

Abstract
Meckel syndrome (MKS) is a lethal malformation disorder characterized classically by encephalocele, polycystic kidneys, and polydactyly. MKS is also one of the major contributors to syndromic neural tube defects (NTDs). Recent findings have shown primary cilia dysfunction in the molecular background of MKS, indicating that cilia are critical for early human development. However, even though four genes behind MKS have been identified to date, they elucidate only a minor proportion of the MKS cases. In this study, instead of traditional linkage analysis, we selected 10 nonrelated affected fetuses and looked for the homozygous regions shared by them. Based on this strategy, we identified the sixth locus and the fifth gene, CC2D2A (MKS6), behind MKS. The biological function of CC2D2A is uncharacterized, but the corresponding polypeptide is predicted to be involved in ciliary functions and it has a calcium binding domain (C2). Immunofluorescence staining of patient's fibroblast cells demonstrates that the cells lack cilia, providing evidence for the critical role of CC2D2A in cilia formation. Our finding is very significant not only to understand the molecular background of MKS, but also to obtain additional information about the function of the cilia, which can help to understand their significance in normal development and also in other ciliopathies, which are an increasing group of disorders with overlapping phenotypes.
Main Text
Meckel syndrome (MKS [MIM 249000]) is an autosomal-recessive lethal disorder characterized by a variety of severe malformations. Minimal diagnostic criteria are cystic dysplasia of the kidneys with fibrotic changes in the liver and occipital encephalocele or some other central nervous system malformation. Additionally, polydactyly is frequently reported in the patients.1 MKS diagnosis can be established by ultrasound already in the end of the first trimester.2 Figures 1A and 1B show an ultrasound scan of a MKS fetus at the 14th week of gestation with encephalocele and a distended stomach because of the enlarged cystic kidneys.
Figure 1.
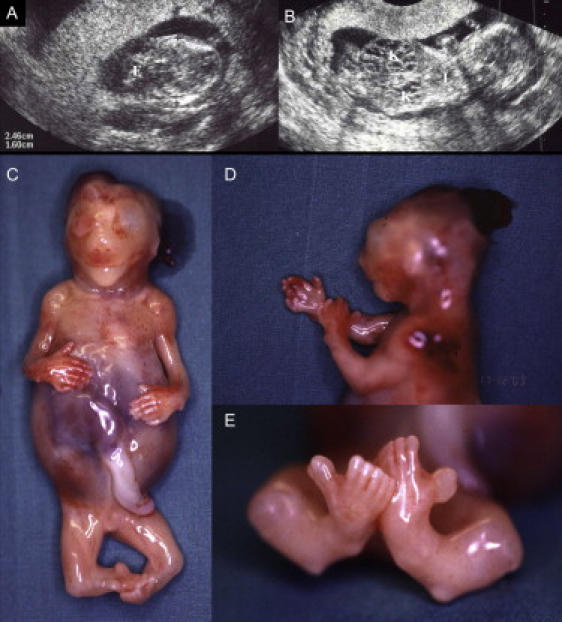
Two MKS6 Fetuses with Typical Features
(A and B) An ultrasound scan of case 3 (Table 1) at the 14th week of gestation. The biparietal measure is 2.46 cm and the occipital encephalocele (E) 1.60 cm in diameter (A). The kidneys (K) are grossly enlarged already with clearly visible cysts. The thorax is shallow and the lungs (L) are small (B). Ultrasound pictures courtesy of Pirkko Ämmalä.
(C–E) Fetus aborted at 15th week of gestation (case 4). Distended stomach because of enlarged kidneys and polydactyly of the hands can be seen in (C), occipital encephalocele in (D), and polydactyly of the feet in (E). Exceptionally, this fetus also has hallux duplex, which is rarely seen in MKS fetuses.
MKS is known to be a heterogenous disease with linkage to five loci and four genes identified so far.3–7 All the genes are associated with ciliary functions. Mutations in ciliary genes are known to cause a number of human monogenic disorders that are collectively known as ciliopathies, disorders with overlapping clinical features. An especially interesting aspect of ciliary diseases is that they range from embryonically lethal Meckel syndrome to less severe multisystem disorders, such as Bardet-Biedl syndrome (BBS [MIM 209900]), where the patients suffer from obesity, retinal degeneration, polydactyly, mental retardation, and cystic kidneys, for example.8
We have earlier reported that in 70% of the Finnish cases, MKS is caused by the Finmajor (IVS15-7_35 del) mutation in a novel MKS1 gene3 that has later been identified also in other populations.3,9–11 In order to find out the genetic defect behind MKS in the remaining Finnish families, we carefully chose 10 out of 17 available fetuses that met the minimal diagnostic criteria. This study has been approved by the ethical committees of the Joint Authority for the Hospital District of Helsinki and Uusimaa, Finland. Because these families had only one affected fetus and no healthy siblings were available, a linkage-based positioning of the underlying locus was not possible. Instead, assuming that in the isolated Finnish population the cases might share a common mutation and surrounding haplotype, we decided to perform a genome-wide single-nucleotide polymorphism (SNP) scan to locate homozygous regions shared by the cases. DNA samples of MKS cases were genotyped according to manufacturer's instructions on Illumina HumanHap300-duo SNP microarrays (Illumina, San Diego, CA) containing 318,237 SNPs. All patient samples had success rates of >99% and were thus included in the study. Illumina Beadstudio v3.1.0 was used to call genotypes, and homozygosity detector option was utilized in the search of extended tracts of homozygosity in each sample using a minimum length of 50 SNPs. The algorithm uses SNP frequencies to calculate the expectation that a single SNP is homozygous in a sample, and it can be used to autobookmark samples with extended tracts of homozygosity (for more information, see Illumina Systems and Software Technical Note).
Six out of ten patients were found to have overlapping homozygous regions on chromosome 4p15. The size of the homozygous regions varied from 730 kb to 6.8 Mb, with all patients sharing a segment of 63 SNPs covering a 565 kb area (Figure 2) (chr4: 14,909,996–15,475,912, UCSC 2006). Illumina GenomeViewer was used to visualize the chromosome 4 region to verify that the region indeed was homozygous and to exclude copy-number changes (data not shown). In addition, one patient shared the same region, but was homozygote for a different allelic haplotype. PLINK v1.0 was used to calculate pairwise identity-by-descent estimates and inbreeding coefficients.12 None of the six affected fetuses were closely related (pairwise IBD sharing estimate maximum was 0.0258 and, for example, third cousins are expected to share 0.031 of their genome by IBD) and the inbreeding coefficients did not suggest hidden relatedness in patients' parents (F < 0.001, data not shown).
Figure 2.
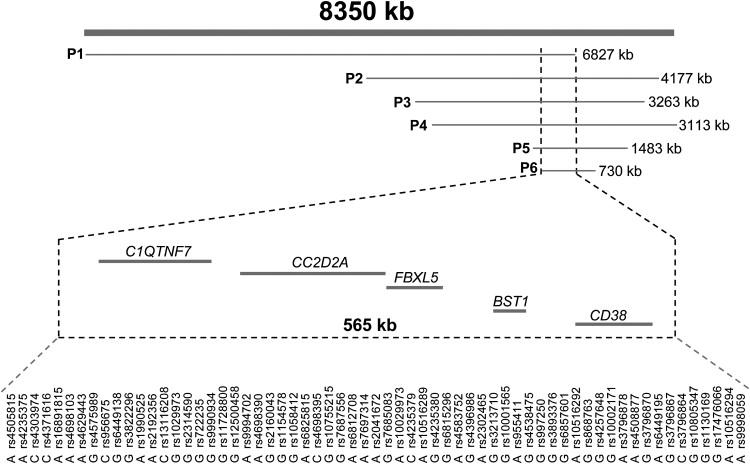
The MKS6 Candidate Region on Chromosome 4
Horizontal lines show the homozygous regions found in six affected MKS fetuses. The candidate region (chr4: 14,909,996-15,475,912, UCSC 2006) contained five genes of which CC2D2A was suggested to have a function in cilia. The fact that the overlapping homozygous region shared by the patients carrying the MKS6 mutation was only 565 kb and 63 SNPs in length gives us an estimation of the region size that could be used in search of genes for both recessive and complex diseases.
To see whether any of the genes in the candidate region were predicted to have a role in ciliary functions, we queried the ciliary proteome database, where all the existing ciliary and basal body proteomics data is gathered. In fact, one of the regional genes, CC2D2A (KIAA1345), was found among the genes of the ciliary proteome, making it an excellent candidate for MKS. CC2D2A contains 37 exons that encode (bp 73–4758) for a 1561 amino acid polypeptide having a coiled-coil and C2 domain, but its biological function is uncharacterized. Bioinformatics analyses carried out with the InterProScan program predicted that CC2D2A has also a centrosomal protein-related domain. Overexpression of CC2D2A-GFP fusion protein in Cos7 cells shows mainly cytoplasmic localization,13 but localization in cilia structures has not been studied.
In order to sequence this candidate gene, we first extracted RNA from fibroblast cell lines of an affected fetus and an age-matched control fetus by Rneasy (QIAGEN) kit. Reverse-transcribed cDNA was done by Advantage RT for PCR kit (Clontech) with oligo(dT) primer. Primers (see Table S1 available online) for sequencing were designed with Primer3 and provided by Sigma-Genosys. The sequence analysis of the cDNA of CC2D2A (NM_001080522) revealed a 4 base pair deletion c.1761-1764 in the patient transcript compared to the control (Figure 3A). Sequence analysis of the genomic DNA revealed that this was due to a C→T substitution (c.1762C→T) in the end of exon 16 (Figure 3B). This mutation creates a new donor splice site that affects the splicing, leading to a defective transcript. The deletion causes a frame shift after valine 587 and results in a stop codon at amino acid 616. Sequence analysis of all the 17 MKS families identified that in 11 families, the parents were heterozygous and affected fetuses homozygous for the mutation. All six fetuses that shared the common haplotype were homozygous for the mutation. We could not identify any mutations in the four cases that had different haplotypes, nor could we find any mutations in the remaining two MKS samples, thus leaving six cases without a mutation. When sequencing 575 healthy control samples, three individuals were found to be carriers of the MKS6 mutation; therefore, the estimated carrier frequency was ∼0.5% in the Finnish population.
Figure 3.
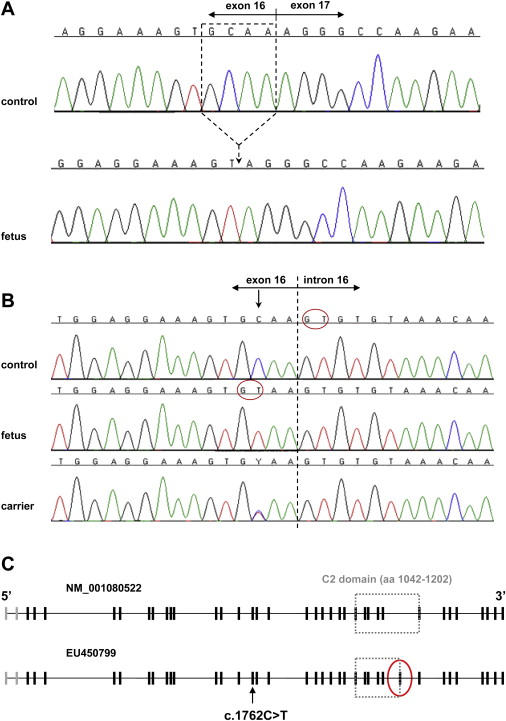
Mutation Analysis and Structure of CC2D2A
(A) Normal cDNA sequence from a control and the cDNA sequence from an affected fetus with the 4 base pair deletion (c.1761-1764) that is located in the end of exon 16.
(B) Genomic sequence of the exon-intron boundary from a control, an affected fetus, and a heterozygous parent. In DNA of the affected fetus, the C→T substitution (c.1762C→T) creates a new splice site leading to the abnormal splicing as seen in (A). Red circles show the splice donor sites in the control's and patient's DNAs.
(C) The gene structure of CC2D2A shows that translation starts from exon 3 and the new exon (marked by a red circle) is located after exon 29.
While sequencing the full-length cDNA from fibroblast cells, we discovered that the sequence was in fact longer than the reference sequence in the database (NM_001080522) in both patient and control samples. To see whether this was just a tissue-specific variant, we also sequenced the cDNAs from a fetal multiple tissue panel (BD Biosciences) and observed that the longer transcript (5067 bp) was present in all the tissues. The longer transcript contains one additional exon that is located after exon 29 of the reference sequence. As a result, the total number of exons of CC2D2A is 38, cDNA being 5067 bp and the corresponding polypeptide 1620 amino acids (EU450799) (Figure 3C). Bioinformatics analyses performed with the longer transcript with the InterProScan program indicated no changes in the predicted protein domains.
To understand the role of CC2D2A in ciliogenesis, we reviewed studies that have used different comparative genomics approaches to identify ciliary components. CC2D2A is listed among the flagellar apparatus basal body proteome genes,14 and expression of the gene is restricted to ciliated cells in transgenic worms, suggesting that the gene has ciliary functions.15 Avidor-Reiss and coworkers16 used comparative genomics analysis to define specialized proteins needed for the formation and function of cilia and identified altogether three novel C2 domain-containing proteins of which CG18631 is the Drosophila melanogaster ortholog for CC2D2A. The C2 domain consists typically of a ∼120 amino acid sequence that functions as a Ca2+-dependent membrane-targeting module found in many cellular proteins involved in signal transduction or membrane trafficking. The C2 domain is thought to be involved in calcium-dependent phospholipid binding and in membrane-targeting processes such as subcellular localization. Because calcium has a central role in regulating cilia function as well as processes as diverse as membrane fusion, protein transport, and protein breakdown, these genes were considered good candidates for sensors of the calcium signals.16,17 Calmodulin (CaM) is the most-studied Ca2+ sensor in eukaryotic cells. Shen and collegues used a mRNA display technique to find CaM-binding partners in the human proteome and CC2D2A was identified as a potential CaM-binding protein, giving further support for the role of CC2D2A in Ca2+-regulated signaling pathways.18
The importance of Ca2+ signaling to development is well recognized, and it has been proposed that Ca2+ signaling, in the form of pulses, waves, and steady gradients, plays a crucial role in the key pattern-forming events in embryonic development.19 Furthermore, Ca2+ has been well linked to the left-right asymmetry and cyst formation in the kidneys,20 characteristic features of MKS fetuses. One of the hallmarks of MKS, neural tube closure, requires the orientation of polarized epithelial cells in a single plane perpendicular to the apical-basal axis as well as convergent extension, which leads to the narrowing and lengthening of tissues during development. Therefore, disruption of the planar cell polarity (PCP) pathway owing to defective cilia-mediated signaling might explain the neural tube defects. It has been suggested that cilia-mediated signaling acts as a switch between canonical and noncanonical (PCP) pathways and that in the absence of fluid flow, canonical Wnt signaling predominates, but on mechanosensation of fluid flow, intracellular Ca2+ release causes increased inversin (INVS) expression, which restrains the canonical Wnt signaling.20
To look for any visible defect in the structure of the cilium or centrioles, we studied primary fibroblast cells of a fetus, homozygote for the MKS6 mutation. Fibroblast cells of the MKS case and a healthy control fetus were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and antibiotics. For acetylated tubulin staining, cells were fixed with 4% paraformaldehyde (PFA) in PBS (pH 7.3) at room temperature for 10 min. PFA-fixed cells were blocked and permeabilized with 0.2% saponin/0.5% BSA in PBS. All steps were carried out with the blocking/permeabilization solution. For γ-tubulin staining, the cells were fixed with ice-cold methanol (−20°C). Methanol-fixed cells were blocked with 0.5% BSA in PBS and the following steps were carried out in the blocking solution. Primary antibody incubation was 1 hr in RT. Both primary antibodies were provided by Sigma and produced in mouse, and the secondary antibody was anti-mouse-TRITC for both protocols. Secondary antibodies were incubated at RT for 45 min. After last washes, cells were mounted onto SuperFrost/Plus microscope slides (Merck, Germany) with Vectashield hard set mounting medium with DAPI (Vector Laboratories) and visualized by Axioplan Z imaging fluorescence microscope with AxioVision Rel. 4.6 program. Pictures were prepared with CorelDraw 9. As a result of this experiment, we found out that the centrioles can be seen in fibroblasts of the MKS case and the healthy control fetus with γ-tubulin antibody (Figures 4A and 4B), but as a striking contrast, no cilia were detected in patient's cells when using acetylated tubulin antibody while cilia were seen in fibroblasts of a control (Figures 4C and 4D). Interestingly, also biliary cells of some MKS fetuses lacked cilia when liver sections were stained with acetylated tubulin. Unfortunately, the genetic background of the studied fetuses was not known.21
Figure 4.
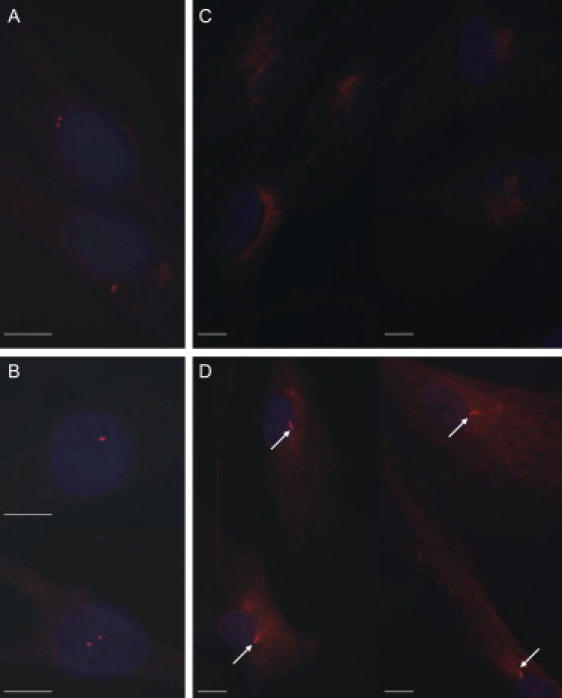
Immunofluorescence Staining of Centrioles and Cilia
Centrioles can be visualized with γ-tubulin staining in fibroblast cells of a patient (A) and a healthy control (B) (magnification 100×). Staining with acetylated tubulin shows that cilia are missing in the patient cells (C) but can be observed in control cells (D) (white arrows, magnification 63×). Scale bar represents 10 μm.
Migration of the centrioles to the apical cell membrane is required for the assembly of the cilia, because they serve as the foundation upon which the cilium is constructed.22 Dawe and coworkers have shown that in MKS1- and TMEM67-silenced cells, centrioles fail to migrate to the apical cell membrane, which might explain the reduced ciliary formation in these cells.23 In order to find out whether the situation is the same in this case, we used Alexa Fluor 488-conjugated wheat germ agglutinin (WGA), which binds to the cell membrane and γ-tubulin to visualize the centrioles. Centriole staining was performed as described previously and 488-WGA was added in the secondary antibody incubation and held in 37°C for 30 min. To assess the localization of the centrioles, we took z-stacks with Leica SP confocal microscope and quantified the positioning of the centrioles in 200 control and MKS cells. The first z-stack was taken when 488-WGA appeared and the last when it started clearly to disappear. In total 12 z-stacks were taken and the separation between stacks was ∼0.20–0.25 μm. Centrioles were located in the apical region in 40%–50% of the cells (first third of the stacks) and the number of the mid-cell positions (the second third of the stacks) was very similar, less than 10% of centrioles being in the basal region. No difference was observed between control and patient cells. It is interesting that the localization of the centrioles seems to be normal in the patient cells, but they are still unable to form cilia.
Based on the immunostaining and data of comparative genomics, we could suggest that CC2D2A appears to have at least two cilia-related functions: the molecule seems to be a part of the basal body complex where the cilium is assembled from and also seems to act as a sensor for the intracellular calcium. An example of unsuccessful Ca2+ sensing comes from polycystic kidney disease (PKD). Experimental data have shown that Ca2+ influx in tubular cells requires polycystin 1 and polycystin 2, the proteins mutated in ADPKD. They form a Ca2+ channel that is activated by fluid-induced ciliary bending. Mutations in PKD1 or PKD2 might disable cilia-mediated mechanosensation and generate false signals that indicate a “lack of flow” that might lead to a compensatory growth of the tubular cells and subsequent cyst.20 Due to the absence of the Ca2+ binding domain in mutant CC2D2A protein and/or loss of cilia in the MKS6 cells, the cells might fail to sense or bind the intracellular calcium and therefore the outcome is similar to the lack of flow.
Prior to this study, four genes behind MKS had been identified3,5–7 and all of them (MKS1 [BBS13], TMEM67 [MKS3, JBTS6], CEP290 [MKS4, JBTS5, SLSN6, LCA10], and RPGRIP1L [MKS5, JBTS7]) are known to be mutated also in other syndromes with overlapping clinical features, making MKS, Joubert syndrome (JS [MIM 610688, 610188, 611560]), Leber congenital amaurosis (LCA [MIM 611755], Senior-Loken syndrome (SLSN [MIM 610189]), and BBS allelic disorders.6,7,24,25 In addition to these syndromes, MKS is now allelic as well with autosomal recessive mental retardation (ARMR) with retinitis pigmentosa because very recently Noor and coworkers identified a disease-causing mutation in CC2D2A in a consanguineous family from Pakistan.13 It will be interesting to see whether more CC2D2A mutations will be found, for example, in BBS patients who also have mental retardation and retinitis pigmentosa.
In order to find out the relative contribution of different MKS genes in reported MKS cases totally, we collected all the previously published MKS mutations3,5–7,9–11,23,26 and added our recent finding to the total number. The MKS1 mutations were found in 52% of the reported cases of which 73% are caused by the Finmajor mutation. The MKS3 gene was mutated in 20%, MKS4 in 13%, MKS5 in 2%, and MKS6 in 13% of the reported cases originating mainly from Europe, the Middle East, and Africa.
After the identification of the MKS6 gene, the DNA-based diagnostics of Meckel syndrome in Finland is excellent given that the genetic defect for MKS is now established in ∼90% of Finnish cases, because 68% have a mutation in the MKS1 gene and 21% have a mutation in the MKS6 gene, leaving only approximately 11% of Finnish MKS cases without a known mutation. Figures 1C–1E show an aborted MKS6 fetus with typical features at the 15th week of gestation. Interestingly, the fetus had, in addition, double halluces that are rarely seen in MKS cases. Detailed clinical features of all MKS6 fetuses are presented in Table 1.
Table 1.
Clinical Features Observed in MKS6 Cases
| Case | Occipital Encephalocele | Other CNS Findings | Large Cystic Kidneys | Hypoplastic Lungs | Fibrotic/Cystic Liver Changes | Cleft Lip/Palate | Polydactyly of Hands | Polydactyly of Feet | Club Feet | Other Findings |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | + | + | + | + | + | − | + | + | ||
| 2 | + | + | + | + | n.r. | + | + | + | gonadal undifferentiation | |
| 3 | + | + | + | n.r. | − | + | + | + | ||
| 4 | + | + | n.r. | + | − | + | + | + | seven toes with double big toes in both feet | |
| 5 | + | anencephaly | + | + | n.r. | n.r. | + | + | + | one large horse-shoe kidney, only two lobes in both lungs |
| 6 | + | + | + | + | n.r. | − | + | + | ||
| 7 | + | + | n.r. | + | n.r. | n.r. | n.r. | n.r. | ||
| 8 | + | anencephaly | + | n.r. | + | n.r. | + | + | n.r. | |
| 9 | + | hydrocephaly | + | n.r. | + | n.r. | + | + | n.r. | |
| 10 | + | + | + | n.r. | n.r. | + | + | n.r. | hypoplastic penis | |
| 11 | + | 2nd hole in the occipital bone | + | n.r. | n.r. | + | + | + | + | aplastic gall bladder and bladder, ureters connect to vagina |
n.r., not recorded.
As a conclusion, we can hypothesize on the function of CC2D2A in the light of evidence, which shows the importance of both cilia- and Ca2+-mediated signaling in development. The amount of signaling that goes through the cilium may be the key question, and further studies will show whether the calcium signaling is disturbed in MKS6 fetuses. It is important to recognize the complexity of the cilia in order to understand why the range of symptoms varies so much between different ciliopathies. They perform multiple functions, and breaking one piece of the machinery can leave some functions intact while destroying others. Further, some gene products involved in ciliary assembly and function may have additional functions.27 The complex function and structure of cilia is still a puzzle, but this finding adds yet another piece of information that will help us to solve the structure and function of the cilia and their critical significance for early human development.
Acknowledgments
The genotyping of SNP markers was performed by the Finnish Genome Center, Institute for Molecular Medicine Finland (FIMM), University of Helsinki, Finland. We thank the families for participating in the study and clinicians and genetic counselors for the sample collection. Katriina Hautaviita and Kaija Antila are thanked for the technical assistance. Vesa Olkkonen, Markku Lehto, and Heli Honkala are acknowledged for their advice in cell biology studies. We thank Vesa Olkkonen also for 488-WGA. The work was supported by the Research Foundation of the University of Helsinki (grant for young researchers) to J. Tallila, by the Center of Excellence of Disease Genetics and the Academy of Finland (grants 64334 and 202887) to L. Peltonen, by the US National Institutes of Health grant PO1 ES11253-03 (L. Peltonen), by the Academy of Finland (grants 211124 and 118468) to M. Kestilä, and by the Sigrid Juselius Foundation (M. Kestilä). The Department of Medical Genetics, Väestöliitto, is funded by Finland's Slot Machine Association (RAY) (R. Salonen).
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Ciliary proteome database, http://www.ciliaproteome.org
Genome Browser, http://genome.ucsc.edu/
InterProScan, http://www.ebi.ac.uk/InterProScan/
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/
Primer3, http://frodo.wi.mit.edu/cgi-bin/primer3/primer3_www.cgi
References
- 1.Salonen R. The Meckel syndrome: clinicopathological findings in 67 patients. Am. J. Med. Genet. 1984;18:671–689. doi: 10.1002/ajmg.1320180414. [DOI] [PubMed] [Google Scholar]
- 2.Ickowicz V., Eurin D., Maugey-Laulom B., Didier F., Garel C., Gubler M.C., Laquerriere A., Avni E.F. Meckel-Gruber syndrome: sonography and pathology. Ultrasound Obstet. Gynecol. 2006;27:296–300. doi: 10.1002/uog.2708. [DOI] [PubMed] [Google Scholar]
- 3.Kyttala M., Tallila J., Salonen R., Kopra O., Kohlschmidt N., Paavola-Sakki P., Peltonen L., Kestila M. MKS1, encoding a component of the flagellar apparatus basal body proteome, is mutated in Meckel syndrome. Nat. Genet. 2006;38:155–157. doi: 10.1038/ng1714. [DOI] [PubMed] [Google Scholar]
- 4.Roume J., Genin E., Cormier-Daire V., Ma H.W., Mehaye B., Attie T., Razavi-Encha F., Fallet-Bianco C., Buenerd A., Clerget-Darpoux F. A gene for Meckel syndrome maps to chromosome 11q13. Am. J. Hum. Genet. 1998;63:1095–1101. doi: 10.1086/302062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Smith U.M., Consugar M., Tee L.J., McKee B.M., Maina E.N., Whelan S., Morgan N.V., Goranson E., Gissen P., Lilliquist S. The transmembrane protein meckelin (MKS3) is mutated in Meckel-Gruber syndrome and the wpk rat. Nat. Genet. 2006;38:191–196. doi: 10.1038/ng1713. [DOI] [PubMed] [Google Scholar]
- 6.Baala L., Audollent S., Martinovic J., Ozilou C., Babron M.C., Sivanandamoorthy S., Saunier S., Salomon R., Gonzales M., Rattenberry E. Pleiotropic effects of CEP290 (NPHP6) mutations extend to Meckel syndrome. Am. J. Hum. Genet. 2007;81:170–179. doi: 10.1086/519494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Delous M., Baala L., Salomon R., Laclef C., Vierkotten J., Tory K., Golzio C., Lacoste T., Besse L., Ozilou C. The ciliary gene RPGRIP1L is mutated in cerebello-oculo-renal syndrome (Joubert syndrome type B) and Meckel syndrome. Nat. Genet. 2007;39:875–881. doi: 10.1038/ng2039. [DOI] [PubMed] [Google Scholar]
- 8.Tobin J.L., Beales P.L. Bardet-Biedl syndrome: beyond the cilium. Pediatr. Nephrol. 2007;22:926–936. doi: 10.1007/s00467-007-0435-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Consugar M.B., Kubly V.J., Lager D.J., Hommerding C.J., Wong W.C., Bakker E., Gattone V.H., 2nd, Torres V.E., Breuning M.H., Harris P.C. Molecular diagnostics of Meckel-Gruber syndrome highlights phenotypic differences between MKS1 and MKS3. Hum. Genet. 2007;121:591–599. doi: 10.1007/s00439-007-0341-3. [DOI] [PubMed] [Google Scholar]
- 10.Khaddour R., Smith U., Baala L., Martinovic J., Clavering D., Shaffiq R., Ozilou C., Cullinane A., Kyttala M., Shalev S. Spectrum of MKS1 and MKS3 mutations in Meckel syndrome: a genotype-phenotype correlation. Mutation in brief #960. Online. Hum. Mutat. 2007;28:523–524. doi: 10.1002/humu.9489. [DOI] [PubMed] [Google Scholar]
- 11.Frank V., Ortiz Bruchle N., Mager S., Frints S.G., Bohring A., du Bois G., Debatin I., Seidel H., Senderek J., Besbas N. Aberrant splicing is a common mutational mechanism in MKS1, a key player in Meckel-Gruber syndrome. Hum. Mutat. 2007;28:638–639. doi: 10.1002/humu.9496. [DOI] [PubMed] [Google Scholar]
- 12.Purcell S., Neale B., Todd-Brown K., Thomas L., Ferreira M.A., Bender D., Maller J., Sklar P., de Bakker P.I., Daly M.J. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Noor A., Windpassinger C., Patel M., Stachowiak B., Mikhailov A., Azam M., Irfan M., Siddiqui Z.K., Naeem F., Paterson A.D. CC2D2A, encoding a coiled-coil and C2 domain protein, causes autosomal recessive mental retardation with retinitis pigmentosa. Am. J. Hum. Genet. 2008;82:1011–1018. doi: 10.1016/j.ajhg.2008.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li J.B., Gerdes J.M., Haycraft C.J., Fan Y., Teslovich T.M., May-Simera H., Li H., Blacque O.E., Li L., Leitch C.C. Comparative genomics identifies a flagellar and basal body proteome that includes the BBS5 human disease gene. Cell. 2004;117:541–552. doi: 10.1016/s0092-8674(04)00450-7. [DOI] [PubMed] [Google Scholar]
- 15.Blacque O.E., Perens E.A., Boroevich K.A., Inglis P.N., Li C., Warner A., Khattra J., Holt R.A., Ou G., Mah A.K. Functional genomics of the cilium, a sensory organelle. Curr. Biol. 2005;15:935–941. doi: 10.1016/j.cub.2005.04.059. [DOI] [PubMed] [Google Scholar]
- 16.Avidor-Reiss T., Maer A.M., Koundakjian E., Polyanovsky A., Keil T., Subramaniam S., Zuker C.S. Decoding cilia function: defining specialized genes required for compartmentalized cilia biogenesis. Cell. 2004;117:527–539. doi: 10.1016/s0092-8674(04)00412-x. [DOI] [PubMed] [Google Scholar]
- 17.Tamm S. Ca2+ channels and signalling in cilia and flagella. Trends Cell Biol. 1994;4:305–310. doi: 10.1016/0962-8924(94)90226-7. [DOI] [PubMed] [Google Scholar]
- 18.Shen X., Valencia C.A., Szostak J.W., Dong B., Liu R. Scanning the human proteome for calmodulin-binding proteins. Proc. Natl. Acad. Sci. USA. 2005;102:5969–5974. doi: 10.1073/pnas.0407928102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Webb S.E., Miller A.L. Ca2+ signaling and early embryonic patterning during the blastula and gastrula periods of zebrafish and Xenopus development. Biochim. Biophys. Acta. 2006;1763:1192–1208. doi: 10.1016/j.bbamcr.2006.08.004. [DOI] [PubMed] [Google Scholar]
- 20.Fliegauf M., Benzing T., Omran H. When cilia go bad: cilia defects and ciliopathies. Nat. Rev. Mol. Cell Biol. 2007;8:880–893. doi: 10.1038/nrm2278. [DOI] [PubMed] [Google Scholar]
- 21.Clotman F., Libbrecht L., Killingsworth M.C., Loo C.C., Roskams T., Lemaigre F.P. Lack of cilia and differentiation defects in the liver of human foetuses with the Meckel syndrome. Liver Int. 2008;28:377–384. doi: 10.1111/j.1478-3231.2007.01617.x. [DOI] [PubMed] [Google Scholar]
- 22.Marshall W.F. What is the function of centrioles? J. Cell. Biochem. 2007;100:916–922. doi: 10.1002/jcb.21117. [DOI] [PubMed] [Google Scholar]
- 23.Dawe H.R., Smith U.M., Cullinane A.R., Gerrelli D., Cox P., Badano J.L., Blair-Reid S., Sriram N., Katsanis N., Attie-Bitach T. The Meckel-Gruber Syndrome proteins MKS1 and meckelin interact and are required for primary cilium formation. Hum. Mol. Genet. 2007;16:173–186. doi: 10.1093/hmg/ddl459. [DOI] [PubMed] [Google Scholar]
- 24.Baala L., Romano S., Khaddour R., Saunier S., Smith U.M., Audollent S., Ozilou C., Faivre L., Laurent N., Foliguet B. The Meckel-Gruber syndrome gene, MKS3, is mutated in Joubert syndrome. Am. J. Hum. Genet. 2007;80:186–194. doi: 10.1086/510499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Leitch C.C., Zaghloul N.A., Davis E.E., Stoetzel C., Diaz-Font A., Rix S., Al-Fadhel M., Lewis R.A., Eyaid W., Banin E. Hypomorphic mutations in syndromic encephalocele genes are associated with Bardet-Biedl syndrome. Nat. Genet. 2008;40:443–448. doi: 10.1038/ng.97. [DOI] [PubMed] [Google Scholar]
- 26.Frank V., den Hollander A.I., Bruchle N.O., Zonneveld M.N., Nurnberg G., Becker C., Du Bois G., Kendziorra H., Roosing S., Senderek J. Mutations of the CEP290 gene encoding a centrosomal protein cause Meckel-Gruber syndrome. Hum. Mutat. 2008;29:45–52. doi: 10.1002/humu.20614. [DOI] [PubMed] [Google Scholar]
- 27.Marshall W. The cell biological basis of ciliary disease. J. Cell Biol. 2008;180:17–21. doi: 10.1083/jcb.200710085. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.