

Abstract
Our recent studies have shown that deregulated expression of R2, the rate-limiting component of ribonucleotide reductase, enhances transformation and malignant potential by cooperating with activated oncogenes. We now demonstrate that the R1 component of ribonucleotide reductase has tumor-suppressing activity. Stable expression of a biologically active ectopic R1 in ras-transformed mouse fibroblast 10T½ cell lines, with or without R2 overexpression, led to significantly reduced colony-forming efficiency in soft agar. The decreased anchorage independence was accompanied by markedly suppressed malignant potential in vivo. In three ras-transformed cell lines, R1 overexpression resulted in abrogation or marked suppression of tumorigenicity. In addition, the ability to form lung metastases by cells overexpressing R1 was reduced by >85%. Metastasis suppressing activity also was observed in the highly malignant mouse 10T½ derived RMP-6 cell line, which was transformed by a combination of oncogenic ras, myc, and mutant p53. Furthermore, in support of the above observations with the R1 overexpressing cells, NIH 3T3 cells cotransfected with an R1 antisense sequence and oncogenic ras showed significantly increased anchorage independence as compared with control ras-transfected cells. Finally, characteristics of reduced malignant potential also were demonstrated with R1 overexpressing human colon carcinoma cells. Taken together, these results indicate that the two components of ribonucleotide reductase both are unique malignancy determinants playing opposing roles in its regulation, that there is a novel control point important in mechanisms of malignancy, which involves a balance in the levels of R1 and R2 expression, and that alterations in this balance can significantly modify transformation, tumorigenicity, and metastatic potential.
Ribonucleotide reductase is the only enzyme in the cell that is responsible for converting ribonucleotides into deoxyribonucleotides, the eventual substrates for DNA polymerase (1–3). In mammalian cells, this enzyme contains two dissimilar protein components, called R1 and R2, which are encoded by two different genes located on different chromosomes (4). Protein R1 is a homodimeric structure, with a molecular mass of 168 kDa, and has substrate sites and allosteric effector sites that control enzyme activity and substrate specificity (3, 5). Protein R2 is a homodimer, with a molecular mass of 88 kDa, and forms two equivalent dinuclear iron centers that stabilize a tyrosyl free radical required for the initiation of electron transformation during catalysis (5, 6). R1 and R2 proteins interact at their C-terminal ends to form an active holoenzyme (5, 7).
Expression of ribonucleotide reductase is highly regulated (3, 5, 8). Nonproliferating cells do not contain ribonucleotide reductase activity. In proliferating cells the R2 protein is found primarily in S phase of the cell cycle (9). Although the R1 protein can be detected throughout the cell cycle, synthesis of R1 mRNA, like R2 mRNA, appears to occur mainly during S phase (9, 10). The broader distribution of the R1 protein during the cell cycle is attributed to its longer half-life as compared with the R2 protein (8). Ribonucleotide reductase serves other biological functions in addition to providing substrates for DNA replication. For example, its activity can be induced outside the S phase by DNA crosslinking agents such as chlorambucil, indicating a role for the enzyme in the DNA repair process (11). Ribonucleotide reductase has long been suspected of playing a role in malignancy, because higher levels of enzyme activity have been observed in cultured malignant cells when compared with nonmalignant cells (12, 13), a positive correlation between the growth rate of tumors in mice and the levels of ribonucleotide reductase activity has been described (12), and increased levels of R2 protein and R2 mRNA have been found in premalignant and malignant human tissues as compared with normal control tissue samples (14, 15). Furthermore, previous studies have shown that regulation of ribonucleotide reductase, and in particular the R2 component, is significantly elevated in transformed cells exposed to tumor promoters, or to transforming growth factor β in growth factor-mediated mechanisms of tumor progression (16–19). Recently, we have directly tested a possible role for ribonucleotide reductase in malignancy by analyzing malignancy-related characteristics of cells containing deregulated R2 expression achieved by gene transfer techniques (20). Interestingly, overexpression of R2 led to an increased frequency of transformed foci formation by mouse fibroblasts after transfection with activated H-ras. In addition, expression of recombinant R2 in ras-transformed cells resulted in enhanced colony-forming efficiency in soft agar and markedly elevated tumorigenic and metastatic potential in vivo (20). Furthermore, deregulated R2 expression can cooperate with other oncogenes like rac-1 in mechanisms of transformation (ref. 20 and unpublished observations). These results demonstrated that R2 is a novel tumor progressor gene, which can play an important role in mechanisms leading to cellular transformation and malignant progression.
Having observed the malignancy-promoting activity of the R2 component of ribonucleotide reductase, we decided to investigate a possible role in malignancy for the R1 component, alone or in combination with R2. We reasoned that because R2-deregulated expression enhanced tumorigenic and metastatic potential, an increase in R1 expression through its interaction with the R2 component may play an opposite role in the malignant process, and lead to a reduction in malignancy-related characteristics. Interestingly, we show that overexpression of R1 strongly suppresses cellular transformation as well as malignant progression in vivo.
MATERIALS AND METHODS
Expression Vectors.
The human Myc epitope-tagged mouse R1 cDNA was obtained by PCR by using the 5′-primer ACCGCTCGAGCCACCATGGAACAAAAGCTTATTTCTGAAGAAGACTTGATGCATGTGATCAAGCGAGA [where the Kozak sequence, a possible ribosomal binding signal (21), is in italics; the sequence encoding the human Myc epitope is underlined; and the natural ATG initiation codon is in bold], and the 3′-primer CCGCTCGAATCAGGATCCACACATCAG (where the termination codon is in bold), and the template plasmid pcD-M1 (22). The PCR product was treated with proteinase K, digested with XhoI, gel-purified, and ligated to dephosphorylated XhoI-digested pLXSHD plasmid (23, 24), to generate the retroviral vector pSHD/mR1. Packaging of retroviral vector and preparation of viral stock was accomplished by using Ψ2 and PA317 cell lines, as we have described (23), except that PA317-derived stable packaging lines were obtained by selection with histidinol for 15 days. To obtain an expression vector for R1 in the antisense orientation, mouse R1 cDNA was prepared by PCR that used the primers GCCTCGAGCTGACAGTCGTCTCTGTCCCT and TAAAGCTTATCACTTAGAAATGTTTATTTCAAAAT, digested with XhoI and HindIII, and inserted into the mammalian expression plasmid pcDNA3 (Invitrogen), to give the plasmid pASR1. The construction of both pSHD/mR1 and pASR1 was confirmed by sequencing analysis as well as restriction by endonuclease digestions.
Cell Lines and Cell Culture.
Cell lines used in this study and related information are shown in Table 1. Cells were routinely cultured in α-minimal essential medium (MEM) supplemented with 8% calf serum (Fetalclone III, HyClone). To generate cells expressing recombinant R1, cells were infected with SHD/mR1 viral stock, prepared from the stable packaging line PA/mR1, in the presence of polybrene (24). Stable infectants (≥500 clones) were obtained by selection with 4–15 mM histidinol depending on the cell lines and were pooled and expanded. Control cell lines were generated in parallel by using LXSHD virus lacking the R1 sequence. R1 antisense-expressing cells were generated by transfection of NIH 3T3 cells by using a LipofectAmine kit (Life Technologies) followed by selection with G418. Cell growth rates were assessed by measuring absorbance at 260 nm with cell extracts prepared in 1.0 N NaOH (27), and/or by counting cells at different time points after seeding. Growth in soft agar was estimated in 10-cm tissue culture plates containing 15 ml of base agar (0.5% Bacto agar in MEM containing 10% calf serum), and 10 ml of growth agar (0.33% agar in MEM containing 10% calf serum). Cells were obtained from subconfluent cultures, and colonies were scored 14–21 days later (20).
Table 1.
Information about cell lines
| Designate | Parental line | Related characteristics and transgene expression | Reference or source |
|---|---|---|---|
| BHK | ATCC | ||
| Ψ2 | Retroviral packaging | ATCC | |
| PA317 | Retroviral packaging | ATCC | |
| PA/SHD | PA317 | Packaging control virus LXSHD | This study |
| PA/mR1 | PA317 | Packaging SHD/mR1* virus | This study |
| SC2 | L60 | R2 is in excess as compared to R1 | McClarty et al. (6) |
| SC2/SHD | SC2 | hisD† | This study |
| SC2/mR1 | SC2 | mR1, hisD† | This study |
| CIRAS-1 | 10T½ | T24 H-ras | Egan et al. (25) |
| C1/SHD | CIRAS-1 | hisD†, T24 H-ras | This study |
| C1/mR1 | CIRAS-1 | mR1*, hisD†, T24 H-ras | This study |
| C1/mR2 | CIRAS-1 | mR2*, T24 H-ras | Fan et al. (20) |
| C1/mR2/SHD | C1/mR2 | hisD†, mR2*, T24 H-ras | This study |
| C1/mR2/mR1 | C1/mR2 | mR1, hisD†, mR2*, T24 H-ras | This study |
| C1/mR2a/SHD | C1/mR2a‡ | hisD†, mR2*, T24 H-ras | This study |
| C1/mR2a/mR1 | C1/mR2a‡ | mR1*, hisD†, mR2*, T24 H-ras | This study |
| ras-3 (or R-3) | 10T½ | T24 H-ras | Taylor et al. (26) |
| ras-3/SHD | ras-3 | hisD†, T24 H-ras | This study |
| ras-3/mR1 | ras-3 | mR1*, hisD†, T24 H-ras | This study |
| RMP-6 | 10T½ | T24 H-ras, c-myc, mutant p53 | Taylor et al. (26) |
| RMP/SHD | RMP-6 | hisD†, T24 H-ras, c-myc, mutant p53 | This study |
| RMP/mR1 | RMP-6 | mR1*, hisD†, T24 H-ras, c-myc, mutant p53 | This study |
| Colo 320HSR | Human colon carcinoma | ATCC | |
| Colo/SHD | Colo 320HSR | hisD† | This study |
| Colo/mR1 | Colo 320HSR | mR1*, hisD† | This study |
| NIH 3T3 | ATCC | ||
| N/ras | NIH3T3 | T24 H-ras, neo† | This study |
| N/ras&ASR1 | NIH3T3 | R1 antisense, T24 H-ras, neo† | This study |
ATCC, American Type Culture Collection.
mR1 and mR2, human Myc epitoped-tagged R1 and R2 proteins, respectively.
Selective marker genes, hisD (histidinol dehydrogenase), and neo (neomycin phosphotransferase).
C1/mR2a is a subclone derived from C1/mR2.
Assays for Tumorigenicity and Metastasis.
C3H/HeN syngeneic mice (Charles River Breeding Laboratories) were used in these assays as described (20). Cells were prepared from subconfluent, logarithmically growing cultures, collected by gentle treatment with trypsin/EDTA solution, and adjusted to appropriate concentration. Tumor latency was determined by injecting cells subcutaneously and recording the time required to form a tumor (2 × 2 mm) detectable by palpation. Tumors were removed from the mice, and tumor weight was recorded 21 days later. In the case of no tumor formation, mice were kept for 2 months after injection and then sacrificed. For metastasis assays, cells were injected into the tail veins of 6- to 8-week-old C3H/HeN syngeneic mice and an estimate of the number of lung tumors was made 21 days later, as described (20). To confirm that equal numbers of test and control cells were injected, duplicate culture plates containing growth medium were inoculated with 100 cells per plate. After 10 days in culture, plates were stained with methylene blue, and colonies were scored.
Detection of Recombinant R1 Protein Expression.
An indirect immunofluorescence assay was used to detect transient expression of recombinant R1 protein in BHK cells. Seventy percent confluent cultures growing on coverslips were transfected with pSHD/mR1 plasmid by using a LipofectAmine reagent. At 20 hr after transfection, cells were fixed with 3% formaldehyde prepared in PBS, pH 7.2, permeabilized with 0.1% Triton X-100 (in PBS), incubated with anti-Myc epitope monoclonal 9E10 antibody (American Type Culture Collection), washed, reacted with goat anti-mouse IgG (full molecule) fluorescein isothiocyanate conjugate (Sigma), washed again, and finally examined under a fluorescence microscope (28). Immunoprecipitation of recombinant R1, by the 9E10 antibody, from [35S]methionine/cysteine-labeled cells was performed by using previously described procedures (6). In some experiments R1 protein levels were determined by Western blot analysis by using the AD203 anti-R1 mAb (Accurate Chemicals) as we have described (6, 20).
Ribonucleotide Reductase Assay.
Ribonucleotide reductase activity in crude extracts prepared from SC2/mR1 and control SC2/SHD cell lines was assayed as we have described (11). In some experiments, enzyme assays were performed by combining purified recombinant R2 protein with 9E10 antibody-precipitated R1 protein. Pansorbin cells (formaldehyde fixed Staphylococci, Calbiochem) carrying surface protein A and rabbit anti-mouse IgG was prepared as described (29). This conjugate was further incubated with an excess amount of 9E10 antibody and washed five times. Twenty microliters of this complex (10% suspension) was added to 1.0 ml of extract prepared from 5 × 107 cells and placed on a rocker at 4°C for 2 hr, washed three times with PBS containing 10 mg/ml BSA, and assayed for ribonucleotide reductase activity after the addition of 1.0 μg of purified recombinant R2 protein (30).
RESULTS AND DISCUSSION
Expression of Recombinant R1.
To overexpress the R1 protein in cells, we constructed a mammalian expression vector pSHD/mR1. In this vector, the expression of the human Myc-epitope tagged R1 cDNA is under the control of a retroviral promoter, long terminal repeat sequence (23). The expression of recombinant R1 first was analyzed in BHK cells after transient transfection. An indirect immunofluorescence assay that used the anti-Myc monoclonal 9E10 antibody revealed cytoplasmic expression of the recombinant R1 protein in pSHD/mR1 transfected cells (Fig. 1A). As a control, nontransfected or empty vector pLXSHD transfected cells did not show any specific fluorescence (data not shown). After demonstrating that the recombinant R1 protein can be expressed in mammalian cells, we then converted the expressible DNA into an infectious, but replication-deficient, vector virus that has a high delivery efficiency, by using retroviral packaging cells (Table 1). The expression of recombinant R1 in the stable packaging line PA/mR1 was analyzed again. Immunoprecipitation with the 9E10 antibody detected a single, approximately 88-kDa protein from extract prepared from PA/mR1 cells, which had been metabolically labeled with [35S]methionine/cysteine (Fig. 1B). As expected, no protein was precipitated from the stable control virus packaging cell line PA/SHD (Fig. 1B). These results indicated that stable expression of the recombinant R1 protein could be achieved.
Figure 1.

Analysis of Myc-tagged R1 expression from pSHD/mR1 transiently transfected BHK cells by the indirect immunofluorescence assay (A), and from the stable retroviral packaging cell line PA/mR1 by radioimmunoprecipitation (B). Anti-Myc epitope antibody 9E10 was used for both assays.
We then determined if cell-expressed recombinant R1 is biologically active. For this we infected a hydroxyurea-resistant mouse L cell line, SC2, with SHD/mR1 or LXSHD viral vectors and selected stable infectants (Table 1). Because SC2 cells express more R2 relative to the R1 subunit, expression of biologically active R1 protein in this cell line would result in increased ribonucleotide reductase activity (6). In four experiments, the cytidine 5′-diphosphate reductase activity in the crude extract prepared from SC2/mR1 cells was 13.2 ± 0.7 nmols/mg of protein per hr, which is approximately 30% higher than that in extract prepared from control SC2/SHD cells (10.1 ± 0.2 nmols/mg per hr). Furthermore, we immunoprecipitated the recombinant R1 from C1/mR1 cells by using the 9E10 antibody, and assayed ribonucleotide reductase activity by combining the washed immunoprecipitate with purified recombinant R2 protein. In three independent experiments, enzyme activity of 15.4 ± 2.0 pmols/mg per hr was detected when C1/mR1 cells (Table 1) were used as a source of recombinant R1, and as expected no activity was found when control C1/SHD (Table 1) cells were used.
Reduced Anchorage Independence by Cells that Overexpress R1.
Cellular transformation frequently is accompanied by anchorage independent growth in vitro, which often correlates with tumorigenic potential in vivo, and can be evaluated by the ability to proliferate and form colonies in medium containing agar (20, 25). To investigate the role that R1 may play in cellular transformation we infected CIRAS-1 cells with the SHD/mR1 vector virus or the control LXSHD virus without the R1 insert. CIRAS-1 cells were derived from wild-type, nonmalignant mouse 10T½ cells by transfection with oncogenic T24 H-ras (25). Previous studies have shown that it is a moderately malignant cell line and can serve as a good model for analyzing transformation and malignancy-related characteristics (20, 31). Stable infectants obtained after histidinol selection were evaluated for soft agar growth abilities. We found that the efficiency in forming colonies in soft agar by C1/mR1 cells that contained elevated levels of R1 was significantly decreased as compared with control C1/SHD cells (Fig. 2). We also tested the effects of R1 expression on soft agar growth with cells containing deregulated R2 expression. C1/mR2 is a CIRAS-1 derivative expressing recombinant R2 and has acquired an increased malignant potential (20). Again we observed that colony-forming efficiency of C1/mR2/mR1 cells with elevated R1 was significantly reduced when compared with control C1/mR2 cells (Fig. 2). To exclude the possibility (albeit unlikely) that the reduced soft agar growth efficiencies observed with R1 cells may have resulted from selection of cells with intrinsically lower growth efficiencies from a relatively heterogeneous cell population, we isolated a subclone named C1/mR2a from C1/mR2 cells. We then derived two cell lines (C1/mR2a/mR1 and control C1 mR2a/SHD) from this subclone population (Table 1). Consistent with the observations with the parental line, C1/mR2a/mR1 cells exhibited similar reductions in growth efficiencies in soft agar, when compared with control C1/mR2a/SHD cells (Fig. 2). This strongly suggests that the reduced anchorage-independence is brought about by recombinant R1 expression.
Figure 2.

Reduced growth efficiency in soft agar with recombinant R1 expressing cells. Cell lines stably infected with the R1 viral vector were compared with appropriate empty vector-infected control cell lines (see Table 1 and below). Data presented were obtained from at least three independent experiments, each consisting of triplicate plates per cell line. Inoculum sizes (cells/plate) were as follows: 5 × 105 for C1/SHD and C1/mR1 cells, 1 × 105 for C1/mR2 and C1/mR2/mR1 cells, 1 × 104 for C1/mR2a/SHD and C1/mR2a/mR1 cells, ras-3/SHD and ras-3/mR1 cells, and 1 × 103 for Colo/SHD and Colo/mR1 cells. In all cases, the difference in numbers of colonies formed between recombinant R1 expressing cells and the control cells were statistically significant (P < 0.001).
The above tested cell lines are of mouse origin. To determine if expression of recombinant R1 in human tumor cells also alters transformation characteristics as demonstrated by changes in soft agar growth ability, we used the human colon adenocarcinoma Colo 320HSR cell line. These cells were infected with the viral vector containing the R1 sequence to obtain the Colo/mR1 cell line (Table 1). Interestingly, growth efficiency in soft agar with Colo/mR1 cells was decreased by about 50% when compared with Colo/SHD cells containing the empty vector (Fig. 2).
Suppression of Tumorigenic and/or Metastatic Potential in Vivo with Cells Containing Elevated R1.
To further evaluate the role that alterations in R1 expression may play in malignant progression, tumorigenic and metastatic properties of C1/mR1 cells were analyzed in in vivo models. C1/mR1 cells exhibited a dramatic reduction in malignant potential when compared with control C1/SHD cells (Table 2). Whereas all mice injected subcutaneously with C1/SHD cells developed tumors at about 11 days after injection, none of the animals injected with C1/mR1 cells formed detectable tumors even up to 2 months after injection. In addition, experimental metastasis assays showed that C1/mR1 cells were much less efficient at forming lung metastases as compared with the control C1/SHD cells (Table 2). We also carried out similar experiments with another independently selected T24 H-ras transfected mouse 10T1/2 cell line called ras-3, which has been described (26). Although the tumor-suppressing activity of the recombinant R1 in ras-3 cells was not as great as was observed in C1RAS-1 derived cells, tumorigenicity with ras-3/mR1 cells, as compared with control ras-3/SHD cells, was significantly reduced as judged by an extended tumor latency and smaller tumor sizes. Similar to the CIRAS-1 derived cells, ras-3/mR1 cells exhibited a markedly reduced metastatic potential when compared with the control ras-3/SHD cells (Table 2).
Table 2.
Decreased tumorigenic and metastatic potential with cells expressing recombinant R1 protein
| Cell line | Tumorigenicity assay* (subcutaneous tumors)
|
Metastasis assay† (lung tumors)
|
||||
|---|---|---|---|---|---|---|
| Frequency | Latency (days) | Weight (g) | Frequency | Number of tumors | P | |
| C1/SHD | 5/5 | 11 ± 2 | 0.3 ± 0.1 | 5/5 | 44 ± 13 | |
| C1/mR1 | 0/5 | — | — | 5/5 | 6 ± 3 | <0.001 |
| ras-3/SHD | 5/5 | 9 ± 2 | 0.9 ± 0.3 | 5/5 | 120 ± 22 | |
| ras-3/mR1 | 4/5 | 14 ± 2 | 0.3 ± 0.2 | 4/5 | 4 ± 2 | <0.001 |
| C1/mR2 | 10/10 | 8 ± 1 | >1.0‡ | 10/10 | 195 ± 34 | |
| C1/mR2/mR1 | 2/10‡ | 19 ± 4 | <0.1‡ | 10/10 | 20 ± 6 | <0.001 |
| RMP/SHD | 4/4 | 7 ± 1 | 1.1 ± 0.3 | 4/4 | 61 ± 8 | |
| RMP/mR1 | 4/4 | 7 ± 1 | 1.0 ± 0.2 | 4/4 | 27 ± 7 | <0.001 |
Number of cells injected subcutaneously were as the follows: 1 × 105 for RMP/SHD and RMP/mR1 cells, and 3 × 105 for all other cell lines.
Number of cells injected intravenously were: 2 × 105 for RMP/SHD and RMP/mR1 cells, and 1 × 106 for all other cell lines.
Mice injected with C1/mR2 cells were sacrificed 21 days after injection. Tumors from these mice were estimated to be at least 1.0 g. One of the mice injected with C1/mR2/mR1 developed tumor on day 15; when this mouse was killed on day 21, the tumor was estimated to be less than 0.1 g. Another mouse in this group had an extremely slow-growing tumor, which became detectable on day 23 and was estimated to be less than 0.1 g on day 60.
We also tested in vivo the impact of recombinant R1 expression on malignant potential with cells that contained deregulated R2 expression. Consistent with previous observations, C1/mR2 cells showed higher tumorigenic and metastatic potential than control C1/SHD cells, confirming the malignancy promoting function of R2 (20). Also in agreement with the data obtained in in vitro experiments (Fig. 2), C1/mR2/mR1 cells were much less malignant than control C1/mR2 cells (Table 2). Further, both the tumorigenic and the metastatic potential of C1/mR2/mR1 cells were reduced to significantly lower levels than that of C1/SHD cells (Table 2).
We next tested the potential ability of recombinant R1 expression to modify the malignant properties of a highly malignant cell line that contains multiple oncogene alterations. The RMP-6 mouse 10T1/2 line, which has been transfected with a combination of activated H-ras, c-myc, and a mutated oncogenic form of p53, has been well characterized (26, 32) and was used in these studies. Unlike the above R1 overexpressing cell lines studied, RMP/mR1 cells (Table 1) did not show changes in tumorigenicity when compared with control RMP/SHD cells (Table 2). In agreement with these in vivo results, we observed that RMP/mR1 and RMP/SHD cells had approximately the same colony-forming efficiencies in soft agar growth experiments (data not shown). Interestingly, however, RMP/mR1 cells formed significantly fewer lung tumors in syngeneic mice than control RMP/SHD cells in experimental metastasis assays (Table 2). The difference in numbers of lung metastases shown in Table 2 actually may have been underestimated, because lung tumors generally were larger in mice that had received RMP/SHD cells than those that developed in the lungs of mice injected with RMP/mR1 cells. These results indicate that overexpression of R1 in these highly malignant RMP-6 cells can suppress metastatic potential without obviously affecting tumor growth properties. Similar effects on tumorigenicity and metastasis have been reported previously with several tumor suppressors (33, 34).
Increased Anchorage Independence with Oncogenic ras-Transfected Cells Expressing R1 in the Antisense Orientation.
The results obtained from both in vitro and in vivo experiments with multiple malignant cell lines of both mouse and human origins have shown that R1 can suppress transformation and malignant progression when it is overexpressed. Human tumor suppressors mostly have been identified by studies linking increased tumor incidence and/or accelerated tumor progression to lost expression of functional molecules as a result of mutations. Interestingly, it recently has been reported that a deletion of the coding sequence in the R1 mRNA can be detected in some human colorectal carcinomas (35). Because we observed that the levels of R1 expression can determine malignant potential, it is certainly possible that an R1 deletion would decrease R1 protein levels and enhance tumor progression. This prompted us to analyze the malignancy-related properties of cells with down-regulated R1 expression. Antisense approaches are commonly used to achieve decreased gene expression (36), therefore we constructed an expression vector in which the R1 sequence has been placed in an antisense orientation relative to the vector promoter. NIH 3T3 cells were cotransfected with the antisense vector and the pH06Ti plasmid that expresses the T24 H-ras oncogene (20). H-ras expression transforms mammalian cells so that they are often capable of colony formation in soft agar containing growth medium (20, 25). Stable cotransfectants obtained after selection with G418 were evaluated for anchorage independent growth. Interestingly, N/ras & ASR1 cells containing R1 antisense exhibited a dramatically higher colony-forming efficiency in soft agar, when compared with control N3/ras cells that contained the H-ras oncogene without the R1 antisense sequence (Fig. 3). Western blot analysis indicated, as expected, lower expression of R1 in N/ras & ASR1 cells than in N/ras cells (Fig. 4). These data together with a R1 deletion found in human colorectal carcinomas (35) suggest that decreased R1 expression is important in tumorigenesis and support the concept that R1 can act as a tumor suppressor activity. However, it is unlikely that a complete lack of R1 expression would occur in the development of malignancies, because the R1 protein is essential for an enzyme activity that is required for DNA synthesis (1–3), and a lack of R1 expression would be incompatible with cell proliferation. Therefore, R1 differs from some other tumor suppressors, in which inactivating mutations to both alleles may occur (37). In a proliferating cell there must be at least one R1 allele that encodes an active R1 protein. These observations suggest that a reduction in R1 levels as opposed to a complete lack of R1 protein is important in malignant development. Studies aimed at determining the relative levels of R1 protein in human cancers, and especially an extensive search for possible R1 gene mutations, are required to further test this hypothesis.
Figure 3.

Increased colony-forming efficiency in soft agar with N/ras & ASR1 cells (b) as compared with control N/ras cells (a). Each plate was inoculated with 1 × 104 cells. N/ras & ASR1 cells formed at least four times more colonies, which were generally larger than those formed by N/ras cells. The colonies shown in a developed after 3 weeks and those shown in b developed after 2 weeks of incubation. When data from six experiments each consisting of four plates per cell line were analyzed, the difference in colony-forming efficiencies exhibited by the two cell lines were found to be highly significant (P < 0.0001).
Figure 4.
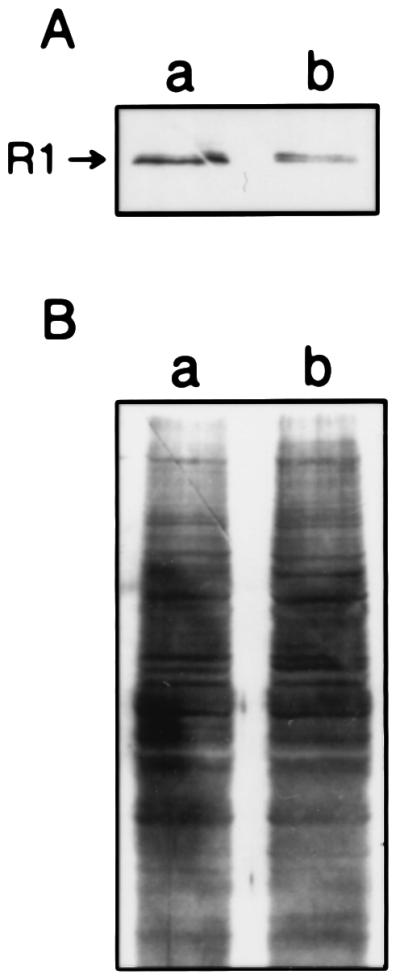
(A) Western blot analysis showing reduced R1 protein in N/ras & ASR1 (b) as compared with control N/ras cells (a). (B) India ink staining of the nitrocellulose membrane shown in A, demonstrating approximately equal loading of cell extracts. Densitometric analysis indicated a decrease of 3.2-fold in R1 protein level in N/ras & ASR1 cells when compared with N/ras cells.
Unaltered Growth Rate and Plating Efficiency for Cells with Deregulated R1 Expression Grown on a Solid Surface.
Whereas tumor suppressors such as p53 when overexpressed suppress cell growth in vitro (37), others represented by nucleoside diphosphate kinase nm-23 suppress malignancy without affecting cell growth in culture (33). Furthermore, the RB tumor suppressor has been shown to suppress growth of some cell lines (38), but not of others (39). Therefore, growth suppression is not necessarily an indicator of tumor suppressor potential. Cell growth rates and plating efficiencies were determined at least two times for all the cell lines listed in Table 1, except the retroviral packaging lines, and we found that cell lines containing the R1 sense or antisense vectors had essentially the same or very similar growth rates and plating efficiencies as their corresponding control lines. These observations indicate that deregulated R1 expression can occur without significantly modifying in vitro growth properties as determined in growth studies performed on the surface of plastic culture plates.
Malignant Potential Modified by the Levels of R1 and R2 Gene Expression.
The present study and a previous report (20) show that R1 and R2 can play opposing roles in the regulation of cellular transformation, tumorigenicity, and metastatic potential. We suggest that there is a delicate balance between R1 and R2 levels in cells, and that alterations in this balance brought about by R2 overexpression and/or decreased R1 expression can promote transformation and malignancy, whereas alterations that decrease R2 expression and/or elevate R1 expression will favor malignant suppression. As a working model, we suggest that in addition to functioning as subunits for ribonucleotide reductase activity (1–3), R1 and R2 have novel, but yet undefined, properties that affect signal pathways involved in determining transformation and malignant potential. Support for this view comes from observations that deregulated R2 expression activates a Ras/Raf/Erk pathway, which is important in mediating the expression of genes that regulate such important biological functions as cell cycle progression and cellular differentiation (20). We propose that regulation in the expression of the two components of ribonucleotide reductase provides a novel control point in mechanisms of malignant progression, a concept that is potentially useful when considering new strategies to intervene in this process.
Acknowledgments
We thank Arthur Chan and Gao Chen for helpful discussions and technical assistance, and Sabine Mai for providing the Colo 320HSR cell line and for helpful discussions. This study was supported by the National Cancer Institute of Canada and the Natural Sciences and Engineering Research Council of Canada (to J.A.W.). H.F. has been the recipient of a Medical Research Council of Canada Postdoctoral Fellowship and a Medical Research Council of Canada-Pfizer Industrial Fellowship. A.H. is a former recipient of Postdoctoral Fellowships from the George H. Sellers Endowment Fund, Manitoba Cancer Treatment and Research Foundation, and the Manitoba Health Research Council. J.A.W. is a Terry Fox Senior Scientist of the National Cancer Institute of Canada.
Footnotes
This paper was submitted directly (Track II) to the Proceedings Office.
References
- 1.Lewis W H, Kuzik B A, Wright J A. J Cell Physiol. 1978;94:287–298. doi: 10.1002/jcp.1040940306. [DOI] [PubMed] [Google Scholar]
- 2.Reichard P. Science. 1993;260:1773–1777. doi: 10.1126/science.8511586. [DOI] [PubMed] [Google Scholar]
- 3.Wright J A. Int Encyclop Pharmacol Therapeut. 1989;128:89–111. [Google Scholar]
- 4.Tonin P N, Stallings R L, Carman M D, Bertino J R, Wright J A, Srinivasan P R, Lewis W H. Cytogenet Cell Genet. 1987;45:102–108. doi: 10.1159/000132438. [DOI] [PubMed] [Google Scholar]
- 5.Wright J A, Chan A K, Choy B K, Hurta R A R, McClarty G A, Tagger A Y. Biochem Cell Biol. 1990;68:1364–1371. doi: 10.1139/o90-199. [DOI] [PubMed] [Google Scholar]
- 6.McClarty G A, Chan A K, Choy B K, Wright J A. J Biol Chem. 1990;265:7539–7547. [PubMed] [Google Scholar]
- 7.Davis R, Thelander M, Mann G J, Behravan G, Soucy F, Beaulieu P, Lavallee P, Gräslund A, Thelander L. J Biol Chem. 1994;269:23171–23176. [PubMed] [Google Scholar]
- 8.Choy B K, McClarty G A, Chan A K, Thelander L, Wright J A. Cancer Res. 1988;48:2029–2035. [PubMed] [Google Scholar]
- 9.Eriksson S, Gräslund A, Skog A, Thelander L, Tribukait B. J Biol Chem. 1984;259:11695–11700. [PubMed] [Google Scholar]
- 10.Björklund S, Skog S, Tribukait B, Thelander L. Biochemistry. 1990;29:5452–5458. doi: 10.1021/bi00475a007. [DOI] [PubMed] [Google Scholar]
- 11.Hurta R A R, Wright J A. J Biol Chem. 1992;267:7066–7071. [PubMed] [Google Scholar]
- 12.Weber G. Cancer Res. 1983;43:3466–3492. [PubMed] [Google Scholar]
- 13.Wright J A, McClarty G A, Lewis W H, Srinivasan P R in. In: Drug Resistance in Mammalian Cells. Gupta R S, editor. Vol. 1. Boca Raton, FL: CRC; 1989. pp. 15–27. [Google Scholar]
- 14.Saeki T, Takashashi T, Okabe M, Furuya A, Hanai N, Yamagami K, Mandai K, Moriwaki S, Doihara H, Takashima S, Salomon D S. Int J Oncol. 1995;6:523–533. doi: 10.3892/ijo.6.3.523. [DOI] [PubMed] [Google Scholar]
- 15.Jensen R A, Page D L, Holt J T. Proc Natl Acad Sci USA. 1994;91:9257–9261. doi: 10.1073/pnas.91.20.9257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Amara F M, Sun J, Wright J A. J Biol Chem. 1996;271:20126–20131. doi: 10.1074/jbc.271.33.20126. [DOI] [PubMed] [Google Scholar]
- 17.Chen F Y, Amara F M, Wright J A. EMBO J. 1993;12:3977–3986. doi: 10.1002/j.1460-2075.1993.tb06075.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Amara F M, Chen F Y, Wright J A. Nucleic Acids Res. 1995;23:1461–1467. doi: 10.1093/nar/23.9.1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hurta R A R, Samuel S K, Greenberg A H, Wright J A. J Biol Chem. 1991;266:24097–24100. [PubMed] [Google Scholar]
- 20.Fan H, Villegas C, Wright J A. Proc Natl Acad Sci USA. 1996;93:13036–14040. doi: 10.1073/pnas.93.24.14036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kozak M. Nucleic Acids Res. 1987;20:8125–8148. doi: 10.1093/nar/15.20.8125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Thelander L, Berg P. Mol Cell Biol. 1986;6:3433–3442. doi: 10.1128/mcb.6.10.3433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Miller A D, Miller D G, Garcia J V, Lynch C M. Methods Enzymol. 1993;217:581–599. doi: 10.1016/0076-6879(93)17090-r. [DOI] [PubMed] [Google Scholar]
- 24.Fan H, Villegas C, Wright J A. FEBS Lett. 1996;382:145–148. doi: 10.1016/0014-5793(96)00143-3. [DOI] [PubMed] [Google Scholar]
- 25.Egan S E, McClarty G A, Jarolim L, Wright J A, Spiro I, Hager G, Greenberg A H. Mol Cell Biol. 1987;7:830–837. doi: 10.1128/mcb.7.2.830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Taylor W R, Egan S E, Mowat M, Greenberg A H, Wright J A. Oncogene. 1992;7:1383–1390. [PubMed] [Google Scholar]
- 27.Kempe T D, Swaryl E A, Bruist M, Stark G R. Cell. 1976;9:541–550. doi: 10.1016/0092-8674(76)90036-2. [DOI] [PubMed] [Google Scholar]
- 28.Leonhardt, U., Page, A. W., Weier, H.-U. & Bestor, T. H. Cell 71, 865–873. [DOI] [PubMed]
- 29.Goding J W. J Immunol Methods. 1978;20:241–253. doi: 10.1016/0022-1759(78)90259-4. [DOI] [PubMed] [Google Scholar]
- 30.Mann G J, Gräslund A, Ochiai E-I, Ingermarson R, Thelander L. Biochemistry. 1991;30:1939–1947. doi: 10.1021/bi00221a030. [DOI] [PubMed] [Google Scholar]
- 31.Wright J A, Turley E A, Greenberg A H. Crit Rev Oncog. 1993;4:473–492. [PubMed] [Google Scholar]
- 32.Huang A, Jin H, Taylor W R, Wright J A. Int J Oncol. 1995;7:57–63. doi: 10.3892/ijo.7.1.57. [DOI] [PubMed] [Google Scholar]
- 33.MacDonald N J, De La Rosa A, Steeg P S. Eur J Cancer. 1995;31A:1096–1100. doi: 10.1016/0959-8049(95)00152-9. [DOI] [PubMed] [Google Scholar]
- 34.Welch D R, Chen P, Miele M E, McGary C T, Bower J M, Stanbridge E J, Weissman B E. Oncogene. 1994;9:255–262. [PubMed] [Google Scholar]
- 35.Mader R M, Schmidt W, Rizovski B, Sedivy R, Steger G G, Jakesz R, Rainer H, Mueller M W. Proceedings of the Eighty-Seventh Annual Meeting, American Association of Cancer Research. Vol. 37. abstr.; 1996. p. 547. [Google Scholar]
- 36.Wright J A, Anazodo M. Cancer J. 1995;8:185–189. [Google Scholar]
- 37.Levine A J. Cell. 1997;88:323–331. doi: 10.1016/s0092-8674(00)81871-1. [DOI] [PubMed] [Google Scholar]
- 38.Connell-Crowley L, Harper J W, Doodrich D W. Mol Biol Cell. 1997;8:287–301. doi: 10.1091/mbc.8.2.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bookstein R, Shew J Y, Chen P L, Scully P, Lee W-H. Science. 1990;247:712–715. doi: 10.1126/science.2300823. [DOI] [PubMed] [Google Scholar]