation starting with the A-form structure in water. The relaxation times are calculated on the basis of exponential fit of the decay in the autocorrelation function of these parameters (Table 2). Neglecting the outlier observed in sugar pucker relaxation time of the G10 nucleotide, we observe that, on average, the sugar transition from C3′-endo to C2′-endo conformations relax faster than XDP and INC. With respect to sequence effects, the AATT tract converts to B form first, followed at a later point by the GC tracts. These observations are consistent with those obtained from UVRR spectroscopy, described above, which indicate that the mechanism of the A-to-B transition is sequential, with sugars converting from A form to B form, followed later by the changes in base stacking. The structure passes through an A/B hybrid form, with the AT tract converting at lower relative hydration than the GC tracts. The MD results point to populations of mixed ribose conformation for A DNA in ethanol and B DNA in water but also indicate that small fractions of other ribose conformations, not resolved in the UVRR spectra, may be present. Interestingly, MD simulations predict that even A DNA in ethanol/water contains ∼20% of the C2′-endo configuration and that the ribose conformation is more heterogeneous in this conformation than in B DNA (Fig. 5 b). In summary, the MD and UVRR results agree well on the issue of a sequential mechanism, i.e., one in which changes happen in sequence as opposed to “all or none.” Since water activity is the agent of change, we now look to the simulation results to obtain an understanding of the manner by which solvation effects the transition.
Our analysis of solvation examines the general nature of the A and B forms of d(CGCGAATTCGCG) based on calculation of solvent accessibility, and is followed by a detailed analysis of the MD results using the proximity method. Calculations of the solvent-accessible surface area of the four nucleotides in the A- and B-form structures, computed from the MD trajectories, are shown in Fig. 6. The solvent accessibility analysis is broken down in terms of the constituent atoms in the sugar, phosphate, and major and minor groove regions. Although the grooves together contribute ∼20% of the net solvent-accessible surface area, the related change on going from the A- to the B-form structures is large, and comparable to that found for the phosphate and sugar groups (Fig. 6 c). There is a net reduction in the solvent-exposed surface area for both the AT and GC basepairs in the A-form structures, compared to the B-form, consistent with the idea of reduced hydration in A DNA. Moreover, the change in solvent-accessible surface area for the two forms is distinctly different for the AT and GC basepairs. Specifically, the solvent accessibility of AT basepairs in the major groove in the A-form structure is greatly reduced. This finding is indicative of a higher level of water rearrangement in the immediate proximity of the AT basepairs, compared to the GC basepairs, during the B-to-A transition. This finding is in good qualitative agreement with experimental results, which indicates that AT-rich regions convert from the A to B form at a lower level of relative hydration. Thus, the differences in accessible surface area for GC and AT basepairs can be correlated with their relative differences in energy in the transition, and these differences are reflected in the experimental data.
FIGURE 6.

(a and b) Percentage composition of the solvent-accessible surface areas of the G, A, C, and T nucleotides in the MD simulation of the central 10 basepairs of the CGCGAATTCGCG sequence in A-form DNA (a) and B-form DNA (b). (c) Difference in the average solvent-accessible surface area (in Å2) between the A and B conformations for the four nucleotides and the AT and GC basepairs.
In the B-form structures, phosphate groups are the most solvent-accessible components of the nucleotides, contributing 49.5% of the total, followed by sugars at 30.5%. The corresponding contribution of the phosphates and sugars in the A-form structure is 47.25 and 34.25%, respectively. For all the nucleotides, the solvent-accessible surface area of the sugar and minor groove regions is larger in the A form, but is consistently lower for the phosphate group and the major groove. The hydrophobic sugar moieties are more solvent-exposed in A-form structures, whereas the charged phosphate groups experience decreased solvent exposure since the interphosphate distance is only sufficient to accommodate one water molecule. The increased accessibility of the surface to the solvent is a result of the shallow nature of the minor groove region in the A-form structure, whereas the concomitant decrease in major groove accessibility is a result of the deeper, narrower groove dimension (81).
The details of the MD-calculated water structure around the A and B forms of DNA have been examined based on proximity analysis of water molecules in MD trajectories of A-form DNA in an ethanol/water mixture and of B-form DNA in water (82). The analysis is presented in Table 3 in terms of the four structural moieties, i.e., sugar, phosphate, and major and minor grooves, for each of the central eight basepairs in the dodecamer DNA. In particular, we report the volume of the first solvation shell, the corresponding coordination numbers, and binding energies for the stable A- and B-form structures. Since the ethanol/water mixture contained 528 water molecules, the number of water molecules closest to that in the B-DNA simulation in aqueous solution was included in this proximity analysis. The ions are assumed to be part of the solute in the binding-energy calculations. The first shell volume of the phosphate and major-groove functional group proximity region is much smaller in the A-form structure, whereas the sugar moiety has higher accessibility in the A-form structure. This high accessibility of the sugar moiety is likely to be an important factor in destabilizing this structure in aqueous medium, in which the A-form spontaneously transitions to a B-form structure due at least in part to hydrophobic pressure. The coordination numbers indicate that the number density of water molecules in the proximity region of the functional groups is much smaller in the A-DNA structures. On average, 16 water molecules are coordinated in the first two shells of the A-form structure, compared to an average of 34 water molecules observed in the B-form structure. The reported first-shell solvation energy is the interaction energy between the solute functional groups and the water molecules in their corresponding proximity region, averaged over the 5-ns MD trajectory. It is interesting to note that although the average coordination number is much lower, the average interaction energy of the water molecules in the major groove of the A-form DNA structure, especially in the central region, is more favorable compared to the B-DNA trajectory structures. Localization of sodium ions also occurs within the major groove of A DNA. The localization of water is in good accord with Raman spectroscopic results, which suggests that the water molecules are more tightly bound in the major groove of A-form DNA than in the B-form structure. The water molecules in the first shell of the B-DNA structure are bound much more strongly in the minor groove compared to the major groove, which can be attributed to the presence of the conserved spine of hydration. In the B-DNA structure, the proximity analysis reveals that, on average, the GC basepairs bind to water molecules more strongly than the AT basepairs, in accord with experimental volumetric results (83).
TABLE 3.
Proximity analysis of A- and B-form DNA trajectories
| Volume of first solvation shell*
|
Coordination number 〈k〉†
|
|||||
|---|---|---|---|---|---|---|
| Basepair position | Basepair | B DNA | A DNA | B DNA | A DNA | |
| 3 | CG | |||||
| Sugar | 315.3 | 335.9 | 1.6 | 1.2 | ||
| Phosphate | 321.8 | 301.3 | 9.5 | 4.7 | ||
| Major groove | 282.0 | 256.4 | 2.7 | 1.6 | ||
| Minor groove | 234.3 | 243.5 | 4.3 | 1.9 | ||
| 4 | GC | |||||
| Sugar | 327.0 | 342.9 | 2.1 | 0.3 | ||
| Phosphate | 319.4 | 290.5 | 9.2 | 4.5 | ||
| Major groove | 275.0 | 242.8 | 2.7 | 1.4 | ||
| Minor groove | 229.6 | 276.0 | 3.7 | 1.1 | ||
| 5 | AT | |||||
| Sugar | 307.7 | 345.2 | 1.6 | 0.2 | ||
| Phosphate | 308.7 | 276.1 | 9.0 | 3.4 | ||
| Major groove | 325.5 | 295.1 | 3.8 | 2.4 | ||
| Minor groove | 229.9 | 245.4 | 3.1 | 0.6 | ||
| 6 | AT | |||||
| Sugar | 302.7 | 359.1 | 1.5 | 0.2 | ||
| Phosphate | 322.4 | 297.5 | 9.9 | 3.7 | ||
| Major groove | 321.5 | 269.3 | 4.2 | 2.3 | ||
| Minor groove | 225.8 | 258.4 | 3.1 | 1.3 | ||
| 7 | TA | |||||
| Sugar | 299.0 | 340.4 | 1.5 | 0.4 | ||
| Phosphate | 326.1 | 287.6 | 9.4 | 4.2 | ||
| Major groove | 327.4 | 274.7 | 4.7 | 2.1 | ||
| Minor groove | 231.0 | 234.4 | 3.2 | 1.2 | ||
| 8 | TA | |||||
| Sugar | 311.8 | 325.0 | 1.6 | 1.0 | ||
| Phosphate | 320.4 | 269.4 | 8.8 | 3.2 | ||
| Major groove | 335.5 | 268.8 | 3.6 | 1.6 | ||
| Minor groove | 236.6 | 255.6 | 3.0 | 1.5 | ||
| 9 | CG | |||||
| Sugar | 321.3 | 334.3 | 2.5 | 0.7 | ||
| Phosphate | 328.2 | 299.3 | 9.6 | 3.5 | ||
| Major groove | 266.3 | 225.6 | 2.4 | 0.7 | ||
| Minor groove | 242.6 | 264.1 | 4.4 | 1.5 | ||
| 10 | GC | |||||
| Sugar | 308.5 | 328.2 | 1.6 | 0.6 | ||
| Phosphate | 331.0 | 311.0 | 10.1 | 2.9 | ||
| Major groove | 292.2 | 231.0 | 3.0 | 0.5 | ||
| Minor groove | 235.4 | 254.9 | 4.5 | 0.8 | ||
Volume of primary solvent shell (Å3) derived by the proximity method.
Number of water molecules in the primary proximity region.
DISCUSSION
The CD studies have established that A/B interconversion does occur in each of the sequences studied. The average transition midpoint is consistent with the general idea of preferential stability of the B form at high water activity and of the A-form at low water activity, as well as of A/B philicity as a function of sequence. The UVRR results make it possible to monitor the ribose conformations, as well as the base-stacking of dG, dA, and dT. Examination of a series of equilibrium states as a function of increasing hydration shows that the sugar moieties change to C2′-endo, followed by A- to B-type changes in base-stacking interactions. Intermediate forms involve A/B hybrid structures, with AT-rich tracts assuming the B form, whereas the CG tracts are still A-like. In the experimental case, the reduced water activity in the solution was achieved through addition of TFE, whereas in the computations, the cosolvent used was ethanol. Since ethanol can precipitate DNA, TFE was used in solution experiments. Although hydrogen-bonding properties of the two solvents are different, the changes observed are mainly attributed to a reduction in water activity. This conclusion is supported by our analyses, which indicates that the MD and UVRR results agree well on the issue of a sequential mechanism for the A-to-B transition in DNA and the detailed nature of it at the molecular level. The UVRR and MD results thus collectively support the idea of a cooperative but sequential mechanism for the B-to-A transition, as opposed to an all-or-none, simple two-state mechanism.
In comparing experimentally observed and theoretically calculated results, it is important to note that the experimental results report the A/B ratios for a series of equilibrium mixtures, whereas the MD simulation models a kinetic pathway between the calculated solution structures of the A and B forms. The features of the A-to-B transition indicated by the UVRR spectroscopy and the MD simulations are consistent with the diverse experimental results reviewed in Background, which pertain to the general aspects of conformational stability. At a more detailed level, we have compared the UVRR/MD sequential model of the transition to the mechanism proposed on the basis of x-ray diffraction of various derivatized intermediates (see Fig 9 for our MD-calculated results). A comparison of the equilibrium model from experiment and the kinetic model from MD in terms of the order of events agrees well with the idea of a sequential mechanism. Comparison with the crystal structure model indicates that the results obtained on the crystalline solid carry over to the solution state.
FIGURE 9.

Molecular dynamics snapshots of the A-to-B transition. As indicated, the sugar puckers and the helical parameter INC transition to B-form values within 500–750 ps. The basepair XDP converts from A to B values in the 750–1250 ps time range. The A-to-B interconversion is complete by 1500 ps of MD. NP and SP correspond to C3′-endo and C2′-endo ribosyl conformations, respectively.
A new perspective on the A-to-B transition was indicated by the observations that H-bonding to the dT C4=O group is stronger in the A form and the transition midpoint occurs at a high molar hydration (87.5% MH) (Fig. 4; Table 2). The increased H-bonding strength was attributed to greater solvent interaction and MD simulations point to increased localization of water and ions in the major groove in the A form. These results and the accessible surface area analyses support the idea that reorganization of the solvent is a major determinant in the transition.
We turn now to a methodological issue relevant to MD simulations on DNA. Many groups have independently shown that MD simulations in water using the popular AMBER parm94 force field equilibrate in a B-form-like structure regardless of whether the simulation starts in an A- or B-form structure (4,65). In an ethanol/water solvent mixture of 30% relative MH, a simulation started from the canonical A-form structure remains A-like (4,65). According to conventional wisdom, a B-form structure in such a low-water-activity state should transition to an A-like structure, but in this case it was originally reported, and confirmed in this study, that the MD structure remains B-like in this case. This discrepancy has raised questions about the quality of the force field, the length of the simulation, and the sequence dependence of the transition (4,65). To pursue this issue, we have extended our MD solvent analysis using a numerical procedure to determine the average solvent positions, taking into consideration the diffusional interchanges that would occur as a result of solvent motion. These so-called generic sites (84) represent the locations of the peaks in the 3D density distribution of the solvent. Solvation sites computed from the trajectories of A- and B-DNA structures in the 30% MH mixture are shown in Fig. 7. The stable A-form structure in the ethanol/water mixture shows a strong preference to localize water and ions in the major groove, in accord with our Raman spectroscopic results and the solute-solvent interaction energy calculated using the proximity analysis. However, the solvent analysis for the corresponding B-form structure in the ethanol/water mixture indicates that the solvent distribution in the groove region has not achieved localization of water and ions in the major groove equivalent to that observed for the A-form structure.
FIGURE 7.

Representation of generic solvent site analysis (84) of the MD trajectories of A- and B-form d(CGCGAATTCGCG)2 DNA in the ethanol/water mixture with the water locations color-coded by fractional occupancy. A gradient coloring scheme is employed for the generic water positions, with the sites of high occupancy (>0.9) shaded blue, those of low occupancy (∼0.3) shaded red, and those of intermediate occupancy shaded green. The C, G, T, and A nucleotides are colored yellow, green, blue, and red, respectively. Sodium ions are colored magenta. The figures have been generated using the program PyMOL (DeLano Scientific, Palo Alto, CA).
To assess the importance of water structure in the major groove of the DNA in facilitating the transition from B to A structures, we constructed a simulation “experiment” in the ethanol/water mixture that began with a small cluster of 30 water molecules restrained to the vicinity of major-groove surface atoms of the B-form starting structure. The results of this “computer experiment” are shown in Fig. 8. The XDP of the 10 central nucleotide pairs in the dodecamer sequence moves rapidly toward more negative values, which is indicative of a transition toward A-like structures. Reversible transitions of the sugar pseudorotation phase angle from the C2′-endo toward the C3′-endo conformations is also observed, indicating that the selective hydration of the major groove does lead to an A-like sugar puckering. (Fig. S3 in Data S1). We also observe that in this simulation of the CGCGAATTCGCG sequence, the sugars attached to C and G bases are more likely to be in the C3′-endo state than are those attached to the central A and T bases.
FIGURE 8.

x-displacement of basepairs 2–11 in the dodecamer sequence CGCGAATTCGCG. The x-displacement of basepairs in B DNA simulated in ethanol/water mixture is shown in green. The data in red represent the x-displacement of B DNA in ethanol/water mixture simulated with a cluster of water molecules restrained in the major groove. The blue and pink horizontal lines at 0 and −4 Å correspond to the values of x-displacement in canonical B- and A-form DNA structures.
This result suggests that the problem with converting B to A at reduced water activity, observed in MD simulations, may be a matter of sampling rather than, as previously suspected, a force field problem. The idea is that the B-to-A transition evidently proceeds if the solvent is structured properly in the major groove, but this is a highly improbable event on the nanosecond timescale, and thus the transition is not observed in the MD simulations. On the timescale of the simulation, this is a quasiergodic problem. In time frames from microseconds to seconds, even improbable events at the nanosecond level can occur and nucleate the conformational change, analogous to what happens in the observation of particles undergoing Brownian motion. Although this is only a leading idea, and not unequivocally demonstrated by the analysis, our results do indicate that quasiergodic problems deserve serious consideration in assessing future MD studies of the B-to-A transition at reduced water activity.
The time actually required for the reorganization of the solvent remains an open question. The measurements of Jose and Porschke (29) suggest that the timescale of the A-to-B transition is 10 μs and that a significant transition barrier exists. The results presented here indicate that the barrier for the transition lies in the organization of the solvent. Mazur had concluded that occupation of ions in the major groove was required for the transition (11); however, the results of this study suggest that reorganization of water is equally important in this transition. The ultrafast dynamic measurements of Zewail and co-workers (85) demonstrate that solvent reorganization can occur on a timescale of 20 ps. However, these measurements were confined to a relatively localized area, and a complete reorganization of solvent in the major groove may require a longer timescale. Further studies are needed to appropriately address this issue.
SUMMARY AND CONCLUSIONS
A combination of methodologies has been utilized to examine the A-to-B transition. The comparison of UVRR results with information gleaned from MD simulations has proven illuminating with respect to the nature and mechanism of the DNA A-to-B transition. Of significance are the correlations between UVRR, MD, and crystallography results, which all point to a sequential mechanism for the A-to-B transition (Fig. 9) (8). In addition, UVRR and MD results also reveal that sugar conformation may not be the most reliable measure of A-to-B DNA conformation, as suggested in earlier NMR studies (6). Finally, these measurements highlight the significance of water and ion positions in facilitating the A-to-B transition.
SUPPLEMENTARY MATERIAL
To view all of the supplemental files associated with this article, visit www.biophysj.org.
Acknowledgments
This work was supported by National Science Foundation grant MCB 0316625 (I.M.) and an investigator grant from the Patrick and Catherine Weldon Donaghue Medical Research Foundation (I.M.). D.L.B. acknowledges support from National Institute of General Medical Sciences 7909. National Institutes of Health Molecular Biophysics training grant GM 008272 and the Howard Hughes Medical Institute supported K.M.K. and C.E.A., respectively.
Kelly M. Knee and Surjit B. Dixit contributed equally to this work.
Editor: Jonathan B. Chaires.
References
- 1.Olson, W. K., A. A. Gorin, X. J. Lu, L. M. Hock, and V. B. Zhurkin. 1998. DNA sequence-dependent deformability deduced from protein-DNA crystal complexes. Proc. Natl. Acad. Sci. USA. 95:11163–11168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cheatham 3rd, T. E., and P. A. Kollman. 1996. Observation of the A-DNA to B-DNA transition during unrestrained molecular dynamics in aqueous solution. J. Mol. Biol. 259:434–444. [DOI] [PubMed] [Google Scholar]
- 3.Cheatham 3rd, T. E., and P. A. Kollman. 1997. Insight into the stabilization of A-DNA by specific ion association: spontaneous B-DNA to A-DNA transitions observed in molecular dynamics simulations of d(ACCCGCGGGT)2 in the presence of hexaamminecobalt(III). Structure. 5:1297–1311. [DOI] [PubMed] [Google Scholar]
- 4.Sprous, D., M. A. Young, and D. L. Beveridge. 1998. Molecular dynamics studies of the conformational preferences of a DNA double helix in water and an ethanol/water mixture: theoretical considerations of the A to B transition. J. Phys. Chem. B. 102:4658–4667. [Google Scholar]
- 5.Peticolas, W. L., and E. Evertsz. 1992. Conformation of DNA in vitro and in vivo from laser Raman scattering. Methods Enzymol. 211:335–353. [DOI] [PubMed] [Google Scholar]
- 6.Trantirek, L., R. Stefl, M. Vorlickova, J. Koca, V. Sklenar, and J. Kypr. 2000. An A-type double helix of DNA having B-type puckering of the deoxyribose rings. J. Mol. Biol. 297:907–922. [DOI] [PubMed] [Google Scholar]
- 7.Vermulen, A., H. Zhou, and A. Pardi. 2000. Determining DNA global structure and DNA bending by application of NMR residual dipolar couplings. J. Am. Chem. Soc. 122:9638–9647. [Google Scholar]
- 8.Vargason, J. M., K. Henderson, and P. S. Ho. 2001. A crystallographic map of the transition from B-DNA to A-DNA. Proc. Natl. Acad. Sci. USA. 98:7265–7270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cheatham 3rd, T. E., M. F. Crowley, T. Fox, and P. A. Kollman. 1997. A molecular level picture of the stabilization of A-DNA in mixed ethanol-water solutions. Proc. Natl. Acad. Sci. USA. 94:9626–9630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Feig, M., and B. M. Pettitt. 1998. A molecular simulation picture of DNA hydration around A- and B-DNA. Biopolymers. 48:199–209. [DOI] [PubMed] [Google Scholar]
- 11.Mazur, A. K. 2003. Titration in silico of reversible B ↔ A transitions in DNA. J. Am. Chem. Soc. 125:7849–7859. [DOI] [PubMed] [Google Scholar]
- 12.Pastor, N. 2005. The B- to A-DNA transition and the reorganization of solvent at the DNA surface. Biophys. J. 88:3262–3275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lavery, R., and H. Sklenar. 1988. The definition of generalized helicoidal parameters and of axis curvature for irregular nucleic acids. J. Biomol. Struct. Dyn. 6:63–91. [DOI] [PubMed] [Google Scholar]
- 14.Franklin, R. E., and R. G. Gosling. 1953. Molecular configuration in sodium thymonucleate. Nature. 171:740–741. [DOI] [PubMed] [Google Scholar]
- 15.Leslie A. G., S. Arnott, R. Chandrasekaran, and R. L. Ratliff. 1980. Polymorphism of DNA double helices. J. Mol. Biol. 143:49–72. [DOI] [PubMed] [Google Scholar]
- 16.Ivanov, V. I., L. E. Minchenkova, A. K. Schyolkina, and A. I. Poletayev. 1973. Different conformations of double stranded nucleic acid in solution as revealed by circular dichroism. Biopolymers. 12:98–110. [DOI] [PubMed] [Google Scholar]
- 17.Malenkov, G., L. Minchenkova, E. E. Minyat, A. Schyolkina, and V. I. Ivanov. 1975. The nature of B-A transition of DNA in solution. FEBS Lett. 51:38–42. [DOI] [PubMed] [Google Scholar]
- 18.Alden, C. J., and S. H. Kim. 1979. Solvent accessible surfaces of nucleic acids. J. Mol. Biol. 132:411–434. [DOI] [PubMed] [Google Scholar]
- 19.Hunter, C. A. 1993. Sequence dependent DNA structure: the role of base-stacking interactions. J. Mol. Biol. 230:1025–1054. [DOI] [PubMed] [Google Scholar]
- 20.Saenger, W., W. N. Hunter, and O. Kennard. 1986. DNA conformation is determined by economics in the hydration of phosphate groups. Nature. 324:385–388. [DOI] [PubMed] [Google Scholar]
- 21.Kopka, M. L., C. Yoon, D. Goodsell, P. Pjura, and R. E. Dickerson. 1985. The molecular origin of DNA-drug specificity in netropsin and distamycin. Proc. Natl. Acad. Sci. USA. 82:1376–1380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pjura, P. E., K. Grzeskowiak, and R. E. Dickerson. 1987. Binding of Hoechst 33258 to the minor groove of B-DNA. J. Mol. Biol. 197:257–271. [DOI] [PubMed] [Google Scholar]
- 23.Hanlon, S., S. Brudno, T. T. Wu, and B. Wolf. 1975. Structural transitions of deoxyribonucleic acid in aqueous elecrolyte solutions. I. Reference spectra of conformational limits. Biochemistry. 14:1648–1660. [DOI] [PubMed] [Google Scholar]
- 24.Minyat, E. E., V. I. Ivanov, A. M. Kritzyn, L. E. Minchenkova, and A. K. Schyolkina. 1979. Spermine and spermidine-induced B to A transition of DNA in solution. J. Mol. Biol. 128:397–409. [DOI] [PubMed] [Google Scholar]
- 25.Xu, Q., R. K. Shoemaker, and W. H. Braunlin. 1993. Induction of B-A transitions of deoxyoligonucleotides by multivalent cations in dilute aqueous solution. Biophys. J. 65:1039–1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Robinson, H., and A. H. Wang. 1996. Neomycin, spermine and hexaamminecobalt (III) share common structural motifs in converting B- to A-DNA. Nucleic Acids Res. 24:676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jayaram, B., D. Sprous, M. A. Young, and D. L. Beveridge. 1998. Free energy analysis of the conformational preferences of A and B forms of DNA in solution. J. Am. Chem. Soc. 120:10629–10633. [Google Scholar]
- 28.Ng, H. L., M. L. Kopka, and R. E. Dickerson. 2000. The structure of a stable intermediate in the A ↔ B DNA helix transition. Proc. Natl. Acad. Sci. USA. 97:2035–2039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jose, D., and D. Porschke. 2004. Dynamics of the B-A transition of DNA double helices. Nucleic Acids Res. 32:2251–2258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Drew, H. R., and R. E. Dickerson. 1981. Structure of a B-DNA dodecamer. III. Geometry of hydration. J. Mol. Biol. 151:535–556. [DOI] [PubMed] [Google Scholar]
- 31.Minchenkova, L. E., A. Schyolkina, B. K. Chernov, and V. I. Ivanov. 1986. CC/GG contacts facilitate the B to A transition of DNA in solution. J. Biomol. Struct. 4:463–476. [DOI] [PubMed] [Google Scholar]
- 32.Braunlin, W. H., and Q. Xu. 1992. Hexaamminecobalt(III) binding environments on double-helical DNA. Biopolymers. 32:1703–1711. [DOI] [PubMed] [Google Scholar]
- 33.Basham, B., G. P. Schroth, and P. S. Ho. 1995. An A-DNA triplet code: thermodynamic rules for predicting A- and B-DNA. Proc. Natl. Acad. Sci. USA. 92:6464–6468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Peticolas, W. L., Y. Wang, and G. A. Thomas. 1988. Some rules for predicting the base-sequence dependence of DNA conformation. Proc. Natl. Acad. Sci. USA. 85:2579–2583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Arnott, S., and D. W. Hukins. 1972. Optimised parameters for A-DNA and B-DNA. Biochem. Biophys. Res. Commun. 47:1504–1509. [DOI] [PubMed] [Google Scholar]
- 36.Lindqvist, M., and A. Graslund. 2001. An FTIR and CD study of the structural effects of G-tract length and sequence context on DNA conformation in solution. J. Mol. Biol. 314:423–432. [DOI] [PubMed] [Google Scholar]
- 37.Pohle, W., and H. Fritzsche. 1980. A new conformation-specific infrared band of A-DNA in films. Nucleic Acids Res. 8:2527–2536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pichler, A., S. Rudisser, R. H. Winger, K. R. Liedl, A. Halibrucker, and E. Mayer. 2000. Nonoriented d(CGCGAATTCGCG)2 dodecamer persists in the B-form even at low water activity. J. Am. Chem. Soc. 122:716–717. [Google Scholar]
- 39.Thomas, G. J., Jr., and M. Tsuboi. 1993. Raman spectroscopy of nucleic acids and their complexes. Adv. Biophys. Chem. 3:1–70. [Google Scholar]
- 40.Grygon, C. A., and T. G. Spiro. 1990. UV resonance Raman spectroscopy of nucleic acid duplexes containing A-U and A-T base pairs. Biopolymers. 29:707–715. [Google Scholar]
- 41.Fodor, S. P. A., and T. G. Spiro. 1986. Ultraviolet resonance Raman spectroscopy of DNA with 200–266-nm laser excitation. J. Am. Chem. Soc. 108:3198–3205. [Google Scholar]
- 42.Mukerji, I., M. C. Shiber, J. R. Fresco, and T. G. Spiro. 1996. A UV resonance Raman study of hairpin dimer helices of d(A-G)10 at neutral pH containing intercalated dA residues and alternating dG tetrads. Nucleic Acids Res. 24:5013–5020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mukerji, I., L. Sokolov, and M.-R. Mihailescu. 1998. A UV resonance Raman investigation of poly (rI): evidence for cation dependent structural perturbations. Biopolymers. 46:475–487. [Google Scholar]
- 44.Sokolov, L., K. Wojtuszewski, E. Tsukroff, and I. Mukerji. 2000. Nucleic acid structure investigated by UV resonance Raman spectroscopy: protonation effects and A-tract structure. J. Biomol. Struct. Dyn. 11:327–334. [DOI] [PubMed] [Google Scholar]
- 45.Mukerji, I., and A. P. Williams. 2002. UV resonance Raman and circular dichroism studies of a DNA duplex containing an A(3)T(3) tract: evidence for a premelting transition and three-centered H-bonds. Biochemistry. 41:69–77. [DOI] [PubMed] [Google Scholar]
- 46.Serban, D., J. M. Benevides, and G. J. Thomas, Jr. 2003. HU protein employs similar mechanisms of minor-groove recognition in binding to different B-DNA sites: demonstration by Raman spectroscopy. Biochemistry. 42:7390–7399. [DOI] [PubMed] [Google Scholar]
- 47.Dostal, L., C.-Y. Chen, A. H.-J. Wang, and H. Welfle. 2004. Partial B-to-A DNA transition upon minor groove binding of protein Sac7d monitored by Raman spectroscopy. Biochemistry. 43:9600–9609. [DOI] [PubMed] [Google Scholar]
- 48.Wojtuszewski, K., and I. Mukerji. 2004. The HU-DNA binding interaction probed with UV resonance Raman spectroscopy: structural elements of specificity. Protein Sci. 13:2416–2428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Beveridge, D. L., S. B. Dixit, G. Barreiro, and K. M. Thayer. 2004. Molecular dynamics simulations of DNA curvature and flexibility: helix phasing and premelting. Biopolymers. 73:380–403. [DOI] [PubMed] [Google Scholar]
- 50.Cheatham 3rd, T. E. 2004. Simulation and modeling of nucleic acid structure, dynamics and interactions. Curr. Opin. Struct. Biol. 14:360–367. [DOI] [PubMed] [Google Scholar]
- 51.Mackerell, A. D., Jr. 2004. Empirical force fields for biological macromolecules: overview and issues. J. Comput. Chem. 25:1584–1604. [DOI] [PubMed] [Google Scholar]
- 52.Orozco, M., A. Perez, A. Noy, and F. J. Luque. 2003. Theoretical methods for the simulation of nucleic acids. Chem. Soc. Rev. 32:350–364. [DOI] [PubMed] [Google Scholar]
- 53.Giudice, E., and R. Lavery. 2002. Simulations of nucleic acids and their complexes. Acc. Chem. Res. 35:350–357. [DOI] [PubMed] [Google Scholar]
- 54.Norberg, J., and L. Nilsson. 2002. Molecular dynamics applied to nucleic acids. Acc. Chem. Res. 35:465–472. [DOI] [PubMed] [Google Scholar]
- 55.Arthanari, H., S. Basu, T. L. Kawano, and P. H. Bolton. 1998. Fluorescent dyes specific for quadruplex DNA. Nucleic Acids Res. 26:3724–3728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Dixit, S. B., F. Pitici, and D. L. Beveridge. 2004. Structure and axis curvature in two dA6 × dT6 DNA oligonucleotides: comparison of molecular dynamics simulations with results from crystallography and NMR spectroscopy. Biopolymers. 75:468–479. [DOI] [PubMed] [Google Scholar]
- 57.Tjandra, N., S.-i. Tate, A. Ono, M. Kainosho, and A. Bax. 2000. The NMR structure of a DNA dodecamer in an aqueous dilute liquid crystalline phase. J. Am. Chem. Soc. 122:6190–6200. [Google Scholar]
- 58.MacDonald, D., and P. Lu. 2002. Residual dipolar couplings in nucleic acid structure determination. Curr. Opin. Struct. Biol. 12:337–343. [DOI] [PubMed] [Google Scholar]
- 59.Barbic, A., D. P. Zimmer, and D. M. Crothers. 2003. Structural origins of adenine-tract bending. Proc. Natl. Acad. Sci. USA. 100:2369–2373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Young, M. A., and D. L. Beveridge. 1998. Molecular dynamics simulations of an oligonucleotide duplex with adenine tracts phased by a full helix turn. J. Mol. Biol. 281:675–687. [DOI] [PubMed] [Google Scholar]
- 61.Beveridge, D. L., G. Barreiro, K. S. Byun, D. A. Case, T. E. Cheatham III, S. B. Dixit, E. Giudice, F. Lankas, R. Lavery, J. H. Maddocks, R. Osman, E. Seibert, H. Sklenar, G. Stoll, K. M. Thayer, P. Varnai, and M. A. Young. 2004. Molecular dynamics simulations of the 136 unique tetranucleotide sequences of DNA oligonucleotides. I. Research design and results on d(CpG) steps. Biophys. J. 87:3799–3813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Dixit, S. B., D. L. Beveridge, D. A. Case, T. E. Cheatham 3rd, E. Giudice, F. Lankas, R. Lavery, J. H. Maddocks, R. Osman, H. Sklenar, K. M. Thayer, and P. Varnai. 2005. Molecular dynamics simulations of the 136 unique tetranucleotide sequences of DNA oligonucleotides. II: Sequence context effects on the dynamical structures of the 10 unique dinucleotide steps. Biophys. J. 89:3721–3740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Perez, A., I. Marchan, D. Svozil, J. Sponer, T. E. Cheatham III, C. A. Laughton, and M. Orozco. 2007. Refinement of the AMBER force field for nucleic acids: improving the description of α/γ conformers. Biophys. J. 92:3817–3829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Banavali, N. K., and B. Roux. 2005. Free energy landscape of A-DNA to B-DNA conversion in aqueous solution. J. Am. Chem. Soc. 127:6866–6876. [DOI] [PubMed] [Google Scholar]
- 65.Cheatham 3rd, T. E., P. Cieplak, and P. A. Kollman. 1999. A modified version of the Cornell et al. force field with improved sugar pucker phases and helical repeat. J. Biomol. Struct. Dyn. 16:845–862. [DOI] [PubMed] [Google Scholar]
- 66.Jorgensen, W. L. 1981. Transferable intermolecular potential functions for water, alcohols and ethers. Application to liquid water. J. Am. Chem. Soc. 103:335–340. [Google Scholar]
- 67.Jorgensen, W. L. 1986. Optimized intermolecular potential functions for liquid alcohols. J. Phys. Chem. 90:1276–1284. [Google Scholar]
- 68.Jorgensen, W. L. 1982. Revised TIPS for simulations of liquid water and aqueous solutions. J. Chem. Phys. 77:4156–4163. [Google Scholar]
- 69.Aqvist, J. 1990. Ion-water interaction potentials derived from free energy perturbation simulations. J. Phys. Chem. 94:8021–8024. [Google Scholar]
- 70.Cornell, W. D., P. Cieplak, C. I. Bayly, I. R. Gould, K. M. Merz, D. M. Ferguson, D. C. Spellmeyer, T. Fox, J. W. Caldwell, and P. A. Kollman. 1995. A second generation force field for the simulation of proteins, nucleid acids and organic molecules. J. Am. Chem. Soc. 117:5179–5197. [Google Scholar]
- 71.Darden, T. A., D. M. York, and L. G. Pedersen. 1993. Particle mesh Ewald: an N log (N) method for Ewald sums in large systems. J. Chem. Phys. 98:10089–10092. [Google Scholar]
- 72.York, D. M., W. Yang, H. Lee, T. Darden, and L. G. Pedersen. 1995. Toward the accurate modeling of DNA: the importance of long-range electrostatics. J. Am. Chem. Soc. 117:5001–5002. [Google Scholar]
- 73.Ryckaert, J.-P., G. Ciccotti, and H. J. Berendsen. 1977. Numerical integration of the Cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J. Comput. Phys. 23:327–341. [Google Scholar]
- 74.Ben-Naim, A. 1982. Water and Aqueous Solutions. Plenum Press, New York.
- 75.Mehrotra, P. K., and D. L. Beveridge. 1980. Structural analysis of molecular solutions based on quasi-component distribution functions. Application to [H2CO]aq at 25°C. J. Am. Chem. Soc. 102:4287–4294. [Google Scholar]
- 76.Mezei, M. 2006. MMC: Monte Carlo Program For Computer Simulation of Molecular Solutions. New York.
- 77.Tsodikov, O. V., M. T. Record, Jr., and Y. V. Sergeev. 2002. Novel computer program for fast exact calculation of accessible and molecular surface areas and average surface curvature. J. Comput. Chem. 23:600–609. [DOI] [PubMed] [Google Scholar]
- 78.Bloomfield, V. A., D. M. Crothers, and I. Tinoco. 2000. Nucleic Acids: Structures, Properties and Functions. University Science Books, Sausalito, CA.
- 79.Perno, J. R., C. A. Grygon, and T. G. Spiro. 1989. Ultraviolet Raman excitation profiles for the nucleotides and for the nucleic acid duplexes p(rA)-poly(rU) and poly(dG-dC). J. Phys. Chem. 93:5672–5678. [Google Scholar]
- 80.Badger, R. M., and S. H. Bauer. 1937. Spectroscopic studies of the hydrogen bond. II. The shift of the O-H vibrational frequency in the formation of the hydrogen bond. J. Chem. Phys. 5:839–851. [Google Scholar]
- 81.Mitra, S. N., M. C. Wahl, and M. Sundaralingam. 1999. Structure of the side-by-side binding of distamycin to d(GTATATAC). Acta Crystallogr. D Biol. Crystallogr. 55:602–609. [DOI] [PubMed] [Google Scholar]
- 82.Mezei, M., and D. L. Beveridge. 1986. Structural chemistry of biomolecular hydration: the proximity criterion. Methods Enzymol. 127:21–47. [DOI] [PubMed] [Google Scholar]
- 83.Chalikian, T. V., J. Volker, A. R. Srinivasan, W. K. Olson, and K. J. Breslauer. 1999. The hydration of nucleic acid duplexes as assessed by a combination of volumetric and structural techniques. Biopolymers. 50:459–471. [DOI] [PubMed] [Google Scholar]
- 84.Mezei, M., and D. L. Beveridge. 1984. Generic solvation sites in a crystal. J. Comput. Chem. 5:523–527. [Google Scholar]
- 85.Pal, S. K., L. Zhao, T. Xia, and A. H. Zewail. 2003. Site- and sequence-selective ultrafast hydration of DNA. Proc. Natl. Acad. Sci. USA. 100:13746–13751. [DOI] [PMC free article] [PubMed] [Google Scholar]