

Abstract
Production of an extracellular matrix is a hallmark of biofilm formation. In the spore-forming bacterium Bacillus subtilis, the matrix consists of an exopolysaccharide, which is specified by the epsA-O operon, and a secreted protein TasA, which is encoded by the yqxM-sipW-tasA operon. Past and present evidence establish that the epsA-O and yqxM-sipW-tasA operons are controlled by the repressor proteins SinR and AbrB. Here, we report the identification of a novel regulatory protein Slr that promotes transcription of the yqxM-sipW-tasA operon but is not needed for expression of the epsA-O operon. We further show that the gene for Slr is itself under the negative control of SinR and AbrB. These findings reveal that matrix production is governed by an intricate network involving the interplay of negatively- and positively-acting regulatory proteins.
Introduction
The gram-positive soil bacterium Bacillus subtilis is capable of forming surface-attached, multicellular communities or biofilms (Branda et al., 2001; Hamon and Lazazzera, 2001). Some strains of domesticated B. subtilis produce thin, floating biofilms at the liquid-air interface (pellicles) and exhibit some measure of colony architecture on solid medium (Branda et al., 2004; Veening et al., 2006). However, robust pellicle formation and complex colony architecture is primarily observed in wild, undomesticated strains. The pellicles and colonies of these wild strains exhibit wrinkles with tongue-like aerial projections that serve as preferential sites for sporulation (Branda et al., 2001).
Biofilms consist of long chains of cells in parallel arrangement (Branda et al., 2001). The bundled chains are held together by an extracellular matrix that is primarily composed of exopolysaccharide and protein (Branda et al., 2006). The 15-gene operon epsA-O (henceforth simply eps) encodes the biosynthetic machinery required to produce the exopolysaccharide (Branda et al., 2001; Kearns et al., 2005), whereas the protein component is encoded by tasA, the terminal gene in the yqxM-sipW-tasA operon (henceforth simply yqxM) (Branda et al., 2006). The secretion of TasA requires processing of its N-terminal signal sequence by the signal peptidase SipW (Stover and Driks, 1999). Cells lacking both components of the extracellular matrix form flat, featureless colonies and are unable to form pellicles (Branda et al., 2006). Interestingly, only a fraction of the cells in a culture are responsible for producing matrix for the entire community when the eps and yqxM operons are initially induced (Chai et al., 2008). Moreover, fluorescence microscopy reveals that the biofilm is a dynamic community composed of at least three cell types (motile cells, sporulating cells and matrix-producing cells) whose relative proportions change with time (Vlamakis and Aguilar et al., 2008).
Because of the importance of the extracellular matrix to biofilm formation, a central challenge has been to elucidate the regulatory circuitry that governs the expression of the eps and yqxM operons. These operons are under the negative control of SinR, which binds to and blocks transcription of both operons (Kearns et al., 2005; Chu et al., 2006). Derepression is achieved by the action of the antirepressor SinI, which is produced under the control of Spo0A, a response regulator that also governs entry into sporulation (Bai et al., 1993; Hoch 1993; Shafikhani et al., 2002). An additional regulatory protein implicated in biofilm formation is the repressor AbrB. Using laboratory strains of B. subtilis, it was previously shown that the dependence of biofilm formation on Spo0A can be bypassed by a mutation in the gene for AbrB (Hamon and Lazazzera, 2001; Hamon et al., 2004; Veening et al., 2006). This finding suggested that AbrB is a repressor of one or more genes involved in biofilm formation. Indeed, microarray analyses and biochemical experiments identified the yqxM operon as a target of AbrB (Hamon et al., 2004; Strauch et al., 2007).
The initial goal of the present work was to further evaluate potential targets of AbrB in biofilm formation and to elucidate the relative contributions of AbrB and SinR to controlling the expression of the matrix operons. During the course of this investigation, we discovered an additional regulatory protein, called Slr, that contributes to the control of matrix production. We show that Slr is needed for the transcription of the yqxM operon and that the gene for Slr is itself under the negative control of SinR and AbrB. These findings reveal that biofilm formation is governed by a regulatory circuit involving two parallel pathways of repression mediated by SinR and AbrB and an embedded regulatory loop involving an apparent activator protein Slr (Fig. 1).
Figure 1. Regulatory circuit governing biofilm formation.
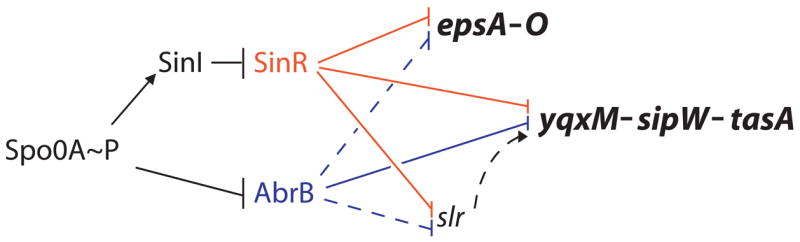
Arrows indicate activation of transcription and T bars indicate repression. Solid lines denote direct regulation and the dashed lines denote cases in which it is unknown if the regulation is direct. Red indicates SinR and its targets and blue denotes AbrB and its targets.
Results
AbrB represses the eps and yqxM operons for matrix production
As a starting point for this work, we took advantage of the capacity of the undomesticated strain 3610 to form robust biofilms on solid medium and in standing liquid medium. Accordingly, we created an insertion/deletion (null) mutation of abrB in strain 3610 by replacing the abrB coding sequence with a drug-resistance gene. The resulting mutant formed hyper-wrinkled colonies on solid medium and very thick, architecturally complex pellicles (Fig. 2A).
Figure 2. Effect of regulatory mutations on biofilm formation.

‘Colony’ column depicts 16 × images of colonies grown on MSgg semi-solid agar medium for 3 days at 22°C. Scale bar is 1 mm. ‘Pellicle’ column shows top-down images of 6-well microtitre plates in which cells have been grown statically in MSgg medium for 3 days at 22°C. Scale bar is 1 cm. A) Opposing phenotypes of abrB and slr mutations. The indicated wild type and mutant strains were as follows: wild type (3610), abrB (FC280), slr (FC525), slr; amyE::slr (FC536). B) The slr mutant phenotype is rescued by yqxM operon overexpression. IPTG was added to MSgg medium at the indicated concentrations. The strain (FC600) was mutant for slr and contained yqxMΩPIPTG-yqxM-sipW-tasA.
The hyper-wrinkled phenotype of the abrB mutant was similar to that of a sinR mutant (Kearns et al., 2005). We therefore hypothesized that, like SinR, AbrB represses transcription of the eps and yqxM operons. Indeed, sipW, a member of the yqxM operon, was one of two genes previously identified as targets of AbrB and reported to be involved in biofilm formation (Hamon et al., 2004). The other gene so identified, yoaW, encodes a protein of unknown function. We created a null mutation of yoaW in strain 3610, but in our hands the resulting mutant formed biofilms that were indistinguishable from those of the wild type strain on both solid and in liquid medium (Fig. S1). Therefore, we restricted our analysis to the eps and yqxM operons.
Next, we measured the effect of the abrB mutation on transcription from the promoters for the eps and yqxM operons using fusions to lacZ. The absence of AbrB results in constitutive bundling, which interferes with accurate measurements of cell number. We therefore introduced a mutation in epsH (a member of the eps operon) to block exopolysaccharide production and hence alleviate bundling (Kearns et al., 2005; Chu et al., 2006). The results of Table 1 show that the absence of AbrB did indeed derepress the eps and yqxM operons. Furthermore, a mutation in abrB bypassed the requirement for Spo0A for eps and yqxM operon expression (Table 1).
Table 1.
AbrB represses the yqxM and eps operons
| Activity (MU)b | ||
|---|---|---|
| Genotypea | Peps-lacZ | PyqxM-lacZ |
| Wild type | 40 ± 1 | 130 ± 9 |
| spo0A | 2.8 ± 0.2 | 4 ± 0.8 |
| abrB | 150 ± 2 | 1100 ± 31 |
| sinR | 1100 ± 13 | 670 ± 47 |
| spo0A abrB | 150 ± 10 | 1000 ± 28 |
| sinR abrB | 1300 ± 20 | 1600 ± 32 |
The following Peps-lacZ containing strains were used FC13, FC413, FC415, FC14, FC444, and FC480. The following PyqxM-lacZ containing strains were used FC134, FC414, FC416, FC135, FC445, and FC481.
β-Galactosidase activity, calculated in Miller Units (MU), was measured from late-exponential phase cells grown in MSgg and is the average of at least three replicates.
We wondered whether this derepression was an indirect effect on the level of expression of sinI, which encodes an antagonist of the SinR repressor (Bai et al., 1993). SinR is known to bind to the promoter regions for, and inhibit the transcription of, both matrix operons (Kearns et al., 2005; Chu et al., 2006). However, the results shown in Table S1 indicate that the absence of AbrB failed to increase the level of expression of lacZ fused to the sinI promoter. We also performed immunoblotting assays using antibodies against SinI and failed to observe an increase in SinI levels in the absence of AbrB (data not shown).
AbrB and SinR act independently in repressing the matrix operons
Next, we asked whether AbrB and SinR exert their repressive effects independently. To do so, we created double mutants lacking both repressors. The results of Table 1 show that the contributions of the two repressors was approximately additive. That is, the level of expression of each operon for the double mutant was approximately equal to the sum of the expression levels for the single mutants. Interestingly, the contribution of SinR to the repression of the eps operon was substantially greater than the contribution of AbrB. Conversely, the contribution of SinR to the repression of the yqxM operon was somewhat less than that for AbrB.
AbrB binds to the promoter region for the yqxM operon
To investigate whether AbrB binds to the eps and yqxM promoter regions, we performed electrophoretic mobility shift assays (EMSA) using purified AbrB. As shown in Fig. 3A, AbrB retarded the electrophoretic mobility of a DNA fragment containing the yqxM promoter region (PyqxM) with a similar binding affinity to a known target of AbrB, the comK promoter region (Hamoen et al., 2003). However, we were unable to detect any change in the electrophoretic mobility of a DNA fragment containing the eps promoter region. This result was similar to that seen using a DNA fragment containing a promoter known to not be under the control of AbrB (recA).
Figure 3. AbrB binds to the yqxM but not to the eps promoter.

A) EMSA in which radiolabeled DNA’s (denoted in the upper left corner of each panel) were mixed with purified His6-AbrB at the indicated concentrations. The sizes of the DNA fragments containing PcomK, PyqxM, PepsA, and PrecA were 475, 284, 250, and 435 base pairs, respectively. The horizontal arrow indicates the position of free probe. B) Footprinting experiment in which a 223 bp fragment of DNA containing PyqxM was 5′ end labeled at the terminus downstream of the promoter, mixed with purified AbrB at the indicated concentrations, and subjected to treatment with DNase I. The vertical bar indicates the region of AbrB protection. Ticks to the left of the panel indicate the AbrB-protected bands. The corresponding DNA sequence is shown to the right of the panel and was determined from a DNA sequencing ladder. The asterisk corresponds to the site of hypersensitivity. A bent arrow indicates the yqxM operon transcription start site. The number to the left of the sequence indicates the yqxM transcription start site. The number to the right of the sequence indicates the position of the AbrB binding site relative to the transcription start site.
DNase I footprinting experiments using radiolabeled PyqxM-containing DNA were employed to map the AbrB binding site in the yqxM upstream regulatory region, which was centered at position -133 relative to the transcription start site (Fig. 3B). This region was relatively refractory to DNase I digestion, but we detected two sites of weak but reproducible protection (indicated by the ticks) and a site of pronounced hypersensitivity at position -119. AbrB binding sites in the yqxM promoter region were independently examined by Strauch et al. (2007). The length of -133 AbrB binding site detected in the present work was consistent with (although slightly smaller; see dotted box in Fig. 4A) than that mapped by Strauch et al. (2007). Strauch et al. (2007) additionally detected an upstream binding site centered at position –182, which was absent in the PyqxM-containing DNA fragment used in our analysis.
Figure 4. Transcription of yqxM requires an upstream, direct repeat sequence.

A) The horizontal line depicts the upstream regulatory region of PyqxM. Open boxes denote the indicated repressor binding sites. Dashed lines indicate the AbrB binding sites mapped by Strauch et al. (2007). The bent arrow denotes the transcription start site. Numbers above the horizontal line indicate the positions of the repressor binding sites relative to the transcription start site. Black bars represent the region of PyqxM included in each construct. Numbers to the left of each black bar denote the 5′ end point of the respective construct. ‘FL’ indicates the full-length PyqxM reporter construct. The relative level of β-galactosidase activity for each lacZ reporter construct is indicated at the right. The ‘+’ symbol denotes high levels of β-galactosidase activity and the ‘−‘ symbol indicates background levels of β-galactosidase activity. The following lacZ reporter strains were used: full-length (FC591), -112 (FC488), -100 (ALM16), and -64 (FC495). B) Shown is the sequence of the wild type and mutant versions of the direct repeat in the upstream regulatory region of PyqxM. Upper case letters are the sequence of the wild type or mutant versions of the direct repeat. Lower case letters are the surrounding sequence. Numbers above the sequence indicate the positions of each site relative to the yqxM transcription start site. Colonies to the right were grown on MSgg semi-solid agar supplemented with X-gal to detect β-galactosidase activity. The following lacZ reporter strains were used: wild type (FC591), 3′ mutated (ALM36), 5′ mutated (ALM35), 3′, 5′ sites mutated (ALM37).
Our results together with those of Strauch et al. (2007) and Hamon et al. (2004) indicate that both operons for matrix production are under the negative control of AbrB. In the case of eps, we failed to detect binding of the repressor to the operon’s promoter region, and hence we do not know whether this control is direct or indirect. In contrast, the promoter region for the yqxM operon does contain binding sites for AbrB.
yqxM operon transcription requires an activator
To test the functional significance of the AbrB binding sites, we created a deletion of the PyqxM regulatory region extending from the upstream direction to position -113 and fused this to a promoterless lacZ gene (Fig. 4A). The resulting deletion-mutated fusion retained the -10 and -35 sequence elements of the promoter but not the AbrB binding sites. Rather than observing an increase in transcription, as had been expected, the deletion-mutated construct exhibited a slightly lower level of expression than that of the full-length lacZ fusion. Based on this observation, we suspected that the downstream endpoint of the -113 deletion was in or near a sequence that was needed for efficient transcription from the promoter. Indeed, the -10 (TACTCT) and -35 (TTTAAA) sequences for the yqxM promoter are poor matches to the canonical sequence elements (TATAAT and TTGACA, respectively) recognized by RNA polymerase containing the house-keeping sigma factor σA (Chu et al., 2006; Moran et al., 1982), a frequent characteristic of promoters that are under positive control. Thus, we hypothesized that in addition to being subject to negative regulation by AbrB and SinR, PyqxM is subject to positive regulation from an activator sequence located in the vicinity of the -113 deletion.
To investigate this hypothesis further, we created two additional deletions with downstream endpoints at positions -100 and -64. Strikingly, both deletion-mutated fusions were severely impaired in transcription (Fig. 4A). We infer that the region between -113 and -100 contained a sequence element that was required for transcription from PyqxM.
This possibility was reinforced by the observation that the region in question contained a direct repeat of the sequence TGAGCAA centered at positions -100 and -110. We hypothesized that this direct repeat was the binding site for an unknown activator. We refer to the upstream heptad as the 5′ repeat and the downstream heptad as the 3′ repeat. To investigate the significance of the repeats, we created nucleotide substitution mutations of each repeat separately and together. We observed a slight decrease in lacZ expression levels when either the 5′ or the 3′ repeat alone was mutated and a dramatic decrease in lacZ expression when both were mutated (Fig. 4B). All together, the above results suggest that PyqxM is under the positive control of an activator protein that binds to direct repeat sequences upstream of PyqxM to stimulate transcription.
An attractive candidate for the hypothesized activator protein was Slr for the following reasons. First, Slr is highly similar to SinR, a known, direct (negative) regulator of yqxM. Slr contains a putative helix-turn-helix domain in its N-terminal region whose amino acid sequence is 41% identical and 73% similar to that of SinR (Fig. 5A). In fact, SinR is antigenically similar to Slr as antibodies directed against SinR also detected Slr protein in immunoblot assays (Fig. S2). In addition, the C-terminal domain of Slr contains a region of similarity to SinI, the antagonist of SinR (28% identical, 65% similar) (Fig. 5A). Second, the gene encoding slr is located adjacent to, and in divergent orientation from, the eps operon (Fig. 5B).
Figure 5. The slr gene encodes a SinR-like protein and shares a common regulatory region with the eps operon.

A) The amino acid sequence alignment of Slr and SinR and SinI. Asterisks indicate identical amino acids and the dots indicate similar amino acids. Numbers indicate the amino acid position. Sequence alignment information was obtained from http://www.ebi.ac.uk/Tools/clustalw/. B) A schematic of the shared upstream regulatory region between the slr and epsA coding regions. Open arrows indicate the open reading frames. Black, horizontal arrows indicate the mapped SinR binding sites. The bent arrow denotes the eps transcription start site. The three direct repeats of SinR binding sites on the right are centered at 31, 39, and 60 base pairs upstream from the slr coding region.
The SinR-like protein Slr stimulates transcription from PyqxM
To investigate whether Slr is involved in biofilm formation, we replaced the slr coding sequence with a drug-resistance gene and found that the resulting null mutant formed flat, featureless colonies and thin, flat pellicles (Fig. 2A). This mutant could be complemented with a copy of slr inserted into the chromosome at the amyE locus (Fig. 2A). That an Slr mutant exhibits a defect in biofilm formation was independently noted by Kobayashi (2007) in a large-scale survey of the effect of transcriptional regulator mutants on pellicle formation.
Next, we investigated whether Slr is needed for transcription from the promoters for eps or yqxM using lacZ fusions to Peps and PyqxM. Interestingly, expression of Peps-lacZ exhibited little or no dependence on Slr, whereas expression of PyqxM-lacZ was markedly impaired in the absence of the putative regulatory protein (Fig. 6). Whereas SinR is a repressor, its homolog Slr acts to stimulate transcription in a manner that is selective for yqxM and not eps.
Figure 6. Slr is required for transcription of yqxM but not eps.
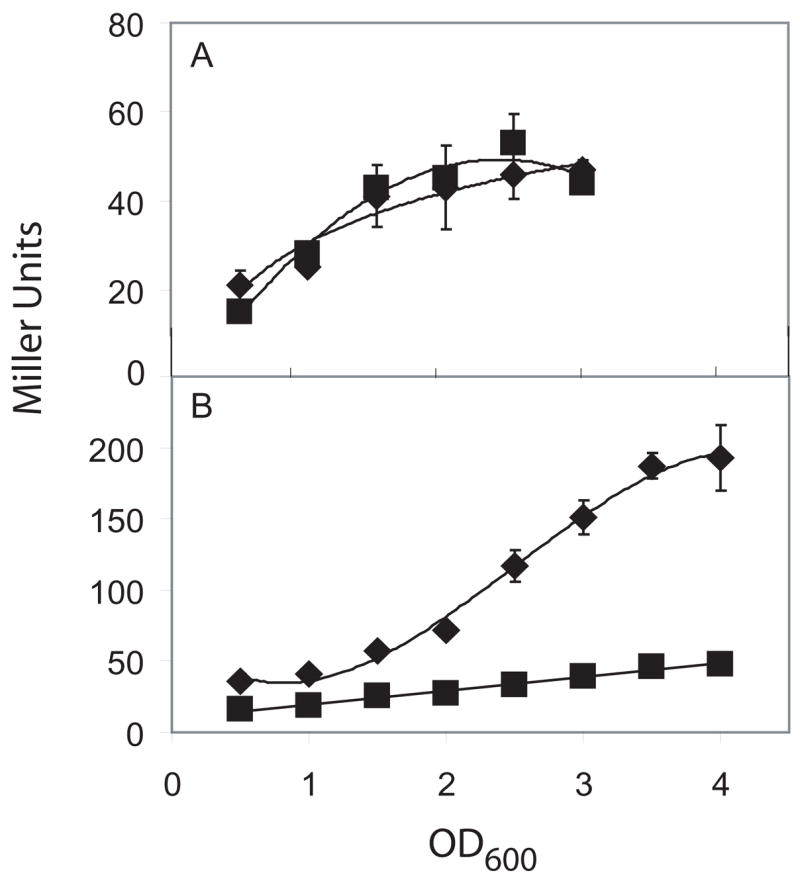
The indicated strains were grown at 30°C in MSgg and β-galactosidase was measured in Miller Units. A) β-galactosidase expression of Peps-lacZ in wild type (◆)(FC13) and slr (■) (FC526). B) β-galactosidase expression of PyqxM-lacZ in wild type (◆) (FC591) and slr (■) (FC593).
We next addressed the question of whether yqxM is the only operon required for biofilm formation that depends on Slr for its transcription. To do so, we overexpressed the yqxM operon using an IPTG-inducible promoter in an slr mutant background. As shown in Fig. 2B, the induction of the yqxM operon largely restored complex colony architecture and pellicle formation to the slr mutant. Thus, the yqxM operon is the major, if not the only, target of the Slr activator required for biofilm formation.
Interestingly, the dependence both of yqxM transcription and of biofilm formation on Slr could be partially bypassed by the absence of SinR but not by the absence of AbrB (Fig. S3). That is, a sinR mutation (but not an abrB mutation) was partially epistatic to the effects of an slr mutation. We infer from this that PyqxM is able to function to a significant extent when Slr is absent under circumstances when it is entirely freed of the repressive effects of SinR.
A simple interpretation of the results so far is that Slr binds to, and stimulates transcription from, PyqxM. In an effort to test this idea, we attempted to perform EMSA experiments using purified Slr protein. However, the full-length Slr protein was insoluble and efforts to solubilize and renature the protein failed. We were able to obtain soluble versions of Slr using only the N-terminal DNA-binding portion of Slr as well as a GST-tagged version of full-length Slr. We performed EMSA experiments using these two versions of Slr and found that both proteins bound DNA in a non-specific manner (Fig. S4 and data not shown). Thus, Slr is indeed a DNA-binding protein but we were unable to determine whether it acts directly on the PyqxM promoter region.
If Slr does act directly on PyqxM, it is conceivable that it does so by binding to the above identified heptad repeat. If so, it is pleasing to note that the heptad sequences are absent from the regulatory region of eps, whose transcription, as we have seen, is not dependent on Slr. On the other hand, mutations of both of the 5′ and 3′ heptad sequences more severely impaired yqxM transcription (Fig. 4B) than did the absence of Slr (Fig. 6). It is conceivable, therefore, that an additional unknown regulatory protein also binds to the heptad sequences or that the heptad sequences additionally act in cis to promote transcription.
slr is under the negative control of SinR and AbrB
Previously, we mapped SinR binding sites in the intergenic region between the divergently oriented eps operon and the slr gene (Kearns et al., 2004). We found that SinR bound to two regions: a pair of inverted repeat sequences located proximal to eps and three direct repeat sequences located just upstream of slr (Fig 5B). The direct repeats are centered at positions -31, -39, and -60 upstream from the slr open reading frame (Fig. 5B). We wondered whether these SinR binding sites were responsible for repressing the transcription of the slr gene. To test this hypothesis, we fused the slr promoter to a promoterless lacZ gene and monitored lacZ expression in a sinR mutant. Again, a mutation in the epsH gene was introduced to relieve constitutive cell bundling. The epsH mutation had little or no effect on the transcription from the slr promoter (Table 2). In the absence of SinR, we observed a marked increase in lacZ expression, suggesting that SinR is a repressor of slr (Table 2). In confirmation of this, we observed a decidedincrease in Slr levels in the absence of SinR (Fig. S2).
Table 2.
AbrB and SinR repress slr
| Genotypea | Pslr-lacZ activity(MU)b |
|---|---|
| Wild type | 7 ± 0.3 |
| epsH | 7 ± 0.2 |
| sinR epsH | 180 ± 2 |
| abrB epsH | 16 ± 1 |
| abrB sinR epsH | 133 ± 7 |
The following Pslr-lacZ containing strains were used FC518, FC519, FC520, FC522, and FC606.
β-Galactosidase activity, calculated in Miller Units (MU), was measured from late-exponential phase cells grown in MSgg and is the average of at least three replicates.
AbrB and SinR both repress at least two operons, eps and yqxM, required for biofilm formation. Therefore, we wondered if AbrB, like SinR, also represses the transcription of slr. As shown in Table 2, the absence of AbrB resulted in the derepression of slr. The extent of derepression was, however, much less than that caused by the absence of SinR, and the level of expression of slr in a sinR abrB double mutant was no higher (indeed somewhat lower) than in a sinR single mutant. We therefore conclude both SinR and AbrB contribute to the control of slr but that SinR plays a larger role in repessing slr than does AbrB. All together, we have shown that SinR, directly, and AbrB, directly or indirectly, repress at least three transcription units required for biofilm formation (Fig. 1).
Discussion
The present investigation has revealed important new features of the regulatory circuitry governing the expression of the eps operon, which specifies exopolysaccharride production, and the yqxM operon, which encodes the matrix protein TasA. The most important contribution is the discovery of an additional regulatory protein, Slr, involved in matrix production. In addition, we have have identified additional targets of the repressor AbrB, which was previously known to be involved in biofilm formation (Hamon et al., 2004). Finally, we show that AbrB and SinR act in parallel to control the expression of the eps and yqxM operons and that all three regulators, Slr, AbrB, and SinR, converge in governing the production of the matrix protein TasA (Fig. 1).
We were alerted to the possibility that Slr is involved in biofilm formation by the following considerations. First, the gene for Slr is located immediately adjacent to, and shares a common regulatory region with, the eps operon. Second, Slr is strikingly similar in amino acid sequence to SinR, which, as we have argued, is largely dedicated to the expression of genes and operons involved in biofilm formation (Kearns et al., 2005). Thus, the N-terminal region of Slr is homologous to the helix-turn-helix in the N-terminal, DNA-binding region of SinR. Also, the C-terminal region of Slr resembles SinI, the antirepressor for SinR (Bai et al., 1993). Finally, our dissection of the regulatory region for the yqxM operon revealed an apparent requirement for an unknown activator.
Indeed, our present results, and the independent finding of Kobayashi (2007) reveal that Slr is required for biofilm formation. In addition, we have shown that the slr gene is under the negative control of SinR and that it is required for induction of the yqxM operon. Interestingly, and even though slr is located adjacent to eps, it is not required for the expression of the exopolysaccharide operon. In fact, our results suggestthat yqxM is the principal, if not the exclusive, target of Slr’s action in promoting biofilm formation. We were unable to settle the issue of whether Slr acts directly on yqxM, but the simplest interpretation of our results is that it activates transcription by binding to the promoter. If so, it is conceivable that its target is the 5′ and 3′ heptad repeat sequences, although with the important caveat that mutation of the heptads impaired transcription more severely than did the absence of Slr. Finally, and reinforcing the view that both SinR and Slr are largely dedicated to the control of genes involved in biofilm formation, it is pleasing to note that sinR and slr each flank one of the two operons governing the production of extracellular matrix.
We have also integrated AbrB into the circuitry that governs biofilm formation. We have shown that AbrB represses both the eps operon and the slr gene, although we do not know whether this effect is direct or indirect. In addition, our present findings together with those of two other groups indicate that AbrB represses the yqxM operon (Hamon et al., 2004; Strauch et al., 2007). Furthermore, our results and those of Strauch et al., (2007) indicate that AbrB acts directly on yqxM. Finally, we have shown that AbrB and SinR act independently in repressing their targets. That is, the effects of the absence of both repressors was not significantly greater than the sum of the effects of the absence of either AbrB or SinR alone.
In toto, these findings lead to the view that SinR and AbrB represent parallel pathways, both controlled by the response regulator, Spo0A. Thus, activation of Spo0A sets in motion events (induction of the antirepressor gene sinI and repression of abrB) that relieve AbrB- and SinR-mediated repression. The ultimate targets of this repression are the eps and yqxM operons, which govern extracellular matrix production and are the only genes and operons directly involved in formation of the extracellular matrix so far identified.
This notion that Spo0A sets in motion two parallel pathways might at first glance appear to be at odds with epistasis experiments involving the abrB gene. Thus, an arbB mutation is known to be epistatic (Hamon and Lazazzera, 2001) to a spo0A mutation but not to a sinI mutation (Kearns et al., 2005). That is, an abrB spo0A double mutant makes hyper-wrinkled colonies whereas an abrB sinI double mutant makes smooth colonies. Yet, spo0A is required for the expression of sinI (Shafikhani et al., 2002; Fujita et al., 2005). Therefore, in the abrB spo0A double mutant, the sinI gene should be off and hence an abrB spo0A double mutant should be equivalent to an abrB sinI double mutant. How are we to resolve this paradox? We propose that sinI is not totally silent in the absence of Spo0A. Rather, it is transcribed at a low but significant level by read-through transcription from the upstream operon (yqhH-yqhG). In keeping with this hypothesis, we note that the intergenic region between the upstream operon and sinI does not contain a conspicuous transcriptional terminator, and a drug-resistance cassette inserted in this region (with its promoter oriented away from the sinI sinR operon) impairs biofilm formation (Y. Chai, unpublished results). We propose that read-through transcription from the upstream operon augments transcription of sinI and that in a spo0A abrB double mutant, a small amount of SinI is produced, which suffices to relieve SinR-mediated repression partially. We propose that when partially inhibited by a low level of SinI, SinR is unable to silence the eps and yqxM operons without the additional contribution of AbrB.
In summary, the present works reveals that the circuitry for extracellular matrix production is unexpectedly intricate. It involves four transcriptional regulatory proteins (Spo0A, SinR, AbrB and Slr), two of which (SinR and Slr) are largely, if not exclusively, dedicated to the control of the two operons involved in matrix production. In addition, each operon is the direct or indirect target of more than one regulatory protein, two in the case of eps and three in the case of yqxM. We infer that this high level of regulation serves to ensure tight regulation of the operons, such that the matrix is produced only under the appropriate circumstances.
Materials and Methods
Strains and growth conditions
Strain 3610 and its derivatives were grown at 37°C in Luria-Bertani broth (LB; 10 g tryptone, 5 g yeast extract, and 5 g NaCl per L) or on solid medium containing LB supplemented with 1.5% agar. For formation of architecturally complex colonies, cells were toothpick-inoculated onto MSgg medium (Branda et al., 2001) containing 1.5% Bacto agar and incubated at 22°C for 72 hours. For pellicle formation, 10 μl of mid-log phase culture was inoculated into 10 ml minimal MSgg medium and incubated statically at 22°C for 72 hours. When appropriate, antibiotics and supplements were included at the following concentrations: 100 μg ml−1 X-gal, 10 μg ml−1 tetracycline, 100 μg ml−1 spectinomycin, 5 μg ml−1 chloramphenicol, 5 μg ml−1 kanamycin, and 1 μg ml−1 erythromycin plus 25 μg ml−1 linomycin.
Strain construction
All insertion/deletion mutations were generated using long flanking homology PCR using primers indicated in Table S3. The spo0A::spec and epsH::tet constructs were previously generated (Fawcett et al., 2000; Kearns et al., 2005). The abrB::kan construct was a gift from E. Hobbs. The strains were transformed into competent PY79 cells and mutations were transferred into the 3610 background using SPP1-mediated generalized transduction (Yasbin and Young, 1974; Kearns et al., 2004). All strains used in this study are listed in Table S2.
Complementation and reporter expression constructs
All primers used in the construction of plasmids are listed in Table S3. To generate the Pslr-lacZ reporter construct, a PCR product containing the Pslr promoter was amplified from 3610 chromosomal DNA using primers Pslr-F1 and Pslr-R1. The PCR product was cloned into the EcoRI and BamHI sites of plasmid pDG268 (Antoniewski et al., 1990). To generate the PsinI-lacZ reporter construct, a PCR product containing the PsinI promoter was amplified from 3610 chromosomal DNA using primers -499sinI and -2sinI. The resulting PCR product was cloned into the EcoRI and BamHI sites of pDG268.
To generate the deletion constructs of PyqxM-lacZ, PCR products containing various versions PyqxM were amplified from 3610 chromosomal DNA using the primer -26yqxM in combination with the following forward primers: for the -113 construct, -175yqxM; for the -100 construct, -160yqxM; and for -64 construct, -126yqxM. The PCR products were then cloned into the EcoRI and BamHI sites of plasmid pDG268. To generate the mutation of the 3′ site of the heptad repeats, two PCR products were amplified from the primers -302yqxM/ALM3 and -26yqxM/ALM1. The two PCR products were ligated together using XhoI sites and cloned into the EcoRI and BamHI sites of pDG268. To generate the mutation of the 5′ site of the heptad repeats two PCR products were amplified from the primers -302yqxM/ALM5 and -26yqxM/ALM4. The two PCR products were ligated together using XhoI sites and cloned into the EcoRI and BamHI sites of pDG268. To generate the mutations in both sites of the heptad repeats two PCR products were amplified from the primers -302yqxM/ALM2 and -26yqxM/ALM1. The two PCR products were ligated together using XhoI sites and cloned into the EcoRI and BamHI sites of pDG268.
To generate the slr complementation construct, a PCR product containing the promoter and open reading frame of slr was amplified using the primers slrcomp_F and slrcomp_R. The PCR product was cloned into EcoRI and BamHI sites of plasmid pDG364 (Karmazyn-Campelli et al., 1992).
β-galactosidase assay
Cells were harvested from shaking cultures growing at 30°C in MSgg broth. Cells were collected in 1 ml aliquots and suspended in an equal volume of Z buffer (40 mM NaH2PO4, 60 mM Na2HPO4, 1 mM MgSO4, 10 mM KCl, and 38 mM 2-mercaptoethanol). Lysozyme was added to each sample to a final concentration of 0.2 mg ml−1 and incubated at 30°C for 15 minutes. Each sample was diluted in Z-buffer to a final volume of 1 ml and the reaction was started with 200 μl of 4 mg ml−1 2-nitrophenyl β-D-galactopyranoside in Z buffer and stopped with 500 μl 1 M Na2CO3. After centrifuging the samples for 20 minutes at 15 000 × g, the OD420 of the reaction mixture was measured. The β-galactosidase specific activity was calculated according to the equation: [OD420/(time × OD600)] × dilution factor × 1000.
Protein expression constructs
To generate plasmids for the expression of N-terminal 6-histidine translation fusions to AbrB and the N-terminus of Slr (pEH213 and pFC588, respectively), PCR products were generated using the primers ECH318/ECH336 and +1slr/+339slr, respectively. The PCR products were cloned into the NdeI and XhoI sites of plasmids pET15b (for AbrB) and pET21b (for Slr) (Novagen). The plasmids were then transformed into Escherichia coli BL21(DE3) RIL-CodonPlus cells (Stratagene).
Protein purification
E. coli strains were grown at 30°C in 1 L of LB supplemented with 100 μl ml−1 ampicillin and 50 μg ml−1 chloramphenicol until OD600 reached ~1.0. IPTG was added to a final concentration of 1 mM and the culture grew for two additional hours at 30°C. The cells were harvested, resuspended in 30 ml of lysis/binding buffer [20 mM Tris-HCl (pH8.0), 20% (v/v) glycerol, 100 mM NaCl, and 20 mM imidazole], and lysed by sonication. The lysate was centrifuged at 32, 000 × g and the resulting supernatant was loaded onto a column containing 1 ml of Ni2+-NTA agarose beads (Qiagen) that had been equilibrated in lysis/binding buffer. The beads were washed five times with 2 ml aliquots of lysis/binding buffer. A stepwise elution was performed with 2 ml aliquots of buffer [20 mM Tris-HCl (pH8.0), 20% (v/v) glycerol, 100 mM NaCl] supplemented with 50 mM, 100 mM, 200 mM, and 400 mM imidazole. The fractions containing purified protein were pooled and dialyzed against 4 L dialysis buffer [20 mM Tris-HCl (pH8.0), 50% (v/v) glycerol, 100 mM NaCl, 1mM EDTA, 1mM DTT] overnight at 4°C. The dialyzed protein was stored at −20°C for AbrB and −80°C for Slr.
Electrophoretic mobility shift assay (EMSA)
Radiolabeled probes were generated by PCR using 3610 chromosomal DNA and the following primer combinations: −246epsA/+4epsA (Peps), ECH341/ECH342 (PrecA), -270yqxM/+14yqxM (PyqxM), and ECH343/ECH344 (PcomK). The gfp probe was generated by PCR using plasmid pKL147 DNA as a template and primers JSK362/JSK445. Each probe was PCR purified and 5′ end labeled with 10 μCi of [γ-32P]-ATP (NEG002A, New England Nuclear) using polynucleotide kinase (New England Biolabs). Various concentrations of purified AbrB or N-Slr were added to approximately 100 nM of radiolabeled probe (Kearns et al., 2004). N-Slr-DNA binding reactions were carried out in 30 μl volumes, including binding buffer (10 mM Tris HCl, 50 mM NaCl, 1mM EDTA, 5% glycerol, 1mM dithiothreitol (DTT), 10 μg ml−1 bovine serum albumin) and 25 μg ml−1 polydeoxyinosinic-deoxycytidlyic acid (poly dI-dC), at room temperature for 20 minutes. A 6% polyacrylamide 0.5× TBE gel was loaded with 10 μl of each binding reaction and resolved for 1 hour at 100 mV. AbrB-DNA binding reactions were carried out in 30 μl volumes including binding buffer [20mM Tris-HCl (pH8.0), 100 mM KCl, 5mM MgCl2, 0.5 mM DTT, 10% glycerol, 0.05% Nonidet P40, 0.05 μg μl−1 bovine serum albumin] containing 25 μg ml−1 poly dI-dC, at 37°C for 20 minutes. A 6% polyacrylamide 1× TAE gel was loaded with 10 μl of each binding reaction and resolved for 2.5 hours at 50 mV.
DNase I footprinting assay
Primer -21yqxM_R was 5′ end labeled with [γ-32P]-ATP and polynucleotide kinase. A PCR product of the PyqxM region was generated using 3610 chromosomal DNA as a template and 50 pmol of labeled primer -21yqxM_R and unlabeled primer -244yqxM_F. The PCR product was then PCR purified and the c.p.m. was measured using a scintillation counter. Approximately 30 000 c.p.m. of radiolabeled PCR product was added to varying concentrations of AbrB protein in 100 μl footprinting buffer (20 mM Tris pH 8.0, 5 mM MgCl2, 5 mM CaCl2, 0.1 mM DTT, 0.1 mM EDTA, 50 μg ml−1 BSA) with 5 μg ml−1 poly dI-dC and incubated at room temperature for 20 minutes. Each sample was then incubated for 30 seconds with 2 μl DNaseI (1:25 dilution of 1 U μl−1 stock, Invitrogen) before digestion was stopped by adding 25 μl of stop solution (1.5 M sodium acetate pH 5.3, 20 mM EDTA, and 400 μg ml−1 glycogen). The reactions were then ethanol precipitated and resuspended in 8 μl of formamide loading dye (80% deionized formamide, 10 mM EDTA, 1 mg ml−1 xylene cyanol, and 1 mg ml−1 bromophenol blue). A total of 6 μl of each reaction was loaded on an 8% sequencing gel and resolved before being visualized using phosphoimaging.
Western blot assay
Cells of each strain were grown to 1 OD600 in LB broth at 37°C, harvested, resuspended to 11.5 OD600, and lysed for 30 minutes in lysis buffer (20 mM Tris pH 7.0, 10 mM EDTA, 10mg/ml lysozyme, 1 mg/ml DNase, and 10 mg/ml RNase A). An amount of 6X sample buffer (0.5 M Tris pH 6.8, 15% glycerol, 0.1g/ml SDS, 1.2 mg/ml bromophenol blue, 60 μl/ml β-mercaptoethanol) was added to the cell suspension to a final concentration of 1X and the samples were boiled for 1 minute. 10 μl of each sample (0.1 OD600 equivalent) was loaded per lane and proteins were resolved by SDS-polyacrylamide gel electrophoresis (15%). Proteins were transferred onto nitrocellulose membrane by electroblotting for 1 hour at 400 mA. The membrane was blocked for 1 hour at room temperate in TBSTM buffer (20 mM Tris pH 8.0, 200 mM NaCl, 0.1% Tween 20, 5% instant non-fat dry milk). The membrane was probed with a 1:20,000 dilution of anti-SinR primary antibody in TBSTM buffer overnight at 4°C. The membrane was washed three times in TBSTM and probed with a 1:10,000 HRP conjugated goat anti-rabbit secondary antibody in TBSTM buffer for one hour at room temperature. The blot was developed with an ECL chemiluminescence detection kit (Biorad).
Supplementary Material
Acknowledgments
We thank M. Strauch for advice on the manuscript and members of the Losick and Kolter labs, and in particular, E. Hobbs, J. Kain, A. Chastenet, and K. Ramamurthi for strains, helpful discussions, and advice. This work was supported by NIH grants GM58213 to R.K. and GM18568 to R.L.
References
- Antoniewski C, Savelli B, Stragier P. The spoIIJ gene, which regulates early developmental steps in Bacillus subtilis, belongs to a class of environmentally responsive genes. J Bacteriol. 1990;172:86–93. doi: 10.1128/jb.172.1.86-93.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai U, Mandic-Mulec I, Smith I. SinI modulates the activity of SinR, adevelopmental switch protein of Bacillus subtilis, by protein-protein interaction. Genes Dev. 1993;7:139–148. doi: 10.1101/gad.7.1.139. [DOI] [PubMed] [Google Scholar]
- Branda SS, González-Pastor JE, Ben-Yehuda S, Losick R, Kolter R. Fruiting body formation by Bacillus subtilis. Proc Natl Acad Sci USA. 2001;98:11621–11626. doi: 10.1073/pnas.191384198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Branda SS, González-Pastor JE, Dervyn E, Ehrlich SD, Losick R, Kolter R. Genes involved in the formation of structured multicellular communities by Bacillus subtilis. J Bacteriol. 2004;186:3970–3979. doi: 10.1128/JB.186.12.3970-3979.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Branda SS, Chu F, Kearns DB, Losick R, Kolter R. A major protein component of the Bacillus subtilis biofilm matrix. Mol Microbiol. 2005;59:1229–1238. doi: 10.1111/j.1365-2958.2005.05020.x. [DOI] [PubMed] [Google Scholar]
- Chu F, Kearns DB, Branda SS, Kolter R, Losick R. Targets of the master regulator of biofilm formation in Bacillus subtilis. Mol Microbiol. 2005;59:1216–1228. doi: 10.1111/j.1365-2958.2005.05019.x. [DOI] [PubMed] [Google Scholar]
- Chai Y, Chu F, Losick R, Kolter R. Bistability and biofilm formation in Bacillus subtilis. Mol Microbiol. 2008;67:254–263. doi: 10.1111/j.1365-2958.2007.06040.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fawcett P, Eichenberger P, Losick R, Youngman P. The transcriptional profile of early to middle sporulation in Bacillus subtilis. Proc Natl Acad Sci USA. 2000;5:8063–8. doi: 10.1073/pnas.140209597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita M, González-Pastor JE, Losick R. High- and low-threshold genes in theSpo0A regulon of Bacillus subtilis. J Bacteriol. 2005;187:1357–68. doi: 10.1128/JB.187.4.1357-1368.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamoen LW, Kausche D, Marahiel MA, van Sinderen D, Venema G, Serror P. The Bacillus subtilis transition state regulator AbrB binds to the -35 promoter region of comK. FEMS Microbiol Lett. 2003;218:299–304. doi: 10.1111/j.1574-6968.2003.tb11532.x. [DOI] [PubMed] [Google Scholar]
- Hamon MA, Lazazzera BA. The sporulation transcription factor Spo0A is required for biofilm development in Bacillus subtilis. Mol Microbiol. 2001;42:1199–1209. doi: 10.1046/j.1365-2958.2001.02709.x. [DOI] [PubMed] [Google Scholar]
- Hamon MA, Stanley NR, Britton RA, Grossman AD, Lazazzera BA. Identification of AbrB-regulated genes involved in biofilm formation by Bacillus subtilis. Mol Microbiol. 2004;52:847–860. doi: 10.1111/j.1365-2958.2004.04023.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoch JA. Regulation of the phosphorelay and the initiation of sporulation in Bacillus subtilis. Annu Rev Microbiol. 1993;47:442–65. doi: 10.1146/annurev.mi.47.100193.002301. [DOI] [PubMed] [Google Scholar]
- Karmazyn-Campelli C, Fluss L, Leighton T, Stragier P. The spoIIN279(ts) mutation affects the FtsA protein of Bacillus subtilis. Biochimie. 1992;74:689–694. doi: 10.1016/0300-9084(92)90141-z. [DOI] [PubMed] [Google Scholar]
- Kearns DB, Chu F, Rudner R, Losick R. Genes governing swarming in Bacillus subtilis and evidence for phase variation mechanism controlling surface motility. Mol Microbiol. 2004;52:357–369. doi: 10.1111/j.1365-2958.2004.03996.x. [DOI] [PubMed] [Google Scholar]
- Kearns DB, Chu F, Branda SS, Kolter R, Losick R. A masterregulator for biofilm formation by Bacillus subtilis. Mol Microbiol. 2005;55:739–749. doi: 10.1111/j.1365-2958.2004.04440.x. [DOI] [PubMed] [Google Scholar]
- Kobayashi K. Bacillus subtilis pellicle formation proceeds through genetically defined morphological changes. J Bacteriol. 2007;189:4920–31. doi: 10.1128/JB.00157-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moran CP, Jr, Lang N, LeGrice SF, Lee G, Stephens M, Sonenshein AL, Pero J, Losick R. Nucleotide sequences that signal the initiation of transcription and translation in Bacillus subtilis. Mol Gen Genet. 1982;186:339–346. doi: 10.1007/BF00729452. [DOI] [PubMed] [Google Scholar]
- Shafikhani SH, Mandic-Mulec I, Strauch MA, Smith I, Leighton T. Postexponential regulation of sin operon expression in Bacillus subtilis. J Bacteriol. 2002;184:564–71. doi: 10.1128/JB.184.2.564-571.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stover AG, Driks A. Secretion, localization, and antibacterial activity of TasA, a Bacillus subtilis spore-associated protein. J Bacteriol. 1999;181:1664–72. doi: 10.1128/jb.181.5.1664-1672.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strauch MA, Bobay BG, Cavanagh J, Yao F, Wilson A, Le Breton Y. Abh and AbrB control of Bacillus subtilis antimicrobial gene expression. J Bacteriol. 2007;189:7720–32. doi: 10.1128/JB.01081-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veening JW, Kuipers OP, Brul S, Hellingwerf KJ, Kort R. Effects of phosphorelay perturbations on architecture, sporulation, and spore resistance in biofilms of Bacillus subtilis. J Bacteriol. 2006;188:3099–109. doi: 10.1128/JB.188.8.3099-3109.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vlamakis H, Aguilar C, Losick R, Kolter R. Control of cell fate by theformation of an architecturally complex bacterial community. Genes Dev. 2008 doi: 10.1101/gad.1645008. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasbin RE, Young FE. Transduction in Bacillus subtilis by bacteriophage SPP1. J Virol. 1974;14:1343–1348. doi: 10.1128/jvi.14.6.1343-1348.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.