

In recent years, the homogeneous catalysis by gold complexes received increasing attention. Particularly, cyclizations of alkynes and allenes mediated by gold complexes have been widely explored.1 Along this line, the Gevorgyan group has recently reported a gold-catalyzed regiodivergent cycloisomerization of bromoallenyl ketones a into isomeric bromofurans b and c (Scheme 1).2 According to the originally proposed mechanism, products b and c result from different activation modes of the substrate by gold complexes.2 It was thought that more oxophilic Au(III) catalyst3 coordinates to the carbonyl oxygen (d), which, via halirenium intermediate e, produces the 3-bromofuran b, a product of 1,2-Br migration. Alternatively, the more π-philic Au(I) catalyst coordinates to the distal double bond of the allene (f), leading to the gold carbenoid intermediate g. Subsequent 1,2-hydride shift in the latter forms 2-bromofuran c (Scheme 1).2 Great interest has been aroused by this important discovery, since not only it opens an unprecedented access to different types of halofurans, but also represents the very first example of reaction, in which employment of gold catalyst in different oxidation states leads to different products.1a,c,4,5,6 A more detailed mechanistic investigation of origins of the observed regioselectivity of this cycloisomerization, properties of the key intermediates, as well as profiles of the overall catalytic cycles, may give better understanding on the important transition metal-catalyzed chemistry of alkynes and allenes. This in turn, may provide guide for future design of new catalytic transformations. Herein, we wish to report our theoretical and experimental results on the mechanism of Au(III)- and Au(I)-catalyzed cycloisomerization of bromoallenyl ketones, with the emphasis on the origins of the regiochemistry of 1,2-migrations. Our study indicates that the 1,2- H migration could be assisted by the ligand L of the Au(PR’3)L complex, thus, the regioselectivity of 1,2-migrations can be controlled by ligands at Au-complexes.
Scheme 1.
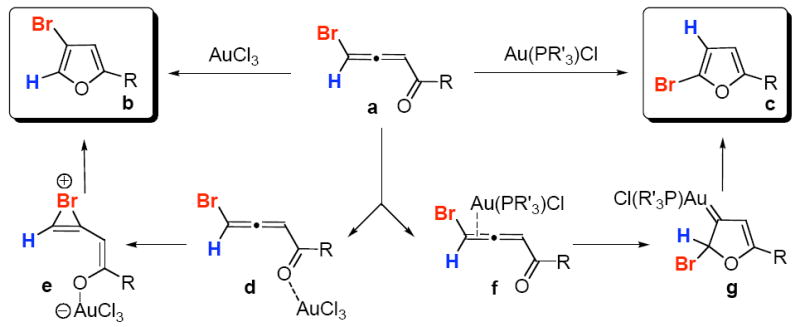
Regiodivergent Au(III)- and Au(I)-catalyzed cycloisomerizations of bromoallenyl ketones.
Thus, DFT calculations to investigate the above mentioned cycloisomerization have been performed.7,8 The computed energy surfaces for the AuCl3 and Au(PH3)Cl-catalyzed cycloisomerizations of bromoallenyl ketone 1 are provided in Figure 1. Results of the Au(III)-catalyzed reaction (I, Figure 1) indicate that the gold catalyst, upon coordination to the distal double bond of the allene 1,9 instantly gives the cyclic intermediate 2 via a barrierless and highly exergonic process with relative free energies of 32.6 and 33.6 kcal/mol, in the gas phase and in toluene solution, respectively. Next, intermediate 2 undergoes more favorable 1,2-Br migration via the transition state TS1 to generate the product complex 3 rather than 1,2-H shift through TS2 to give 4. Thus, the computed free energy barriers difference of 14.3 kcal/mol for the latter processes is in a good agreement with the experimentally observed nearly exclusive 1,2-Br migration.2 Although, the formation of the complex 3 from intermediate 2 is endergonic (Figure 1), the latter will be transformed into the product b irreversibly via a highly exergonic (30.3 kcal/mol) ligand exchange process of 3 with another molecule of substrate, thus, completing the catalytic cycle.10
Figure 1.
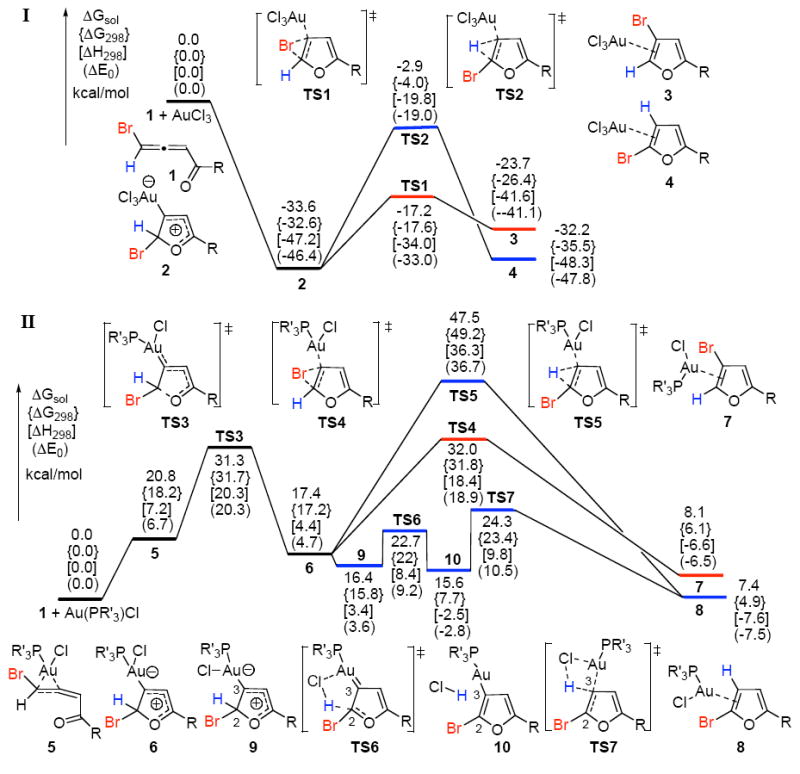
Potential energy surfaces for AuCl3- and Au(PR’3)Cl-catalyzed cycloisomerizations (R = CH3, R’= H).7
Remarkably, it was found that the AuCl3-catalyzed cycloisomerization of 1 in wet toluene11 leads to the decreased ratio of 1,2-Br vs 1,2-H migration products. This implies that the H-shift could be catalyzed by a trace amount of water present in the system, analogously to the recently reported H2O-assisted formal 1,2-H shift in the Au+-catalyzed carbocyclization.8c This hypothesis was unambiguously confirmed by the calculations and further by reactions performed in the presence of D2O.10 Thus, DFT calculations revealed the H2O-assisted H-shift from 2 to 4 is a stepwise H-abstraction/donation process with an activation free energy of 17.8 kcal/mol in toluene, which is only about 1.4 kcal/mol higher than that for the 1,2-Br migration.10
The energy surface for the reaction catalyzed by Au(I) (II, Figure 1) is quite different from that for Au(III). Coordination of the less electrophilic Au(PH3)Cl to the distal double bond of 1 gives π-complex 5 with the free energy increased by 20.8 kcal/mol in toluene solution. The latter, via the TS3 with 10.5 kcal/mol activation energy barrier, transforms into the zwitterionic intermediate 6. From intermediate 6, the reaction may proceed through the direct 1,2-migration steps to generate complexes 7 and 8. Calculations showed that the activation barrier for the direct 1,2-H shift (TS5) is 30.1 kcal/mol, while that for the 1,2-Br migration (TS4) is only 14.6 kcal/mol, thus contradicting with the experimental results on the observed predominant 1,2-H shift.2 However, the major 1,2-H migration pathway can reasonably be rationalized in terms of the stepwise ligand-assisted H-shift process. Thus, as shown in II (Figure 1), 6 rotamerizes to a relatively more stable intermediate 9. Geometries of 6 and 9, revealed the remarkably prolonged Au-Cl distances, implying quite weak Au-Cl interactions. 12 Consequently, the weakly coordinated chloride is now capable of hydrogen abstraction at C2 via TS6 with a barrier of 6.3 kcal/mol only to give intermediate 10. Notably, the HCl moiety is associated with the gold-substituted furan through Cl-H---C3 hydrogen bonding interaction.10 Gas phase calculations showed that the formation of 10 is slightly exergonic, though a solvation by toluene substantially destabilizes this intermediate. Protiodeauration of 10 at C3 via TS7 leads to the product complex 8. The activation free energy of this step is 15.7 and 8.7 kcal/mol in the gas phase and in toluene, respectively. Dissociation of 8 is quite favorable with an exergonicity of 26.3 kcal/mol in toluene. Thus, the overall barrier for the chloride-assisted H-migration process from 9 to the complex 8 is 8.7 kcal/mol only.
Based on these results, we hypothesized that if no labile chloride ligand present in a Au(I)-catalyzed cycloisomerization (e.g. using cationic Au(I) or AuCl complexes), reaction may proceed via a predominant 1,2-Br migration. As predicted, our experiments indicated that employment of AuCl or cationic catalysts, such as Au(PPh3)BF4 and Au(PPh3)SbF6, produced the 1,2-Br migration product A as a major or sole isomer (Scheme 2).10 Unexpectedly, use of Au(PPh3)OTf lead to exclusive formation of the 1,2-H shift product B, thus, exhibiting a dramatic counterion effect on the regioselectivity of the reaction. The DFT calculations shed light on this seeming controversy.8d,10 The computations revealed that in both Au(PR3)+ and AuCl-catalyzed reactions, the 1,2-Br migration is more favorable with the activation barrier about 13 kcal/mol. However, 1,2-H shift in Au(PR3)+-catalyzed reactions can be assisted by the counterions. Thus, it was found that the OTf--assisted 1,2-H shift requires an activation free energy of 9.6 kcal/mol only, whereas those for BF4- and SbF6- are much higher (20.2 and 29 kcal/mol, respectively), which is in a good agreement with the experiments.
Scheme 2.
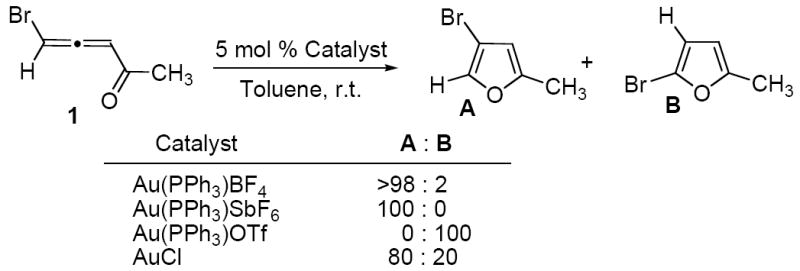
Experimental results of Au(I)-catalyzed reations.10
In summary, the mechanism of Au-catalyzed cycloisomerization of bromoallenyl ketones has been investigated through DFT computational and experimental studies. It was found that both Au(I) and Au(III) catalysts13 activate distal double bond of the allene to produce cyclic zwitterionic intermediates, which undergo a kinetically favored 1,2-Br migration. However, in the cases of Au(PR3)L (L = Cl, OTf) catalysts, the counterion-assisted H-shift is the major process, indicating that the regioselectivity of the Au-catalyzed 1,2-H vs 1,2-Br migration is ligand dependent. The present study may provide better understanding on the reactions of allenes and alkynes activated by Au(I) and Au(III) complexes,14,15 different catalytic performance of Au-complexes in different oxidation states, and particularly the ligand effect, which has received limited attention only.1c,8d,16
Supplementary Material
Computational and experimental details. This material is available free of charge via the Internet at http://pubs.acs.org.
Acknowledgments
This work was supported by Hundreds of Talents Program of CAS, Natural Science Foundation of Jiangsu Province (BK2005030), and NIH of the US (GM-64444). We thank Prof. Z.-X. Yu of Peking University for helpful discussion.
References
- 1.For recent reviews, see: Hashmi ASK, Hutchings GJ. Angew Chem Int Ed. 2006;45:7896. doi: 10.1002/anie.200602454.Zhang L, Sun J, Kozmin SA. Adv Synth Catal. 2006;348:2271.Gorin DJ, Toste FD. Nature. 2007;446:395. doi: 10.1038/nature05592.Hashmi ASK. Chem Rev. 2007;107:3180. doi: 10.1021/cr000436x.
- 2.(a) Sromek AW, Rubina M, Gevorgyan V. J Am Chem Soc. 2005;127:10500. doi: 10.1021/ja053290y. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Dudnik AS, Sromek AW, Rubina M, Kim JT, Kel’in AV, Gevorgyan V. J Am Chem Soc. 2008;130:1440. doi: 10.1021/ja0773507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.For higher oxophilicity of AuCl3 compared to that of AuCl, see: Yamamoto Y. J Org Chem. 2007;72:7817. doi: 10.1021/jo070579k.
- 4.(a) Kirsch SF. Org Biomol Chem. 2006;4:2076. doi: 10.1039/b602596j. [DOI] [PubMed] [Google Scholar]; (b) Fürstner A, Davies PW. Angew Chem Int Ed. 2007;46:3410. doi: 10.1002/anie.200604335. [DOI] [PubMed] [Google Scholar]
- 5.Hashmi ASK. Angew Chem Int Ed. 2005;44:6990. doi: 10.1002/anie.200502735. [DOI] [PubMed] [Google Scholar]
- 6.Ferrer C, Echavarren AM. Angew Chem Int Ed. 2006;45:1105. doi: 10.1002/anie.200503484. [DOI] [PubMed] [Google Scholar]
- 7.B3LYP/6-31G*(LANL2DZ for Au and Br) method was used for all the calculations, and solvation effect was calculated by CPCM model. See Supporting Information for details.
- 8.For selected examples of DFT studies in gold chemistry, see: Nevado C, Echavarren AM. Chem Eur J. 2005;11:3155. doi: 10.1002/chem.200401069.Comas-Vives A, González-Arellano C, Corma A, Iglesias M, Sánchez F, Ujaque G. J Am Chem Soc. 2006;128:4756. doi: 10.1021/ja057998o.Shi F-Q, Li X, Xia Y, Zhang L, Yu Z-X. J Am Chem Soc. 2007;129:15503. doi: 10.1021/ja071070+.Kovács G, Ujaque G, Lledós A. J Am Chem Soc. 2008;130:853. doi: 10.1021/ja073578i.Nieto-Oberhuber C, López S, Muñoz MP, Cárdenas DJ, Buñuel E, Nevado C, Echavarren AM. Angew Chem Int Ed. 2005;44:6146. doi: 10.1002/anie.200501937.Faza ON, López CS, Álvarez R, de Lera AR. J Am Chem Soc. 2006;128:2434. doi: 10.1021/ja057127e.Correa A, Marion N, Fensterbank L, Malacria M, Nolan SP, Cavallo L. Angew Chem Int Ed. 2008;47:718. doi: 10.1002/anie.200703769.
- 9.Our computational studies confirmed possible formation of the originally proposed Au(III)-coordination complex d (Scheme 1),2 however, neither cyclization transition state nor halirenium intermediate e were located.
- 10.See Supporting Information for experimental and computational details.
- 11.Possible involvement of Brønsted acid as a true catalyst was ruled out by control experiments. Besides, addition of TTBP, a proton scavenger, did not inhibit the Au-catalyzed cycloisomerization.10
- 12.The Au-Cl distances in Au(PH3)Cl, 5, 6, and 9 are 2.327, 2.481, 2.573, and 2.561 Å, respectively. See Supporting Information for bond critical point analysis of the weak interactions.
- 13.Possible involvement of AuCl as a true catalyst in AuCl3-catalyzed reactions via an in-situ redox process was ruled out by the observed different reactivity patterns of these complexes.10
- 14.For selected examples of allene activation by Au(I) and Au(III), see: Hashmi ASK, Schwarz L, Choi J-H, Frost TM. Angew Chem Int Ed. 2000;39:2285. doi: 10.1002/1521-3773(20000703)39:13<2285::aid-anie2285>3.0.co;2-f.Gockel B, Krause N. Org Lett. 2006;8:4485. doi: 10.1021/ol061669z.Morita N, Krause N. Eur J Org Chem. 2006:4634.Zhou C-Y, Chan PWH, Che C-M. Org Lett. 2006;8:325. doi: 10.1021/ol052696c.Zhang L. J Am Chem Soc. 2005;127:16804. doi: 10.1021/ja056419c.Morita N, Krause N. Angew Chem Int Ed. 2006;45:1897. doi: 10.1002/anie.200503846.Hoffmann-Röder A, Krause N. Org Lett. 2001;3:2537. doi: 10.1021/ol016205+.Morita N, Krause N. Org Lett. 2004;6:4121. doi: 10.1021/ol0481838.Kang J-E, Lee E-S, Park SI, Shin S. Tetrahedron Lett. 2005;46:7431.Luzung MR, Mauleon P, Toste FD. J Am Chem Soc. 2007;129:12402. doi: 10.1021/ja075412n.Huang X, Zhang L. Org Lett. 2007;9:4627. doi: 10.1021/ol7021356.Dudnik AS, Gevorgyan V. Angew Chem Int Ed. 2007;46:5195. doi: 10.1002/anie.200701128.
- 15.For examples of alkyne activation by gold proceeding via allenyl intermediates, see: ref 2b and: Marion N, Nolan SP. Angew Chem Int Ed. 2007;46:2750. doi: 10.1002/anie.200604773.Zhang L, Wang S. J Am Chem Soc. 2006;128:1442. doi: 10.1021/ja057327q.Wang S, Zhang L. Org Lett. 2006;8:4585. doi: 10.1021/ol0618151.Wang S, Zhang L. J Am Chem Soc. 2006;128:8414. doi: 10.1021/ja062777j.Buzas A, Istrate F, Gagosz F. Org Lett. 2006;8:1957. doi: 10.1021/ol0606839.Buzas A, Gagosz F. J Am Chem Soc. 2006;128:12614. doi: 10.1021/ja064223m.
- 16.(a) Markham JP, Staben ST, Toste FD. J Am Chem Soc. 2005;127:9708. doi: 10.1021/ja052831g. [DOI] [PubMed] [Google Scholar]; (b) Zhang Z, Liu C, Kinder RE, Han X, Qian H, Widenhoefer RA. J Am Chem Soc. 2006;128:9066. doi: 10.1021/ja062045r. [DOI] [PubMed] [Google Scholar]; (c) Marion N, Díez-González S, de Frémont P, Noble AR, Nolan SP. Angew Chem Int Ed. 2006;45:3647. doi: 10.1002/anie.200600571. [DOI] [PubMed] [Google Scholar]; (d) Sherry BD, Toste FD. J Am Chem Soc. 2004;126:15978. doi: 10.1021/ja044602k. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Computational and experimental details. This material is available free of charge via the Internet at http://pubs.acs.org.