

Abstract
We have performed a kinetic and thermodynamic analysis of 3Dpol derivatives containing substitutions in the ribose-binding pocket with ATP analogues containing correct and incorrect sugar configurations. We find that Asp-238, a residue in structural motif A that is conserved in all RNA-dependent RNA polymerases, is a key determinant of polymerase fidelity. Alterations in the position of the Asp-238 side chain destabilize the catalytically competent 3Dpol-primer/template-NTP complex and reduce the efficiency of phosphoryl transfer. The reduction in phosphoryl transfer may be a reflection of increased mobility of other residues in motif A that are required for stabilizing the triphosphate moiety of the nucleotide substrate in the active conformation. We present a structural model to explain how Asp-238 functions to select nucleotides with a correct sugar configuration and a correct base. We propose that this mechanism is employed by all RNA-dependent RNA polymerases. We discuss the possibility that all nucleic acid polymerases with the canonical “palm”-based active site employ a similar mechanism to maximize fidelity.
In our studies of the kinetic mechanism for nucleotide incorporation catalyzed by the RNA-dependent RNA polymerase (RdRP)1 from poliovirus, 3Dpol, we showed that the catalytic cycle is comprised of at least five steps that can be evaluated experimentally (Scheme 1) (1, 2). Nucleotide substrate binds to the enzyme-primer/template complex (step 1), and this complex isomerizes into a catalytically active complex (step 2). Phosphoryl transfer occurs (step 3), followed by a second conformational change (step 4) and pyrophosphate release (step 5). When Mg2+ is employed as the divalent cation cofactor, both the first conformational change and phosphoryl-transfer steps are partially rate limiting for nucleotide incorporation (1, 2). Moreover, both of these steps are critical for the enzyme to distinguish between a correct ribonucleotide and a nucleotide with an incorrect base or sugar configuration (1, 2).
Scheme 1.

Complete Kinetic Mechanism for 3Dpol-Catalyzed Nucleotide Incorporation
We have proposed that the partially rate limiting conformational change reflects reorientation of the triphosphate moiety of the incoming nucleotide from its ground-state conformation into an orientation appropriate for phosphoryl transfer (1, 2). Moreover, we have suggested that the active conformation of the triphosphate requires interactions with residues located in the ribose-binding pocket (1-3). Therefore, changes in these interactions caused by binding of a nucleotide with an incorrect base or sugar configuration should decrease the stability of the actively oriented triphosphate (step 2) and preclude optimal orientation of the triphosphate, causing a decrease in the rate constant for phosphoryl transfer (step 3).
Interrogation of the 3Dpol nucleotide-binding pocket revealed at least six residues that are in the vicinity of the nucleotide substrate: Asp-233, Asp-238, Ser-288, Thr-293, Asn-297, and Asp-328 (3). The location of each residue relative to the others, as observed in the unliganded crystal structure of 3Dpol (3, 4), is shown in Figure 1A. We have used the crystal structure of human immunodeficiency virus reverse transcriptase in complex with primer/template and nucleotide (5) to develop a model for the corresponding 3Dpol complex (Figure 1B) (3). To accommodate the nucleotide substrate, Asp-238 and Asn-297 required repositioning. The new arrangement permitted the Asp-238 side chain to hydrogen bond to the Ser-288 and Thr-293 side chains and the Asn-297 side chain to hydrogen bond to the 2′-OH of the nucleotide substrate (Figure 1B) (3).
FIGURE 1.
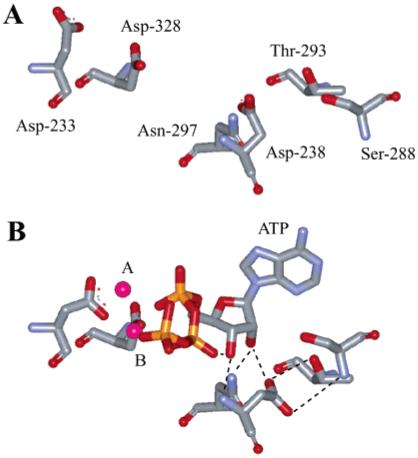
Nucleotide-binding pocket of 3Dpol. (A) Residues located in the NTP-binding pocket as observed in the unliganded structure of 3Dpol (4). Asp-233 and Asp-238 are from structural motif A, Ser-288, Thr-293, and Asn-297 are from motif B, and Asp-328 is from motif C. (B) Model for interaction of 3Dpol with bound nucleotide (3). ATP and metal ions required for catalysis are labeled. In this model, the side chains for Asp-233 and Asp-238 have been rotated to permit interactions with ATP. Asp-238, Ser-288, and Thr-293 have been positioned to interact. The image was created by using the program WebLab Viewer (Molecular Simulations Inc., San Diego, CA).
3Dpol derivatives that contain alanine at position 238 or 297 were incapable of distinguishing between ATP and 2′-dATP (3). While the loss of the Asn-297 side chain caused a 20-fold reduction in the observed rate constant for correct nucleotide incorporation, the loss of the Asp-238 side chain caused a 2000-fold reduction (3). Interestingly, by using Mn2+ as the divalent cation cofactor, the activity of both 3Dpol derivatives could be brought to within 3-14-fold of the wild-type enzyme, respectively. The kinetic behavior of the N297A and D238A derivatives with ATP (3) was very similar to the behavior of wild-type 3Dpol with 2′-dATP and GTP, respectively (1, 2). This relationship even held true for the effects of Mn2+ on the activity of 3Dpol derivatives (3). Together, these data support a role for residues in the ribose-binding pocket in both nucleotide selection and catalytic efficiency that can be modulated by the divalent cation cofactor employed.
The objective of this study was to elucidate the mechanistic basis for the phenotypes observed for the D238A and N297A derivatives of 3Dpol with the goal of establishing a link between these residues and the capacity of this enzyme to catalyze ribonucleotide incorporation with high fidelity. We have accomplished this goal and present a structural model for 3Dpol fidelity that likely extends to all animal virus RdRPs. In addition, we discuss the implications of this model on our understanding of fidelity of nucleotide incorporation for other classes of nucleic acid polymerases.
EXPERIMENTAL PROCEDURES
Materials
[γ-32P]ATP (>7000 Ci/mmol) was from ICN; nucleoside 5′-triphosphates and 2′,3′-dideoxyadenosine 5′-triphosphate (ultrapure solutions) were from Amersham Pharmacia Biotech, Inc.; 3′-deoxyadenosine 5′-triphosphate (cordycepin) was from Sigma; all DNA oligonucleotides and T4 DNA ligase were from Life Technologies, Inc.; RNA oligonucleotides were from Dharmacon Research, Inc. (Boulder, CO); T4 polynucleotide kinase was from New England Biolabs, Inc., and all other reagents were of the highest grade available from Sigma, Fisher, or VWR.
Construction of the D238A/N297A Expression Vector
The D238A/N297A expression vector was constructed by introducing the N297A-encoding mutation into the D238A 3Dpol gene by using PCR and subcloning into the pET26-Ub-3D expression plasmid as described previously (3). The authenticity of the construct was verified by sequencing.
Expression and Purification of Wild-Type 3Dpol and 3Dpol Derivatives
Wild-type 3Dpol and 3Dpol derivatives were expressed and purified as described previously (3, 6).
Purification, 5′-32P Labeling, and Annealing of sym/sub
RNA oligonucleotides were purified, labeled, and annealed as described previously (7).
3Dpol Assays
Reactions contained 50 mM HEPES, pH 7.5, 10 mM 2-mercaptoethanol, 5 mM MgCl2 or MnCl2, 60 μM ZnCl2, nucleotide, sym/sub, and 3Dpol. Reactions were performed at 30 °C. 3Dpol was diluted immediately prior to use in 50 mM HEPES, pH 7.5, 10 mM 2-mercaptoethanol, 60 μM ZnCl2, and 20% glycerol. Zn2+ was added to increase the stability of the enzyme; however, the level of Zn2+ employed is insufficient to support nucleotide incorporation. The volume of enzyme added to any reaction was always less than or equal to one-tenth the total volume. Reactions were quenched by addition of EDTA to a final concentration of either 50 mM or 0.3 M or by addition of HCl to a final concentration of 1 M. Immediately after the addition of HCl, the solution was neutralized by addition of 1 M KOH and 300 mM Tris (final concentration). Specific concentrations of primer/template and 3Dpol, along with any deviations from the above, are indicated in the appropriate figure legend.
Rapid Chemical-Quench-Flow Experiments
Rapid mixing/quenching experiments were performed by using a Model RQF-3 chemical-quench-flow apparatus (KinTek Corp., Austin, TX). Experiments were performed at either 20 or 30 °C by using a circulating water bath. 3Dpol-sym/sub complexes were assembled by mixing 3Dpol and sym/sub for 3 min at room temperature and then rapidly mixed with the nucleotide substrate. After mixing, reactant concentrations were reduced by 50%. Reactions were quenched either by addition of EDTA to a final concentration of 0.3 M or by addition of HCl to a final concentration of 1 M. Immediately after the addition of HCl, the solution was neutralized by addition of 1 M KOH and 300 mM Tris (final concentration).
Product Analysis: Denaturing PAGE
An equal volume of loading buffer (90% formamide, 0.025% bromophenol blue, and 0.025% xylene cyanol) was added to 10 μL of the quenched reaction mixtures and heated to 70 °C for 2-5 min prior to loading 5 μL on a denaturing 23% polyacrylamide gel containing 1× TBE and 7 M urea. Electrophoresis was performed in 1× TBE at 90 W. Gels were visualized by using a phosphorimager and quantified by using the Imagequant software (Molecular Dynamics).
Data Analysis
Data were fit by nonlinear regression using the program KaleidaGraph (Synergy Software, Reading, PA). Time courses at fixed nucleotide concentration were fit to
| (1) |
where A is the amplitude of the burst, kobs is the observed first-order rate constant describing the burst, t is the time, and C is a constant. The apparent binding constant (Kd,app) and maximal rate constant for nucleotide incorporation (kpol) were determined using the equation:
| (2) |
The value for the equilibrium constant across the conformational-change step was determined using the equations (9):
| (3) |
| (4) |
| (5) |
kchem is the rate constant for the chemical step, kpol is the maximal rate constant for nucleotide incorporation, σ is the maximal elemental effect, Eobs is the observed elemental effect, and k+2 and k-2 are the forward and reverse rate constants for the conformational-change step, respectively.
Calculation of ΔΔG
Experimental ΔΔG values for 3Dpol derivatives containing single and double substitutions were determined by using kpol/Kd values (Tables 1 and 2) and eq 6 as described by Fersht (10, 11) and Mildvan et al. (12). Predicted ΔΔG values were calculated as described by eq 7.
Table 1.
Kinetic and Thermodynamic Constants for 3Dpol-Catalyzed Nucleotide Incorporation at 20 °C in the Presence of Mg2+ a
| Mg2+ |
|||
|---|---|---|---|
| kpol (s-1) | Kd,app (μM) | kpol/Kd,app (μM-1 s-1) | |
| ATP | |||
| WT | 17 ± 1 | 98 ± 10 | 0.18 ± 0.02 |
| D238A | (8.0 ± 1.0) × 10-3 | 28 ± 2 | (3.0 ± 0.5) × 10-4 (600)b |
| N297A | 0.60 ± 0.03 | 175 ± 20 | (3.0 ± 0.1) × 10-3 (60) |
| D238A/N297A | (6.0 ± 1.0) × 10-3 | 46 ± 6 | (1.0 ± 0.1) × 10-4 (1800) |
| 2′-dATP | |||
| WT | 0.051 ± 0.001 | 83 ± 8 | (1.0 ± 0.1) × 10-3 |
| D238A | (9.0 ± 1.0) × 10-3 | 27 ± 2 | (3.0 ± 0.5) × 10-4 (3) |
| N297A | 0.033 ± 0.001 | 134 ± 13 | (3.0 ± 0.2) × 10-4 (3) |
| D238A/N297A | (7.0 ± 1.0) × 10-3 | 18 ± 3 | (3.0 ± 0.5) × 10-4 (3) |
| 3′-dATP | |||
| WT | 0.118 ± 0.004 | 160 ± 14 | (1.0 ± 0.1) × 10-3 |
| D238A | NDc | ND | ND |
| N297A | ND | ND | ND |
| D238A/N297A | ND | ND | ND |
Experiments were performed as described under Experimental Procedures.
Numbers in parentheses indicate the fold difference from the WT value.
ND = kpol and Kd values could not be determined due to the substantial decrease in the rate constant for nucleotide incorporation under the given reaction conditions.
Table 2.
Kinetic and Thermodynamic Constants for 3Dpol-Catalyzed Nucleotide Incorporation at 20 °C in the Presence of Mn2+ a
| Mn2+ |
|||
|---|---|---|---|
| kpol (s-1) | Kd,app (μM) | kpol/Kd,app (μM-1 s-1) | |
| ATP | |||
| WT | 7.8 ± 0.2 | 7.0 ± 1.0 | 1.1 ± 0.1 |
| D238A | 0.24 ± 0.02 | 18.1 ± 4.2 | 0.013 ± 0.003 (85)b |
| N297A | 1.9 ± 0.2 | 19.4 ± 3.0 | 0.10 ± 0.01 (11) |
| D238A/N297A | 0.083 ± 0.003 | 10.3 ± 1.2 | 0.010 ± 0.001 (1100) |
| 2′-dATP | |||
| WT | 1.0 ± 0.02 | 15.0 ± 1.1 | 0.060 ± 0.005 |
| D238A | 0.29 ± 0.04 | 19.7 ± 3.2 | 0.020 ± 0.003 (3) |
| N297A | 0.64 ± 0.05 | 21.0 ± 5.1 | 0.030 ± 0.007 (2) |
| D238A/N297A | 0.12 ± 0.002 | 14.3 ± 1.3 | 0.010 ± 0.001 (6) |
| 3′-dATP | |||
| WT | 0.14 ± 0.07 | 24.5 ± 4.4 | (5.0 ± 0.1) × 10-3 |
| D238A | 0.027 ± 0.001 | 20.3 ± 0.8 | (1.0 ± 0.1) × 10-3 (5) |
| N297A | 0.029 ± 0.002 | 28.4 ± 2.8 | (1.0 ± 0.1) × 10-3 (5) |
| D238A/N297A | 0.021 ± 0.003 | 18.2 ± 3.1 | (9.0 ± 0.2) × 10-4 (6) |
| 2′,3′-ddATP | |||
| WT | 0.022 ± 0.002 | 39.1 ± 3.2 | (1.0 ± 0.1) × 10-3 |
| D238A | 0.013 ± 0.003 | 21.3 ± 2.5 | (1.0 ± 0.2) × 10-3 |
| N297A | 0.017 ± 0.002 | 36.7 ± 1.9 | (1.0 ± 0.1) × 10-3 |
| D238A/N297A | 0.010 ± 0.001 | 25.2 ± 4.3 | (5.0 ± 1.0) × 10-4(2) |
Experiments were performed as described under Experimental Procedures.
Numbers in parentheses indicate the fold difference from the WT value.
experimental
| (6) |
predicted
| (7) |
Kinetic Simulation
Kinetic simulations were performed by using KinTekSim Version 2.03 (KinTek Corp., Austin, TX). The agreement between the experimental data and kinetic simulations was determined by visual inspection.
RESULTS AND DISCUSSION
The Structural Model for the 3Dpol-sym/sub-ATP Complex Is Supported by Kinetic and Thermodynamic Analysis of the Nucleotide Specificity of 3Dpol Derivatives with Substitutions in the Ribose-Binding Pocket
In addition to the D238A and N297A derivatives constructed previously (3), we have constructed a derivative containing both substitutions, referred to as D238A/N297A, to determine whether the function of each residue in nucleotide selection is independent of the other (see Experimental Procedures). For these studies, we have employed the symmetrical primer/template substrate (sym/sub) developed by us to study the kinetic mechanism of 3Dpol-catalyzed nucleotide incorporation (7). This substrate is shown in Figure 2; this particular substrate templates incorporation of AMP. Assembly of 3Dpol-sym/sub complexes is slow and suffers from the complication that unbound polymerase is sensitive to thermal inactivation when glycerol and/or nucleotide are (is) omitted during complex assembly (7). Under the conditions employed in this study, the combination of 2 μM 3Dpol and 1 μM sym/sub duplex yields 0.42 μM complex prior to mixing with nucleotide substrate. Importantly, the half-life of this complex is 2 h (7). Therefore, neither complex dissociation nor polymerase inactivation need to be considered in our interpretation of the kinetic experiments described herein.
FIGURE 2.
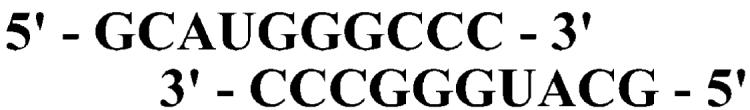
Symmetrical primer/template substrate (sym/sub) employed in this study.
The kinetics of AMP, 2′-dAMP, and 3′-dAMP incorporation into sym/sub were evaluated over a range of nucleotide concentrations at 20 °C for the wild-type, D238A, N297A, and D238/N297A enzymes in order to determine the apparent dissociation constant (Kd,app) and maximal rate constant for nucleotide incorporation (kpol) for each enzyme and nucleotide substrate in the presence of either Mg2+ (Table 1) or Mn2+ (Table 2). We have shown that the use of Mn2+ permits determination of kinetic constants that are inaccessible when Mg2+ is employed as the divalent cation cofactor (1, 2). This information facilitates calculation of key kinetic constants for reactions performed in the presence of Mg2+ that are required to complete the mechanism (1, 2). In the presence of Mg2+,3′-dAMP incorporation could not be evaluated with any of the 3Dpol derivatives because the rate constant for nucleotide incorporation was very similar to the rate constant for dissociation of the polymerase-sym/sub complex. Again, at the concentrations of sym/sub employed in these studies, any polymerase that dissociates during the course of the experiment will not rebind quantitatively to sym/sub owing to thermal inactivation of the enzyme (7). However, in the presence of Mn2+, analysis of 3′-dAMP incorporation was not a problem (Table 2). In fact, by using this divalent cation cofactor, we were also able to evaluate 2′,3′-ddAMP incorporation into sym/sub by the various enzymes (Table 2).
We calculated values for kpol/Kd,app for each enzyme with each substrate (Tables 1 and 2) in order to permit a comparison of the various polymerase-substrate pairs by construction of thermodynamic boxes (10-12) (see Experimental Procedures). The analysis of each enzyme with ATP is shown in Figure 3. On the basis of the structural model shown in Figure 1B, we assume that most of the critical interactions in the complex originate from hydrogen bonding. While the exact value for a hydrogen bond is not known and is context dependent, reported values range from 0.5 to 1.5 kcal/mol (11). We have employed the average value of 1 kcal/mol to estimate the number of hydrogen-bonding interactions occurring in the catalytically competent complex.
FIGURE 3.

Thermodynamic analysis of AMP incorporation into sym/sub by 3Dpol and 3Dpol derivatives. (A) Analysis in the presence of Mg2+. (B) Analysis in the presence of Mn2+. The ΔΔG values are given in kcal/mol and were calculated as described under Experimental Procedures by using appropriate kpol/Kd values from Tables 1 or 2.
Asp-238 contributed on average four hydrogen bonds (3.7 kcal/mol) and Asn-297 contributed on average two hydrogen bonds (2.4 kcal/mol) to the catalytically competent complex (Figure 3A). However, these two residues did not function independently because the double mutant only lost four hydrogen bonds (4.4 kcal/mol) (Figure 3A), which is 1.7 kcal/mol less than the sum of effects on the single mutants. Interactions formed by Asp-238 likely facilitate interaction (s) between Asn-297 and ATP because removal of the Asn-297 side chain from the D238A derivative was not as deleterious (0.7 kcal/mol) as removal of the Asn-297 side chain from the wild-type enzyme (2.4 kcal/mol) (Figure 3A). These data are consistent with our structural model for ribose-polymerase interactions shown in Figure 1B (3). The side chains of Asp-238 and Asn-297 participate in at least four hydrogen bonds; three of these derive from the Asp-238 side chain.
Mn2+ suppressed the observed defect of each mutant by 0.5-1.1 kcal/mol (compare values reported in Figure 3A to those reported in Figure 3B). This difference may be a reflection of the fact that Mn2+ has the capacity to suppress defects to phosphoryl transfer that might contribute to differences observed in values for kpol obtained in the presence of Mg2+ (2).
By using Mn2+ as the divalent cation cofactor, we could construct thermodynamic boxes for ATP, 2′-dATP, 3′-dATP, and 2′,3′-ddATP with the various enzymes to obtain additional insight into the importance of these ribose substituents for ribonucleotide selection by 3Dpol and to scrutinize further our model for the ternary complex of 3Dpol (Figure 1B). The 2′-OH contributed two hydrogen bonds (1.7 kcal/mol) and the 3′-OH contributed three hydrogen bonds (3.1 kcal/mol) to the stability of the catalytically competent complex, and each hydroxyl group functioned independently, based upon the finding that the observed reduction for 2′,3′-ddATP (4.1 kcal/mol) was essentially the sum of 2′-dATP and 3′-dATP (Figure 4A). Please note that, given the error associated with the kpol/Kd,app values used to calculate ΔΔG values, differences between ΔΔG values must exceed 20% in order to be considered significant. The capacity of each hydroxyl group to function independently was observed with all of the 3Dpol derivatives (Figure 4B-D).
FIGURE 4.

Thermodynamic analysis of ribose specificity of 3Dpol and 3Dpol derivatives in the presence of Mn2+: (A) wild type; (B) D238A; (C) N297A; (D) D238A/N297A. The ΔΔG values are given in kcal/mol and were calculated as described under Experimental Procedures by using appropriate kpol/Kd values from Table 2.
In the absence of the Asp-238 side chain, none of the interactions with the 2′-OH occurred (0 kcal/mol difference between ATP and 2′-dATP in Figure 4B), not even the interaction known to occur with the Asn-297 side chain (0.7 kcal/mol difference between ATP and 2′-dATP in Figure 4C). This observation suggests that, in order for Asn-297 to be in a position to permit interaction with the 2′-OH, Asp-238 must first adopt the appropriate conformation. Because Asn-297 only contributed one hydrogen bond (0.7 kcal/mol difference between ATP and 2′-dATP in Figure 4C) of the two hydrogen bonds to the 2′-OH (1.7 kcal/mol difference between ATP and 2′-dATP in Figure 4A) and both were dependent upon the Asp-238 side chain (0 kcal/mol difference between ATP and 2′-dATP in Figure 4B), it is possible that the Asp-238 carboxylate either has a direct interaction with the 2′-OH as shown in Figure 1B or is required for some unidentified residue in the ribose-binding pocket to interact with the 2′-OH.
Of the three interactions between the enzyme and the 3′-OH (3.1 kcal/mol difference between ATP and 3′-dATP in Figure 4A), at least one of these interactions required the Asp-238 side chain (1.5 kcal/mol difference between ATP and 3′-dATP in Figure 4B). However, given the position of this side chain in the ribose-binding pocket (Figure 1B), it is likely that the 3′-OH actually interacts with the backbone nitrogen of Asp-238 and proper positioning of the Asp-238 side chain must facilitate this interaction. The remaining interactions with the 3′-OH did not require Asn-297 (2.7 kcal/mol difference between ATP and 3′-dATP in Figure 4C was essentially identical to wild type), suggesting that these interactions originated from outside of the ribose-binding pocket. As shown in Figure 1B, this interaction could occur with an oxygen of the β-phosphate of the triphosphate moiety of the nucleotide substrate.
The Asp-238 Side chain Permits Communication between the Ribose-Binding Pocket and the Catalytic Center of 3Dpol
In the presence of either Mg2+ or Mn2+, the primary defect observed for all of the 3Dpol derivatives evaluated was a reduction in the observed rate constant for nucleotide incorporation without any deleterious effect on nucleotide binding (Tables 1 and 2). The observation that Mn2+ could compensate for the substantial reduction in the rate constant for nucleotide incorporation observed in the presence of Mg2+ (compare Table 1 to Table 2) suggested that these derivatives with changes in the ribose-binding pocket caused problems with phosphoryl transfer when Mg2+ was employed as the cofactor. This conclusion is based upon our recent finding that in the presence of Mn2+ the rate constant for phosphoryl transfer is independent of the architectural integrity of the ribose-binding pocket (2). However, empirical evidence to support the validity of this conclusion was required.
The goal of the following experiments was to provide empirical evidence for a change in kchem (k+3 in Scheme 1) caused by the amino acid substitutions in the ribose-binding pocket. By using eqs 3-5 (see Experimental Procedures), we can calculate kchem for the mutants by knowing the values for the observed rate constant for nucleotide incorporation (kpol), the observed phosphorothioate effect (Eobs), the theoretical maximum for the phosphorothioate effect (σ), and the equilibrium constant for the first conformational-change step (K2).
We have shown that in the presence of Mn2+ for wild-type 3Dpol phosphoryl transfer is the rate-limiting step, thus permitting determination of the maximal phosphorothioate effect for AMP incorporation into sym/sub (7.9) (1). These data were obtained at 30 °C; therefore, we evaluated the kinetics of AMP and AMPαS incorporation into sym/sub by the D238A and N297A derivatives at 30 °C in the presence of Mg2+ and Mn2+ in order to obtain values for kpol, Kd,app, and Eobs under the different experimental conditions (Table 3). For 3Dpol, we have established that the upper limit for K2,Mg2+ is set by the following quotient: K2,Mn2+/5 (2). Therefore, if we can establish K2,Mn2+ experimentally, then we can set a limit on K2,Mg2+ that will permit calculation of kchem in the presence of Mg2+.
Table 3.
Observed Phosphorothioate Effect for 3Dpol-Catalyzed Nucleotide Incorporation at 30 °C in the Presence of Mg2+ or Mn2+
| WTa |
D238A |
N297A |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Mg2+ |
Mn2+ |
Mg2+ |
Mn2+ |
Mg2+ |
Mn2+ |
|||||||
| kpol (s-1) | Kd,app (μM) | kpol (s-1) | Kd,app (μM) | kpol (s-1) | Kd,app (μM) | kpol (s-1) | Kd,app (μM) | kpol (s-1) | Kd,app (μM) | kpol (s-1) | Kd,app (μM) | |
| ATP | 86.7 ± 3.7 | 134 ± 18 | 21.4 ± 0.6 | 4.1 ±0.5 | 0.047 ± 0.001 | 12 ± 1 | 1.5 ± 0.4 | 9 ± 1 | 4.6 ± 0.2 | 176 ± 31 | 5.9 ± 0.4 | 33 ± 4 |
| ATPαS | 20.6 ± 1.6 | 89 ± 24 | 2.7 ± 0.1 | 1.0 ± 0.2 | 0.024 ± 0.001 | 83 ± 10 | 0.68 ± 0.08 | 61 ± 16 | 1.1 ± 0.1 | 300 ± 85 | 1.3 ± 0.1 | 29 ± 6 |
| Eobs | 4.2 ± 0.4 | 7.9 ± 0.4 | 2.0 ± 0.1 | 2.2 ± 0.2 | 4.2 ± 0.1 | 4.5 ± 0.1 | ||||||
We calculated values for K2 in the presence of Mn2+ for the D238A and N297A derivatives by using eqs 3-5 and the data reported in Table 3, assuming a value of 30 s-1 for kchem (2). K2 values of 0.4 and 0.6 were obtained for the D238A and N297A derivatives, respectively (Table 4). The corresponding value for wild-type 3Dpol was 3 (2). As long as the value for K2 in the presence of Mn2+ is accurate, the calculated value for K2 in the presence of Mg2+ (Table 4) should be reliable (2). Because assumptions are made in any experiment leading to a value for K2 in the presence of Mn2+ for the 3Dpol derivatives, at best, a range for the value of K2 in the presence of Mn2+ can be obtained. Therefore, to establish this range and verify the magnitude of the defect on the conformational-change step of the D238A and N297A derivatives, we performed two additional experiments.
Table 4.
Comparison of K2 and k+3 Values for AMP Incorporation into sym/sub-U for Wild-Type 3Dpol and 3Dpol Derivatives at 30 °C in the Presence of Mg2+ or Mn2+
|
K2 |
k+3 (s-1) |
|||
|---|---|---|---|---|
| Mn2+ | Mg2+ | Mn2+ | Mg2+ | |
| WT | 3a | 0.6b | 30a | 520b |
| D238A | 0.4c | 0.08d | 30 | 4.4e |
| N297A | 0.6c | 0.12d | 30 | 92e |
The K2 value was taken from ref 2.
The K2 value was taken from ref 1.
The K2 value in the presence of Mn2+ was calculated by using eqs 3-5 under Experimental Procedures, the values for the observed and maximal phosphorothioate effects listed in Table 3, and a kpol value of 30 s-1.
K2,Mg2+ was calculated using K2,Mg2+ = K2,Mn2+/5 (2).
Isotope-trapping experiments combined with kinetic simulations have been used to determine values for K2 and kchem for wild-type 3Dpol in the presence of Mg2+ or Mn2+ (1, 2). Similar results can be obtained in the presence of Mn2+ by comparing the observed kinetics of nucleotide incorporation of a reaction quenched by using EDTA to those of a reaction quenched by using acid (Figure 5A) and fitting the data by kinetic simulation to the mechanism shown in Scheme 2 (2). EDTA is incapable of quenching the activated ternary complex (*ERnNTP in Scheme 1), causing an apparent increase in product formed relative to reactions quenched by using acid (2). The amount of “additional” product formed when EDTA is employed as a quench is dictated by both K2 and kchem. When this experiment was performed with the D238A and N297A derivatives, values of 0.1 and 0.2 were obtained for K2, respectively, and a value of 30 s-1 for kchem was obtained for both (Figure 5B,C). These values are approximately 3-4-fold lower than calculated by using the phosphorothioate effect data (Table 4). However, the ratio of the K2 values for the D238A and N297A derivatives was within 35% of that obtained by using the phosphorothioate effect data (Table 4).
FIGURE 5.

*ERnATP intermediate accumulation during AMP incorporation into sym/sub by 3Dpol and 3Dpol derivatives. Kinetics of AMP incorporation into sym/sub were evaluated in the presence of Mn2+. The reaction was quenched by using either EDTA (●) or HCl (○). The solid lines represent kinetic simulation of the mechanism shown in Scheme 2. The output for the kinetic simulation of the acid-quench data was *ERn+1PPi (Scheme 2) whereas that for the EDTA-quench data was *ERnATP and *ERn+1-PPi. (A) Wild type, with K2 equal to 3 and k+3 equal to 30 s-1. (B) D238A, with K2 equal to 0.1 and k+3 equal to 30 s-1. (C) N297A, with K2 equal to 0.2 and k+3 equal to 30 s-1
Scheme 2.

Minimal Mechanism for Pulse-Chase Analysis
Incubation of the 3Dpol-sym/sub complex with the non-hydrolyzable ATP analogue, AMPCPP, produces a ternary complex whose overall stability is dictated by K1 and K2 (Scheme 1). Because the Kd,app values for ATP with all of the enzymes studied here are roughly equivalent (Table 2), any observed change in the stability of this complex for 3Dpol derivatives should reflect changes in the value for K2. The apparent stability was measured by forming the 3Dpol-sym/sub-AMPCPP complex, mixing this complex with ATP, and measuring the kinetics of AMP incorporation. The observed rate constant for AMP incorporation will be attenuated by the rate constant for dissociation of AMPCPP from the ternary complex to liberate 3Dpol-sym/sub complexes to which ATP can bind (Scheme 3). The kinetic data can be fit by simulation to the mechanism shown in Scheme 3 to obtain the rate constant for AMPCPP dissociation (designated k1 in Scheme 3). In order for this experiment to be valid as written in Scheme 3, once AMPCPP dissociates, this nucleotide should not be able to rebind to the enzyme. We verified that this condition was satisfied by demonstrating the lack of an effect of AMPCPP on the kinetics of AMP incorporation in the absence of preincubation of AMPCPP with the 3Dpol-sym/sub complex (data not shown). This experiment was performed for wild-type, D238A, and N297A 3Dpol enzymes (Figure 6), yielding k1 values of 0.6, 8, and 3 s-1, respectively. This result confirms that a step prior to phosphoryl transfer is affected by loss of the Asp-238 and/or Asn-297 side chains, causing approximately a one-log reduction in the magnitude of K2 relative to wild-type 3Dpol.
Scheme 3.
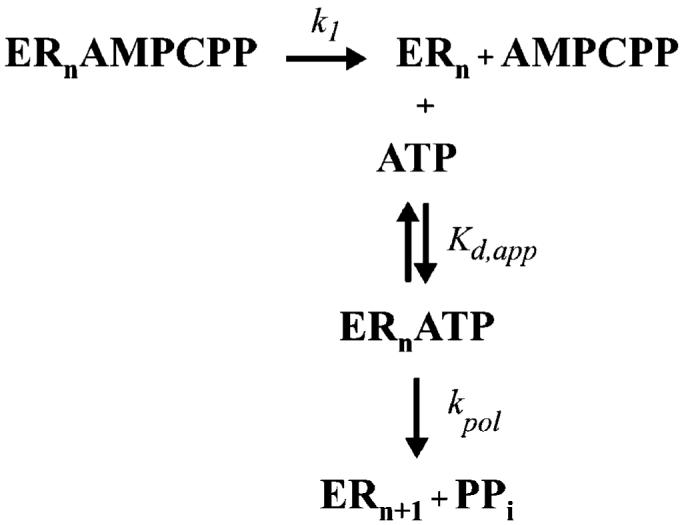
Minimal Kinetic Mechanism for Dissociation of the 3Dpol-sym/sub-AMPCPP Complex Monitored by AMP Incorporation into sym/sub
FIGURE 6.

Stability of ternary complexes of 3Dpol or 3Dpol derivatives with AMPCPP at 30 °C in the presence of Mn2+.2 μM enzyme was assembled with 2 μM sym/sub for 90 s at 22 °C in 50 mM HEPES, pH 7.5, 60 μM ZnCl2, 5 mM MnCl2, and 10 mM β-mercaptoethanol. Either buffer or 200 μM AMPCPP was added, and the sample was loaded into the rapid-mixing/quenching device. Reactions were initiated by addition of an equal volume of 2 mM ATP and permitted to react at 30 °C for the indicated time. The kinetics of AMP incorporation into sym/sub in the presence (○) or absence (●) of AMPCPP was determined as described under Experimental Procedures and employed an acid quench. Solid lines represent kinetic simulation of the mechanism shown in Scheme 3 using the Kd,app and kpol values listed in Table 2 and values for k1 equal to 0.6, 8, and 3 s-1 for wild-type, D238A, and N297A, respectively. The value is the apparent dissociation rate of AMPCPP from the ternary complex. Panels: (A) wild type; (B) D238A; (C) N297A.
Given the reasonable agreement between K2 values in the presence of Mn2+ for the 3Dpol derivatives measured by using the various methods, we chose to continue using the values reported in Table 4 to calculate the K2 values in the presence of Mg2+, which are also reported in Table 4. The value for K2 in the presence of Mg2+ for each derivative was then employed to calculate the corresponding value for kchem by using eqs 3-5. The value for the D238A derivative was 4.4 s-1, and the value for the N297A derivative was 92 s-1, reflecting a reduction of 120-fold and 6-fold, respectively, relative to wild-type 3Dpol (Table 4). These data are consistent with the conclusion that, in the presence of Mg2+, information on the nature of the interactions in the ribose-binding pocket can be disseminated to the catalytic center by using the conformation of the Asp-238 and the corresponding orientation of the triphosphate as the conduit.
Structural Model for 3Dpol-Catalyzed Incorporation of Correct Ribonucleotides
Our current model for correct nucleotide incorporation by 3Dpol is shown in Figure 7A-D. This model integrates the insight gleaned from the comprehensive kinetic and thermodynamic analysis of 3Dpol and derivatives thereof in reactions using ATP analogues with correct and incorrect sugar configurations. All nucleotides, correct and incorrect, bind to the enzyme in a similar ground-state configuration that is directed by the metal-bound triphosphate moiety of the nucleotide substrate (Figure 7A). In this ground-state configuration, the ribose cannot bind in a productive orientation because the interaction between Asp-238 and Asn-297 observed in the unliganded enzyme occludes the ribose-binding pocket (Figure 7A) (4). A conformational change occurs that permits base pairing to occur between the incoming nucleotide and template and that orients the phosphate in a configuration in which the triphosphate is oriented appropriately for phosphoryl transfer (Figure 7B). It is this transition that we propose to be partially rate limiting for nucleotide incorporation (step 2 in Scheme 1) (1, 2). Moreover, the stability of this conformation (K2) will dictate the efficiency of phosphoryl transfer as any movement in the position of the triphosphate will produce either a suboptimal orientation or a suboptimal distance for catalysis. To maintain the triphosphate in the appropriate configuration, an extensive hydrogen-bonding network is involved that can be traced to residues in the ribose-binding pocket. Formation of this network requires reorientation of Asp-238 and Asn-297. The oxygen of the β-phosphate interacts with the 3′-OH of the nucleotide substrate. The position of the ribose is held firmly by interactions between the 3′-OH and the backbone of Asp-238 and by interactions between the 2′-OH and Asn-297. The backbone of Asp-238 is restricted by the interaction of the Asp-238 side chain with other residues in the pocket, perhaps Ser-288 and Thr-293. The appropriate organization of this complex will permit binding and/or alignment of the second divalent cation cofactor (Figure 7C), permitting phosphoryl transfer, translocation, and pyrophosphate release (Figure 7D).
FIGURE 7.
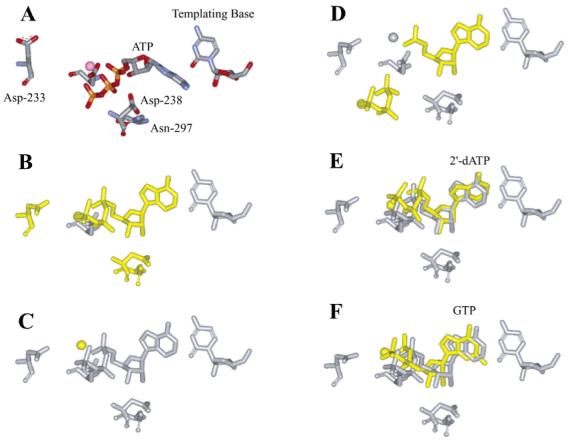
Structural model for fidelity of 3Dpol-catalyzed nucleotide incorporation. Important structural changes from one step to the next are indicated by coloring the pertinent molecules yellow. (A) The correct nucleotide, in this case ATP, binds to the polymerase-primer/template complex driven by the metal-complexed triphosphate moiety. The orientation of the residues of the NTP-binding pocket are as observed in the unliganded crystal structure (4). (B) The nucleotide rearranges into the catalytically competent conformation. A concomitant rearrangement of the NTP-binding pocket occurs (3). Asp-233 moves into place to interact with the nucleotide-bound metal. Asp-238 and Asn-297 move to stabilize the “closed” state of the ribose-binding pocket and to permit interactions between the enzyme and the 2′- and 3′-hydroxyl groups of the ribonucleotide. (C) The second metal ion binds. (D) Phosphoryl transfer occurs followed by translocation of the enzyme into the next register for nucleotide incorporation and pyrophosphate release. (E) Possible conformation of 2′-dATP bound to the NTP-binding pocket. The change here could be caused by the different sugar pucker. (F) Possible conformation of GTP bound to the NTP-binding pocket. The change here could be caused by the nonplanar G,U base pair.
Structural Basis for Fidelity of Ribonucleotide Incorporation by 3Dpol
There is no question that the orientation of the triphosphate is critical for efficient nucleotide incorporation. Therefore, reduced stabilization of the ribose moiety of bound nucleotide caused by deleting interactions with the 2′-OH (e.g., 2′-dATP or N297A) may also alter the interaction between the 3′-OH and the β-phosphate of the nucleotide substrate, resulting in movement of the triphosphate and the consequent reduction in the efficiency of nucleotide incorporation (Figure 7E). This model explains the observation that both the conformational change preceding phosphoryl transfer and phosphoryl transfer are reduced for 2′-dAMP incorporation by 3Dpol (1, 2) or AMP incorporation by the N297A derivative (Tables 1, 2, and 4). Selection against nucleotides with other ribose modifications could employ a similar mechanism.
In the case of incorporation of ribonucleotides with an incorrect base, changes in the orientation of the triphosphate will be caused by formation of the nonplanar G·U pair (Figure 7F). By placing purines opposite purines, changes in the orientation of Asp-238 may occur. Such a change would initiate a cascade of events: movement of the 3′-OH, the β-phosphate, and ultimately the entire triphosphate moiety of the incorrect nucleotide substrate. Again, these structural changes explain the observation that both the conformational change preceding phosphoryl transfer and phosphoryl transfer are reduced for GMP incorporation by 3Dpol (1, 2) for AMP incorporation by the D238A derivative (Tables 1, 2, and 4). Because Asn-297 positioning is dependent upon appropriate positioning of Asp-238, changing the position of Asp-238 will cause effects similar to those described above for alteration of the 2′-OH of the nucleotide substrate.
Selection against pyrimidine-pyrimidine and purine-pyrimidine mispairs by 3Dpol is not as obvious. If this pairing does not support an intrahelical arrangement of both bases, then steric problems should arise that may alter the position of Asp-238 or other residues in the ribose-binding pocket. Alternatively, the increased dynamics of the base due to the lack of hydrogen bonding may increase the dynamics of other portions of the nucleotide substrate, for example, the triphosphate, causing a reduction in the efficiency of incorporation. Unfortunately, neither kinetic nor thermodynamic data evaluating the capacity of 3Dpol to catalyze misincorporation events leading to transversion mutations are available currently to assist in defining a structural model for prevention of this class of mutations.
Fidelity of Ribonucleotide Incorporation by Other RNA-Dependent RNA Polymerases
We have provided kinetic, thermodynamic, and structural evidence that Asp-238 is a key component in the line of communication between the ribose-binding pocket and the catalytic center that functions by modulating the conformation of the triphosphate moiety of the nucleotide substrate. This mechanism for faithful selection of correct nucleotides is likely conserved in all animal virus RdRPs. Asp-238 is a part of conserved structural motif A (4). This structural element is found in all RdRPs for which structural information is available (4, 13-17), and this residue is completely conserved in sequence alignments of all animal virus RdRPs (18).
A Conserved Strategy for Coupling Nucleotide Selection to Phosphoryl-Transfer Efficiency for Polymerases
Inspection of all structures for polymerases poised for or undergoing catalysis (19-24) shows absolute conservation of the orientation of the triphosphate moiety of the nucleotide substrate (Figure 8). Stabilization of the conformation of the triphosphate requires conserved structural motif A (Figure 8). At the top of this motif in all polymerases is one of the two aspartates involved in binding the divalent cation cofactor (Table 5, Figure 8). At the bottom of this motif is a residue that resides in the sugar-binding pocket whose backbone can hydrogen to the 3′-OH of the nucleotide substrate (Table 5, Figure 8). The central region of this motif stabilizes the triphosphate conformation by contributing hydrogen bond donors and acceptors directly to the triphosphate and/or to the divalent cation cofactor; the hydrogen bond donors or acceptors derive from the backbone of these residues rather than the side chains (Figure 8). Therefore, any movement of the motif A side chain located in the sugar-binding pocket will be transmitted through the rest of motif A, consequently perturbing the position of both the sugar and triphosphate and reducing the efficiency of phosphoryl transfer as described above for Asp-238 of 3Dpol.
FIGURE 8.
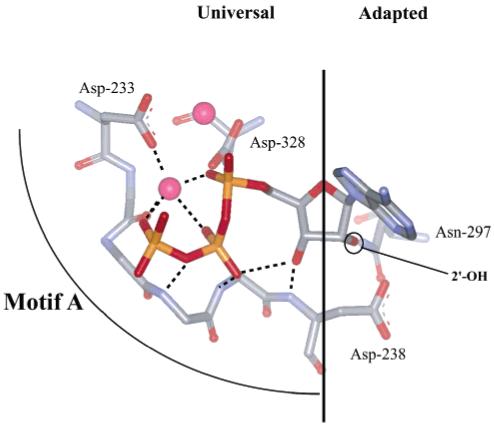
A conserved mechanism for linking binding of a correct nucleotide to the efficiency of phosphoryl transfer? The nucleotide-binding pocket of all nucleic acid polymerases with a canonical “palm”-based active site is highly conserved. The site can be divided into two parts: a region that has “universal” interactions mediated by conserved structural motif A that organize the metals and triphosphate for catalysis and a region that has “adapted” interactions mediated by conserved structural motif B that dictate whether ribo- or 2′-deoxyribonucleotides will be utilized. In the classical polymerase, there is a motif A residue located in the sugar-binding pocket capable of interacting with motif B residue(s) involved in sugar selection. This motif A residue in other polymerases (Table 5) could represent the link between the nature of the bound nucleotide (correct vs incorrect) to the efficiency of phosphoryl transfer as described herein for Asp-238 of 3Dpol.
Table 5.
Polymerase Residues Interacting with Divalent Cation or Sugar
| divalentcation |
sugar |
||||
|---|---|---|---|---|---|
| polymerase | motif A | motif C | motif A | motif B | ref |
| 3Dpol | Asp-233 | Asp-328 | Asp-238 | Asn-297 | 3, 4 |
| HIV-RT | Asp-110 | Asp-185 | Tyr-115 | Phe-160 | 5 |
| T7 RNAP | Asp-537 | Asp-812 | Gly-542/Tyr-639a | His784 | 20, 21 |
| T7 DNAP | Asp-475 | Asp-654 | Glu-480 | Gln-615b | 22 |
| BF | Asp-653 | Asp-830 | Glu-658 | Gln-797b | 19 |
| Klenow | Asp-705 | Asp-882 | Glu-710 | Gln-849b | 23, 31 |
| RB69 | Asp-411 | Asp-623 | Tyr-416 | Gly-393 | 30 |
| Taq | Asp-610 | Asp-785 | Glu-615 | Gln-754 | 24 |
G542 is the structurally analogous residue to Asp-238 of 3Dpol. Tyr-639 is involved in selection for a 2′-OH (28).
These residues are locked in a conformation away from the ribose-binding pocket by the Asp-238 equivalent in these enzymes.
Also apparent from inspection of polymerase structures is the fact that the position of the base will influence the position of the side chain of the motif A residue of the sugar-binding pocket (Figure 8). In addition, selection at the 2′ position of ribose can also clearly influence the position of the side chain of the motif A residue in the sugar-binding pocket (Figure 8). In all polymerases, at least one residue in conserved structural motif B has evolved to sense the presence or absence of a 2′-OH as appropriate for the nucleotide substrate specificity of the enzyme (Table 5). Similar to the Asn-297 (motif B) story for 3Dpol, T7 RNA polymerase uses His-784 (motif B) for hydrogen bonding to the 2′-OH of the NTP substrate. In the absence of this interaction, motif A will move. In HIV-RT, Phe-160 (motif B) has van der Waals interactions with the 2′-H of the 2′-dNTP substrate. In the presence of a 2′-OH, Phe-160 will cause movement of Tyr-115 (motif A). Likewise, the presence of a motif B residue in DNA polymerases (e.g., T7, BF, Klenow, Taq, and RB69 in Table 5) will cause movement of motif A via the motif A residue located in the sugar-binding pocket (Table 5).
We conclude that the nucleotide-binding pocket of all polymerases can be divided into two parts: a universal portion and an adapted portion. Conserved structural motif A mediates the universal functions whereas conserved structural motif B mediates the adapted function. These two motifs intersect in the sugar-binding pocket, providing a mechanism for inappropriate base pairing and/or sugar configuration to be identified and cause the appropriate reduction in phosphoryl-transfer efficiency by moving the triphosphate moiety of the nucleotide substrate into a suboptimal orientation. It should be noted that the nucleotidyl transferases of the polymerase X family do not have structural motif B; these enzymes use a portion of the thumb for sugar selection (25-27). Therefore, it is likely that these enzyme do not follow the rules described here for fidelity.
We propose that the kinetic basis for fidelity of all polymerases containing the canonical palm organization rests in both the conformational-change step preceding phosphoryl transfer and phosphoryl transfer as determined for 3Dpol (1, 28). Recently, there has been a suggestion that the conformational-change step preceding phosphoryl transfer does not influence polymerase fidelity (29). This conclusion was based upon mechanistic studies of the fidelity of DNA polymerase β, a member of the polymerase X family. As suggested above, the polymerase X family is incapable of employing the same structural basis for nucleotide selection as the other polymerase families discussed herein so it is plausible that the kinetic and thermodynamic basis for nucleotide selection in this system is unique.
ACKNOWLEDGMENT
We thank Professors J. Martin Bollinger and Kevin D. Raney for critical review of the manuscript.
Footnotes
- RdRP
- RNA-dependent RNA polymerase
- PAGE
- polyacrylamide gel electrophoresis
- nt
- nucleotide
- EDTA
- ethylene-diaminetetraacetic acid
- NTP
- nucleoside 5′-triphosphate
- 2′-dNTP
- 2′-deoxynuceloside 5′-triphosphate
- 3′-dNTP
- 3′-deoxynuceloside 5′-triphosphate
- ddNTP
- 2′,3′-dideoxynucleoside 5′-triphosphate
- NMP
- nucleoside 5′-monophosphate
- PPi
- pyrophosphate
- AMPCPP
- α,β-methyleneadenosine 5′-triphosphate
This work was supported, in part, by a Howard Temin Award (CA75118) from the NCI, National Institutes of Health, and by a grant (AI45818) from the NIAID, National Institutes of Health (both to C.E.C.). C.E.C. is the recipient of an Established Investigator Award (0340028N) from the American Heart Association.
REFERENCES
- 1.Arnold JJ, Cameron CE. Poliovirus RNA-dependent RNA polymerase (3Dpol): Pre-steady-state kinetic analysis of ribonucleotide incorporation in the presence of Mg2+ Biochemistry. 2004;43:5126–5137. doi: 10.1021/bi035212y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Arnold JJ, Gohara DW, Cameron CE. Poliovirus RNA-dependent RNA polymerase (3Dpol): Pre-steady-state kinetic analysis of ribonucleotide incorporation in the presence of Mn2+ Biochemistry. 2004;43:5138–5148. doi: 10.1021/bi035213q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gohara DW, Crotty S, Arnold JJ, Yoder JD, Andino R, Cameron CE. Poliovirus RNA-dependent RNA polymerase (3Dpol): Structural, biochemical, and biological analysis of conserved structural motifs A and B. J. Biol. Chem. 2000;275:25523–25532. doi: 10.1074/jbc.M002671200. [DOI] [PubMed] [Google Scholar]
- 4.Hansen JL, Long AM, Schultz SC. Structure of the RNA-dependent RNA polymerase of poliovirus. Structure. 1997;5:1109–1122. doi: 10.1016/s0969-2126(97)00261-x. [DOI] [PubMed] [Google Scholar]
- 5.Huang H, Chopra R, Verdine GL, Harrison SC. Structure of a covalently trapped catalytic complex of HIV-1 reverse transcriptase: implications for drug resistance. Science. 1998;282:1669–1675. doi: 10.1126/science.282.5394.1669. [DOI] [PubMed] [Google Scholar]
- 6.Gohara DW, Ha CS, Kumar S, Ghosh B, Arnold JJ, Wisniewski TJ, Cameron CE. Production of “authentic” poliovirus RNA-dependent RNA polymerase (3Dpol) by ubiquitin-protease-mediated cleavage in Escherichia coli. Protein Expression Purif. 1999;17:128–138. doi: 10.1006/prep.1999.1100. [DOI] [PubMed] [Google Scholar]
- 7.Arnold JJ, Cameron CE. Poliovirus RNA-dependent RNA polymerase (3Dpol): Assembly of stable, elongation-competent complexes by using a symmetrical primer-template substrate (sym/sub) J. Biol. Chem. 2000;275:5329–5336. doi: 10.1074/jbc.275.8.5329. [DOI] [PubMed] [Google Scholar]
- 8.Wong I, Patel SS, Johnson KA. An induced-fit kinetic mechanism for DNA replication fidelity: direct measurement by single-turnover kinetics. Biochemistry. 1991;30:526–537. doi: 10.1021/bi00216a030. [DOI] [PubMed] [Google Scholar]
- 9.Patel SS, Wong I, Johnson KA. Pre-steady-state kinetic analysis of processive DNA replication including complete characterization of an exonuclease-deficient mutant. Biochemistry. 1991;30:511–525. doi: 10.1021/bi00216a029. [DOI] [PubMed] [Google Scholar]
- 10.Serrano L, Horovitz A, Avron B, Bycroft M, Fersht AR. Estimating the contribution of engineered surface electrostatic interactions to protein stability by using double-mutant cycles. Biochemistry. 1990;29:9343–9352. doi: 10.1021/bi00492a006. [DOI] [PubMed] [Google Scholar]
- 11.Fersht AR. Dissection of the structure and activity of the tyrosyl-tRNA synthetase by site-directed mutagenesis. Biochemistry. 1987;26:8031–8307. doi: 10.1021/bi00399a001. [DOI] [PubMed] [Google Scholar]
- 12.Mildvan AS, Weber DJ, Kuliopulos A. Quantitative interpretations of double mutations of enzymes. Arch. Biochem. Biophys. 1992;294:327–340. doi: 10.1016/0003-9861(92)90692-p. [DOI] [PubMed] [Google Scholar]
- 13.Ago H, Adachi T, Yoshida A, Yamamoto M, Habuka N, Yatsunami K, Miyano M. Crystal structure of the RNA-dependent RNA polymerase of hepatitis C virus. Struct. Folding Des. 1999;7:1417–1426. doi: 10.1016/s0969-2126(00)80031-3. [DOI] [PubMed] [Google Scholar]
- 14.Lesburg CA, Cable MB, Ferrari E, Hong Z, Mannarino AF, Weber PC. Crystal structure of the RNA-dependent RNA polymerase from hepatitis C virus reveals a fully encircled active site. Nat. Struct. Biol. 1999;6:937–943. doi: 10.1038/13305. [DOI] [PubMed] [Google Scholar]
- 15.Bressanelli S, Tomei L, Roussel A, Incitti I, Vitale RL, Mathieu M, De Francesco R, Rey FA. Crystal structure of the RNA-dependent RNA polymerase of hepatitis C virus. Proc. Natl. Acad. Sci. U.S.A. 1999;96:13034–13039. doi: 10.1073/pnas.96.23.13034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ng KK, Cherney MM, Vazquez AL, Machin A, Alonso JM, Parra F, James MN. Crystal structures of active and inactive conformations of a caliciviral RNA-dependent RNA polymerase. J. Biol. Chem. 2002;277:1381–1387. doi: 10.1074/jbc.M109261200. [DOI] [PubMed] [Google Scholar]
- 17.Butcher SJ, Grimes JM, Makeyev EV, Bamford DH, Stuart DI. A mechanism for initiating RNA-dependent RNA polymerization. Nature. 2001;410:235–240. doi: 10.1038/35065653. [DOI] [PubMed] [Google Scholar]
- 18.Koonin EV. The phylogeny of RNA-dependent RNA polymerases of positive-strand RNA viruses. J. Gen. Virol. 1991;72:2197–2206. doi: 10.1099/0022-1317-72-9-2197. [DOI] [PubMed] [Google Scholar]
- 19.Johnson SJ, Taylor JS, Beese LS. Processive DNA synthesis observed in a polymerase crystal suggests a mechanism for the prevention of frameshift mutations. Proc. Natl. Acad. Sci. U.S.A. 2003;100:3895–3900. doi: 10.1073/pnas.0630532100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yin YW, Steitz TA. Structural basis for the transition from initiation to elongation transcription in T7 RNA polymerase. Science. 2002;298:1387–1395. doi: 10.1126/science.1077464. [DOI] [PubMed] [Google Scholar]
- 21.Cheetham GM, Steitz TA. Structure of a transcribing T7 RNA polymerase initiation complex. Science. 1999;286:2305–2309. doi: 10.1126/science.286.5448.2305. [DOI] [PubMed] [Google Scholar]
- 22.Doublie S, Tabor S, Long AM, Richardson CC, Ellenberger T. Crystal structure of a bacteriophage T7 DNA replication complex at 2.2 A resolution. Nature. 1998;391:251–258. doi: 10.1038/34593. [DOI] [PubMed] [Google Scholar]
- 23.Beese LS, Derbyshire V, Steitz TA. Structure of DNA polymerase I Klenow fragment bound to duplex DNA. Science. 1993;260:352–355. doi: 10.1126/science.8469987. [DOI] [PubMed] [Google Scholar]
- 24.Li Y, Korolev S, Waksman G. Crystal structures of open and closed forms of binary and ternary complexes of the large fragment of Thermus aquaticus DNA polymerase I: Structural basis for nucleotide incorporation. EMBO J. 1998;17:7514–7525. doi: 10.1093/emboj/17.24.7514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pelletier H, Sawaya MR, Wolfle W, Wilson SH, Kraut J. Crystal structures of human DNA polymerase beta complexed with DNA: implications for catalytic mechanism, processivity, and fidelity. Biochemistry. 1996;35:12742–12761. doi: 10.1021/bi952955d. [DOI] [PubMed] [Google Scholar]
- 26.Pelletier H, Sawaya MR, Kumar A, Wilson SH, Kraut J. Structures of ternary complexes of rat DNA polymerase beta, a DNA template-primer, and ddCTP. Science. 1994;264:1891–18903. [PubMed] [Google Scholar]
- 27.Arndt JW, Gong W, Zhong X, Showalter AK, Liu J, Dunlap CA, Lin Z, Paxson C, Tsai M-D, Chan MK. Insight into the catalytic mechanism of DNA polymerase beta: structures of intermediate complexes. Biochemistry. 2001;40:5368–5375. doi: 10.1021/bi002176j. [DOI] [PubMed] [Google Scholar]
- 28.Huang Y, Eckstein F, Padilla R, Sousa R. Mechanism of ribose 2′-group discrimination by an RNA polymerase. Biochemistry. 1997;36:8231–8242. doi: 10.1021/bi962674l. [DOI] [PubMed] [Google Scholar]
- 29.Showalter AK, Tsai M-D. A reexamination of the nucleotide incorporation fidelity of DNA polymerases. Biochemistry. 2002;41:10571–10576. doi: 10.1021/bi026021i. [DOI] [PubMed] [Google Scholar]
- 30.Wang J, Sattar AK, Wang CC, Karam JD, Konigsberg WH, Steitz TA. Crystal structure of a pol alpha family replication DNA polymerase from bacteriophage RB69. Cell. 1997;89:1087–1099. doi: 10.1016/s0092-8674(00)80296-2. [DOI] [PubMed] [Google Scholar]
- 31.Astatke M, Ng K, Grindley ND, Joyce CM. A single side chain prevents Escherichia coli DNA polymerase I (Klenow fragment) from incorporating ribonucleotides. Proc. Natl. Acad. Sci. U.S.A. 1998;95:3402–3407. doi: 10.1073/pnas.95.7.3402. [DOI] [PMC free article] [PubMed] [Google Scholar]