

Abstract
survival of motor neuron 2, centromeric (SMN2) is a gene that modifies the severity of spinal muscular atrophy (SMA), a motor-neuron disease that is the leading genetic cause of infant mortality. Increasing inclusion of SMN2 exon 7, which is predominantly skipped, holds promise to treat or possibly cure SMA; one practical strategy is the disruption of splicing silencers that impair exon 7 recognition. By using an antisense oligonucleotide (ASO)-tiling method, we systematically screened the proximal intronic regions flanking exon 7 and identified two intronic splicing silencers (ISSs): one in intron 6 and a recently described one in intron 7. We analyzed the intron 7 ISS by mutagenesis, coupled with splicing assays, RNA-affinity chromatography, and protein overexpression, and found two tandem hnRNP A1/A2 motifs within the ISS that are responsible for its inhibitory character. Mutations in these two motifs, or ASOs that block them, promote very efficient exon 7 inclusion. We screened 31 ASOs in this region and selected two optimal ones to test in human SMN2 transgenic mice. Both ASOs strongly increased hSMN2 exon 7 inclusion in the liver and kidney of the transgenic animals. Our results show that the high-resolution ASO-tiling approach can identify cis-elements that modulate splicing positively or negatively. Most importantly, our results highlight the therapeutic potential of some of these ASOs in the context of SMA.
Introduction
Premessenger RNA (pre-mRNA) splicing is catalyzed by the spliceosome, a large dynamic ribonucleoprotein complex.1–3 Splicing involves several stepwise assembly and catalytic processes, including exon and intron recognition, excision of intervening introns, and exon joining. Generally, splicing signals at or near the exon-intron junctions of pre-mRNA, including the 5′ splice site, 3′ splice site, polypyrimidine tract, and branchpoint sequence, are necessary but not sufficient for accurate and efficient exon recognition by the spliceosome. Additional positive signals in an exon and/or its flanking introns are also required for efficient exon recognition, particularly when the exon is alternatively spliced or is constitutively spliced but has weak splice sites.4 These positive cis-elements, including exonic splicing enhancers (ESEs) and intronic splicing enhancers (ISEs), are generally binding sites for splicing activators, such as serine-arginine-rich (SR) proteins, or may adopt favorable secondary structures. ESEs and ISEs can counteract negative cis-elements, such as exonic splicing silencers (ESSs) and intronic splicing silencers (ISSs), which generally are the binding sites for splicing repressors, such as certain hnRNP proteins, or adopt unfavorable higher-order structures. The antagonism between SR proteins and hnRNP proteins is one mechanism by which splicing is finely tuned.5 Disruption of cis-elements, inducing exon skipping, such as in the survival of motor neuron 2, centromeric (SMN2) gene (MIM 601627), or tilting the ratio of different mRNA isoforms derived from a single gene, such as the MAPT (microtubule-associated protein tau) gene (MIM 137140) in frontotemporal dementia (MIM 600274), can lead to severe diseases.4,6–11
SMN2 is a modifying gene in spinal muscular atrophy (SMA types I, II, and III [MIMs 253300, 253550, and 253400]), which is caused by loss-of-function mutations or deletions of the closely related survival of motor neuron 1, telomeric (SMN1) gene (MIM 600354).12 Both genes encode identical SMN proteins; however, only SMN1 generates full-length mRNA and protein (UniProt accession number Q16637-1) as predominant products. The majority of SMN2 mRNA lacks exon 7 because of a C6T transition in SMN2 exon 7 (relative to SMN1) that affects exon recognition during splicing, resulting in a defective exon 7 skipped protein isoform (UniProt accession number Q16637-3).13,14 Recently, an A100G transition in SMN2 intron 7 (relative to SMN1) has also been reported to partially contribute to the predominant skipping of SMN2 exon 7.15
The SMN protein, together with several Gemin proteins, forms an SMN complex that functions as a chaperone to facilitate assembly of U snRNPs and possibly other RNPs.16,17 SMN may have additional roles in assisting arginine methylation of some splicing-related proteins18 and transporting axonal mRNAs in motor neurons.19 The 54-nt-long alternatively spliced exon 7 encodes a C-terminal peptide of 16 amino acids, which is essential for SMN protein stability and proper cytoplasmic localization, and possibly comprises a motif that plays specific functions in maintaining growth cones in motor neurons.20–24
Exon 7 in SMN1 and SMN2 has a weak 5′ splice site, reflecting its divergence from the consensus sequence and a stem-loop structure at the exon 7-intron 7 junction that interferes with U1 small nuclear RNA (snRNA) base pairing to the 5′ splice site.25 The drastic difference in exon 7 inclusion between these two genes and the involvement of multiple cis-elements and trans-factors are indicative of the complex regulatory interplay between various splicing signals. For both genes, a Tra2-β1 (Uniprot accession number P62995-1) motif in the central region of exon 7 is crucial in promoting inclusion of the exon.26 It appears that the central element is recognized by a splicing-activating protein complex that includes at least Tra2-β1, SRp30c (UniProt accession number Q13242), and hnRNP G (UniProt accession number P38159).27,28 Interestingly, antagonism between Tra2-β1 and hnRNP G has been proposed to regulate other alternatively spliced exons.29 We predicted the existence of another ESE motif in the central region (+28 to +34) that might bind to another trans-acting protein essential for the central-core protein complex, because antisense oligonucleotides (ASOs) binding to this region potently inhibit exon 7 inclusion.30
In SMN1, an SF2/ASF (UniProt accession number Q07955) motif near the 3′ splice site (+6 to +12) serves as another crucial ESE, which is absent or weakened in the SMN2 gene because of the C6T transition, accounting for the predominant skipping of SMN2 exon 7.6,7 In addition, an ISE in intron 7 (+56 to +79) that comprises a potential stem-loop structure has been characterized in the context of an exon-trapping vector.31
On the other hand, several ESSs and ISSs contribute to the repression of exon 7 splicing. In SMN2, the C6T change, which abrogates the SF2/ASF binding site, has also been purported to create an hnRNP A1 (UniProt accession number P09651-2) motif.7,32,33 We recently identified two inhibitory regions, A (+4 to +21) and B (+34 to +51) in SMN2 exon 7,30 which is consistent with a previous SELEX study pointing to a conserved track in the middle of SMN2 exon 7 flanked on both sides by inhibitory sequences.34 Because for both SMN genes the majority of ASOs binding to these two regions promote exon 7 inclusion, it appears that the net effect of region A is also inhibitory, even in SMN1, in spite of the presence of the SF2/ASF site (+6 to +12). Region B and its downstream 5′ splice site comprise the above-mentioned inhibitory stem-loop structure that hinders U1 binding to the 5′ splice site of intron 7.25 Moreover, two ISSs with unknown mechanisms of action were reported earlier: One, in intron 6 (−75 to −89), named element 1, was identified in the context of an exon-trapping vector and an SMN2 minigene;35 the other one, in intron 7 (+10 to +24), named N1, is a potent ISS.36 Finally, the A100G transition in SMN2 intron 7 was recently reported to create another hnRNP A1 binding site, exacerbating the repression of exon 7 splicing caused by the C6T change.15
cis-elements are useful targets for deliberate manipulation of pre-mRNA splicing with antisense molecules. For example, splice sites, ESEs, and ISEs are ideal ASO targets for promotion of exon skipping; this strategy has been employed, e.g., in the development of a treatment for Duchenne muscular dystrophy.37,38 On the other hand, ESSs and ISSs represent ideal ASO targets for promotion of exon inclusion. Recently, we screened the entire SMN2 exon 7 by tiling ASOs, followed by a higher-resolution ASO walk through two promising regions; we identified two putative ESSs and corresponding ASOs with therapeutic potential for SMA.30
In this study, we applied a similar ASO-tiling method to search for inhibitory cis-acting elements residing in the flanking intron sequences, within 60 nt on each side of exon 7. With the first coarse ASO walk, we identified two ISSs: a new one in intron 6 and one in intron 7 that corresponds to a recently reported ISS.36 With the secondary microwalk, we optimized ASOs with different lengths. By using several assays, we discovered that the intron 7 ISS consists of two weak hnRNP A1 or hnRNP A2 (UniProt accession number P22626-2) motifs, which, working together, strongly inhibit exon 7 inclusion in the SMN2 context. Moreover, on the basis of our results in cell-free extracts and in cultured cells, we further tested selected ASOs that abrogate the intron 7 ISS in hSMN2 transgenic mice, hemizygote or wild-type (WT) at the mouse Smn locus. We demonstrate that these ASOs strongly promote exon 7 inclusion in the transgene mRNA in liver, after intravenous administration. Our results show that the high-resolution ASO-tiling approach can identify exonic and intronic elements or structures that modulate splicing positively or negatively. Furthermore, they highlight the therapeutic potential of some of these ASOs in the context of SMA, provided that they can be effectively delivered to the central nervous system (CNS).
Material and Methods
Oligonucleotide Synthesis
Synthesis and purification of chimeric 2′-O-methoxyethyl-modified oligonucleotides with phosphodiester or phosphorothioate backbone were performed with an Applied Biosystems 380B automated DNA synthesizer as described.39 The oligonucleotides with phosphodiester backbone for experiments in cell culture were dissolved in water, and the ones with phosphorothioate backbone for experiments in mice were dissolved in saline. The sequences of all the oligonucleotides are shown in Table 1.
Table 1.
MOE ASOs Used in the Two-Step Intron Walks
| ASO Walk | ASO # | ASO Sequence | Target |
|---|---|---|---|
| Initial Walk in Intron 6a | |||
| 15–01 | 5′-CTGTAAGGAAAATAA-3′ | −1 to −15 | |
| 20–06 | 5′-AGGAAAATAAAGGAA-3′ | −6 to −20 | |
| 25–11 | 5′-AATAAAGGAAGTTAA-3′ | −11 to −25 | |
| 30–16 | 5′-AGGAAGTTAAAAAAA-3′ | −16 to −30 | |
| 35–21 | 5′-GTTAAAAAAAATAGC-3′ | −21 to −35 | |
| 40–26 | 5′-AAAAAATAGCTATAT-3′ | −26 to −40 | |
| 45–31 | 5′-ATAGCTATATAGATA-3′ | −31 to −45 | |
| 50–36 | 5′-TATATAGATATAGAT-3′ | −36 to −50 | |
| 55–41 | 5′-AGATATAGATAGCTA-3′ | −41 to −55 | |
| 60–46 | 5′-TAGATAGCTATATAT-3′ | −46 to −60 | |
| Initial Walk in Intron 7a | |||
| 01–15 | 5′-TGCTGGCAGACTTAC-3′ | 1 to 15 | |
| 06–20 | 5′-CATAATGCTGGCAGA-3′ | 6 to 20 | |
| 11–25 | 5′-ACTTTCATAATGCTG-3′ | 11 to 25 | |
| 16–30 | 5′-GATTCACTTTCATAA-3′ | 16 to 30 | |
| 21–35 | 5′-AGTAAGATTCACTTT-3′ | 21 to 35 | |
| 26–40 | 5′-ACAAAAGTAAGATTC-3′ | 26 to 40 | |
| 31–45 | 5′-GTTTTACAAAAGTAA-3′ | 31 to 45 | |
| 36–50 | 5′-ATAAAGTTTTACAAA-3′ | 36 to 50 | |
| 41–55 | 5′-AAACCATAAAGTTTT-3′ | 41 to 55 | |
| 46–60 | 5′-TCCACAAACCATAAA-3′ | 46 to 60 | |
| Microwalk in Intron 6a | |||
| 51–37 | 5′-ATATAGATATAGATA-3′ | −37 to −51 | |
| 52–38 | 5′-TATAGATATAGATAG-3′ | −38 to −52 | |
| 53–39 | 5′-ATAGATATAGATAGC-3′ | −39 to −53 | |
| 54–40 | 5′-TAGATATAGATAGCT-3′ | −40 to −54 | |
| 56–42 | 5′-GATATAGATAGCTAT-3′ | −42 to −56 | |
| 57–43 | 5′-ATATAGATAGCTATA-3′ | −43 to −57 | |
| 58–44 | 5′-TATAGATAGCTATAT-3′ | −44 to −58 | |
| 59–45 | 5′-ATAGATAGCTATATA-3′ | −45 to −59 | |
| 48–37 | 5′-ATATAGATATAG-3′ | −37 to −48 | |
| 49–38 | 5′-TATAGATATAGA-3′ | −38 to −49 | |
| 50–39 | 5′-ATAGATATAGAT-3′ | −39 to −50 | |
| 51–40 | 5′-TAGATATAGATA-3′ | −40 to −51 | |
| 52–41 | 5′-AGATATAGATAG-3′ | −41 to −52 | |
| 53–42 | 5′-GATATAGATAGC-3′ | −42 to −53 | |
| 54–43 | 5′-ATATAGATAGCT-3′ | −43 to −54 | |
| 55–44 | 5′-TATAGATAGCTA-3′ | −44 to −55 | |
| 56–45 | 5′-ATAGATAGCTAT-3′ | −45 to −56 | |
| Microwalk in Intron 7a | |||
| 08–25 | 5′-ACTTTCATAATGCTGGCA-3′ | 8 to 25 | |
| 09–26 | 5′-CACTTTCATAATGCTGGC-3′ | 9 to 26 | |
| 10–27 | 5′-TCACTTTCATAATGCTGG-3′ | 10 to 27 | |
| 11–28 | 5′-TTCACTTTCATAATGCTG-3′ | 11 to 28 | |
| 15–29 | 5′-ATTCACTTTCATAAT-3′ | 15 to 29 | |
| 14–28 | 5′-TTCACTTTCATAATG-3′ | 14 to 28 | |
| 13–27 | 5′-TCACTTTCATAATGC-3′ | 13 to 27 | |
| 12–26 | 5′-CACTTTCATAATGCT-3′ | 12 to 26 | |
| 10–24 | 5′-CTTTCATAATGCTGG-3′ | 10 to 24 | |
| 09–23 | 5′-TTTCATAATGCTGGC-3′ | 9 to 23 | |
| 08–22 | 5′-TTCATAATGCTGGCA-3′ | 8 to 22 | |
| 07–21 | 5′-TCATAATGCTGGCAG-3′ | 7 to 21 | |
| 18–29 | 5′-ATTCACTTTCAT-3′ | 18 to 29 | |
| 17–28 | 5′-TTCACTTTCATA-3′ | 17 to 28 | |
| 16–27 | 5′-TCACTTTCATAA-3′ | 16 to 27 | |
| 15–26 | 5′-CACTTTCATAAT-3′ | 15 to 26 | |
| 14–25 | 5′-ACTTTCATAATG-3′ | 14 to 25 | |
| 13–24 | 5′-CTTTCATAATGC-3′ | 13 to 24 | |
| 12–23 | 5′-TTTCATAATGCT-3′ | 12 to 23 | |
| 11–22 | 5′-TTCATAATGCTG-3′ | 11 to 22 | |
| 10–21 | 5′-TCATAATGCTGG-3′ | 10 to 21 | |
| Control Oligonucleotideb | |||
| 00–00 | 5′-TGCATCTCATTGTAG-3′ | None | |
Each ASO designation corresponds to the 5′ and 3′ nucleotide numbers of the intron sense sequence to which the ASO is complementary.
The sequence of the control ASO is unrelated to SMN2 exon 7 or to introns 6 and 7.
Constructs
SMN minigene constructs were pCI-SMN1, pCI-SMN2, pEGFP-SMN1, and pEGFP-SMN2, as described.30 In brief, the two minigenes comprise the 111 nt exon 6, a 200 nt shortened intron 6, the 54 nt exon 7, the 444 nt intron 7, and the first 75 nt of exon 8. A consensus 5′ splice site was placed at the 3′ end of exon 8 in pCI-SMN1 and pCI-SMN2 for the enhancement of exon 7 splicing in vitro. Out of the total four nucleotide differences in these regions that occur naturally between endogenous SMN1 and SMN2, two were carried over to the two minigenes, C6T in exon 7 and G−44A in intron 6 (which is part of the region we targeted with an ASO walk), but not A100G and A215G in intron 7, which until recently were thought to play no role in SMN2 exon 7 skipping;13,14 note that a more recent study reported a contribution of the intron 7 A100G change to the repression of SMN2 exon 7 inclusion.15 Plasmid pCGT7-A1 was generated previously.40 hnRNP A2 complementary DNA (cDNA) was amplified with a set of primers, NheI-F (5′-CCGCTAGCGAGAGAGAAAAGGAACAGTTC-3′) and BclI-R (5′-GATGATCAGTATCGGCTCCTCCCAC-3′), digested with NheI and BclI, and subcloned into the XbaI-BamHI sites of pCGT7-A1.
In Vitro Splicing
Plasmids pCI-SMN1 and pCI-SMN2 were linearized with SalI and then used for in vitro transcription with T7 RNA polymerase (Promega) in the presence of α-32P-UTP and 7Me-GpppG cap analog for the generation of in vitro splicing substrates, which were purified by denaturing polyacrylamide gel electrophoresis (PAGE) and spliced in HeLa cell nuclear extract, as described.6,30,41,42 In brief, we incubated 8 fmol transcript in 10 μl splicing reactions containing 3 μl nuclear extract and 1.6 mM MgCl2 at 37°C for 3.5 hr. RNA was then extracted and separated by 8% denaturing PAGE, and phosphorimage analysis with an Image Reader FLA-5100 (FujiFilm Medical Systems) followed. The extent of exon 7 inclusion was calculated as a percentage of the total amount of spliced mRNA. The signal intensity of each mRNA isoform band was normalized according to its U content.
Cell Culture and Transfection
HEK293 cells, SMA type I homozygous and carrier fibroblasts (3813 and 3814, Coriell Cell Repositories) were cultured in Dulbecco's modified Eagle's medium (DMEM, Invitrogen) containing 10% (v/v) fetal bovine serum and antibiotics (100 U/ml penicillin and 100 μg/ml streptomycin). For transfection of MOE oligonucleotides and plasmids into HEK293 cells, electroporation was carried out as described, and puromycin selection followed.30 For transfection of MOE oligonucleotides into 3813 cells, Lipofectin (Invitrogen) was used according to the manufacturer's instructions.
RT-PCR
The reverse transcriptase-polymerase chain reaction (RT-PCR) primers and procedures for the amplification of transcripts derived from all of the minigene constructs, or from the endogenous SMN1 and SMN2 genes, were as described.30 A pair of human-specific primers was used for the amplification of human SMN2 transcripts in RNA samples from transgenic mouse tissues: E4-33to55-F (5′-AAGTGAGAACTCCAGGTCTCCTG-3′) and E8-15to36-R (5′-GTGGTGTCATTTAGTGCTGCTC-3′). PCR products from the transcripts of endogenous SMN1 and SMN2 genes were digested with DdeI so that SMN1 and SMN2 cDNAs could be separated, as described.30 All PCR products were labeled with α-32P-dCTP and analyzed by 6% or 8% native PAGE, and phosphorimage analysis followed. The extent of exon 7 inclusion was calculated as described,30 and the signal intensity of each cDNA band was normalized according to its G+C content.
RNA-Affinity Chromatography
RNA-affinity chromatography was performed as described.7,43 RNA oligonucleotides WT (5′-CCAGCATTATGAAAGT-3′), A12C (5′-CCCGCATTATGAAAGT-3′), A23C (5′-CCAGCATTATGAACGT-3′), and 2A-2C (5′-CCCGCATTATGAACGT-3′) were purchased from Integrated DNA Technologies (Coralville, IA). Sixty micrograms of each RNA was oxidized with sodium m-periodate in a 24 μl reaction and then mixed with 100 μl (1:1 slurry) of adipic-acid-dihydrazide agarose beads (Sigma) by rotation overnight at 4°C. A 250 μl in vitro splicing reaction mix including 100 μl of HeLa nuclear extract was added to 50 μl of RNA-bound beads equilibrated with buffer D containing 0.1 M KCl. Mixtures were incubated at 30°C for 40 min and divided into two aliquots for washing three times with buffer D containing either 150 mM or 300 mM KCl. After the final wash, the beads were resuspended in 50 μl of 1× Laemmli buffer and heated at 100°C for 5 min for the elution of bound proteins. Twelve microliters of each protein sample was loaded on a 12% sodium dodecyl sulfate (SDS) polyacrylamide gel for Coomassie-Blue staining, and 1 μl was loaded for western blotting.
Western Blotting
Protein samples separated by 12% SDS-PAGE were electroblotted onto nitrocellulose membranes. The blots were then probed with monoclonal antibodies anti-hnRNP A/B family (A1/UP1-62), anti-hnRNP A1 (A1/UP1-55), anti-hnRNP A2/B1 (DP3B3; Abcam), anti-T7 (T7-Tag®, Novagen), anti-SMN (BD Transduction Laboratories) or anti-α-tubulin (Sigma), followed by horseradish-peroxidase-conjugated goat anti-mouse secondary antibody (Pierce). Protein signals were detected with Lumi-Light Western Blotting Substrate (Roche Diagnostics).
Administration of Oligonucleotides to hSMN2 Transgenic Mice
All mouse experiments were performed according to protocols approved by Cold Spring Harbor Laboratory. Thirty-two adult human SMN2 transgenic mice, male or female, hemizygote or WT at the mouse Smn locus, were tested. ASOs, dissolved in 0.9% saline solution, were injected through the tail vein at a dose of 25 mg/kg, twice a week for every mouse. Mice 1–8 were injected with saline alone; mice 9–16 were injected with control ASO 00–00; mice 17–24 were injected with 15-mer ASO 09–23, and mice 25–32 were injected with 18-mer ASO 10–27. Mice 1, 2, 9, 10, 17, 18, 25, and 26 were sacrificed after 1 week; mice 3, 4, 11, 12, 19, 20, 27, and 28 were sacrificed after 2 weeks; mice 5, 6, 13, 14, 21, 22, 29, and 30 were sacrificed after 3 weeks; and mice 7, 8, 15, 16, 23, 24, 31, and 32 were sacrificed after 4 weeks. Mouse tissues and organs, including liver, thigh muscles, kidney, and spinal cord, were snap frozen in liquid N2 and kept at −70°C. For extraction of RNA samples, 0.1 g of mouse tissue was pulverized in liquid N2 with mortar and pestle, and homogenized with 1 ml of Trizol (Invitrogen). Total RNA was then isolated according to the manufacturer's directions.
Results
ASO Walk along the Proximal Intronic Regions Flanking Exon 7
Positive or negative signals, including ISSs residing upstream of a 3′ splice site or downstream of a 5′ splice site, can strongly affect exon recognition. To identify potential ISSs that inhibit SMN2 exon 7 inclusion, we systematically screened 60 nt of intronic sequences on either side of exon 7. We used 15-mer 2′-O-methoxyethyl ribose (MOE)-modified phosphodiester ASOs, with 10 ASOs targeting each flanking intronic region. Neighboring ASOs overlapped by 10 nt (Figure 1, Table 1). ASOs with the MOE modification throughout show nuclease resistance, enhanced affinity for hybridization to complementary RNA, and do not support cleavage of the target mRNA by RNase H;44 this class of compound is highly effective at modifying gene expression by binding to RNA and modifying pre-mRNA splicing patterns.44
Figure 1.

Schematic Representation of the Binding Sites for the 20 MOE ASOs Used in the Initial ASO Walk along the Two Flanking Intronic Regions of Exon 7
(A) ASO walk at the end of intron 6.
(B) ASO walk at the beginning of intron 7.
The position of complementarity of each ASO is indicated by a horizontal line above the sequence.
We first tested each ASO by using a cell-free splicing assay with a radiolabeled SMN2 minigene transcript (Figure 2A).6,30 Two hundred nanomolar of each ASO was included in a standard in vitro splicing reaction.42 The SMN1 minigene transcript without ASO treatment was used as a positive control for exon inclusion, and treatment with an unrelated oligonucleotide, 00–00, was used as a negative control. Compared with the negative control, addition of ASO 55–41 or either of two overlapping ASOs, 11–25 and 16–30, led to a pronounced increase in exon 7 inclusion, with 11–25 giving the strongest effect. These results suggest the existence of two ISSs: one in intron 6 and one in intron 7. ASO 55–41, which is complementary to nt −41 to −55 of intron 6, nearly doubled the extent of exon 7 inclusion. ASO 16–30, whose target overlaps by 10 nt with that of ASO 11–25, was less effective than 11–25, suggesting that the ISS in intron 7 is located within or overlapping ASO 11–25′s target sequences. As expected, four ASOs (15–01, 20–06, 25–10, and 30–16) that target the 3′ splice site, polypyrimidine tract, or branch point sequence in intron 645 strongly inhibited exon 7 inclusion. ASO 01–15 also strongly inhibited exon 7 inclusion, and we presume that it competes with U1 snRNA for binding to the 5′ splice site of intron 7.
Figure 2.

Analysis of the 20 MOE ASOs by Splicing In Vitro and in Cells
The diagrams on the right indicate the mobilities of the various RNA species. The percentage of exon 7 inclusion in each lane was calculated as described in Material and Methods and is indicated below each phosphorimage diagram. For the two in vivo splicing assays, each ASO at a concentration of 10 μM and 2 μg of pBabe-Puro was cotransfected with or without pEGFP-SMN2 into HEK293 cells. Two days after transfection, cells were collected for total RNA extraction, and RT-PCR was performed for the analysis of SMN2 pre-mRNA splicing patterns.
(A) Each ASO at a concentration of 200 nM was tested by in vitro splicing with an SMN2 minigene substrate. ASO 00–00 was used as a negative control, and SMN1 was used as a positive control. The radiolabeled RNAs were analyzed by 8% denaturing PAGE.
(B) The 20 ASOs were cotransfected with a pEGFP-SMN2 minigene, and RT-PCR products were analyzed by 8% native PAGE.
(C) The effects of the 20 ASOs were analyzed with transcripts from the endogenous SMN2 gene in HEK293 cells. RT-PCR products were digested with DdeI so that SMN1 could be distinguished from SMN2 by 6% native PAGE. FL indicates full-length, and Δ7 indicates exon 7 deleted mRNA.
To further examine the effects of individual ASOs on exon 7 inclusion, we measured splicing of SMN2 minigene transcripts, as well as endogenous transcripts, in HEK293 cells.30 We cotransfected the minigene plasmid pEGFP-SMN2 with pBabe-Puro and each of the 20 MOE ASOs by electroporation. pEGFP-SMN2 was chosen because it gives more pronounced exon 7 skipping as compared to pCI-SMN2,30 and therefore the effects of ASOs on exon 7 inclusion can be more readily observed. Transfected cells were selected with puromycin.30 Two days after transfection, both the transiently expressed mRNA (Figure 2B) and the endogenous SMN2 mRNA (Figure 2C) were analyzed by RT-PCR with appropriate primers. For the endogenous transcripts, SMN1 and SMN2 spliced mRNAs can be distinguished from each other after digestion with DdeI.46,47 Both minigene and endogenous-gene assays gave results consistent with those obtained by cell-free splicing (Figure 2A). We also observed that the ASOs affected the endogenous SMN1 transcripts, in addition to the SMN2 transcripts.
In summary, the ASO tiling through the two intronic flanks of exon 7, in combination with three different splicing assays, identified one moderate ISS and one strong ISS in intron 6 and intron 7, respectively. Blocking the intron 7 ISS in SMN2 pre-mRNA promoted exon 7 inclusion to a level comparable to that of the SMN1 pre-mRNA, and therefore, ASO 11–25 has considerable therapeutic potential.
Two Motifs within the Intron 7 ISS Region Mediate Repression of Exon 7 Inclusion
ASO 11–25 displayed the strongest stimulatory effect on exon 7 inclusion, indicating that its target harbors at least one inhibitory cis-element. To determine the mechanism underlying repression, we introduced a series of mutations within and surrounding the ASO-target region, in the context of the pCI-SMN2 minigene plasmid (Figure 3A). Because this region is purine rich and C poor, we individually mutated all As, Gs, and Us into Cs and all Cs into Us. Each plasmid was electroporated into HEK293 cells, and the transiently expressed mRNAs were analyzed by RT-PCR.
Figure 3.

Effects of Mutations in and around the Intron 7 ISS on SMN2 Exon 7 Inclusion
(A and C) WT and mutant intron 7 sequences. Mutations are shaded, and deletions are indicated by dashes.
(B and D) WT pCI-SMN2 and mutant minigene plasmids (5 μg) were electroporated into HEK293 cells. Total RNA was collected two days after transfection and analyzed by radioactive RT-PCR. The radiolabeled PCR products were analyzed by 8% native PAGE and detected and quantitated with a phosphorimager.
Two AG dinucleotides (+12 to +13 and +23 to +24) proved to be critical for silencing: Each of the mutations in these four nucleotides (A12C, G13C, A23C, and G24C) markedly increased exon 7 inclusion compared to the parental (WT) SMN2 minigene (Figure 3B). A21C also noticeably increased exon 7 inclusion (Figure 3B). Interestingly, mutant C11U, which creates a match to the UAG core (+11 to +13) of the hnRNP A1 consensus motif,48–50 further inhibited exon 7 inclusion.
To verify that the two AG dinucleotides are important for silencing—as opposed to the mutations fortuitously creating positive elements—we generated additional mutants that disrupt one or both AG dinucleotides: single mutants A12U, G13U, A23U, and G23U; double mutant 2A-2C (A12C + A23C); single-deletion mutants Δ12A, Δ13G, ΔAG1st (deleting the 12A,13G dinucleotide), Δ23A, Δ24G, ΔAG2nd (deleting the 23A,24G dinucleotide); and double-deletion mutant Δ2AGs (deleting both AG dinucleotides). All of these mutants gave pronounced increases in exon 7 inclusion, and in particular, the two substitution or deletion mutants that target both sites simultaneously (2A-2C and Δ2AGs) resulted in more than 80% exon 7 inclusion. Considering that a hexamer or heptamer sequence is generally sufficient for binding one splicing-repressor molecule,51 these data suggest the existence of two separate motifs within the intron 7 ISS, each encompassing one of the AG dinucleotides that are separated by nine nucleotides. We refer to the upstream motif in the silencer as motif 1, and to the downstream one as motif 2. The increase in exon 7 inclusion in the double-substitution 2A-2C mutant or the double-deletion Δ2AGs mutant is approximately the sum of the effects of the two single-substitution A12C and A23C mutants or of the single-deletion ΔAG1st and ΔAG2nd mutants, respectively (Figure 3), suggesting an additive effect rather than a synergistic effect of the two motifs on splicing repression.
The Intron 7 ISS Is Bound Specifically by hnRNP A1 and A2
The two critical AG dinucletides in the intron 7 ISS provide a hint about the identity of the repressor(s) because previous reports demonstrated that an AG dinucleotide is critical for hnRNP A1 high-affinity binding.49,50,52 On the basis of our mutagenesis analysis, we hypothesized that the two AG dinucleotides are part of two weak hnRNP A1 binding motifs that, owing to their close juxtaposition, make up a strong hnRNP A1 binding element. To test our hypothesis, we conducted RNA-affinity chromatography.43 A WT 16-mer intron 7 RNA fragment (+10 to +25) comprising the two potential weak hnRNP A1 motifs was covalently linked to agarose beads via the 3′ end,43 and incubation with HeLa cell nuclear extract followed. Three mutant RNAs (A12C, A23C, and 2A-2C) were used as controls. Proteins that remained tightly bound to each RNA after washing at two different salt concentrations (150 mM and 300 mM) were analyzed by SDS-PAGE and then Coomassie-blue staining or western blotting (Figure 4). Two strong bands (34 kDa and 36 kDa) were observed in the WT RNA sample by Coomassie-blue staining, and the corresponding bands were weaker with the mutant RNA samples, especially the double mutant. In particular, after the beads were washed in 300 mM KCl, the two bands disappeared in the case of the 2A-2C double-mutant RNA, in which both AG dinucleotides were mutated. These data indicate that both proteins specifically bind the intron 7 ISS, and the binding is dependent on the two potential hnRNP A1 motifs. The lower band has the expected mobility of hnRNP A1, whereas the upper band could be the 36 kDa hnRNP A2 protein, a closely related hnRNP A/B family member with similar effects on SMN2 exon 7 splicing regulation as hnRNP A1.7,32,53
Figure 4.
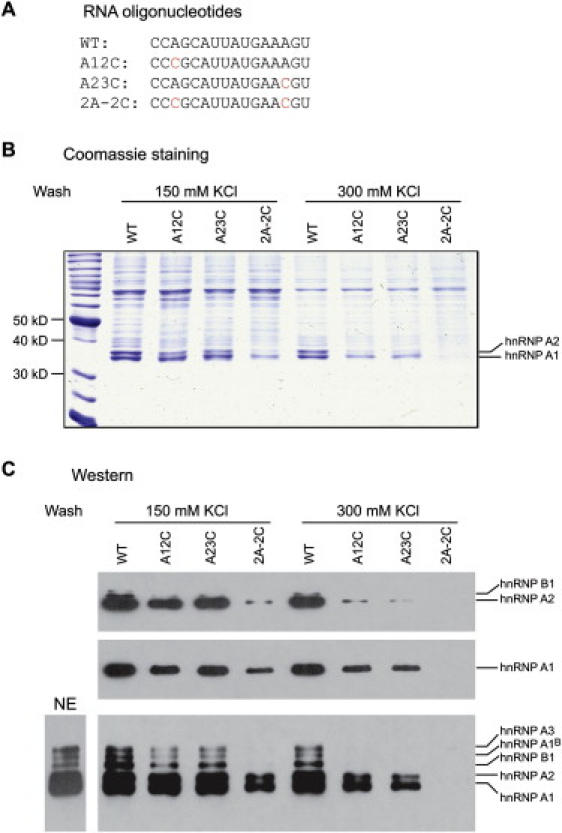
Analysis of Proteins Bound to the Intron 7 ISS by RNA-Affinity Chromatography
(A) Four RNA oligonucleotides corresponding to the WT and three mutant sequences shown were used for RNA-affinity chromatography.
(B) Agarose beads covalently linked to the RNAs shown in (A) were incubated with HeLa cell nuclear extract under splicing conditions, and the beads were washed three times at the indicated salt concentrations. Bound proteins were eluted with SDS and analyzed by SDS-PAGE with Coomassie-Blue staining. The migration of size markers and hnRNP A1 and A2 are indicated.
(C) Western-blotting analysis of the eluted proteins with monoclonal antibodies that recognize only hnRNP A2 and B1 (top), hnRNP A1 (middle), and all the hnRNP A/B family proteins (bottom); HeLa cell nuclear extract (NE) was also analyzed for the determination of the relative signals of the various hnRNP A/B family proteins in the starting material (bottom left).
To verify the identity of the two proteins isolated by RNA-affinity pulldown, we used three different monoclonal antibodies: A1/UP1-55 recognizes only hnRNP A1; DP3B3 recognizes hnRNP A2 and its lower-abundance isoform hnRNP B1 (∼38 kDa); and A1/UP1-62 recognizes all the hnRNP A/B family proteins (Figure 4C). Western blotting clearly showed that the two prominent bands are hnRNP A1 and A2, respectively, and also that the other hnRNP A/B family proteins likewise bind specifically to the silencer. hnRNP B1, A1B, and A3 were not as prominent as A1 and A2 in the pulldown material (Figure 4B), but this may simply reflect their lower abundance in HeLa cell nuclear extract (Figure 4C). These data demonstrate that the intron 7 ISS is recognized specifically by hnRNP A/B proteins, in particular the abundant hnRNP A1 and A2. Other RNA-binding proteins also interacted strongly with the intron 7 RNA fragment, especially a band of approximately 75 kDa (Figure 4B); however, this and other proteins appeared to interact nonspecifically, showing no difference in binding between the WT and all mutant RNAs.
hnRNP A/B family proteins are a group of structurally and functionally similar proteins. In particular, hnRNP A1 and A2 are both abundantly expressed, share approximately 70% amino acid sequence identity, and inhibit both 5′ and 3′ splice-site recognition, or promote distal 5′ splice site selection while suppressing proximal splice site use.53 Several studies showed that hnRNP A2 also binds hnRNP-A1-specific motifs,43,54,55 so it is not surprising that we pulled down both proteins with RNA-affinity chromatography. The UAG motif has been widely described as a critical core of the hnRNP A1 binding motif;43,48,50,56–58 however, CAG has also been found in SELEX winner sequences,49 and especially with equilibrium-binding assays, multiple sequences that contain one or two CAG motifs but no UAG motif have been demonstrated to have strong binding affinity for hnRNP A1.52 It is likely that CAG represents the core of a suboptimal hnRNP A1 binding element in the motif 1 region; this fits with the observation that the mutant C11U, which improves the match to the consensus, displayed stronger inhibition of exon 7 inclusion (Figure 3). In the motif 2 region, the mutant A22C strongly promoted exon 7 skipping, suggesting that CAG (+22 to +24 in mutant A22C) is a stronger motif than AAG in this context for recognition by hnRNP A1/A2. Because 21A in the motif 2 region is also important, it appears that AAAG (+21 to +24) forms another core of a weak hnRNP A1/A2 binding site in the context of the SMN2 intron 7 ISS. In agreement with our result, the 3′ splice site of adenovirus type 2 exon 1a, which has an AAAG motif but neither UAG nor CAG motifs, binds hnRNP A1.52 We conclude that the minimal size of the intron 7 ISS is CAGCTTATGAAAG (+11 to + 24), with one hnRNP A1 motif (CAG) at the 5′ end, and another one (AAAG) at the 3′ end.
Effects of hnRNP A1/A2 Overexpression
Having shown direct binding of hnRNP A1/A2 to the intron 7 ISS, we next tested the prediction that overexpression of hnRNP A1/A2 should lead to stronger inhibition of exon 7 inclusion via the specific intron 7 hnRNP A1 silencer. In fact, previous work demonstrated that overexpression of hnRNP A1 does inhibit exon 7 inclusion for both endogenous SMN1 and SMN2 genes, suggesting the existence of a shared hnRNP-A1-dependent ESS or ISS, though its location remained unknown.7 To examine the interplay between hnRNP A1/A2 overexpression and the intron 7 ISS, we analyzed three of the above mutants, A12C, A23C, and 2A-2C, in the SMN1-minigene context, and compared them to the WT SMN1 minigene; the first two mutants disrupt the first and second motif, respectively, and the third mutant disrupts both motifs. We used an SMN1 minigene rather than an SMN2 minigene because we wanted to minimize the potential influence of other hnRNP A1 binding sites. So far, no hnRNP A1 binding sites have been mapped in SMN1, whereas two such sites have been reported to be present in SMN2.15,32,33
Each mutant or WT SMN1 minigene plasmid, together with an hnRNP A1 or A2 expression plasmid, was electroporated into HEK293 cells, and RNA and protein samples extracted after 48 hr were used for RT-PCR and western-blot assays. Both hnRNP A1 and A2 plasmids express N-terminal T7-tagged proteins to facilitate detection. As shown in Figure 5, when transfected alone, each of the mutant and WT SMN1 minigenes gave a similar extent of exon 7 inclusion; in contrast, when 3 μg of hnRNP A1 plasmid was cotransfected with these minigenes, exon 7 inclusion was reduced to 35% for the WT SMN1 minigene and to 82% for the 2A-2C double mutant, with the A12C and A23C single mutants giving intermediate reductions in exon 7 inclusion (52% and 62%, respectively).
Figure 5.
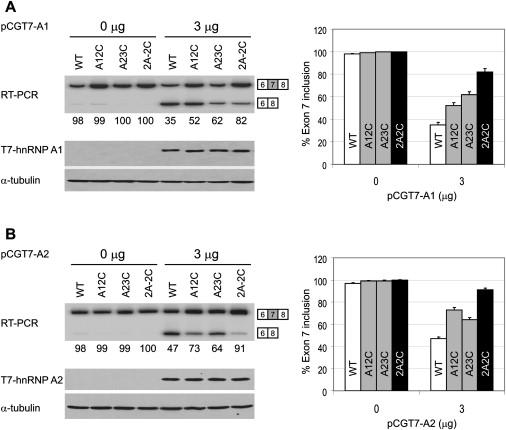
Effects of hnRNP A1 or A2 Overexpression on Exon 7 Inclusion in SMN1 Minigene Transcripts
HEK293 cells were transfected with 5 μg of the indicated WT or mutant pCI-SMN1 plasmids (described in Figures 3A and 3C). The indicated amounts of pCGT7-A1 (A) or pCGT7-A2 (B) expressing N-terminal T7-tagged hnRNP A1 or A2 proteins were cotransfected with the SMN1 reporters. Two days after transfection, total RNA was collected and radioactive RT-PCR was performed to measure the extent of exon 7 inclusion by 8% native PAGE and phosphorimage analysis. The expression of tagged hnRNP A1 or A2 was verified by western blotting with monoclonal antibody against the T7 tag. The histograms on the right show the corresponding quantitation from three independent experiments. Error bars represent the standard deviation.
When 3 μg of hnRNP A2 plasmid was cotransfected, we observed slightly weaker but otherwise similar inhibitory effects for all the WT and mutant minigenes (Figure 5B). These data indicate that the inhibitory effects of hnRNP A1/A2 depend on the intron 7 ISS, and the greater sensitivity of the WT SMN1 minigene to these repressors reflects the presence of the two tandem hnRNP A1/A2 motifs. Interestingly, we reproducibly observed that hnRNP A1 overexpression had a stronger inhibitory effect with the A12C mutant than with the A23C mutant, whereas hnRNP A2 overexpression gave the opposite pattern, suggesting that these two closely related repressors are not completely identical in how they recognize and bind to RNA.
Improvement of hnRNP A1/A2 Motifs and Consequences for Exon 7 Splicing
Having shown that disruption of the weak, tandem hnRNP A1/A2 motifs in the intron 7 ISS abrogates the repression of exon 7 inclusion, we next asked whether improving these two motifs by mutagenesis results in a greater extent of exon 7 skipping (Figure 6). In the motif 1 region, C11U, which should improve hnRNP A1 binding, was already described above (Figures 3A and 3B). We generated three additional mutants of the SMN2 minigene within the motif 1 region: C14G, winner 1(1), and winner 2(1). Winner 1(1) replaces the WT sequence CAGCAU (+11 to +16) with an hnRNP A1 winner sequence, UAGGGU;49 winner 2(1) replaces the same 6 nt sequence with UAGGUC, which is thought to be recognized by RRM2 of hnRNP A149 and is a strong functional hnRNP A1-responsive element (H.O. and A.R.K, unpublished data). In the motif 2 region, we also generated four mutants expected to improve hnRNP A1 binding: A22U, U25G, winner 1(2), and winner 2(2). A22U creates a UAG-containing motif (+22 to +27), but the motif is shifted 3 nt downstream of the original UGAAAG (+19 to +24) motif. U25A creates a hexamer AAGGGA (+22 to +27) that is also shifted 3 nt downstream. All of these seven mutants in the context of the SMN2 minigene displayed greater inhibition of exon 7 inclusion than the parental construct, with the percentage of inclusion ranging from 3% to 37%, compared to 46% for the WT SMN2 minigene (Figure 6B). We also generated these seven mutants, plus mutant C11U, in the context of the SMN1 minigene and observed similar, though, as expected, less pronounced, effects on inhibition of exon 7 inclusion than in the SMN2 minigene context (Figure 6C). The inhibitory effects were stronger when the same motifs were placed in the motif 1 region than in the motif 2 region, perhaps reflecting the shorter distance of motif 1 to the 5′ splice site. Interestingly, the winner 2 sequence UAGGUC resulted in greater inhibition than the winner 1 sequence UAGGGU when placed in the motif 1 region, whereas the relative effects were reversed in the context of the motif 2 region, pointing to the contribution of position and/or context in the activity of each motif.
Figure 6.
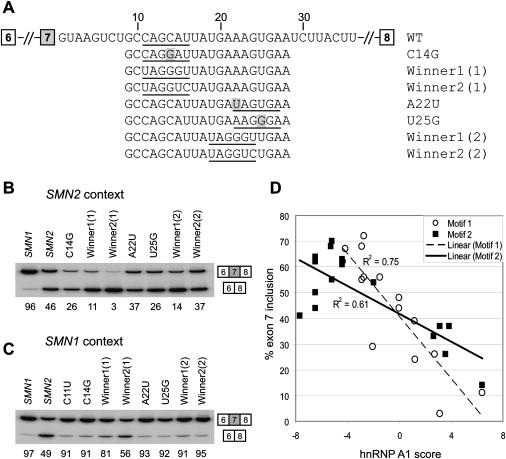
Effect of Improving the hnRNP A1/A2 Motifs in the Intron 7 ISS on Exon 7 Splicing
(A) Sequences of the WT intron 7 ISS and mutants with improved hnRNP A1/A2 motifs. The hnRNP A1/A2 motifs are underlined. Single mutations are shaded.
(B and C) RT-PCR analysis showing the effects of improving the hnRNP A1/A2 motifs in either the motif 1 or motif 2 region. Mutants were tested in both SMN2 and SMN1 minigene contexts. Five micrograms of WT pCI-SMN2, pCI-SMN1, or each mutant plasmid was transfected into HEK293 cells by electroporation. Total RNA was extracted 2 days later and analyzed by radioactive RT-PCR and then 8% native PAGE and phosphorimage quantitation.
(D) Quantitative data of mutagenesis analysis, including previous experiments (Figure 2), are presented as a scatter plot. The percentage of exon 7 inclusion was plotted against hnRNP A1 scores based on a PWM with background correction for the base composition of the winner pool.7 Data points for mutants in the motif 1 and motif 2 regions are shown as open circles and solid squares, respectively. Least-squares lines are shown for each data set (dashed line for motif 1 with R2 = 0.75, and solid line for motif 2 with R2 = 0.61).
To analyze the correlation between hnRNP A1 motif scores of the various wild-type and mutant sequences and their effects on exon 7 inclusion, we took advantage of an hnRNP A1 position weight matrix (PWM) with background correction (A1_winBG), which was previously derived from SELEX data.7,49 The percentage of exon 7 inclusion in the SMN2 minigene context was plotted against the calculated hnRNP A1 scores. Because the same motifs had different effects on exon 7 splicing when they were placed in the motif 1 region versus the motif 2 region (Figures 6B and 6C), we provide both datasets (Figure 6D). We observed a strong negative correlation between the extent of exon 7 inclusion of the SMN2 minigene mutants and the corresponding motif scores: The coefficient of determination (R2) is 0.75 for motif 1 and 0.61 for motif 2. Note that because this PWM is based on hnRNP A1 SELEX winners,49 the scores of weak motifs with an AAAG core are generally negative, whereas the scores of weak motifs with a CAG core can be negative or positive depending on the number of nucleotide matches to the consensus hexamer.
Correction of SMN2 Splicing in Transgenic Mice
After elucidating the exact position and mechanism of the intron 7 ISS, we optimized the most potent ASOs that target this silencer and used them to try to rescue SMN2 splicing in mice harboring a human SMN2 transgene. First, we synthesized 38 ASOs of different lengths and examined their effects on splicing of transcripts of the endogenous SMN2 gene in HEK293 cells. The results are summarized in Figure 7. Four 18-mer ASOs displayed the strongest effects, with ASO 10–27 being slightly better than the other three (08–25, 09–26, and 11–28). The most effective 15-mer was ASO 09–23, which was slightly better than ASOs 10–24 and 11–25; the best 12-mer was ASO 10–21, though it was considerably weaker than the 15-mer ASO 09–23. We also examined ASOs 10–27 and 09–23 in SMA type I patient 3813 fibroblasts and found that both ASOs were more efficient in promoting SMN2 exon 7 splicing and increasing SMN protein levels compared with our best two ASOs targeting exon 7 (Figure S1 available online).30
Figure 7.
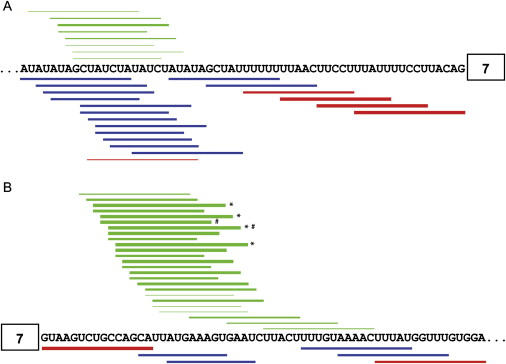
Schematic Diagram of the In Vivo Effects of All Tested Intronic ASOs
Horizontal bars represent ASOs with stimulatory effects (green), inhibitory effects (red), or neutral effects (blue). The thicker the bars, the stronger the effects.
(A) ASOs targeting the 3′ region of intron 6.
(B)ASOs targeting the 5′ region of intron 7. ∗ indicates the four 18-mer ASOs (08–25, 09–26, 10–27, and 11–28) that displayed the strongest stimulatory effects. # indicates the best 15-mer ASO (09–23) and the best 18-mer ASO (10–27) that were tested in hSMN2 transgenic mice (Figure 8).
On the basis of these results in cultured cells, we selected ASOs 10–27 and 09–23 for further work in mice. These ASOs were resynthesized on a larger scale and with a phosphorothioate backbone instead of a phosphodiester backbone, for improved in vivo stability and pharmacokinetics.59 Recipient adult mice of both sexes were transgenic for hSMN2 and hemizygote or WT at the mouse Smn locus.60 ASOs were dissolved in saline and delivered intravenously, twice a week, at 25 mg/kg. Each ASO was administered to eight mice, and tissues and organs were harvested after 1, 2, 3, or 4 weeks of treatment (two mice each). As controls, eight mice received saline only, and another eight mice received a 15-mer scrambled-sequence oligonucleotide, ASO 00–00 (Table 1).
We first analyzed splicing changes of hSMN2 transcripts by RT-PCR with a pair of human-specific primers, with total RNA from liver, skeletal muscle (thigh), kidney, and spinal cord (Figure 8). No increase in exon 7 inclusion was observed after treatment with saline or control ASO 00–00 in any of the tissues. However, we observed a striking increase in exon 7 inclusion in liver samples from mice treated with ASOs 10–27 or 09–23 (Figure 8A). Exon 7 inclusion increased from approximately 21% (the average of all saline- and control-ASO-treated mice) to approximately 45% in the livers of mice treated with ASO 09–23 after 1 week administration, and the rate increased to approximately 69% after 2 week treatment, to approximately 83% after 3 week treatment, and to approximately 91% after 4 week treatment. We also detected a greater than 3-fold increase in exon 7 inclusion in kidney, and an approximately 2-fold increase in muscle samples after 3–4 weeks of treatment, though the effects were not as striking as in liver (Figures 8B and 8C). In contrast, we did not observe any increase in hSMN2 exon 7 inclusion in spinal cord (Figure 8D); this was expected, because these ASOs do not penetrate the blood-brain barrier (BBB).61 These data demonstrate that suitable ASOs, when delivered to mouse tissues, are able to correct the splicing defect of the hSMN2 gene transcripts, indicating that these ASOs have excellent therapeutic potential for SMA.
Figure 8.
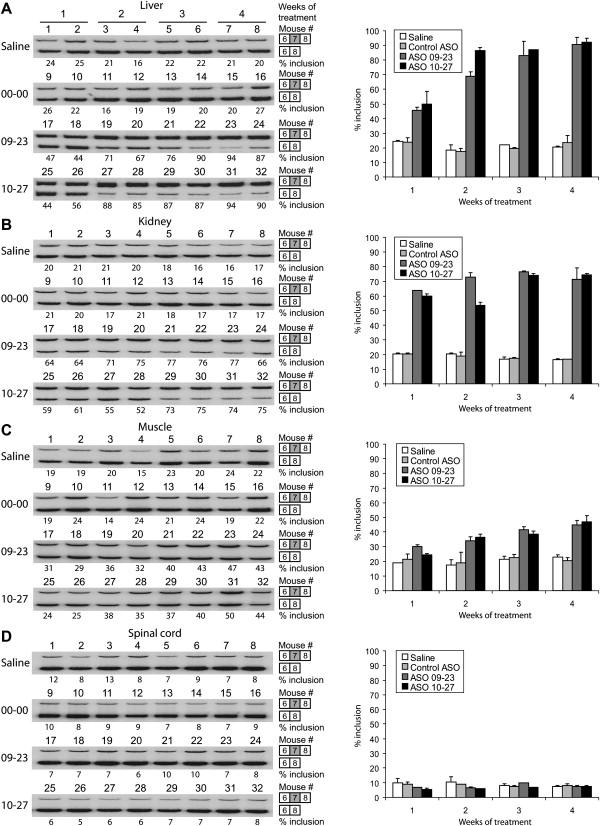
Effects of ASOs 09–23 and 10–27 in hSMN2 Transgenic Mice
ASOs 09–23, 10–27, control ASO 00–00, or saline was delivered intravenously via the tail vein, twice a week, at 25 mg/kg. Each ASO or saline was administered to eight mice, and tissues and organs including liver (A), kidney (B), thigh muscles (C) and spinal cord (D) were harvested after 1, 2, 3, or 4 weeks of treatment (two mice each). Total RNA was extracted from tissues with Trizol reagent, and RT-PCR was carried out with a set of hSMN2-specific primers. Radiolabeled PCR products were analyzed by 8% native PAGE and phosphorimaging. The histograms on the right show the corresponding quantitation. Error bars show standard deviations.
Discussion
Correction of SMN2 exon 7 splicing is an attractive therapeutic approach for SMA because this gene is present in all patients, its exon 7 codes for the correct SMN C-terminal peptide, and there are several ways in which inclusion of this alternative exon can be increased.62 Strategies to promote SMN2 exon 7 inclusion have included cell-based screens for small molecules,63 as well as targeted methods, such as ESSENCE, TOES, and SMaRT.64–66 Antisense technology, which is traditionally used to inhibit gene expression,67 can also be used to modulate pre-mRNA splicing by targeting splice sites or positive or negative elements that affect splice-site selection.30,35,36,38,68–70 In particular, systematic screening for splicing silencers that can be blocked with ASOs is a practical and efficient approach to rescue certain splicing defects. Recently, we used this approach to identify two potential ESSs in SMN2 exon 7 that can be targeted to restore SMN2 exon 7 inclusion.30 Here, we used an analogous ASO-tiling method to systematically map elements within the exon-7-proximal upstream intron 6 and downstream intron 7 sequences, pinpointing the presence of an ISS in each intron. To optimize the length and position of ASOs targeting the intron 7 ISS, which appears to be more potent, we carried out high-resolution tiling in conjunction with cell-based splicing assays. Finally, ASO administration to hSMN2 transgenic mice demonstrated that the optimal ASOs can restore SMN2 exon 7 splicing to a level similar to that of the human endogenous SMN1 gene—about 90% of exon 7 inclusion.60 These data demonstrate that ASOs targeting the intron 7 ISS have significant therapeutic potential.
The intron 7 ISS was recently described and shown to be effective in a heterologous gene context;36 the 15 nt element, dubbed ISS-N1, was further shown to gradually lose its effectiveness when moved farther downstream from the 5′ splice site. 2′-O-methyl-modified ASOs targeting this ISS were shown to correct the SMN2 splicing defect in SMA-patient fibroblasts, increasing SMN protein levels in these cells.36 However, the underlying repression mechanism remained to be defined. Here, by using mutagenesis coupled with cell-based splicing assays, RNA-affinity chromatography, and cDNA overexpression, we demonstrated that splicing repression via the intron 7 ISS is mediated by hnRNP A1 and A2. The ISS not only physically binds hnRNP A1 and the structurally and functionally related protein hnRNP A2, but we further show that it functionally responds to hnRNP A1/A2 protein levels in cells. Although there is a putative hairpin in this region, analysis of mutations that would disrupt or restore the predicted secondary structure failed to uncover any effect (Figure S2).
Pre-mRNA splicing requires the accurate recognition of 5′ and 3′ splice sites. hnRNP A/B family proteins can affect both 5′ and 3′ splice-site selection, in part by antagonizing splicing activators.71 Two potential SMN2-specific hnRNP A1 binding sites have been reported,15,32 whereas no hnRNP A1 binding sites had been identified that repress exon 7 splicing in the context of SMN1. However, knockdown of hnRNP A1 and/or A2 promotes exon 7 inclusion for both SMN2 and SMN1.7,32 In addition, overexpression of hnRNP A1 in cells inhibits both SMN1 and SMN2 exon 7 inclusion.7 These observations suggested the existence of one or more additional hnRNP A1/A2 binding sites present in both SMN1 and SMN2. By using RNA-affinity chromatography, we show that an RNA fragment comprising the intron 7 ISS (+10 to +25) is bound strongly and specifically by hnRNP A1 and A2. This region encompasses two weak hnRNP A1 motifs; two single mutations, A12C and A23C, that disrupt either the first or the second motif reduced the binding of hnRNP A1/A2; and simultaneous mutation of both motifs virtually abrogated binding. The binding of hnRNP A1/A2 to the ISS and its mutants correlated well with the extent of exon 7 skipping in transient transfection experiments. Thus, our data suggest that two juxtaposed weak hnRNP A1/A2 binding sites act additively to form a strong inhibitory element, especially when located near a splice site.
The intron 7 ISS (ISS-N1) was recently reported to comprise nucleotides +10 to +24 (CCAGCATTATGAAAG)36. Our antisense and mutational analyses further sharpen the boundaries of the silencer region (+11 to +24) and establish its constituent motifs. The first hnRNP A1 motif in the element is CAGCAT (+11 to +16), with the core sequence being CAG. Generally, UAG represents a common hnRNP A1 motif core;58,72 however, CAG has also been frequently observed in high-affinity sites identified by SELEX, and/or characterized in equilibrium-binding assays.49,52 The second motif, TGAAAG (+19 to +24), represents a new weak hnRNP A1 motif, with the core nucleotides being AAAG. It has been previously reported that the AG dinucleotide is the only shared feature among various sequences with high affinity for hnRNP A1,52 suggesting that the AG dinucleotide is critical for hnRNP A1 recognition, whereas the individual contributions of the remaining nucleotides of the motif might be context dependent. This notion is consistent with our observation with both SMN2 and SMN1 minigenes that, when we replaced the two natural hnRNP A1 ISS motifs with two SELEX winner hexamers (winner 1: UAGGGU; winner 2: UAGGUC), winner 2 was stronger than winner 1 when placed in the motif 1 region but weaker when placed in the motif 2 region (Figure 6).
The two juxtaposed weak hnRNP A1 motifs make up a strong ISS, illustrating a novel strong splicing silencer pattern: the combination of two or more tandem weak repressor motifs. This type of splicing silencer, which binds two or more repressor molecules and spans at least 14 nt, is unlikely to be captured with previously described computational or cell-based ISS screening methods that assumed ISS lengths of 6–10 nt, but it may represent a common group of strong ISSs, especially taking into account that most 3′ splice sites already comprise one copy of a weak hnRNP A1 motif, such as CAG, or its somewhat stronger version CAGG.
Cooperative binding and propagation of hnRNP A1 along an exon and its flanking introns has been described as a mechanism for antagonizing splicing activation by SR proteins.57,71,73 Therefore, it is reasonable to assume that either of the two hnRNP A1/A2 molecules that bind to the intron 7 ISS and hnRNP A1/A2 molecules bound to other sites, such as the recently reported UAG motif in SMN2 intron 7,15 result in cumulative spreading of hnRNP A1/A2 along the SMN2 exon 7 and its flanking intron sequences, antagonizing the binding of Tra2-β1, SF2/ASF, and other splicing factors that are essential for exon 7 recognition.
Previously, two hnRNP A1 molecules were shown to interact simultaneously with two distant high-affinity sites, such that hnRNP A1 dimerization may loop out segments of the pre-mRNA, affecting splice-site selection.74 However, in the context of the SMN2 gene, we have not thus far observed synergistic effects among different hnRNP A1 motifs (Figures 3 and 5, and Y.H. and A.R.K., unpublished data), as would have been expected from such a bridging model. In the cocrystal structure of the UP1 domain of hnRNP A1 with a 12 nucleotide telomeric single-stranded DNA (ssDNA), d(TTAGGGTTAGGG), UP1 dimerizes and RRM1 and RRM2 within the same protein monomer bind to two separate strands of ssDNA, which are antiparallel.75 Therefore, it is possible that two hnRNP A1 (or A2) molecules bind to the bipartite intron 7 ISS as a dimer.
In the present study, we examined hSMN2 exon 7 inclusion in four transgenic mouse tissues after intravenous administration of ASOs. Liver showed the strongest effects, kidney gave intermediate effects, and muscle gave weak effects, whereas spinal cord showed no change in hSMN2 exon 7 inclusion. These tissue-specific effects are consistent with previous reports that MOE ASOs preferentially distribute to peripheral tissues, and that hepatocytes spontaneously take up these ASOs.61 In addition, the lack of an effect on spinal cord was expected, because of the BBB.
Because of the presence of mouse SMN protein in these mice and the high degree of homology between the murine and human proteins, it is difficult to verify that the increase in full-length mRNA translates into an increase in transgenic SMN protein. The Smn null transgenic mice survive for only a few days in this model,60 which is incompatible with our current delivery protocol. We and others previously showed that ASO-induced increases in full-length hSMN2 mRNA result in increased SMN protein levels, at least in cell culture.30,36 In the future, it may be possible to measure the expected increase in transgenic SMN protein levels in mouse tissues with a suitable human SMN antibody that does not crossreact with the murine protein.
In the context of SMA therapy, these ASOs will either have to be directly administered to the CNS or methods will have to be developed to allow them to efficiently penetrate the BBB. As an example of the first approach, in recent studies of amyotrophic lateral sclerosis (ALS), an MOE ASO designed to inactivate dominant-negative mutant SOD1 transcripts was directly delivered to the CNS of an ALS rat model; spinal-cord motor neurons spontaneously internalized the ASO, resulting in knockdown of the mutant gene.76 Illustrative of the second approach, a 29 amino acid peptide derived from rabies-virus glycoprotein was recently used to facilitate delivery of small interfering RNA (siRNA) across the BBB in mice.77 We plan to explore similar approaches to deliver ASOs that correct hSMN2 exon 7 splicing into transgenic mouse spinal-cord motor neurons.
Supplemental Data
Two figures are available at http://www.ajhg.org/.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/
Universal Protein Resource (UniProt), http://www.pir.uniprot.org/
Acknowledgments
We thank Chaolin Zhang for help with hnRNP A1 PWM analysis and Xavier Roca and Michelle Hastings for useful comments on the manuscript. We also thank A. Burghes for helpful discussions. Y.H. and A.R.K. gratefully acknowledge support for this work from the SMA Foundation, the Muscular Dystrophy Association, the Louis Morin Charitable Trust, and National Institutes of Health grant GM42699. T.A.V. and C.F.B. are employees of Isis Pharmaceutical, the owner of the antisense oligonucleotide chemistry used in this report, and materially benefit either directly or indirectly through stock options. Y.H. and A.R.K., along with their employer, Cold Spring Harbor Laboratory, could materially benefit if a therapeutic for SMA results from this work. A.R.K. serves on the scientific advisory board of two nonprofit SMA foundations.
References
- 1.Hastings M.L., Krainer A.R. Pre-mRNA splicing in the new millennium. Curr. Opin. Cell Biol. 2001;13:302–309. doi: 10.1016/s0955-0674(00)00212-x. [DOI] [PubMed] [Google Scholar]
- 2.Brow D.A. Allosteric cascade of spliceosome activation. Annu. Rev. Genet. 2002;36:333–360. doi: 10.1146/annurev.genet.36.043002.091635. [DOI] [PubMed] [Google Scholar]
- 3.Jurica M.S., Moore M.J. Pre-mRNA splicing: Awash in a sea of proteins. Mol. Cell. 2003;12:5–14. doi: 10.1016/s1097-2765(03)00270-3. [DOI] [PubMed] [Google Scholar]
- 4.Cartegni L., Chew S.L., Krainer A.R. Listening to silence and understanding nonsense: Exonic mutations that affect splicing. Nat. Rev. Genet. 2002;3:285–298. doi: 10.1038/nrg775. [DOI] [PubMed] [Google Scholar]
- 5.Mayeda A., Krainer A.R. Regulation of alternative pre-mRNA splicing by hnRNP A1 and splicing factor SF2. Cell. 1992;68:365–375. doi: 10.1016/0092-8674(92)90477-t. [DOI] [PubMed] [Google Scholar]
- 6.Cartegni L., Krainer A.R. Disruption of an SF2/ASF-dependent exonic splicing enhancer in SMN2 causes spinal muscular atrophy in the absence of SMN1. Nat. Genet. 2002;30:377–384. doi: 10.1038/ng854. [DOI] [PubMed] [Google Scholar]
- 7.Cartegni L., Hastings M.L., Calarco J.A., de Stanchina E., Krainer A.R. Determinants of exon 7 splicing in the spinal muscular atrophy genes, SMN1 and SMN2. Am. J. Hum. Genet. 2006;78:63–77. doi: 10.1086/498853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hutton M., Lendon C.L., Rizzu P., Baker M., Froelich S., Houlden H., Pickering-Brown S., Chakraverty S., Isaacs A., Grover A. Association of missense and 5′-splice-site mutations in tau with the inherited dementia FTDP-17. Nature. 1998;393:702–705. doi: 10.1038/31508. [DOI] [PubMed] [Google Scholar]
- 9.Faustino N.A., Cooper T.A. Pre-mRNA splicing and human disease. Genes Dev. 2003;17:419–437. doi: 10.1101/gad.1048803. [DOI] [PubMed] [Google Scholar]
- 10.Cáceres J.F., Kornblihtt A.R. Alternative splicing: Multiple control mechanisms and involvement in human disease. Trends Genet. 2002;18:186–193. doi: 10.1016/s0168-9525(01)02626-9. [DOI] [PubMed] [Google Scholar]
- 11.Buratti E., Baralle M., Baralle F.E. Defective splicing, disease and therapy: Searching for master checkpoints in exon definition. Nucleic Acids Res. 2006;34:3494–3510. doi: 10.1093/nar/gkl498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lefebvre S., Burglen L., Reboullet S., Clermont O., Burlet P., Viollet L., Benichou B., Cruaud C., Millasseau P., Zeviani M. Identification and characterization of a spinal muscular atrophy-determining gene. Cell. 1995;80:155–165. doi: 10.1016/0092-8674(95)90460-3. [DOI] [PubMed] [Google Scholar]
- 13.Lorson C.L., Hahnen E., Androphy E.J., Wirth B. A single nucleotide in the SMN gene regulates splicing and is responsible for spinal muscular atrophy. Proc. Natl. Acad. Sci. USA. 1999;96:6307–6311. doi: 10.1073/pnas.96.11.6307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Monani U.R., Lorson C.L., Parsons D.W., Prior T.W., Androphy E.J., Burghes A.H., McPherson J.D. A single nucleotide difference that alters splicing patterns distinguishes the SMA gene SMN1 from the copy gene SMN2. Hum. Mol. Genet. 1999;8:1177–1183. doi: 10.1093/hmg/8.7.1177. [DOI] [PubMed] [Google Scholar]
- 15.Kashima T., Rao N., Manley J.L. An intronic element contributes to splicing repression in spinal muscular atrophy. Proc. Natl. Acad. Sci. USA. 2007;104:3426–3431. doi: 10.1073/pnas.0700343104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Battle D.J., Kasim M., Yong J., Lotti F., Lau C.K., Mouaikel J., Zhang Z., Han K., Wan L., Dreyfuss G. The SMN complex: An assembly machine for RNPs. Cold Spring Harb. Symp. Quant. Biol. 2006;71:313–320. doi: 10.1101/sqb.2006.71.001. [DOI] [PubMed] [Google Scholar]
- 17.Meister G., Eggert C., Fischer U. SMN-mediated assembly of RNPs: A complex story. Trends Cell Biol. 2002;12:472–478. doi: 10.1016/s0962-8924(02)02371-1. [DOI] [PubMed] [Google Scholar]
- 18.Cheng D., Côté J., Shaaban S., Bedford M.T. The arginine methyltransferase CARM1 regulates the coupling of transcription and mRNA processing. Mol. Cell. 2007;25:71–83. doi: 10.1016/j.molcel.2006.11.019. [DOI] [PubMed] [Google Scholar]
- 19.Rossoll W., Jablonka S., Andreassi C., Kroning A.K., Karle K., Monani U.R., Sendtner M. Smn, the spinal muscular atrophy-determining gene product, modulates axon growth and localization of beta-actin mRNA in growth cones of motoneurons. J. Cell Biol. 2003;163:801–812. doi: 10.1083/jcb.200304128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hua Y., Zhou J. Modulation of SMN nuclear foci and cytoplasmic localization by its C-terminus. Cell. Mol. Life Sci. 2004;61:2658–2663. doi: 10.1007/s00018-004-4300-z. [DOI] [PubMed] [Google Scholar]
- 21.Wolstencroft E.C., Mattis V., Bajer A.A., Young P.J., Lorson C.L. A non-sequence-specific requirement for SMN protein activity: The role of aminoglycosides in inducing elevated SMN protein levels. Hum. Mol. Genet. 2005;14:1199–1210. doi: 10.1093/hmg/ddi131. [DOI] [PubMed] [Google Scholar]
- 22.Lorson C.L., Androphy E.J. An exonic enhancer is required for inclusion of an essential exon in the SMA-determining gene SMN. Hum. Mol. Genet. 2000;9:259–265. doi: 10.1093/hmg/9.2.259. [DOI] [PubMed] [Google Scholar]
- 23.Carrel T.L., McWhorter M.L., Workman E., Zhang H., Wolstencroft E.C., Lorson C., Bassell G.J., Burghes A.H., Beattie C.E. Survival motor neuron function in motor axons is independent of functions required for small nuclear ribonucleoprotein biogenesis. J. Neurosci. 2006;26:11014–11022. doi: 10.1523/JNEUROSCI.1637-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang H.L., Pan F., Hong D., Shenoy S.M., Singer R.H., Bassell G.J. Active transport of the survival motor neuron protein and the role of exon-7 in cytoplasmic localization. J. Neurosci. 2003;23:6627–6637. doi: 10.1523/JNEUROSCI.23-16-06627.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Singh N.N., Singh R.N., Androphy E.J. Modulating role of RNA structure in alternative splicing of a critical exon in the spinal muscular atrophy genes. Nucleic Acids Res. 2007;35:371–389. doi: 10.1093/nar/gkl1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hofmann Y., Lorson C.L., Stamm S., Androphy E.J., Wirth B. Htra2-beta 1 stimulates an exonic splicing enhancer and can restore full-length SMN expression to survival motor neuron 2 (SMN2) Proc. Natl. Acad. Sci. USA. 2000;97:9618–9623. doi: 10.1073/pnas.160181697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Young P.J., DiDonato C.J., Hu D., Kothary R., Androphy E.J., Lorson C.L. SRp30c-dependent stimulation of survival motor neuron (SMN) exon 7 inclusion is facilitated by a direct interaction with hTra2 beta 1. Hum. Mol. Genet. 2002;11:577–587. doi: 10.1093/hmg/11.5.577. [DOI] [PubMed] [Google Scholar]
- 28.Hofmann Y., Wirth B. hnRNP-G promotes exon 7 inclusion of survival motor neuron (SMN) via direct interaction with Htra2-beta1. Hum. Mol. Genet. 2002;11:2037–2049. doi: 10.1093/hmg/11.17.2037. [DOI] [PubMed] [Google Scholar]
- 29.Nasim M.T., Chernova T.K., Chowdhury H.M., Yue B.G., Eperon I.C. HnRNP G and Tra2beta: Opposite effects on splicing matched by antagonism in RNA binding. Hum. Mol. Genet. 2003;12:1337–1348. doi: 10.1093/hmg/ddg136. [DOI] [PubMed] [Google Scholar]
- 30.Hua Y., Vickers T.A., Baker B.F., Bennett C.F., Krainer A.R. Enhancement of SMN2 exon 7 inclusion by antisense oligonucleotides targeting the exon. PLoS Biol. 2007;5:e73. doi: 10.1371/journal.pbio.0050073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Miyaso H., Okumura M., Kondo S., Higashide S., Miyajima H., Imaizumi K. An intronic splicing enhancer element in survival motor neuron (SMN) pre-mRNA. J. Biol. Chem. 2003;278:15825–15831. doi: 10.1074/jbc.M209271200. [DOI] [PubMed] [Google Scholar]
- 32.Kashima T., Manley J.L. A negative element in SMN2 exon 7 inhibits splicing in spinal muscular atrophy. Nat. Genet. 2003;34:460–463. doi: 10.1038/ng1207. [DOI] [PubMed] [Google Scholar]
- 33.Kashima T., Rao N., David C.J., Manley J.L. hnRNP A1 functions with specificity in repression of SMN2 exon 7 splicing. Hum. Mol. Genet. 2007;16:3149–3159. doi: 10.1093/hmg/ddm276. [DOI] [PubMed] [Google Scholar]
- 34.Singh N.N., Androphy E.J., Singh R.N. In vivo selection reveals combinatorial controls that define a critical exon in the spinal muscular atrophy genes. RNA. 2004;10:1291–1305. doi: 10.1261/rna.7580704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Miyajima H., Miyaso H., Okumura M., Kurisu J., Imaizumi K. Identification of a cis-acting element for the regulation of SMN exon 7 splicing. J. Biol. Chem. 2002;277:23271–23277. doi: 10.1074/jbc.M200851200. [DOI] [PubMed] [Google Scholar]
- 36.Singh N.K., Singh N.N., Androphy E.J., Singh R.N. Splicing of a critical exon of human survival motor neuron is regulated by a unique silencer element located in the last intron. Mol. Cell. Biol. 2006;26:1333–1346. doi: 10.1128/MCB.26.4.1333-1346.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Alter J., Lou F., Rabinowitz A., Yin H., Rosenfeld J., Wilton S.D., Partridge T.A., Lu Q.L. Systemic delivery of morpholino oligonucleotide restores dystrophin expression bodywide and improves dystrophic pathology. Nat. Med. 2006;12:175–177. doi: 10.1038/nm1345. [DOI] [PubMed] [Google Scholar]
- 38.van Deutekom J.C., van Ommen G.J. Advances in Duchenne muscular dystrophy gene therapy. Nat. Rev. Genet. 2003;4:774–783. doi: 10.1038/nrg1180. [DOI] [PubMed] [Google Scholar]
- 39.Baker B.F., Lot S.S., Condon T.P., Cheng-Flournoy S., Lesnik E.A., Sasmor H.M., Bennett C.F. 2′-O-(2-Methoxy)ethyl-modified anti-intercellular adhesion molecule 1 (ICAM-1) oligonucleotides selectively increase the ICAM-1 mRNA level and inhibit formation of the ICAM-1 translation initiation complex in human umbilical vein endothelial cells. J. Biol. Chem. 1997;272:11994–12000. doi: 10.1074/jbc.272.18.11994. [DOI] [PubMed] [Google Scholar]
- 40.Cáceres J.F., Misteli T., Screaton G.R., Spector D.L., Krainer A.R. Role of the modular domains of SR proteins in subnuclear localization and alternative splicing specificity. J. Cell Biol. 1997;138:225–238. doi: 10.1083/jcb.138.2.225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mayeda A., Krainer A.R. Preparation of HeLa cell nuclear and cytosolic S100 extracts for in vitro splicing. Methods Mol. Biol. 1999;118:309–314. doi: 10.1385/1-59259-676-2:309. [DOI] [PubMed] [Google Scholar]
- 42.Mayeda A., Krainer A.R. Mammalian in vitro splicing assays. Methods Mol. Biol. 1999;118:315–321. doi: 10.1385/1-59259-676-2:315. [DOI] [PubMed] [Google Scholar]
- 43.Caputi M., Mayeda A., Krainer A.R., Zahler A.M. hnRNP A/B proteins are required for inhibition of HIV-1 pre-mRNA splicing. EMBO J. 1999;18:4060–4067. doi: 10.1093/emboj/18.14.4060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dean N.M., Butler M., Monia B.P., Manoharan M. Pharmacology of 2′-O-(2-methoxy)ethyl-modified antisense oligonucleotides. In: Crooke S.T., editor. Antisense Drug Technology: Principles, Strategies, and Applications. Marcel Dekker; New York: 2001. pp. 319–338. [Google Scholar]
- 45.Scholl R., Marquis J., Meyer K., Schumperli D. Spinal muscular atrophy: Position and functional importance of the branch site preceding SMN exon 7. RNA Biol. 2007;4:34–37. doi: 10.4161/rna.4.1.4534. [DOI] [PubMed] [Google Scholar]
- 46.Gennarelli M., Lucarelli M., Capon F., Pizzuti A., Merlini L., Angelini C., Novelli G., Dallapiccola B. Survival motor neuron gene transcript analysis in muscles from spinal muscular atrophy patients. Biochem. Biophys. Res. Commun. 1995;213:342–348. doi: 10.1006/bbrc.1995.2135. [DOI] [PubMed] [Google Scholar]
- 47.Parsons D.W., McAndrew P.E., Monani U.R., Mendell J.R., Burghes A.H., Prior T.W. An 11 base pair duplication in exon 6 of the SMN gene produces a type I spinal muscular atrophy (SMA) phenotype: Further evidence for SMN as the primary SMA-determining gene. Hum. Mol. Genet. 1996;5:1727–1732. doi: 10.1093/hmg/5.11.1727. [DOI] [PubMed] [Google Scholar]
- 48.Si Z., Amendt B.A., Stoltzfus C.M. Splicing efficiency of human immunodeficiency virus type 1 tat RNA is determined by both a suboptimal 3′ splice site and a 10 nucleotide exon splicing silencer element located within tat exon 2. Nucleic Acids Res. 1997;25:861–867. doi: 10.1093/nar/25.4.861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Burd C.G., Dreyfuss G. RNA binding specificity of hnRNP A1: Significance of hnRNP A1 high-affinity binding sites in pre-mRNA splicing. EMBO J. 1994;13:1197–1204. doi: 10.1002/j.1460-2075.1994.tb06369.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Si Z.H., Rauch D., Stoltzfus C.M. The exon splicing silencer in human immunodeficiency virus type 1 Tat exon 3 is bipartite and acts early in spliceosome assembly. Mol. Cell. Biol. 1998;18:5404–5413. doi: 10.1128/mcb.18.9.5404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Auweter S.D., Oberstrass F.C., Allain F.H. Sequence-specific binding of single-stranded RNA: Is there a code for recognition? Nucleic Acids Res. 2006;34:4943–4959. doi: 10.1093/nar/gkl620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Abdul-Manan N., Williams K.R. hnRNP A1 binds promiscuously to oligoribonucleotides: Utilization of random and homo-oligonucleotides to discriminate sequence from base-specific binding. Nucleic Acids Res. 1996;24:4063–4070. doi: 10.1093/nar/24.20.4063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mayeda A., Munroe S.H., Cáceres J.F., Krainer A.R. Function of conserved domains of hnRNP A1 and other hnRNP A/B proteins. EMBO J. 1994;13:5483–5495. doi: 10.1002/j.1460-2075.1994.tb06883.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Buratti E., Brindisi A., Giombi M., Tisminetzky S., Ayala Y.M., Baralle F.E. TDP-43 binds heterogeneous nuclear ribonucleoprotein A/B through its C-terminal tail: An important region for the inhibition of cystic fibrosis transmembrane conductance regulator exon 9 splicing. J. Biol. Chem. 2005;280:37572–37584. doi: 10.1074/jbc.M505557200. [DOI] [PubMed] [Google Scholar]
- 55.Hutchison S., LeBel C., Blanchette M., Chabot B. Distinct sets of adjacent heterogeneous nuclear ribonucleoprotein (hnRNP) A1/A2 binding sites control 5′ splice site selection in the hnRNP A1 mRNA precursor. J. Biol. Chem. 2002;277:29745–29752. doi: 10.1074/jbc.M203633200. [DOI] [PubMed] [Google Scholar]
- 56.Bilodeau P.S., Domsic J.K., Mayeda A., Krainer A.R., Stoltzfus C.M. RNA splicing at human immunodeficiency virus type 1 3′ splice site A2 is regulated by binding of hnRNP A/B proteins to an exonic splicing silencer element. J. Virol. 2001;75:8487–8497. doi: 10.1128/JVI.75.18.8487-8497.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Damgaard C.K., Tange T.O., Kjems J. hnRNP A1 controls HIV-1 mRNA splicing through cooperative binding to intron and exon splicing silencers in the context of a conserved secondary structure. RNA. 2002;8:1401–1415. doi: 10.1017/s1355838202023075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Han K., Yeo G., An P., Burge C.B., Grabowski P.J. A combinatorial code for splicing silencing: UAGG and GGGG motifs. PLoS Biol. 2005;3:e158. doi: 10.1371/journal.pbio.0030158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Geary R.S., Watanabe T.A., Truong L., Freier S., Lesnik E.A., Sioufi N.B., Sasmor H., Manoharan M., Levin A.A. Pharmacokinetic properties of 2′-O-(2-methoxyethyl)-modified oligonucleotide analogs in rats. J. Pharmacol. Exp. Ther. 2001;296:890–897. [PubMed] [Google Scholar]
- 60.Monani U.R., Sendtner M., Coovert D.D., Parsons D.W., Andreassi C., Le T.T., Jablonka S., Schrank B., Rossol W., Prior T.W. The human centromeric survival motor neuron gene (SMN2) rescues embryonic lethality in Smn(−/−) mice and results in a mouse with spinal muscular atrophy. Hum. Mol. Genet. 2000;9:333–339. doi: 10.1093/hmg/9.3.333. [DOI] [PubMed] [Google Scholar]
- 61.Geary R.S., Yu R.Z., Leeds J.M., Templin M.V., Watanabe T.A., Henry S.P., Levin A.A. Pharmacokinetic properties in animals. In: Crooke S.T., editor. Antisense Drug Technology: Principles, Strategies, and Applications. Marcel Dekker; New York: 2001. pp. 119–145. [Google Scholar]
- 62.Wirth B., Brichta L., Hahnen E. Spinal muscular atrophy and therapeutic prospects. Prog. Mol. Subcell. Biol. 2006;44:109–132. doi: 10.1007/978-3-540-34449-0_6. [DOI] [PubMed] [Google Scholar]
- 63.Zhang M.L., Lorson C.L., Androphy E.J., Zhou J. An in vivo reporter system for measuring increased inclusion of exon 7 in SMN2 mRNA: potential therapy of SMA. Gene Ther. 2001;8:1532–1538. doi: 10.1038/sj.gt.3301550. [DOI] [PubMed] [Google Scholar]
- 64.Cartegni L., Krainer A.R. Correction of disease-associated exon skipping by synthetic exon-specific activators. Nat. Struct. Biol. 2003;10:120–125. doi: 10.1038/nsb887. [DOI] [PubMed] [Google Scholar]
- 65.Coady T.H., Shababi M., Tullis G.E., Lorson C.L. Restoration of SMN function: Delivery of a trans-splicing RNA re-directs SMN2 pre-mRNA splicing. Mol. Ther. 2007;15:1471–1478. doi: 10.1038/sj.mt.6300222. [DOI] [PubMed] [Google Scholar]
- 66.Skordis L.A., Dunckley M.G., Yue B., Eperon I.C., Muntoni F. Bifunctional antisense oligonucleotides provide a trans-acting splicing enhancer that stimulates SMN2 gene expression in patient fibroblasts. Proc. Natl. Acad. Sci. USA. 2003;100:4114–4119. doi: 10.1073/pnas.0633863100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Crooke S.T. Basic principles of antisense technology. In: Crooke S.T., editor. Antisense Drug Technology: Principles, Strategies, and Applications. Marcel Dekker; New York: 2001. pp. 1–28. [Google Scholar]
- 68.Dominski Z., Kole R. Restoration of correct splicing in thalassemic pre-mRNA by antisense oligonucleotides. Proc. Natl. Acad. Sci. USA. 1993;90:8673–8677. doi: 10.1073/pnas.90.18.8673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sierakowska H., Sambade M.J., Agrawal S., Kole R. Repair of thalassemic human beta-globin mRNA in mammalian cells by antisense oligonucleotides. Proc. Natl. Acad. Sci. USA. 1996;93:12840–12844. doi: 10.1073/pnas.93.23.12840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Scaffidi P., Misteli T. Reversal of the cellular phenotype in the premature aging disease Hutchinson-Gilford progeria syndrome. Nat. Med. 2005;11:440–445. doi: 10.1038/nm1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zhu J., Mayeda A., Krainer A.R. Exon identity established through differential antagonism between exonic splicing silencer-bound hnRNP A1 and enhancer-bound SR proteins. Mol. Cell. 2001;8:1351–1361. doi: 10.1016/s1097-2765(01)00409-9. [DOI] [PubMed] [Google Scholar]
- 72.An P., Grabowski P.J. Exon silencing by UAGG motifs in response to neuronal excitation. PLoS Biol. 2007;5:e36. doi: 10.1371/journal.pbio.0050036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Marchand V., Mereau A., Jacquenet S., Thomas D., Mougin A., Gattoni R., Stévenin J., Branlant C. A Janus splicing regulatory element modulates HIV-1 tat and rev mRNA production by coordination of hnRNP A1 cooperative binding. J. Mol. Biol. 2002;323:629–652. doi: 10.1016/s0022-2836(02)00967-1. [DOI] [PubMed] [Google Scholar]
- 74.Blanchette M., Chabot B. Modulation of exon skipping by high-affinity hnRNP A1-binding sites and by intron elements that repress splice site utilization. EMBO J. 1999;18:1939–1952. doi: 10.1093/emboj/18.7.1939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ding J., Hayashi M.K., Zhang Y., Manche L., Krainer A.R., Xu R.M. Crystal structure of the two-RRM domain of hnRNP A1 (UP1) complexed with single-stranded telomeric DNA. Genes Dev. 1999;13:1102–1115. doi: 10.1101/gad.13.9.1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Smith R.A., Miller T.M., Yamanaka K., Monia B.P., Condon T.P., Hung G., Lobsiger C.S., Ward C.M., McAlonis-Downes M., Wei H. Antisense oligonucleotide therapy for neurodegenerative disease. J. Clin. Invest. 2006;116:2290–2296. doi: 10.1172/JCI25424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kumar P., Wu H., McBride J.L., Jung K.E., Kim M.H., Davidson B.L., Lee S.K., Shankar P., Manjunath N. Transvascular delivery of small interfering RNA to the central nervous system. Nature. 2007;448:39–43. doi: 10.1038/nature05901. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.