

Abstract
Tocochromanols encompass a group of compounds with vitamin E activity essential for human nutrition. Structurally, natural vitamin E includes eight chemically distinct molecules: α-, β-, γ- and δ-tocopherol; and α-, β-, γ- and δ-tocotrienol. Symptoms caused by α-tocopherol deficiency can be alleviated by tocotrienols. Thus, tocotrienols may be viewed as being members of the natural vitamin E family not only structurally but also functionally. Palm oil and rice bran oil represent two major nutritional sources of natural tocotrienol. Taken orally, tocotrienols are bioavailable to all vital organs. The tocotrienol forms of natural vitamin E possesses powerful hypocholesterolemic, anti-cancer and neuroprotective properties that are often not exhibited by tocopherols. Oral tocotrienol protects against stroke-associated brain damage in vivo. Disappointments with outcomes-based clinical studies testing the efficacy of α-tocopherol need to be handled with caution and prudence recognizing the untapped opportunities offered by the other forms of natural vitamin E. Although tocotrienols represent half of the natural vitamin E family, work on tocotrienols account for roughly 1% of the total literature on vitamin E. The current state of knowledge warrants strategic investment into investigating the lesser known forms of vitamin E.
The natural vitamin E family includes eight chemically distinct molecules: α-, β-, γ- and δ-tocopherol; and α-, β-, γ- and δ-tocotrienol. Tocochromanols contain a polar chromanol head group with a long isoprenoid side chain. Depending on the nature of the isoprenoid chain, tocopherols (containing a phytyl chain) or tocotrienols (geranylgeranyl chain) can be distinguished (Dormann 2007). A striking asymmetry in our understanding of the eight-member natural vitamin E tocol family has deprived us of the full complement of benefits offered by the natural vitamin E molecules. Approximately only 1% of the entire literature on vitamin E addresses tocotrienols. A review of the NIH CRISP database shows that funding for tocotrienol research represents less than 1% of all vitamin E research during the last 30+ years. Within the tocopherol literature, the non-α forms remain poorly studied (Dietrich et al. 2006; Hensley et al. 2004; O’Byrne et al. 2000). This represents a major void in vitamin E research. Significance of the void is substantially enhanced by the observation that the biological functions of the different homologues of natural vitamin E are not identical. During the last 5 years, tocotrienol research has gained substantial momentum. More than two-thirds (210/301) of the entire PubMed literature on tocotrienols has been published on or after 2000. This represents a major swing in the overall direction of vitamin E research. The objective of this review is to highlight the potential significance of the tocotrienol half of the vitamin E family in human health and disease in light of current developments. This work focuses on three of the most described biomedical properties of tocotrienols: hypocholesterolemic, anti-cancer and neuroprotective.
Vitamin E Biosynthesis: Tocopherols and Tocotrienols
The condensation of homogentisate, derived from the shikimate pathway, and phytyl pyrophosphate (phytyl-PP), derived from the non-mevalonate pathway, through the action of the homogentisate prenyltransferase (HPT) represent the key committed step of tocopherol biosynthesis (Venkatesh et al. 2006). The product of the above-mentioned reaction is 2-methyl-6-phytylplastoquinone, the first true tocopherol intermediate and common precursor of all tocopherols. Subsequent ring cyclization and methylation reactions result in the formation of the four major (α–,β–,γ–,δ–) tocopherol derivatives. It is probable that the formation of γ– and δ–tocopherol proceeds via a common cyclase. Similarly, the final methylation reaction resulting in α– and β-tocopherol, respectively, is expected to be catalysed by the same methyltransferase (γ-TMT). Tocopherol helps maintain optimal photosynthesis rate under high-light stress (Porfirova et al. 2002). A considerable proportion of tocopherol is synthesized from free phytol suggesting that excess amounts of phytol released from chlorophyll breakdown during stress or senescence might be deposited in the form of tocopherol in chloroplasts (Dormann 2007).
Tocotrienols are the primary form of vitamin E in the seed endosperm of most monocots, including agronomically important cereal grains such as wheat, rice, and barley. Palm oil contains significant quantities of tocotrienol (Sundram et al. 2003). Tocotrienols are also found in the seed endosperm of a limited number of dicots, including Apiaceae species and certain Solanaeceae species, such as tobacco. These molecules are found only rarely in vegetative tissues of plants. Crude palm oil extracted from the fruits of Elaeis guineensis particularly contains a high amount of tocotrienols (up to 800 mg/kg), mainly consisting of γ-tocotrienol and α-tocotrienol. Compared to tocopherols, tocotrienols are considerably less widespread in the plant kingdom (Horvath et al. 2006). In 80 different plant species studied, twenty-four were found to contain significant amounts of tocotrienols. No taxonomic relation was apparent among the 16 dicotyledonous species that were found to contain tocotrienol. Monocotyledonous species (eight species) belonged either to the Poaceae (six species) or the Aracaceae (two species). A more detailed analysis of tocotrienol accumulation revealed the presence of this natural vitamin E in several non-photosynthetic tissues and organs, i.e. seeds, fruits and in latex. No tocotrienols could be detected in mature photosynthetic tissues. Transient accumulation of low levels of tocotrienols is found in the young coleoptiles of plant species whose seeds contained tocotrienols. No measurable tocotrienol biosynthesis was apparent in coleoptiles, or in chloroplasts isolated from coleoptiles. Tocotrienol accumulation in coleoptiles was not associated with chloroplasts. Tocotrienols seem to be transiently present in photosynthetically active tissues, however, it remains to be proven whether they are biosynthesized in such tissues, or imported from elsewhere in the plant (Horvath et al. 2006).
In contrast to tocotrienols, tocopherols occur ubiquitously in plant tissues and are the exclusive form of vitamin E in leaves of plants and seeds of most monocot plants. Transgenic expression of the barley HGGT (homogentisic acid transferase, which catalyzes the committed step of tocotrienol biosynthesis) in Arabidopsis thaliana leaves resulted in accumulation of tocotrienols, which were absent from leaves of non-transformed plants, and a 10- to 15-fold increase in total vitamin E antioxidants (tocotrienols plus tocopherols). Overexpression of the barley HGGT in corn seeds increased tocotrienol and tocopherol content by as much as six-fold. These results provide insight into the genetic basis for tocotrienol biosynthesis in plants and demonstrate the ability to enhance the antioxidant content of crops by introduction of an enzyme that redirects metabolic flux (Cahoon et al. 2003). More recently, another strategy involving genetic engineering of metabolic pathways in plants has proved to be efficient in bolstering tocotrienol biosynthesis (Rippert et al. 2004). In plants, phenylalanine is the precursor of a myriad of secondary compounds termed phenylpropanoids. In contrast, much less carbon is incorporated into tyrosine that provides p-hydroxyphenylpyruvate and homogentisate, the aromatic precursors of vitamin E. The flux of these two compounds has been upregulated by deriving their synthesis directly at the level of prephenate. This was achieved by the expression of the yeast prephenate dehydrogenase gene in tobacco plants that already over-express the Arabidopsis p-hydroxyphenylpyruvate dioxygenase coding sequence. Massive accumulation of tocotrienols was observed in leaves. These molecules, which were undetectable in wild-type leaves, became the major forms of vitamin E in the leaves of the transgenic lines. An increased resistance of the transgenic plants toward the herbicidal p-hydroxyphenylpyruvate dioxygenase inhibitor diketonitril was also observed. Thus, the synthesis of p-hydroxyphenylpyruvate is a limiting step for the accumulation of vitamin E in plants (Rippert et al. 2004).
Natural Sources of Tocotrienols
The identification of α-tocotrienol as a cholesterogenesis-inhibitory factor derived from barley (Hordeum vulgare L.) represents a landmark early discovery highlighting the unique significance of tocotrienols in health and disease (Qureshi et al. 1986). Palm oil represents one of the most abundant natural sources of tocotrienols (Elson 1992). The distribution of vitamin E in palm oil is 30% tocopherols and 70% tocotrienols (Sundram et al. 2003). The oil palm (Elaeis guineensis) is native to many West African countries, where local populations have used its oil for culinary and other purposes. Large-scale plantations, established principally in tropical regions of Asia, Africa and Latin America are mostly aimed at the production of oil (Solomons and Orozco 2003), which is extracted from the fleshy mesocarp of the palm fruit, and endosperm or kernel oil. Palm oil is different from other plant and animal oils in that it contains 50% saturated fatty acids, 40% unsaturated fatty acids, and 10% polyunsaturated fatty acids. Because of its high saturated fat content, palm oil has not been very popular in the United States. Hydrogenated fats contain high levels of trans-fatty acids which are now thought to have adverse health effects. The US Food and Drug Administration’s final ruling on trans-fatty acid labeling issued in 2003 has caused a rapid transformation in the fat and oil industries (Tarrago-Trani et al. 2006). Palm oil is free of trans-fatty acids and is rapidly gaining wider acceptance by the food industry in the country. Primary applications include bakery products, breakfast cereals, wafers and candies.
Rice bran oil, a by-product of the rice milling industry, is a major natural source of γ-tocotrienol but a poor source of α-tocotrienol. In addition, rice bran oil provides desmethyl tocotrienols. Two novel tocotrienols were isolated from stabilized and heated rice bran, apart from the known α-, β-, γ-, and δ-tocopherols and tocotrienols. These new tocotrienols are known as desmethyl tocotrienol [3, 4-dihydro-2-methyl-2-(4,8,12-trimethyltrideca-3′(E),7′(E), 11′-trienyl)-2H-1-benzopyran-6-ol] and didesmethyl tocotrienol [3, 4-dihydro-2-(4,8,12-trimethyltrideca-3′(E),7′(E), 11′-trienyl)-2H-1-benzopyran-6-ol] (Qureshi et al. 2000). Although scientific evidence is relatively limited, rice bran oil is believed to be a healthy vegetable oil in Asian countries (Sugano et al. 1999).
Cereals such as oat, rye and barley contain small amounts of tocotrienol in them. α-Tocotrienol is the predominant form of tocotrienol in oat (Avena sativa L.) and barley (56 and 40 mg/kg of dry weight, respectively). β-Tocotrienol is the major form of tocotrienol found in hulled and dehulled wheats (from 33 to 43 mg/kg of dry weight) (Panfili et al. 2003). Steaming and flaking of dehulled oat groats results in moderate losses of tocotrienols but not of tocopherols (Bryngelsson et al. 2002). Although tocotrienols are present in edible natural products, it is questionable whether these dietary sources could provide sufficient amounts of tocotrienol to humans. For example, the processing of 1000 kg of crude palm oil is necessary to derive 1 kg of the commercial product Tocomin® 50% (Carotech, NJ). Roughly, one would have to consume 100–200g of palm/rice-bran oil or 1.5–4 kg of wheat-germ, barley or oat to achieve doses that have been published to be effective biologically. With this consideration in mind, appropriately configured dietary supplements seem to be a prudent choice. Tocotrienols are synthesized both in edible as well as in inedible plant products. Rubber latex represents a major non-food natural source of tocotrienols (Chow and Draper 1970; Horvath et al. 2006; Whittle et al. 1966).
Bioavailability of tocotrienols taken orally
During the last two decades, efforts to understand how dietary vitamin E is transported to the tissues have focused on α-tocopherol transport (Blatt et al. 2001; Kaempf-Rotzoll et al. 2003; Traber and Arai 1999; Traber et al. 2004). α-Tocopherol transfer protein (TTP) has been identified to mediate α–tocopherol secretion into the plasma while other tocopherol-binding proteins seem to play a less important role (Kaempf-Rotzoll et al. 2003). Tocotrienols have been known for decades but why have they not been studied as well as α–tocopherol? Although there does not seem to be straightforward rational answer to this question, one contributing factor is whether tocotrienol taken orally reaches vital organs of the body. This concern was primarily based on a 1997 finding that the transport system, α-tocopherol transport protein (TTP), responsible to carry α-tocopherol to vital organs has a poorer efficiency to transport tocotrienols to tissues (Hosomi et al. 1997). The lack of relative specific affinity of TTP for tocotrienols led to the notion that availability of dietary tocotrienol to vital organs is negligible.
Although TTP is known to bind to α–tocotrienol with 8.5-fold lower affinity than that for α–tocopherol (Hosomi et al. 1997), it has not been clear whether, or to what extent, the delivery of orally supplemented α–tocotrienol to vital organs is dependent on TTP. Previously it has been reported that TTP-deficient female mice are infertile presumably because of vitamin E deficiency (Terasawa et al. 2000). This important observation was confirmed in a lineage of TTP deficient mice. Placenta of pregnant TTP deficient female mice were severely impaired with marked reduction of labyrinthine trophoblasts, and the embryos died at mid-gestation even when fertilized eggs of TTP-containing wild-type mice were transferred into TTP-deficient recipients (Jishage et al. 2001). Even in the presence of dietary α-tocopherol, TTP knock-out mice are known to suffer from α-tocopherol deficiency (Jishage et al. 2001; Terasawa et al. 2000). Recently it has been noted that oral supplementation of female mice with α–tocotrienol restored fertility of TTP knock-out mice suggesting that tocotrienol was successfully delivered to the relevant tissues and that tocotrienol supported reproductive function under conditions of α–tocopherol deficiency (Khanna et al. 2005a). This observation was consistent with another line of evidence from rats where tocotrienol supplementation spared loss of fertility caused by long-term vitamin E deficiency in the diet (Khanna et al. 2005a). TTP continues to be a key transport mechanism for the delivery of α-tocopherol to tissues. The significance of TTP in the transport of other forms of vitamin E remains unclear at present. It is clear, however, that natural isomers of vitamin E do get transported to vital organs even in the absence of TTP. Identification and characterization of TTP-independent vitamin E transport mechanisms in vivo is warranted.
Current findings support that oral tocotrienol (Carotech Inc., NJ) not only reaches the brain (Khanna et al. 2005a; Khanna et al. 2005b; Roy et al. 2002) but it does so in amounts sufficient to protect against stroke (Khanna et al. 2005b). The standard laboratory chow contains excessive amounts of α-tocopherol (Khosla et al. 2006; van der Worp et al. 1998) but negligible amounts of tocotrienol. Long-term lack of tocotrienol in the diet may repress any putative tocotrienol transport mechanism in vivo. Thus, long-term supplementation studies are needed. In light of the knowledge that natural analogs of vitamin E may compete for specific transporting mechanisms (Hosomi et al. 1997), it is important that tocotrienol supplementation be performed under conditions of minimized co-presence of tocopherols. Another related consideration is that although incorporation of orally supplemented vitamin E into tissues is a slow and progressive process, rapid incorporation of the supplement into tissues of newborns may occur in response to gavaging of pregnant mother rats (Roy et al. 2002). Thus, an experimental design incorporating long-term tocotrienol supplementation under conditions of minimal dietary co-presence of tocopherols and breeding of the supplemented colony would be a valuable approach to generate proof of principle testing whether dietary α–tocotrienol is capable of being transported to vital organs in vivo. In a recent study, rats were maintained on vitamin E deficient diet and gavaged with α–tocotrienol alone, α–tocopherol alone or in combination. Five generations of rats were studied over sixty weeks (Khanna et al. 2005a). Skin, adipose, heart, lungs, skeletal muscle brain, spinal cord, liver and blood were studied. Oral tocotrienol was delivered to all vital organs. In some tissues, the level of tocotrienol exceeded that of tocopherols indicating the presence of an efficient tocotrienol transport system in vivo. Baseline levels of α–tocotrienol in the skin of tocopherol-fed rats that never received any tocotrienol supplementation were negligible. Orally supplemented tocotrienol was rapidly taken up by the skin. Already in second generation rats, α–tocotrienol levels in the skin of tocotrienol supplemented rats exceeded twice the α–tocopherol levels in that organ. Of note, the α–tocotrienol level in the skin matched the α–tocotrienol level in the skin of rats fed with a comparable amount of tocopherol. When tocotrienol and tocopherol were co-supplemented, the uptake of α–tocotrienol by the skin was clearly blunted. In this group, α–tocotrienol levels were lower than α–tocotrienol levels in the skin suggesting a direct competition between orally taken tocotrienol and tocopherol for delivery to the skin. Longer supplementation resulted in a marked increase in the α–tocotrienol levels in the skin of tocotrienol-fed rats indicating a build-up of α–tocotrienol over time. Interestingly, the levels of α–tocotrienol in the skin of these rats were folds higher than the α–tocopherol level in the skin of tocopherol-fed rats. This observation suggests the presence of an effective transport mechanism delivering α–tocotrienol to the skin and efficient retention of α–tocotrienol in the skin over time. Co-supplementation of tocotrienol and tocopherol demonstrated favorable uptake of α–tocopherol over α–tocotrienol. Adipose tissue serves as storage organ for vitamin E (Adachi et al. 1990). Analysis of adipose tissue vitamin E content of fifth generation rats revealed substantially more accumulation of α–tocotrienol in that tissue than α–tocopherol.
In the case of tocotrienol as well as of tocopherol feeding, results from third and fifth generation rats indicate higher levels of vitamin E in the skin of female compared to that of male rats. This gender-specific effect suggesting better transport of tocotrienol in females than in males was noted as a general trend across all organs studied. Gender-based differences in the transport of dietary vitamins are known to exist in specific cases (Garry et al. 1987). Although the effect of several physiological factors on vitamin E transport has been studied, the gender factor remains to be specifically addressed (Lodge et al. 2004). Recently it has been demonstrated that γ-tocopherol is more rapidly metabolized in women than in men (Leonard et al. 2005). The level of α–tocotrienol in the ovary was over five-fold higher than that in the testes from the corresponding males rats (Khanna et al. 2005a). In the ovary, tocopherol is known to accumulate via a lipoprotein receptor dependent mechanism (Aten et al. 1994). Whether tocotrienol shares that mechanism remain to be tested.
Vitamin E enters the circulation from the intestine in chylomicrons. The conversion of chylomicrons to remnant particles results in the distribution of newly absorbed vitamin E to all of the circulating lipoproteins and ultimately to tissues. This enrichment of lipoproteins with vitamin E is a key mechanism by which vitamin E is delivered to tissues (Traber et al. 2004). In the liver, newly absorbed dietary lipids are incorporated into nascent very low density lipoproteins. The liver is responsible for the control and release of α-tocopherol into blood plasma. In the absence of TTP, α-tocopherol is not secreted back into the plasma. Excess vitamin E is not accumulated in the liver, but is metabolized and excreted, mostly in bile (Traber et al. 2004). Recently it has been noted that α–tocotrienol levels in the liver of rats and of TTP-deficient mice was much lower than the levels of this vitamin E isoform in most peripheral tissues studied (Khanna et al. 2005a). Such observation argues against a central role of the liver in delivering oral α–tocotrienol to peripheral tissues. TTP has the ability to bind to both α–tocopherol as well as α–tocotrienol. The affinity to bind α–tocopherol is several-fold higher than that for α–tocotrienol (Hosomi et al. 1997). Thus, under conditions of co-existence, α–tocopherol is expected to out-compete α–tocotrienol for binding. Although studies with the TTP-deficient mice (Khanna et al. 2005a) indicate the existence of a TTP-independent mechanisms for the tissue delivery of oral α–tocotrienol, observations in the rat (Khanna et al. 2005a) indicate that the mechanisms for transporting α–tocopherol and α–tocotrienol seem to compete such that transport of α–tocopherol is favored. Thus, co-supplementation of α–tocopherol and α–tocotrienol is likely to compromise tissue delivery of α–tocotrienol (Khanna et al. 2005a).
Few studies have specifically looked at the fate of oral tocotrienol supplementation in humans. In a study investigating the pharmacokinetics and bioavailability of α-, γ- and δ-tocotrienols under fed and fasted conditions in eight healthy volunteers, subjects were administered a single 300 mg oral dose of mixed tocotrienols under fed or fasted conditions. The peak concentration of α-tocotrienol in the blood plasma was just over 1μM (Yap et al. 2001). The fed state increased the onset as well as the extent of absorption of tocotrienols by more than two folds. In addition, the mean apparent elimination half-life of α-, γ- and δ-tocotrienols was estimated to be 4.4, 4.3 and 2.3 h, respectively, being between 4.5- to 8.7-fold shorter than that reported for α-tocopherol (Yap et al. 2001). In another study, human subjects took tocotrienyl acetate supplements (250 mg/d) for eight weeks while being on low-fat diet. Under basal conditions, the concentration of tocotrienol in the blood plasma of Americans is in a range (undetectable to nM) close to detection limits of HPLC analyses. In response to supplementation, the concentrations of tocotrienol in the mean blood plasma were as follows: α-tocotrienol, 0.98 μM; γ-tocotrienol, 0.54 μM; and δ-tocotrienol 0.09 μM (O’Byrne et al. 2000). Thus, tocotrienyl acetate supplements were observed to be hydrolyzed, absorbed, and detectable in human plasma. Recently, a novel formulation for improved absorption of tocotrienols has been developed (Ho et al. 2003). Emulsions are known to increase absorption of fat-soluble drugs. This invention is based on SEDDS (self-emulsifying drug delivery systems) technology (Araya et al. 2006; Gao and Morozowich 2006; Hong et al. 2006). Soft-gelatin capsules (Tocovid Suprabio™, Carotech Inc., NJ) containing tocotrienol have been produced. Once ingested, the tocotrienols form emulsion when the contents are released and mixed with human gastrointestinal fluid. In a recent study using Tocovid Suprabio™ the post-absorptive fate of tocotrienol isomers and their association with lipoprotein subfractions was examined in humans (Khosla et al. 2006). The peak α-tocotrienol concentrations in supplemented individuals averaged approximately 3 microM in blood plasma, 1.7 microM in LDL, 0.9 microM in triglyceride-rich lipoprotein, and 0.5 microM in HDL. This peak plasma concentration of α-tocotrienol is 2–3 times more than the peak concentration reported in previous studies using generic supplements not based on SEDDS (O’Byrne et al. 2000; Yap et al. 2001).
Functional Uniqueness of Vitamin E Family Members
All eight tocols in the natural vitamin E family share close structural homology and hence possess comparable antioxidant efficacy. Yet, current studies of the biological functions of vitamin E continue to indicate that members of the vitamin E family possess unique biological functions often not shared by other family members. One of the earliest observations suggesting that α-tocopherol may have functions independent of its antioxidant property came from the observation that α-tocopherol strongly inhibited platelet adhesion. The antiadhesive effect of α-tocopherol appeared to be related to a reduction in the number and size of pseudopodia upon platelet activation and this finding led to the hypothesis that within the human body vitamin E may exert functions beyond its antioxidant property (Steiner 1993). That members of the tocopherol family may have functions independent of their antioxidant properties gained more prominence when vitamin E molecules with comparable antioxidant properties exhibited contrasting biological effects (Boscoboinik et al. 1991). At the post-translational level, α-tocopherol inhibits protein kinase C, 5-lipoxygenase and phospholipase A2 and activates protein phosphatase 2A and diacylglycerol kinase. Some genes (e.g. scavenger receptors, α-tocopherol transfer protein, α-tropomyosin, matrix metalloproteinase-19 and collagenase) are specifically modulated by α-tocopherol at the transcriptional level. α-Tocopherol also inhibits cell proliferation, platelet aggregation and monocyte adhesion. These effects have been characterized to be unrelated to the antioxidant activity of vitamin E, and possibly reflect specific interactions of α-tocopherol with enzymes, structural proteins, lipids and transcription factors (Zingg and Azzi 2004). γ-Tocopherol represents the major form of vitamin E in the diet in the USA, but not in Europe. Desmethyl tocopherols, such as γ-tocopherol and specific tocopherol metabolites, most notably the carboxyethyl-hydroxychroman (CEHC) products, exhibit functions that are not shared by α-tocopherol. The activities of these other tocopherols do not map directly to their chemical antioxidant behavior but rather reflect anti-inflammatory, antineoplastic, and natriuretic functions possibly mediated through specific binding interactions (Hensley et al. 2004). Metabolites of γ-tocopherol (2,7,8-trimethyl-2-(β-carboxyethyl)-6-hydroxychroman), but not that of α-tocopherol, provides natriuretic activity. Moreover, a nascent body of epidemiological data suggests that γ-tocopherol is a better negative risk factor for certain types of cancer and myocardial infarction than is α-tocopherol (Wagner et al. 2004).
Structurally, tocotrienols differ from tocopherols by the presence of three trans double-bonds in the hydrocarbon tail. Because of these unsaturations in the isoprenoid side-chain, tocotrienols are thought to assume a unique conformation (Atkinson 2006). While the proposed curved structure has not been confirmed by NMR, α-tocotrienol is very likely much more flexible in the sidechain and it puts a greater curvature stress on phospholipid membranes. This is a dynamic issue (Fig. 1) and has been confirmed in scanning calorimetry data (personal communication by Dr. Jeffrey Atkinson). Indeed, α-tocotrienol possesses numerous functions that are not shared by α-tocopherol (Sen et al. 2006). For example, nanomolar concentrations of α-tocotrienol uniquely prevent inducible neurodegeneration by regulating specific mediators of cell death (Khanna et al. 2006; Khanna et al. 2003; Sen et al. 2000). Oral supplementation of tocotrienol protects against stroke (Khanna et al. 2005b). Micromolar amounts of tocotrienol suppress the activity of 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase, the hepatic enzyme responsible for cholesterol synthesis (Pearce et al. 1994; Pearce et al. 1992). Tocopherols do not share the cholesterol-lowering properties of tocotrienol (Qureshi et al. 1986; Qureshi et al. 2002). Sterol-regulated ubiquitination marks HMG-CoA reductase for endoplasmic reticulum (ER)-associated degradation by 26S proteasomes. This degradation, which results from sterol-induced binding of reductase to ER membrane proteins called Insigs, contributes to the complex, multivalent feedback regulation of the enzyme. Recently it has been demonstrated that δ-tocotrienol stimulates ubiquitination and degradation of reductase and blocks processing of sterol regulatory element-binding proteins (SREBPs), another sterol-mediated action of Insigs. The γ-tocotrienol analog is more selective in enhancing reductase ubiquitination and degradation than blocking SREBP processing. Other forms of vitamin E neither accelerate reductase degradation nor block SREBP processing (Song and Debose-Boyd 2006).
Figure 1.
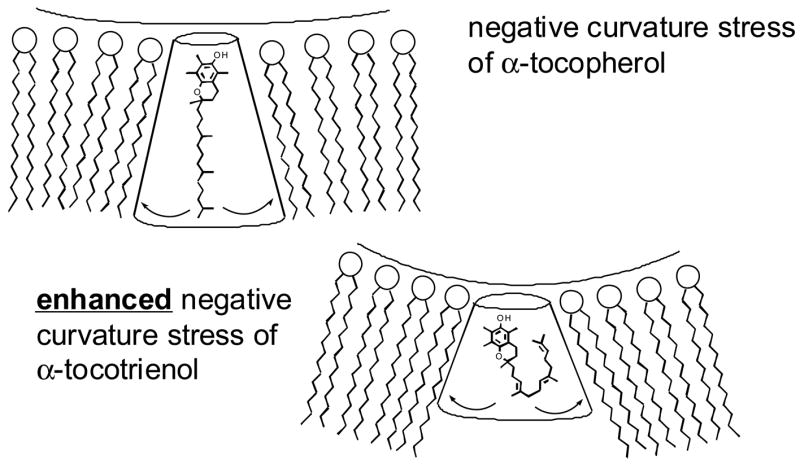
α-Tocotrienol versus α-tocopherol: Ramifications for membrane location and dynamics. Credit: personal communication by collaborator Dr. Jeffrey Atkinson, Brock University.
Tocotrienol, not tocopherol, administration reduces oxidative protein damage and extends the mean life span of C. elegans (Adachi and Ishii 2000). Tocotrienols are thought to have more potent antioxidant properties than α-tocopherol (Serbinova et al. 1991; Serbinova and Packer 1994). Reportedly, the unsaturated side chain of tocotrienol allows for more efficient penetration into tissues that have saturated fatty layers such as the brain and liver (Suzuki et al. 1993). Experimental research examining the antioxidant, free radical scavenging effects of tocopherol and tocotrienols revealed that tocotrienols appear superior due to their better distribution in the fatty layers of the cell membrane (Suzuki et al. 1993). Furthermore, tocotrienol but not tocopherol, suppresses growth of human breast cancer cells (Nesaretnam et al. 1995).
In humans, tocotrienol supplementation results in peak blood plasma level of α-tocotrienol that is over an order of magnitude higher than that required to protect neurons against a range of neurotoxic insults (Khanna et al. 2005a; Khanna et al. 2006; Khanna et al. 2003; Khanna et al. 2005b; Khosla et al. 2006; Sen et al. 2000). Despite such promising potential, tocotrienol research accounts for roughly 1% of all vitamin E research published in PubMed. The unique vitamin action of α-tocopherol, combined with its prevalence in the human body and the similar efficiency of tocopherols as chain-breaking antioxidants, led biologists to almost completely discount the “minor” vitamin E molecules as topics for basic and clinical research. Recent discoveries warrant a serious reconsideration of this conventional wisdom.
Hypocholesterolemic Effects of Tocotrienol
Purification of an oily, non-polar fraction of high protein barley flour by high pressure liquid chromatography yielded ten major components. Two of these components were identified as potent inhibitors of cholesterogenesis both in vivo as well as in vitro. Addition of the purified inhibitor I (2.5–20 ppm) to chick diets significantly decreased hepatic cholesterogenesis and serum total and low density lipoprotein cholesterol and concomitantly increased lipogenic activity. The high resolution mass spectrometric analysis and measurement of different peaks of inhibitor I gave a molecular ion at m/e 424 (C29H44O2) and main peaks at m/e 205, 203, and 165 corresponding to C13H17O2, C13H15O2, and C10H13O2 moieties, respectively. Based on these results, d-α-tocotrienol was identified as the active principle. This identification was confirmed against synthetic samples (Qureshi et al. 1986). That the α-tocotrienol form of natural vitamin E, not tocopherol, may have significant cholesterol-lowering properties represents one of the early findings describing the unique biological properties of tocotrienol that was reported two decades ago (Qureshi et al. 1986). The endoplasmic reticulum enzyme 3-hydroxy-3-methylglutaryl (HMG-CoA) CoA reductase produces mevalonate, which is converted to sterols and other products. It is proposed that tocotrienols are effective in lowering serum total and LDL-cholesterol levels by inhibiting the hepatic enzymic activity of HMG-CoA reductase through a post-transcriptional mechanism. α-Tocopherol, however, had an opposite effect (induces) on this enzyme activity (Qureshi et al. 2002). This contrast is of outstanding significance and requires further characterization. Evidence that the tocotrienol-rich fraction (TRF) of palm oil may indeed lower plasma cholesterol in mammals came from a study of normolipemic and genetically hypercholesterolemic pigs of defined lipoprotein genotype (Qureshi et al. 1991a). The pigs were fed a standard diet supplemented with 50 micrograms/g TRF isolated from palm oil. Hypercholesterolemic pigs fed the TRF supplement showed a 44% decrease in total serum cholesterol, a 60% decrease in LDL-cholesterol, and significant decreases in levels of apolipoprotein B (26%), thromboxane-B2 (41%), and platelet factor 4 (PF4; 29%). It was also noted that TRF had a marked protective effect on the endothelium and platelet aggregation. The effect of the lipid-lowering diet persisted only in the hypercholesterolemic swine after 8 week feeding of the control diet (Qureshi et al. 1991a). These interesting observations were quickly put to test in humans by means of a double-blind, crossover, 8-week study (Qureshi et al. 1991b). The goal was to compare effects of the tocotrienol-enriched fraction of palm oil (200 mg palmvitee capsules/day) with those of 300 mg corn oil/d on serum lipids of hypercholesterolemic human subjects (serum cholesterol 6.21–8.02 mmol/L). Concentrations of serum total cholesterol (-15%), LDL cholesterol (-8%), Apo B (-10%), thromboxane (-25%), platelet factor 4 (-16%), and glucose (-12%) decreased significantly only in the 15 subjects given palmvitee during the initial four weeks. Results from the crossover study established that the noted beneficial effects were indeed caused by palmvitee. A carry-over effect of palmvitee was also reported. Serum cholesterol concentrations of seven hypercholesterolemic subjects (>7.84 mmol/L) decreased 31% during a four-week period in which they were given 200 mg γ-tocotrienol/d. These results suggest that γ-tocotrienol could be the active principle cholesterol inhibitor in palmvitee capsules (Qureshi et al. 1991b). Experimental data from the study of hamsters are in agreement (Raederstorff et al. 2002). What added to the interest in tocotrienol as a cholesterol-lowering nutrient in humans was a concurrent independent study reporting the hypocholesterolemic effects of palmvitee (Tan et al. 1991). Each palmvitee capsule contained approximately 18, 42, and 240 mg of tocopherols, tocotrienols, and palm olein, respectively. All volunteers took one palmvitee capsule per day for 30 consecutive days. Overnight fasting blood was recorded from each volunteer before and after the experiment. Palmvitee lowered both serum total cholesterol and low-density-lipoprotein cholesterol concentrations in all subjects. The magnitude of reduction of serum total cholesterol ranged from 5.0% to 35.9% whereas the reduction of low-density-lipoprotein cholesterol values ranged from 0.9% to 37.0% when compared with their respective baseline values (Tan et al. 1991). In another study, the cholesterol-lowering effects of palmvitee and γ-tocotrienol were examined in hypercholesterolemic subjects after acclimation to the American Heart Association Step 1 dietary regimen for four and eight weeks, respectively (Qureshi et al. 1995). The four-week dietary regimen alone caused a 5% significant decrease in the cholesterol level of thirty-six subjects. Subjects continuing on the dietary regimen for a second four-week period benefited from an additional 2% decrease in their cholesterol levels. The subjects experienced significant palmvitee- and γ-tocotrienol-mediated decreases in plasma cholesterol. The group of subjects acclimated to the dietary regimen for four weeks responded to palmvitee with a 10% statistically significant decrease in cholesterol. Of interest, α-tocopherol attenuated the cholesterol-suppressive action of the tocotrienols. This antagonism between tocopherol and tocotrienol warrants further research. The second group of subjects acclimated to the dietary regimen for eight weeks, received 200 mg γ-tocotrienol/d for four weeks. The cholesterol-suppressive potency of this α-tocopherol-free preparation was calculated to be equivalent to that of the mixture of tocotrienols (220 mg) used in the prior study. Cholesterol levels of the sixteen subjects in the second group significantly decreased by 13% during the four-week trial. Plasma apolipoprotein B and ex vivo generation of thromboxane B2 were similarly responsive to the tocotrienol preparations, whereas neither preparation had an impact on high density lipoprotein cholesterol and apolipoprotein A-1 levels (Qureshi et al. 1995).
Tocotrienol, not only of palm oil origin, but also isolated from rice bran show cholesterol-lowering properties (Chen and Cheng 2006; Qureshi et al. 2001a). Amaranth oil, containing tocotrienol, possesses hypocholesterolemic properties as well (Martirosyan et al. 2007). A human study with 28 hypercholesterolemic subjects has been executed in 5 phases of 35 days each. The goal was to check the efficacy of a TRF preparation from rice bran alone and in combination with lovastatin. After placing subjects on the American Heart Association (AHA) Step-1 diet (phase II), the subjects were divided into two groups, A and B. The AHA Step-1 diet was continued in combination with other treatments during phases III to V. Group A subjects were given 10 mg lovastatin, 10 mg lovastatin plus 50 mg TRF, 10 mg lovastatin plus 50 mg α-tocopherol per day, in the third, fourth, and fifth phases, respectively. Group B subjects were treated exactly according to the same protocol except that in the third phase, they were given 50 mg TRF instead of lovastatin. The TRF or lovastatin plus AHA Step-1 diet effectively lowered serum total cholesterol (14%, 13%) and LDL-cholesterol (18%, 15%), respectively. The combination of TRF and lovastatin plus AHA Step-1 diet significantly reduced the lipid parameters by 20–25%. Especially significant were the increase in the HDL/LDL ratio to 46% in group A and 53% in group B. None of the subjects reported any side-effects throughout the study of 25 weeks (Qureshi et al. 2001b). Consistent results were obtained using rice-bran derived TRF in another human study (Qureshi et al. 2002). A dose of 100 mg/day of TRF decreased the level of serum total cholesterol, LDL-cholesterol, apolipoprotein B and triglycerides compared with the baseline values. The work led to the suggestion that a dose of 100 mg/day TRF plus AHA Step-1 diet could control the risk of coronary heart disease in hypercholesterolemic humans (Qureshi et al. 2002).
Mechanistic evidence supporting the cholesterol-lowering properties of tocotrienol is gradually mounting. Tocotrienols cause post-transcriptional suppression of HMG-CoA reductase by a process distinct from other known inhibitors of cholesterol biosynthesis (Pearce et al. 1992). In addition, γ-tocotrienol may stimulate cholesterol catabolism (Chen and Cheng 2006). In vitro, γ-tocotrienol possesses 30-fold greater activity toward cholesterol biosynthesis inhibition compared to α-tocotrienol. The synthetic (racemic) and natural (chiral) tocotrienols exhibited nearly identical inhibition of cholesterol biosynthesis and HMG-CoA reductase suppression properties (Pearce et al. 1992). Incubation of several cell-types with γ-tocotrienol inhibited the rate of [14C]acetate but not [3H] mevalonate incorporation into cholesterol in a concentration- and time-dependent manner, with 50% inhibition at approximately 2 microM and maximum approximately 80% inhibition observed within 6 h in HepG2 cells (Parker et al. 1993). Both HMG-CoA reductase activity and protein expression are sensitive to tocotrienol. In vivo studies lend support to that in vitro observation (Iqbal et al. 2003). Tocotrienols influence the mevalonate pathway in mammalian cells by post-transcriptional suppression of HMG-CoA reductase, and specifically modulate the intracellular mechanism for controlled degradation of the reductase protein, an activity that mirrors the functional properties of the putative non-sterol isoprenoid regulators derived from mevalonate (Parker et al. 1993). It is suggested that the farnesyl side chain and the methyl/hydroxy substitution pattern of γ-tocotrienol deliver a high level of HMG-CoA reductase suppression, unsurpassed by synthetic analogues studied (Pearce et al. 1994). HMG-CoA reductase activity in tumor tissues differs from that of liver in being resistant to sterol feedback regulation. Tumor reductase activity retains sensitivity to the post-transcriptional regulation. As a consequence, tocotrienol is effective in suppressing mevalonate synthesis. By doing so, tocotrienol can deplete tumor tissues of two intermediate products, farnesyl pyrophosphate and geranylgeranyl pyrophosphate, which are incorporated post-translationally into growth control-associated proteins (Elson and Qureshi 1995).
Ubiquitination followed by rapid degradation by 26S proteasomes represents a key mechanism to silence HMG-CoA reductase. This pathway is activated when sterols and non-sterol end products of mevalonate metabolism accumulate in cells. Sterol-accelerated ubiquitination of HMG-CoA reductase requires Insig-1 and Insig-2, membrane-bound proteins of the endoplasmic reticulum (Sever et al. 2003). Recently it has been demonstrated that δ-tocotrienol stimulates the ubiquitination and degradation of HMG-CoA reductase and blocks processing of sterol regulatory element-binding proteins, another sterol-mediated action of Insigs. The γ-tocotrienol analog was noted to be more selective in enhancing reductase ubiquitination and degradation than blocking the processing of sterol regulatory element-binding proteins. Interestingly, other forms of vitamin E neither accelerate reductase degradation nor block the processing of sterol regulatory element-binding proteins. δ- and γ-Tocotrienol triggered reductase ubiquitination directly and did not require further metabolism for their activity (Song and Debose-Boyd 2006).
Anti-Cancer Effects of Tocotrienol
Pure and mixed isoprenoids are known to possess potent anti-cancer activity (Mo and Elson 1999). Tocotrienols are isoprenoids but tocopherols are not. Unlike in the case of neuroprotection where α-tocotrienol has emerged to be the most potent isoform (Khanna et al. 2006; Khanna et al. 2005b; Sen et al. 2004; Sen et al. 2006), there seems to somewhat of a consensus that γ- and δ-tocotrienols are the most potent anti-cancer isoform of all natural existing tocotrienols. One of the first studies addressing the role of tocotrienols in neoplastic disorders was reported in 1989 (Komiyama et al. 1989). The effects of intraperitoneally injected α- and γ-tocotrienol, as well as that of α-tocopherol, were examined. Both tocotrienols were effective against sarcoma 180, Ehrlich carcinoma, and invasive mammary carcinoma. γ-Tocotrienol showed a slight life-prolonging effect in mice with Meth A fibrosarcoma, but the tocotrienols had no antitumor activity against P388 leukemia at doses of 5–40 mg/kg/d (Komiyama et al. 1989). Compared to tocotrienols, α-tocopherol was not as effective. The antitumor activity of γ-tocotrienol was higher than that of α-tocotrienol. In contrast to α-tocopherol, tocotrienols caused growth-inhibition of human and mouse tumor cells when the cells were exposed to these agents for 72 h in vitro (Komiyama et al. 1989). In an independent study published in the same year the anti-carcinogenic properties of palm oil, a rich source of tocotrienols, was reported (Sundram et al. 1989). In this study, young female Sprague-Dawley rats were treated with a single dose of 5 mg of 7,12-dimethylbenz(a)anthracene intragastrically. Three days after carcinogen treatment, the rats were put on semisynthetic diets containing 20% by weight of corn oil, soybean oil, crude palm oil, refined, bleached, deodorized palm oil and metabisulfite-treated palm oil for 5 months. During the course of experiments, rats fed on different dietary fats had similar rate of growth. Rats fed 20% corn oil or soybean oil diet had marginally higher tumor incidence than rats fed on palm oil diets. At autopsy, rats fed on high corn oil or soybean oil diets had significantly more tumors than rats fed on the three palm oil diets. Palm oil is different from corn oil and soybean oil in many ways. In addition to possessing higher levels of tocotrienol, palm oil has a contrasting fatty acid profile and also much higher levels of tocopherol and carotenes. As such, the favorable anti-carcinogenic effects noted in this study cannot be directly associated with tocotrienols (Sundram et al. 1989). The antioxidant or redox property of tocotrienol is not responsible for its anti-cancer property. Results in support of this hypothesis show that a redox-silent analogue of α-tocotrienol, 6-O-carboxypropyl-α-tocotrienol is cytotoxic against A549 cells, a human lung adenocarcinoma cell line (Yano et al. 2005). Although the phenolic antioxidant group in tocotrienol may not be implicated in its anticancer property, it is apparent that the side-chain has some antioxidant property which prevents against carcinogenesis (Yu et al. 2005).
Breast cancer has been most extensively studied in cell culture and rodent in vivo models for the efficacy of tocotrienols. Tocopherol and tocotrienol have been tested side-by-side for chemopreventive activity in a chemically induced rat mammary-tumor model. When mammary tumors were induced by 7,12-dimethylbenz(a)anthracene, only the tocotrienol group showed enhanced tumor latency (Gould et al. 1991). The TRF of palm oil is not only rich in tocotrienols but also contains some α-tocopherol. The effects of TRF and α-tocopherol on the proliferation, growth, and plating efficiency of the MDA-MB-435 estrogen-receptor-negative human breast cancer cells have been examined (Nesaretnam et al. 1995). TRF inhibited the proliferation of these cells with a concentration required to inhibit cell proliferation by 50% of 180 microgram/mL whereas α-tocopherol had no effect at concentrations up to 1000 microgram/mL. The effects of TRF and α-tocopherol were also tested in longer-term experiments, using concentrations of 180 and 500 microgram/mL. TRF, but not α-tocopherol, inhibited the growth as well as plating efficiency of the cells. These findings point towards the hypothesis that α-tocopherol contained in the TRF does not account for its beneficial effects and that tocotrienols may have been the active principle responsible for the observed effects of TRF (Nesaretnam et al. 1995). It is now known that TRF, α-, γ- and δ-tocotrienols inhibits proliferation of estrogen receptor-negative MDA-MB-435 human breast cancer cells with IC50 of 180, 90, 30 and 90 microg/mL, respectively. In contrast, α-tocopherol is not effective at concentrations up to 500 microg/mL. Tocotrienols inhibit the proliferation of estrogen receptor-positive MCF-7 cells. The IC50 for TRF, α-, γ- and δ-tocotrienols have been estimated to be 4, 6, 2 and 2 microg/mL, respectively. In sharp contrast, the efficiency of α-tocopherol under comparable conditions is 20–50 times lower with a IC50 of 125 microg/mL (Guthrie et al. 1997).
Tamoxifen, a widely used synthetic anti-estrogen inhibits the growth of MCF-7 cells with an IC50 of 0.04 microg/mL. In MCF-7 cells, only 1:1 combinations of γ- or δ-tocotrienol with tamoxifen showed a synergistic inhibitory effect on the proliferative rate and growth of the cells. α-Tocopherol did not exhibit this beneficial synergistic effect with tamoxifen (Guthrie et al. 1997). The inhibition by tocotrienols was not overcome by addition of excess estradiol to the culture medium suggesting that tocotrienols are effective inhibitors of both estrogen receptor-negative and -positive cells and that combinations with tamoxifen may be useful for breast cancer therapy (Guthrie et al. 1997). Subsequent studies demonstrated that TRF inhibits growth of MCF7 cells in both the presence and absence of estradiol such that complete suppression of growth is achieved at 8 microg/mL. MDA-MB-231 cells are also inhibited by TRF such that 20 microg/mL of TRF is needed for complete growth suppression. Study of the individual component tocotrienols in TRF revealed that all fractions inhibit growth of both estrogen-responsive as well as estrogen-nonresponsive cells and of estrogen-responsive cells in both the presence and absence of estradiol. This estradiol-independent effect of tocotrienols is of clinical interest (Nesaretnam et al. 2000; Nesaretnam et al. 1998). γ- and δ-Tocotrienol fractions were most potent inhibitors of breast cancer cell growth. Complete inhibition of MCF7 cell growth was achieved at 6 microg/mL of γ-tocotrienol/δ-tocotrienol in the absence of estradiol and 10 microg/mL of δ-tocotrienol in the presence of estradiol. In contrast, complete suppression of MDA-MB-231 cell growth was not achieved even at concentrations of 10 microg/mL of δ-tocotrienol. Of note, unlike tocotrienols α-tocopherol does not inhibit MCF7, MDA-MB-231 or ZR-75-1 cell growth in either the presence or the absence of estradiol (Mo and Elson 1999; Nesaretnam et al. 2000; Nesaretnam et al. 1998). Studies examining the mechanisms by which tocotrienols check the growth of breast cancer cells have identified that tocotrienols do not act via an estrogen receptor-mediated pathway and must therefore act differently from estrogen antagonists. Furthermore, tocotrienols did not increase the levels of growth-inhibitory insulin-like growth factor binding proteins in MCF7 cells, implying a different mechanism from that proposed for retinoic acid inhibition of estrogen-responsive breast cancer cell growth (Nesaretnam et al. 1998).
Unlike α-tocopherol, δ-tocopherol seems to be more promising albeit much less so than the tocotrienols. The apoptosis-inducing properties of RRR-α-, β-, γ-, and δ-tocopherols, and α-, γ-, and δ-tocotrienols have been compared in estrogen-responsive MCF7 and estrogen-nonresponsive MDA-MB-435 human breast cancer cell lines. Vitamin E succinate, a known inducer of apoptosis in several cell lines, including human breast cancer cells, served as a positive control. The estrogen-responsive MCF7 cells was found to be more susceptible than the estrogen-nonresponsive MDA-MB-435 cells, with concentrations for half-maximal response for tocotrienols (α, γ, and δ) and RRR-δ-tocopherol of 14, 15, 7, and 97 micrograms/ml, respectively. The tocotrienols (α, γ, and δ) and RRR-δ-tocopherol induced MDA-MB-435 cells to undergo apoptosis, with concentrations for half-maximal response of 176, 28, 13, and 145 micrograms/ml, respectively. With the exception of RRR-δ-tocopherol, the tocopherols (α, β, and γ) and the acetate derivative of RRR-α-tocopherol (RRR-α-tocopheryl acetate) were ineffective in the induction of apoptosis in both cell lines when tested within the range of their solubility, i.e., 10–200 micrograms/ml (Yu et al. 1999)
Mammary tissue homeostasis depends upon dynamic interactions between the epithelial cells, their microenvironment (including the basement membrane and the stroma), and the tissue architecture, which influence each other reciprocally to regulate growth, death and differentiation in the gland. The study of normal mammary epithelial cells isolated from mid-pregnant mice grown in collagen gels and maintained on serum-free media showed that treatment with 0–120 microM α- or γ-tocopherol had no effect, whereas 12.5–100 microM TRF, 100–120 microM δ-tocopherol, 50–60 microM α-tocotrienol, and 8–14 microM γ- or δ-tocotrienol significantly inhibited cell growth in a dose-responsive manner. In acute studies, 24h exposure to 0–250 microM α-, γ-, and δ-tocopherol had no effect, whereas similar treatment with 100–250 microM TRF, 140–250 microM α-, 25–100 microM γ- or δ-tocotrienol significantly reduced cell viability. The observed growth-inhibitory doses of TRF, δ-tocopherol, and α-, γ-, and δ-tocotrienol induced apoptosis in these cells. Mammary epithelial cells preferentially took up tocotrienols as compared to tocopherols, suggesting that at least part of the reason tocotrienols display greater potency than tocopherols is because of greater cellular uptake. These observations suggest that the highly biopotent γ- and δ-tocotrienol isoforms may play a physiological role in modulating normal mammary gland growth, function, and remodeling (McIntyre et al. 2000b). A subsequent study identified that highly malignant cells are specifically more sensitive, whereas the pre-neoplastic cells are least sensitive to the antiproliferative and apoptotic effects of tocotrienols (McIntyre et al. 2000a). The comparative effects of tocopherols and tocotrienols were examined using preneoplastic (CL-S1), neoplastic (−SA), and highly malignant (+SA) mouse mammary epithelial cells. Over a five-day culture period, treatment with 0–120 microM α- and γ-tocopherol had no effect on cell proliferation, whereas cell growth was inhibited 50% (IC50) as compared with controls by treatment with the following: 13, 7, and 6 microM tocotrienol-rich-fraction of palm oil (TRF); 55, 47, and 23 microM δ-tocopherol; 12, 7, and 5 microM α-tocotrienol; 8, 5, and 4 microM γ-tocotrienol; or 7, 4, and 3 microM δ-tocotrienol in CL-S1, −SA and +SA cells, respectively. Acute 24-hr exposure to 0–250 microM α- or γ-tocopherol (CL-S1, −SA, and +SA) or 0–250 microM δ-tocopherol (CL-S1) had no effect on cell viability, whereas cell viability was reduced 50% (LD50) as compared with controls by treatment with 166 or 125 microM δ-tocopherol in −SA and +SA cells, respectively. Additional LD50 doses were determined as the following: 50, 43, and 38 microM TRF; 27, 28, and 23 microM α-tocotrienol; 19, 17, and 14 microM γ-tocotrienol; or 16, 15, or 12 microM δ-tocotrienol in CL-S1, −SA, and +SA cells, respectively. Treatment-induced cell death resulted from activation of apoptosis. Consistent with previous observations, CL-S1, −SA, and +SA cells preferentially accumulated tocotrienols as compared with tocopherols. Highly malignant +SA cells were the most sensitive, whereas the pre-neoplastic CL-S1 cells were the least sensitive to the anti-proliferative and apoptotic effects of tocotrienols (McIntyre et al. 2000a).
The molecular mechanisms by which tocotrienols selectively kill breast cancer cells are in the process of being characterized. δ-Tocotrienol induces TGF-β receptor II expression and activates TGF-β-, Fas- and JNK-signaling pathways (Shun et al. 2004). Are the caspase-3,8,9 pathways involved in tocotrienol-induced death of cancer cells? In search for the answer, highly malignant +SA mouse mammary epithelial cells were grown in culture and maintained on serum-free media. Treatment with TRF or γ-tocotrienol, but not α-tocopherol, induced a dose-dependent decrease in +SA cell viability (Shah et al. 2003). TRF- and γ-tocotrienol-induced cell death resulted from apoptosis. Treatment of cells with TRF or γ-tocotrienol increased intracellular activity and levels of processed caspase-8 and -3 but not caspase-9. Furthermore, treatment with specific caspase-8 or -3 inhibitors, but not caspase-9 inhibitor, completely blocked tocotrienol-induced apoptosis in +SA cells suggesting that tocotrienol-induced apoptosis in +SA mammary cancer cells is mediated by activation of the caspase-8 signaling pathway and is independent of caspase-9 activation (Shah et al. 2003).
Tocotrienol-induced caspase 8 activation is not associated with death receptor apoptotic signaling (Shah and Sylvester 2004). γ-Tocotrienol significantly decreases the relative intracellular levels of phospho-phosphatidylinositol 3-kinase (PI3K)-dependent kinase 1 (phospho-PDK-1 active), phospho-Akt (active), and phospho-glycogen synthase kinase3. It also decreases the intracellular levels of FLICE-inhibitory protein (FLIP), an anti-apoptotic protein that inhibits caspase-8 activation. Because stimulation of the PI3K/PDK/Akt mitogenic pathway is associated with increased FLIP expression, enhanced cellular proliferation, and survival, these observations suggest that tocotrienol-induced caspase-8 activation and apoptosis in malignant +SA mammary epithelial cells is associated with a suppression in PI3K/PDK-1/Akt mitogenic signaling and subsequent reduction in intracellular FLIP levels (Shah and Sylvester 2004). More recently it has been reported that the antiproliferative effects of γ-tocotrienol results, at least in part, from a reduction in Akt and NFkappaB activity in neoplastic +SA mammary epithelial cells (Shah and Sylvester 2005a). α-Tocotrienol (20 microM) seems to share some of the cytotoxic effects on cancer cells by inducing caspase-8 and caspase-3 activity (Sylvester and Shah 2005). Combined treatment with specific caspase-8 or caspase-3 inhibitors completely blocked α-tocotrienol-induced apoptosis and caspase-8 or caspase-3 activity, respectively. In contrast, α-tocotrienol treatment had no effect on caspase-9 activation, and combined treatment with a specific caspase-9 inhibitor did not block α-tocotrienol-induced apoptosis in +SA cells. α-Tocotrienol-induced caspase-8 activation and apoptosis is not mediated through death receptor activation in malignant +SA mammary epithelial cells. Tocotrienol-induced caspase-8 activation and apoptosis in malignant +SA mammary epithelial cells is not mediated through the activation of death receptors, but appears to result from the suppression of the PI3K/PDK/Akt mitogenic signaling pathway, and subsequent reduction in intracellular FLIP expression (Sylvester and Shah 2005).
Because the NF-kappaB pathway has a central role in tumorigenesis, the effect of γ-tocotrienol on the NF-kappaB pathway has been examined (Ahn et al. 2007). γ-Tocotrienol (25 μM), but not γ-tocopherol, completely abolished tumor necrosis factor alpha (TNF)-induced NF-kappaB activation. Besides TNF, γ-tocotrienol also abolished NF-kappaB activation induced by a wide range of agonists including phorbol myristate acetate, okadaic acid, lipopolysaccharide, cigarette smoke, interleukin-1beta, and epidermal growth factor. Of note, constitutive NF-kappaB activation expressed by certain tumor cells was also abrogated by γ-tocotrienol. γ–Tocotrienol blocked TNF-induced phosphorylation and degradation of IkappaBalpha through the inhibition of IkappaBalpha kinase activation, thus leading to the suppression of the phosphorylation and nuclear translocation of p65. γ-Tocotrienol also suppressed NF-kappaB-dependent reporter gene transcription induced by TNF, TNFR1, TRADD, TRAF2, TAK1, receptor-interacting protein, NIK, and IkappaBalpha kinase but not that activated by p65. Additionally, the expressions of NF-kappaB-regulated gene products associated with antiapoptosis (IAP1, IAP2, Bcl-xL, Bcl-2, cFLIP, XIAP, Bfl-1/A1, TRAF1, and Survivin), proliferation (cyclin D1, COX2, and c-Myc), invasion (MMP-9 and ICAM-1), and angiogenesis (vascular endothelial growth factor) were down-regulated by γ-tocotrienol. This correlated with potentiation of apoptosis induced by TNF, paclitaxel, and doxorubicin. Overall, although such high dose of γ-tocotrienol is not likely to be found in humans, the study provides interesting mechanistic insight. In sum, γ-tocotrienol inhibits the NF-kappaB activation pathway, leading to down-regulation of various gene products and potentiation of apoptosis (Ahn et al. 2007).
Bcl-2 family proteins tightly control apoptosis by regulating the permeabilization of the mitochondrial outer membrane and, hence, the release of cytochrome c and other proapoptotic factors. Is tocotrienol-induced apoptosis of cancer cells dependent on mitochondrial mechanisms? Incubation of MDA-MB-231cells with γ-tocotrienol causes membrane blebbing, formation of apoptotic bodies, chromatin condensation/fragmentation, and phosphatidylserine externalization (Takahashi and Loo 2004). These are all hallmarks of apoptosis. In γ-tocotrienol-treated cells, mitochondria were disrupted. Collapse of the mitochondrial membrane potential was followed by the release of mitochondrial cytochrome c. However, the expression of Bax and Bcl-2 mRNA and protein did not change. In contrast to other studies reporting that tocotrienol-induced cell death is caspase-dependent (Shah et al. 2003; Shah and Sylvester 2004), it was noted that in this model caspases were not involved in γ-tocotrienol-induced apoptosis (Takahashi and Loo 2004). In a study of +SA cells it was noted that although γ-tocotrienol induced apoptosis, it did not disrupt mitochondrial membrane potential or cause the release of mitochondrial cytochrome c into the cytoplasm. Tocotrienol-treated apoptotic +SA cells showed a paradoxical decrease in mitochondrial levels of pro-apoptotic proteins Bid, Bax, and Bad, and a corresponding increase in mitochondrial levels of anti-apoptotic proteins, Bcl-2 and Bcl-xL, suggesting that mitochondrial membrane stability and integrity might actually be enhanced for a limited period of time following acute tocotrienol exposure. The significance of this finding remains to be recognized (Shah and Sylvester 2005b).
Over the past 30 years, a relatively simple growth factor and its cognate receptor have provided seminal insights into the understanding of the genetic basis of cancer, as well as growth factor signaling. The epidermal growth factor (EGF), its cognate receptor (EGFR) and related family members have been shown to be important in normal, as well as the malignant growth of many cell types including breast cancer. EGF is a potent mitogen for normal and neoplastic mammary epithelial cells. Initial events in EGFR mitogenic-signaling are G-protein activation, stimulation of adenylyl cyclase and cyclic AMP (cAMP) production. Do the antiproliferative effects of tocotrienols associate with reduced EGF-induced G-protein and cAMP-dependent mitogenic signaling? To answer this question, preneoplastic CL-S1 mouse mammary epithelial cells were grown in culture and maintained on serum-free media containing 0–25 micro mol/L tocotrienol-rich fraction of palm oil and/or different doses of pharmacological agents that alter intracellular cAMP levels. Tocotrienol-induced effects on EGF-receptor levels of tyrosine kinase activity, as well as EGF-dependent mitogen-activated pathway kinase (MAPK) and Akt activation, were examined. It was noted that the anti-proliferative effects of tocotrienols in pre-neoplastic mammary epithelial cells do not reflect a reduction in EGF-receptor mitogenic responsiveness, but rather, result from an inhibition in early post-receptor events involved in cAMP production upstream from EGF-dependent MAPK and phosphoinositide 3-kinase/Akt mitogenic signaling (Sylvester et al. 2002).
7,12-Dimethylbenz[a]anthracene (DMBA) is a potent inducer of breast cancer in rats. The anti-tumour and anti-cholesterol effects of tocotrienols have been examined in rats treated with the chemical carcinogen DMBA, which is known to induce mammary carcinogenesis and hypercholesterolemia. DMBA induced multiple tumors on mammary glands after 6 months. Feeding of TRF (10 mg/kg body weight/day) for 6 months, isolated from rice bran oil, to DMBA-administered rats, attenuated the severity and extent of neoplastic transformation in the mammary glands. Consistently, plasma and mammary alkaline phosphatase activities increased during carcinogenesis, were significantly decreased in TRF-treated rats. TRF treatment to rats maintained low levels of glutathione S-transferase activities in liver and mammary glands, which is consistent with the anti-carcinogenic properties of TRF (Iqbal et al. 2003). Administration of DMBA also caused a significant increase of 30% in plasma total cholesterol and 111% in LDL-cholesterol levels compared with normal control levels. Feeding of TRF to rats caused a significant decline of 30% in total cholesterol and 67% in LDL-cholesterol levels compared with the DMBA-administered rats. The experimental hypercholesterolaemia caused a significant increase in enzymatic activity (23%) and protein mass (28%) of hepatic 3-hydroxy-3-methylglutaryl co-enzyme A (HMG-CoA) reductase. Consistent with TRF-mediated reduction in plasma lipid levels, enzymatic activity and protein mass of HMG-CoA reductase was significantly reduced. These observations support that TRF is a promising anti-cancer and anti-cholesterol agent in rats (Iqbal et al. 2003).
Tocotrienols act on cell proliferation in a dose-dependent manner and can induce programmed cell death in breast cancer cells. To elucidate the molecular basis of the effect of tocotrienols, MCF-7 breast cancer cells were injected into athymic nude mice. Feeding quite large amounts (1mg/d) of TRF for 20 weeks delayed the onset, incidence, and size of tumors. At autopsy, the tumor tissue was excised and cDNA array analysis was performed. Thirty out of 1176 genes were significantly affected by TRF. Ten genes were down-regulated and 20 genes up-regulated with respect to untreated animals. Expression of the interferon-inducible transmembrane protein-1 gene was significantly up-regulated in tumors excised from TRF-treated animals compared with control mice. Within the group of genes related to the immune system, CD59 glycoprotein precursor gene was up-regulated. Among the functional class of intracellular transducers/effectors/modulators, the c-myc gene was significantly down-regulated in tumors in response to TRF treatment. This work on the survey of TRF-sensitive genes in the tumor in vivo provides key insight (Nesaretnam et al. 2004).
Unlike the literature on breast cancer cells, work on prostate cancer cells investigating the effect of tocotrienol is scanty. In a model where prostate cancer was induced by injecting PC3 cells into nude BALB/c mice, the radiotherapy efficacy of prostate cancer could be increased with γ-tocotrienol (Kumar et al. 2006). When the tumors were about 5 mm in diameter, mice were injected subcutaneously with 400 mg/kg γ-tocotrienol and irradiated 24 h later at the site of the tumor with a dose of 12 Gy (60) Cobalt. The size of the tumors was reduced by almost 40%, but only in tocotrienol-treated and irradiated mice (Kumar et al. 2006). The growth-inhibitory and apoptotic effects of TRF has been tested on normal human prostate epithelial cells (PrEC), virally transformed normal human prostate epithelial cells (PZ-HPV-7), and human prostate cancer cells (LNCaP, DU145, and PC-3) (Srivastava and Gupta 2006). TRF selectively inhibited the growth of cancer cells but not of normal cells. In response to TRF, cancer cells underwent G0/G1 phase arrest and sub-G1 accumulation. Colony formation by all three prostate cancer cell lines studied was clearly arrested by TRF. The IC50 after 24h TRF treatment in LNCaP, PC-3, and DU145 cells were in the order 16.5, 17.5, and 22.0 microg/ml. Interestingly, TRF treatment resulted in significant apoptosis of cancer cells but not of normal cells (Srivastava and Gupta 2006).
Inhibition of tumor promotion by tocopherols and tocotrienols has been also examined utilizing an in vitro assay involving the activation of Epstein-Barr virus early antigen expression in Epstein-Barr virus genome-carrying human lymphoblastoid cells. γ- and δ-Tocotrienols derived from palm oil exhibited strong activity against tumor promotion by inhibiting Epstein-Barr virus early antigen expression in Raji cells induced by 12-O-tetradecanoylphorbol-13-acetate. In contrast, the corresponding tocopherols lacked this activity (Goh et al. 1994).
Consistent with observation made in other in vitro systems, tocotrienol inhibits the growth of hepatoma cells but not that of hepatocytes from healthy rat liver (Sakai et al. 2004). Consistently, tocotrienol killed murine liver cancer cells but not normal cells (Har and Keong 2005). Again, this interesting function of tocotrienol is not shared by tocopherol. Tocotrienol induced apoptosis of hepatoma cells was mediated by caspase 3 activation. In addition, tocotrienol induced caspase 8 activity. An inhibitor of caspase 8 suppressed the induction of apoptosis in hepatoma by tocotrienol. Compared to tocopherol, tocotrienol was more quickly taken up by the cancer cells suggesting that this could be one reason why tocotrienol was so effective in specifically killing the hepatoma cells (Har and Keong 2005; Sakai et al. 2004). γ-Tocotrienol has been noted to inhibit the proliferation of human hepatoma Hep3B cells at lower concentrations and shorter treatment times than α-tocotrienol. γ-Tocotrienol induces poly (ADP-ribose) polymerase (PARP) cleavage activating caspase-3. In addition, γ-tocotrienol activates caspase-8 and caspase-9 and up-regulates Bax and fragments of Bid (Sakai et al. 2005). In human hepatocellular carcinoma HepG2 cells, δ-tocotrienol exerts more significant anti-proliferative effect than α-, β-, and γ-tocotrienols. δ-Tocotrienol induces apoptosis, and also tends to induce S-phase arrest. The phase I enzyme CYP1A1 was induced by δ-tocotrienol (Wada et al. 2005).
2-Acetylaminofluorene (AAF) is a potent hepatocarcinogen. Prolonged feeding of rats with 2-acetylaminofluorene causes hepatocellular damage. Such damage is prevented by tocotrienol supplementation (Ngah et al. 1991). 2-Acetylaminofluorene significantly increased the activities of both plasma and liver microsomal γ-glutamyltranspeptidase (GGT) and liver microsomal UDP-glucuronyltransferase (UDP-GT). Tocotrienols administered together with AAF significantly decrease the activities of plasma GGT after 12 and 20 wk and liver microsomal UDP-GT after 20 wk when compared with matched controls (Ngah et al. 1991). In a scenario of stronger chemical carcinogen insult caused by 2-acetylaminofluorene in conjunction with diethylnitrosamine the effects of tocotrienol turned out to be more encouraging. In response to challenge by the chemical carcinogens, all ten rats in the group showed the presence of two grayish white nodules in the liver. Rats subjected to long-term administration of tocotrienol were protected (Rahmat et al. 1993).
The anticancer efficacy of TRF has been evaluated during diethylnitrosamine (DEN)/2-acetylaminofluorene (AAF)-induced hepatocarcinogenesis in male Sprague-Dawley rats. TRF treatment was carried out for 6 months, and was started 2 weeks before the initiation phase of hepatocarcinogenesis. Morphological examination of the livers from DEN/AAF rats showed numerous off-white patches and few small nodules, which were significantly reduced by TRF treatment. DEN/AAF caused a two-fold increase in the activity of alkaline phosphatase in the plasma as compared with normal control rats. This increase of tissue damage marker was prevented significantly by TRF treatment. Hepatic activity of glutathione S-transferase was also increased (3.5-fold) during the induction of hepatic carcinogenesis. Lipid peroxidation and low-density lipoprotein oxidation increased three-fold following initiation by DEN/AAF as compared with healthy control rats. TRF treatment to DEN/AAF-treated rats substantially decreased (62–66%) the above parameters and thus limited the action of DEN/AAF. Thus, TRF exhibited clear protective properties in this model of chemical carcinogenesis (Iqbal et al. 2004).
RKO, a poorly differentiated colon carcinoma cell line, represents a commonly used in vitro model for human colon carcinoma. RKO cells contain wild-type p53 but lack endogenous human thyroid receptor nuclear receptor (h-TRbeta1). In a dose- and time- dependent manner TRF inhibited the growth and colony formation of RKO. In addition, TRF induced WAF1/p21 which appeared to be independent of cell cycle regulation and was transcriptionally up-regulated in a p53-dependent manner. TRF treatment also resulted in alteration in Bax/Bcl2 ratio in favor of apoptosis, which was associated with the release of cytochrome c and induction of apoptotic protease-activating factor-1. Such altered expression of Bcl2 family members triggered the activation of initiator caspase-9 followed by activation of effector caspase-3. Thus, in RKO cells the pathways involved in TRF-induced apoptosis is fairly well characterized (Agarwal et al. 2004). Since the discovery that telomerase is repressed in most normal human somatic cells but strongly expressed in most human tumors, telomerase emerged as an attractive target for diagnostic, prognostic and therapeutic purposes to combat human cancer (Shay and Wright 2006). Tocotrienol inhibits telomerase activity of DLD-1 human colorectal adenocarcinoma cells in a time- and dose-dependent manner. δ-Tocotrienol demonstrated the highest inhibitory activity. Tocotrienol inhibited protein kinase C activity, resulting in down-regulation of c-myc and human telomerase reverse transcriptase (hTERT) expression, thereby reducing telomerase activity. Of note, tocopherol does not share the potent activity of tocotrienol in this regard (Shay and Wright 2006).
How much tocotrienol is needed to inhibit the increase in population of murine B16(F10) melanoma cells during a 48-h incubation by 50% (IC50)? The IC50 estimated for farnesol, the side-chain analog of the tocotrienols (50 micromol/L) falls midway between that of α-tocotrienol (110 micromol/L) and those estimated for γ- (20 micromol/L) and δ- (10 micromol/L) tocotrienol. Experimental diets were fed to weanling C57BL female mice for 10 d prior to and 28 d following the implantation of the aggressively growing and highly metastatic B16(F10) melanoma. The isomolar (116 micromol/kg diet) and the vitamin E-equivalent (928 micromol/kg diet) substitution of d-γ-tocotrienol for dl-α-tocopherol in the AIN-76A diet produced 36 and 50% retardations, respectively, in tumor growth. Thus, in this skin melanoma model both tocotrienol as well as tocopherol were significantly effective (He et al. 1997). The growth suppressive effects of γ-tocotrienol on murine B16(F10) melanoma cells have been independently reproduced (Mo and Elson 1999).
Recent studies have indentified the anti-angiogenic properties of tocotrienols. Tocotrienol, but not tocopherol, inhibited both the proliferation and tube formation of bovine aortic endothelial cells (Inokuchi et al. 2003). Consistently, in a cell culture model tocotrienol diminished VEGF-induced tube formation by human umbilical vein endothelial cells (HUVEC). Among the tocotrienol isomers investigated, δ-tocotrienol showed the highest activity (Mizushina et al. 2006). Tocotrienol, but not tocopherol, inhibited new blood vessel formation on the growing chick embryo chorioallantoic membrane. In endothelial cells, tocotrienol specifically down-regulated the expression of VEGF receptor (Nakagawa et al. 2004). Consistent results were obtained in another study where tocotrienol, but not tocopherol, inhibited the proliferation of bovine aortic endothelial cells in dose dependent manner at half-maximal concentrations in the low micromolar range. Tocotrienol also significantly inhibited the formation of networks of elongated endothelial cells within 3D collagen gels (Miyazawa et al. 2004). The anti-angiogenic properties of tocotrienol could contribute to the anti-cancer effects of tocotrienol in vivo. Further studies investigating the effects of tocotrienol on tumor growth/regression in vivo is warranted.
Neuroprotective Effects of Tocotrienol
On a concentration basis, the neuroprotective effects of nM tocotrienol represent the most potent biological function of all natural forms of vitamin E. Glutamate-toxicity is a major contributor to neurodegeneration. It includes excitotoxicity and an oxidative stress component also known as oxytosis (Schubert and Piasecki 2001; Tan et al. 2001). Murine HT hippocampal neuronal cells, lacking intrinsic excitotoxicity-pathway, have been used as a standard model to characterize the oxidant-dependent component of glutamate toxicity. In 1999, we conducted a side by side comparison of all eight forms of natural vitamin E in a model of glutamate-induced neurodegeneration of HT neural cells. In subsequent experiments it was observed that the neuroprotective property of tocotrienol applies not only to neural cell lines but also to primary cortical neurons. This line of experimentation led to an observation that eventually turned out to be the most potent function of any natural form of vitamin E on a concentration basis reported. Until then, all biological functions of vitamin E studied in vitro were observed at micromolar concentration. Our studies led to the first evidence that α-tocotrienol was the most potent neuroprotective form of vitamin E in glutamate-induced degeneration of HT4 hippocampal neurons (Sen et al. 2000). What was striking in this study was the observation that nanomolar concentrations of α-tocotrienol, not α-tocopherol, provide complete neuroprotection. At such low dose, tocotrienol was not protective against direct oxidant insult suggesting that the observed neuroprotective effects of nanomolar tocotrienol was not dependent on the widely known antioxidant property of vitamin E. That tocotrienol-dependent neuroprotection includes a significant antioxidant-independent mechanism has been now established (Khanna et al. 2006). The neuroprotective property of tocotrienol holds good not only in response to glutamate challenge but also in response to other insults such as homocysteic acid, glutathione deficiency, and linoleic acid induced oxidative stress (Khanna et al. 2006; Sen et al. 2000). It is now evident that at micromolar concentrations tocotrienol protects neural cells by virtue of its antioxidant property. At nanomolar concentrations, however, tocotrienol regulates specific neurodegenerative signaling processes.
The major tocotrienol-sensitive signaling pathways which are known to be involved in glutamate-induced neurodegeneration include c-Src and 12-lipoxygenase (Khanna et al. 2006; Khanna et al. 2003; Khanna et al. 2005b; Sen et al. 2004; Sen et al. 2000). In our initial search for signaling pathways that are sensitive to tocotrienol and play a decisive role in neurodegeneration we were led to c-Src kinase (Sen et al. 2000). c-Src and the structurally related members of the Src family are non-receptor tyrosine kinases that reside within the cell associated with cell membranes and appear to transduce signals from transmembrane receptors to the cell interior. SH2 and SH3 domains are known to play a central role in regulating the catalytic activity of src protein tyrosine kinase. High-resolution crystal structures of human Src, in their repressed state, have provided a structural explanation for how intramolecular interactions of the SH3 and SH2 domains stabilize the inactive conformation of Src (Thomas and Brugge 1997).
Our hypothesis that tocotrienol prevents neurodegeneration by regulating specific signaling processes involved in neurotoxicity led to screening for potential tocotrienol-sensitive candidate death pathways in HT4 cells. During such screening studies, inhibitors of the protein tyrosine kinase activity completely prevented glutamate-induced cell death. Herbimycin and geldanamycin potently inhibit c-Src tyrosine kinase activity (Hall et al. 1994; Yoneda et al. 1993) whereas lavendustin A is an inhibitor of extracellular growth factor receptor protein tyrosine kinase activity (Hsu et al. 1991). The observation that herbimycin and geldanamycin, but not lavendustin A prevented glutamate-induced death of HT4 neuronal cells hinted the involvement of c-Src kinase activity in the death pathway. Immunoprecipitation of tyrosine phosphorylated protein from cellular extracts confirmed that protein tyrosine phosphorylation reactions were indeed triggered by exposure of cells to elevated levels of glutamate, and that such reactions were inhibited by nanomolar concentrations of α-tocotrienol (Sen et al. 2000). These results, however, did not provide any information regarding the specific kinases involved. The involvement of c-Src kinase activity in the death pathway was verified by experiments involving the over-expression of catalytically active or inactive Src kinase. Indeed, over-expression of catalytically active Src-kinase markedly sensitized the cells to HT4 induced death. Tocotrienol treatment completely prevented glutamate-induced death even in active c-Src kinase over-expressing cells indicating that it either inhibited c-Src kinase activity or regulated one or more events upstream of c-Src kinase activation. Further evidence supporting this contention was provided by results obtained from the determination of c-Src kinase activity in HT4 cells. Glutamate treatment resulted in marked enhancement of c-Src kinase activity, and this change was completely blocked in cells treated with nanomolar amounts of α-tocotrienol. Further evidence establishing that signal transduction processes related to the cell death pathway are involved in glutamate-induced cytotoxicity was obtained from the study of ERK1 and ERK2 activation. Mitogen-activated/extracellular response kinase kinase (MEK) kinase (MEKK) is a serine-threonine kinase that regulates sequential protein phosphorylation pathways, leading to the activation of mitogen- activated protein kinases (MAPK), including members of the extracellular signal-regulated kinases (ERKs). MEKK selectively regulates signal transduction pathways that contribute to the apoptotic response (Johnson et al. 1996). When activated, p44 and p42 MAPK (ERK1 and ERK2) are phosphorylated at specific threonine and tyrosine residues. ERK has been implicated in mediating the signaling events that precede apoptosis. ERK2 plays an active role in mediating anti-IgM-induced apoptosis of B cells (Lee and Koretzky 1998). It has been also shown that H2O2 induces the activation of multiple MAPKs in oligodendrocyte progenitors and that the activation of ERK is associated with oxidant- mediated cytotoxicity (Bhat and Zhang 1999). Our studies showed that ERK1 and 2 are sensitive to elevated levels of extracellular glutamate. Rapid activation of ERK, particularly ERK2, was observed in response to glutamate treatment. Such response of ERK was completely inhibited in cells treated with α-tocotrienol suggesting that α-tocotrienol influences an early event in the glutamate-induced death pathway (Sen et al. 2000). In some cases Src kinase activity is known to be required for the activation of ERK (Aikawa et al. 1997). Thus, it is likely tocotrienol inhibits inducible ERK activation by down-regulating Src kinase activity (Sen et al. 2000).
c-Src is heavily expressed in the brain (Soriano et al. 1991) and in human neural tissues (Pyper and Bolen 1989). Differentiating rodent neurons are known to express high levels of c-Src. In neurons and astrocytes, c-Src is present at 15–20 times higher levels than that found in fibroblasts. The specific activity of the c-Src protein from neuronal cultures is 6–12-times higher than that from the astrocyte cultures suggesting a key function of this protein in neurons (Brugge et al. 1985). Initially, c-Src was identified as being important in growth cone-mediated neurite extension and synaptic plasticity (Maness et al. 1988) and in neuronal differentiation (Ingraham et al. 1989). Targeted disruption of c-Src, however, did not cause any abnormality in the brain (Soriano et al. 1991). Our pursuit for the neuroprotective mechanisms of tocotrienols led to the first evidence demonstrating that rapid c-Src activation (Khanna et al. 2002; Sen et al. 2000) plays a central role in executing neurodegeneration. Consistently, it was demonstrated in a subsequent report that Src deficiency or blockade of Src activity in mice provides cerebral protection following stroke (Paul et al. 2001). Further support of our claim that c-Src is a key player in neurodegeneration is provided by observation that the Src family kinase-inhibitor PP2 reduces focal ischemic brain injury (Lennmyr et al. 2004). Our observation that tocotrienol-dependent inhibition of c-Src is beneficial for neuroprotection has now been extended to the heart. A recent study showed that c-Src mediates post-ischemic cardiac injury and dysfunction. Tocotrienol supplementation inhibited c-Src activation and protected the heart (Das et al. 2005). Many intracellular pathways can be stimulated upon Src activation, and a variety of cellular consequences can result. High c-Src is tightly associated with carcinogenesis. c-Src inhibitors are being actively studied for cancer therapy (Alper and Bowden 2005; Ishizawar and Parsons 2004; Lau 2005; Shupnik 2004). Based on the inducible c-Src inhibitory properties of tocotrienol one may postulate that tocotrienol has anti-cancer properties. The anti-cancer properties of tocotrienol have been discussed in a separate section below.
GSH is the major cellular thiol present in mammalian cells and is critical for maintenance of redox homeostasis (Sun et al. 2006). GSH is a key survival factor in cells of the nervous system and lowered [GSH]i is one of the early markers of neurotoxicity induced by a variety of agonists (Bains and Shaw 1997; Dringen et al. 2000). We observed that α-tocotrienol clearly protects primary cortical neurons against a number of GSH-lowering neurotoxins (Khanna et al. 2003). Of interest, the neurons survived even in the face of GSH loss. These observations led to the hypothesis that loss of [GSH]i alone is not lethal (Khanna et al. 2003). Given that pro-GSH agents are known to be neuroprotective in a variety of scenarios (Bains and Shaw 1997; Han et al. 1997; Schulz et al. 2000) it becomes reasonable to hypothesize that glutamate-induced lowering of [GSH]i triggers downstream responses that execute cell death. Our works led to the identification of 12-lipoxygenase (12-Lox) as a key tocotrienol-sensitive mediator of neurodegeneration (Khanna et al. 2003). Specific inhibition of 12-Lox by BL15 protected neurons from glutamate-induced degeneration although [GSH]i is compromised by 80%. Similar protective effects of BL15 were noted when BSO, a specific inhibitor of GSH synthesis, was used as the agonist. Importantly, neurons isolated from mice lacking the 12- lipoxygenase gene were observed to be resistant to glutamate-induced loss of viability (Khanna et al. 2003). This key piece of evidence established that indeed 12-Lox represents a critical checkpoint in glutamate-induced neurodegeneration.
Understanding of the intracellular regulation of 12-lipoxygenase requires knowledge of the distribution of both enzyme protein and its activity. For example, in human erythroleukemia cells, the membrane fraction contains about 90% of the total cellular 12-Lox activity, whereas only 10% of 12-Lox activity resides in the cytosol. However, the majority of cellular 12-Lox protein is found in the cytosol (Hagmann et al. 1993). Upon activation, 12-Lox may translocate to the membrane (Hagmann et al. 1993). Consistently, we have observed the decreased presence of 12-Lox in the cytosol and increased presence in the membrane of glutamate-treated cells. For 5-lipoxygenase, both catalytic function and translocation of the enzyme from the cytosol to the membrane are known to be regulated by tyrosine kinases (Lepley et al. 1996). Recently we have noted that 12-Lox is subject to rapid tyrosine phosphorylation in neuronal cells challenged with glutamate or GSH-lowering agents. Such rapid phosphorylation coincides with the timeline of c-Src activation (Khanna et al. 2005b). Inhibitors of c-Src abrogated inducible 12-Lox tyrosine phosphorylation supporting the notion that c-Src may directly phosphorylate 12-Lox in challenged neurons. To test this hypothesis we utilized genetic approaches of over-expressing kinase-active, kinase-dead or dominant negative c-Src in neuronal cells. Findings from cell biology studies as well as from the study of c-Src and 12-Lox in cell-free systems indicate that in response to challenge by glutamate or GSH-lowering agents, c-Src is rapidly activated and phosphorylates 12-Lox.(Khanna et al. 2005b).
Neurons and the brain are rich in arachidonic acid (AA; 20:4ω-6). Massive amounts of AA are released from the membranes in response to brain ischemia or trauma (Bazan 1976; Bazan 1970; Bazan 1971a; Bazan 1971b; Bazan and Rakowski 1970). Subsequent work has established that AA and its metabolites may be neurotoxic. There are three major pathways of AA metabolism: lipoxygenases, cycloxygenases and cytochrome P450. The cycloxygenase pathway has been preliminarily ruled out from being a contributor to neurodegeneration (Kwon et al. 2005). In the lipoxygenase pathway, metabolites of 12-Lox seem to be the major metabolite of arachidonic acid in the brain (Adesuyi et al. 1985; Carlen et al. 1994) as well as in cultured cortical neurons(Ishizaki and Murota 1991; Miyamoto et al. 1987a; Miyamoto et al. 1987b). Lipoxygenases, mainly 5-, 12- and 15-Lox, are named for their ability to insert molecular oxygen at the 5, 12, or 15-carbon atom of arachidonic acid forming a distinct hydroperoxy-eicosatetraenoic (HPETE) acid (Yamamoto 1992). 12-Lox produces 12(S)-HPETE which is further metabolized into four distinct products: an alcohol [12(S)-HETE], a ketone (12-keto-eicosatetraenoic acid), or two epoxy alcohols (hepoxilin A3 and B3). Immunohistochemical studies revealed the occurrence of 12-Lox in neurons; particularly in hippocampus, striatum, olivary nucleus, as well as in glial and in cerebral endothelial cells (Nishiyama et al. 1992; Nishiyama et al. 1993). Using immature cortical neurons and HT cells, it has been shown that a decrease in [GSH]i triggers the activation of neuronal 12-Lox, which leads to the production of peroxides, the influx of Ca2+, and ultimately to cell death (Li et al. 1997; Tan et al. 2001). The 12-Lox metabolite 12-HPETE proved to be capable of causing cell death (Gu et al. 2001). Inhibition of 12-Lox protected cortical neurons from β-amyloid induced toxicity (Lebeau et al. 2001). Intracellular calcium chelation delayed cell death by lipoxygenase-mediated free radicals in mouse cortical cultures (Wie et al. 2001). In sum, 12-Lox poses clear threat to neuronal survival especially under GSH-deficient conditions.
Lipoxygenase activity is sensitive to vitamin E. α-Tocopherol strongly inhibits purified 5-Lox with a IC50 of 5 μM. The inhibition is independent of the antioxidant property of tocopherol. Tryptic digestion and peptide mapping of the 5-Lox-tocopherol complex indicated that tocopherol binds strongly to a single peptide (Reddanna et al. 1985). Another study reported inhibition of 15-Lox by tocopherol via specific interaction with the enzyme protein (Grossman and Waksman 1984). Of interest, inhibitors specific for cycloxygenase or 5-Lox are not effective in protecting neuronal cells against glutamate-induced death suggesting a specific role of 12-Lox in glutamate-induced death (Khanna et al. 2003; Khanna et al. 2005b). Our studies addressing the effect of α-tocotrienol on pure 12-Lox indicate that α-tocotrienol directly interacts with the enzyme to suppress arachidonic acid metabolism. In silico studies examining possible docking sites of α-tocotrienol to 12-Lox support the presence of a α-tocotrienol binding solvent cavity close to the active site. Previously it has been demonstrated in 15-Lox that COOH terminal of arachidonic acid enters this solvent cavity while accessing the catalytic site (Borngraber et al. 1999). It is therefore plausible that the binding position of α-tocotrienol prevents access of the natural substrate arachidonic acid to the active site of 12-Lox (Khanna et al. 2003). Does 12-Lox have a tangible impact on neurodegenerative processes in vivo? In 1992, it was reported that a mixed lipoxygenase/cyclooxygenase inhibitor SK&F 105809 reduced cerebral edema after closed head injury in rat (Shohami et al. 1992). We noted that 12-Lox, but not 5-Lox (Kitagawa et al. 2004), deficient mice were significantly protected against stroke-related injury of the brain (Khanna et al. 2005b). The case for 12-Lox as an important mediator of neurodegeneration in vivo is gaining additional support from independent studies (Musiek et al. 2006). 12-Lox has been also implicated in the pathogenesis of Alzheimer’s (Yao et al. 2005). α-Tocotrienol is capable of resisting neurodegeneration in vivo by opposing the c-Src and 12-Lox pathways.
Conclusion
Members of the natural vitamin E family possess overlapping as well as unique functional properties. Our knowledge about the non-α-tocopherol isoforms of natural vitamin E is scanty. Among the natural vitamin E molecules, d-α-tocopherol (RRR-α-tocopherol) has the highest bioavailability and is the standard against which all the others are compared. However, it is only one out of eight natural forms of vitamin E. Interestingly, symptoms caused by α-tocopherol deficiency can be alleviated by tocotrienols. Thus, tocotrienols may be viewed as being members of the natural vitamin E family not only structurally but also functionally. Disappointments with outcomes-based studies investigating α-tocopherol (Friedrich 2004; Greenberg 2005) need to be cautiously handled recognizing the untapped opportunities offered by the other forms of natural vitamin E. Recently it has been suggested that the safe dose of various tocotrienols for human consumption is 200–1000 mg/d (Yu et al. 2006). Vitamin E represents one of the most fascinating natural resources that have the potential to influence a broad range of mechanisms underlying human health and disease. The current state of knowledge warrants strategic investment into the lesser known forms of vitamin E with emphasis on uncovering the specific conditions that govern the function of vitamin E molecules in vivo. Outcome studies designed in light of such information have the clear potential of returning dividends that are more lucrative than findings of the current clinical outcomes studies.
Acknowledgments
Tocotrienol research in the laboratory is supported by NIH RO1NS42617. Tocotrienol used in the laboratory was natural palm-oil derived (either purified or Tocomin® SupraBio™) and provided as gift from Carotech Inc., NJ. Studies on human tissue distribution of orally fed tocotrienol are supported by the Malaysian Palm Oil Board.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adachi H, Ishii N. Effects of tocotrienols on life span and protein carbonylation in Caenorhabditis elegans. Journals of Gerontology Series A, Biological Sciences & Medical Sciences. 2000;55(6):B280–5. doi: 10.1093/gerona/55.6.b280. [DOI] [PubMed] [Google Scholar]
- Adachi K, Miki M, Tamai H, Tokuda M, Mino M. Adipose tissues and vitamin E. J Nutr Sci Vitaminol (Tokyo) 1990;36(4):327–37. doi: 10.3177/jnsv.36.4-supplementi_327. [DOI] [PubMed] [Google Scholar]
- Adesuyi SA, Cockrell CS, Gamache DA, Ellis EF. Lipoxygenase metabolism of arachidonic acid in brain. J Neurochem. 1985;45(3):770–6. doi: 10.1111/j.1471-4159.1985.tb04059.x. [DOI] [PubMed] [Google Scholar]
- Agarwal MK, Agarwal ML, Athar M, Gupta S. Tocotrienol-rich fraction of palm oil activates p53, modulates Bax/Bcl2 ratio and induces apoptosis independent of cell cycle association. Cell Cycle. 2004;3(2):205–11. doi: 10.4161/cc.3.2.654. [DOI] [PubMed] [Google Scholar]
- Ahn KS, Sethi G, Krishnan K, Aggarwal BB. Gamma-tocotrienol inhibits nuclear factor-kappaB signaling pathway through inhibition of receptor-interacting protein and TAK1 leading to suppression of antiapoptotic gene products and potentiation of apoptosis. J Biol Chem. 2007;282(1):809–20. doi: 10.1074/jbc.M610028200. [DOI] [PubMed] [Google Scholar]
- Aikawa R, Komuro I, Yamazaki T, Zou Y, Kudoh S, Tanaka M, Shiojima I, Hiroi Y, Yazaki Y. Oxidative stress activates extracellular signal-regulated kinases through Src and Ras in cultured cardiac myocytes of neonatal rats. Journal of Clinical Investigation. 1997;100(7):1813–21. doi: 10.1172/JCI119709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alper O, Bowden ET. Novel insights into c-Src. Curr Pharm Des. 2005;11(9):1119–30. doi: 10.2174/1381612053507576. [DOI] [PubMed] [Google Scholar]
- Araya H, Tomita M, Hayashi M. The novel formulation design of self-emulsifying drug delivery systems (SEDDS) type O/W microemulsion III: the permeation mechanism of a poorly water soluble drug entrapped O/W microemulsion in rat isolated intestinal membrane by the Ussing chamber method. Drug Metab Pharmacokinet. 2006;21(1):45–53. doi: 10.2133/dmpk.21.45. [DOI] [PubMed] [Google Scholar]
- Aten RF, Kolodecik TR, Behrman HR. Ovarian vitamin E accumulation: evidence for a role of lipoproteins. Endocrinology. 1994;135(2):533–9. doi: 10.1210/endo.135.2.8033800. [DOI] [PubMed] [Google Scholar]
- Atkinson J. Chemical investigations of tocotrienols: isotope substitution, fluorophores and a curious curve. In: Nesaretnam K, editor. Kuching, Malaysia: COSTAM; 2006. p. 22. [Google Scholar]
- Bains JS, Shaw CA. Neurodegenerative disorders in humans: the role of glutathione in oxidative stress-mediated neuronal death. Brain Research - Brain Research Reviews. 1997;25(3):335–58. doi: 10.1016/s0165-0173(97)00045-3. [DOI] [PubMed] [Google Scholar]
- Bazan NG. Free arachidonic acid and other lipids in the nervous system during early ischemia and after electroshock. Adv Exp Med Biol. 1976;72:317–35. doi: 10.1007/978-1-4684-0955-0_26. [DOI] [PubMed] [Google Scholar]
- Bazan NG., Jr Effects of ischemia and electroconvulsive shock on free fatty acid pool in the brain. Biochim Biophys Acta. 1970;218(1):1–10. doi: 10.1016/0005-2760(70)90086-x. [DOI] [PubMed] [Google Scholar]
- Bazan NG., Jr Changes in free fatty acids of brain by drug-induced convulsions, electroshock and anaesthesia. J Neurochem. 1971a;18(8):1379–85. doi: 10.1111/j.1471-4159.1971.tb00002.x. [DOI] [PubMed] [Google Scholar]
- Bazan NG., Jr Phospholipases A 1 and A 2 in brain subcellular fractions. Acta Physiol Lat Am. 1971b;21(2):101–6. [PubMed] [Google Scholar]
- Bazan NG, Jr, Rakowski H. Increased levels of brain free fatty acids after electroconvulsive shock. Life Sci. 1970;9(9):501–7. doi: 10.1016/0024-3205(70)90205-5. [DOI] [PubMed] [Google Scholar]
- Bhat NR, Zhang P. Hydrogen peroxide activation of multiple mitogen-activated protein kinases in an oligodendrocyte cell line: role of extracellular signal-regulated kinase in hydrogen peroxide-induced cell death. Journal of Neurochemistry. 1999;72(1):112–9. doi: 10.1046/j.1471-4159.1999.0720112.x. [DOI] [PubMed] [Google Scholar]
- Blatt DH, Leonard SW, Traber MG. Vitamin E kinetics and the function of tocopherol regulatory proteins. Nutrition. 2001;17(10):799–805. doi: 10.1016/s0899-9007(01)00637-2. [DOI] [PubMed] [Google Scholar]
- Borngraber S, Browner M, Gillmor S, Gerth C, Anton M, Fletterick R, Kuhn H. Shape and specificity in mammalian 15-lipoxygenase active site. The functional interplay of sequence determinants for the reaction specificity. J Biol Chem. 1999;274(52):37345–50. doi: 10.1074/jbc.274.52.37345. [DOI] [PubMed] [Google Scholar]
- Boscoboinik D, Szewczyk A, Hensey C, Azzi A. Inhibition of cell proliferation by alpha-tocopherol. Role of protein kinase C. J Biol Chem. 1991;266(10):6188–94. [PubMed] [Google Scholar]
- Brugge JS, Cotton PC, Queral AE, Barrett JN, Nonner D, Keane RW. Neurones express high levels of a structurally modified, activated form of pp60c-src. Nature. 1985;316(6028):554–7. doi: 10.1038/316554a0. [DOI] [PubMed] [Google Scholar]
- Bryngelsson S, Dimberg LH, Kamal-Eldin A. Effects of commercial processing on levels of antioxidants in oats (Avena sativa L.) J Agric Food Chem. 2002;50(7):1890–6. doi: 10.1021/jf011222z. [DOI] [PubMed] [Google Scholar]
- Cahoon EB, Hall SE, Ripp KG, Ganzke TS, Hitz WD, Coughlan SJ. Metabolic redesign of vitamin E biosynthesis in plants for tocotrienol production and increased antioxidant content. Nat Biotechnol. 2003;21(9):1082–7. doi: 10.1038/nbt853. [DOI] [PubMed] [Google Scholar]
- Carlen PL, Gurevich N, Zhang L, Wu PH, Reynaud D, Pace-Asciak CR. Formation and electrophysiological actions of the arachidonic acid metabolites, hepoxilins, at nanomolar concentrations in rat hippocampal slices. Neuroscience. 1994;58(3):493–502. doi: 10.1016/0306-4522(94)90075-2. [DOI] [PubMed] [Google Scholar]
- Chen CW, Cheng HH. A rice bran oil diet increases LDL-receptor and HMG-CoA reductase mRNA expressions and insulin sensitivity in rats with streptozotocin/nicotinamide-induced type 2 diabetes. J Nutr. 2006;136(6):1472–6. doi: 10.1093/jn/136.6.1472. [DOI] [PubMed] [Google Scholar]
- Chow CK, Draper HH. Isolation of gamma-tocotrienol dimers from Hevea latex. Biochemistry. 1970;9(2):445–50. doi: 10.1021/bi00804a036. [DOI] [PubMed] [Google Scholar]
- Das S, Powell SR, Wang P, Divald A, Nesaretnam K, Tosaki A, Cordis GA, Maulik N, Das DK. Cardioprotection with palm tocotrienol: antioxidant activity of tocotrienol is linked with its ability to stabilize proteasomes. Am J Physiol Heart Circ Physiol. 2005;289(1):H361–7. doi: 10.1152/ajpheart.01285.2004. [DOI] [PubMed] [Google Scholar]
- Dietrich M, Traber MG, Jacques PF, Cross CE, Hu Y, Block G. Does {gamma}-Tocopherol Play a Role in the Primary Prevention of Heart Disease and Cancer? A Review. J Am Coll Nutr. 2006;25(4):292–9. doi: 10.1080/07315724.2006.10719538. [DOI] [PubMed] [Google Scholar]
- Dormann P. Functional diversity of tocochromanols in plants. Planta. 2007;225(2):269–76. doi: 10.1007/s00425-006-0438-2. [DOI] [PubMed] [Google Scholar]
- Dringen R, Gutterer JM, Hirrlinger J. Glutathione metabolism in brain metabolic interaction between astrocytes and neurons in the defense against reactive oxygen species. European Journal of Biochemistry. 2000;267(16):4912–6. doi: 10.1046/j.1432-1327.2000.01597.x. [DOI] [PubMed] [Google Scholar]
- Elson CE. Tropical oils: nutritional and scientific issues. Critical Reviews in Food Science & Nutrition. 1992;31(1–2):79–102. doi: 10.1080/10408399209527562. [DOI] [PubMed] [Google Scholar]
- Elson CE, Qureshi AA. Coupling the cholesterol- and tumor-suppressive actions of palm oil to the impact of its minor constituents on 3-hydroxy-3-methylglutaryl coenzyme A reductase activity. Prostaglandins Leukotrienes & Essential Fatty Acids. 1995;52(2–3):205–7. doi: 10.1016/0952-3278(95)90024-1. [DOI] [PubMed] [Google Scholar]
- Friedrich MJ. To “E” or not to “E,” vitamin E’s role in health and disease is the question. Jama. 2004;292(6):671–3. doi: 10.1001/jama.292.6.671. [DOI] [PubMed] [Google Scholar]
- Gao P, Morozowich W. Development of supersaturatable self-emulsifying drug delivery system formulations for improving the oral absorption of poorly soluble drugs. Expert Opin Drug Deliv. 2006;3(1):97–110. doi: 10.1517/17425247.3.1.97. [DOI] [PubMed] [Google Scholar]
- Garry PJ, Hunt WC, Bandrofchak JL, VanderJagt D, Goodwin JS. Vitamin A intake and plasma retinol levels in healthy elderly men and women. Am J Clin Nutr. 1987;46(6):989–94. doi: 10.1093/ajcn/46.6.989. [DOI] [PubMed] [Google Scholar]
- Goh SH, Hew NF, Norhanom AW, Yadav M. Inhibition of tumour promotion by various palm-oil tocotrienols. International Journal of Cancer. 1994;57(4):529–31. doi: 10.1002/ijc.2910570415. [DOI] [PubMed] [Google Scholar]
- Gould MN, Haag JD, Kennan WS, Tanner MA, Elson CE. A comparison of tocopherol and tocotrienol for the chemoprevention of chemically induced rat mammary tumors. American Journal of Clinical Nutrition. 1991;53(4 Suppl):1068S–1070S. doi: 10.1093/ajcn/53.4.1068S. [DOI] [PubMed] [Google Scholar]
- Greenberg ER. Vitamin E supplements: good in theory, but is the theory good? Ann Intern Med. 2005;142(1):75–6. doi: 10.7326/0003-4819-142-1-200501040-00112. [DOI] [PubMed] [Google Scholar]
- Grossman S, Waksman EG. New aspects of the inhibition of soybean lipoxygenase by alpha-tocopherol. Evidence for the existence of a specific complex. International Journal of Biochemistry. 1984;16(3):281–9. doi: 10.1016/0020-711x(84)90101-0. [DOI] [PubMed] [Google Scholar]
- Gu J, Liu Y, Wen Y, Natarajan R, Lanting L, Nadler JL. Evidence that increased 12-lipoxygenase activity induces apoptosis in fibroblasts. J Cell Physiol. 2001;186(3):357–65. doi: 10.1002/1097-4652(200103)186:3<357::AID-JCP1034>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- Guthrie N, Gapor A, Chambers AF, Carroll KK. Inhibition of proliferation of estrogen receptor-negative MDA-MB-435 and -positive MCF-7 human breast cancer cells by palm oil tocotrienols and tamoxifen, alone and in combination. Journal of Nutrition. 1997;127(3):544S–548S. doi: 10.1093/jn/127.3.544S. [DOI] [PubMed] [Google Scholar]
- Hagmann W, Kagawa D, Renaud C, Honn KV. Activity and protein distribution of 12-lipoxygenase in HEL cells: induction of membrane-association by phorbol ester TPA, modulation of activity by glutathione and 13-HPODE, and Ca(2+)-dependent translocation to membranes. Prostaglandins. 1993;46(6):471–7. doi: 10.1016/0090-6980(93)90066-g. [DOI] [PubMed] [Google Scholar]
- Hall TJ, Schaeublin M, Missbach M. Evidence that c-src is involved in the process of osteoclastic bone resorption. Biochem Biophys Res Commun. 1994;199(3):1237–44. doi: 10.1006/bbrc.1994.1363. [DOI] [PubMed] [Google Scholar]
- Han D, Sen CK, Roy S, Kobayashi MS, Tritschler HJ, Packer L. Protection against glutamate-induced cytotoxicity in C6 glial cells by thiol antioxidants. American Journal of Physiology. 1997;273(5 Pt 2):R1771–8. doi: 10.1152/ajpregu.1997.273.5.R1771. [DOI] [PubMed] [Google Scholar]
- Har CH, Keong CK. Effects of tocotrienols on cell viability and apoptosis in normal murine liver cells (BNL CL.2) and liver cancer cells (BNL 1ME A.7R.1), in vitro. Asia Pac J Clin Nutr. 2005;14(4):374–80. [PubMed] [Google Scholar]
- He L, Mo H, Hadisusilo S, Qureshi AA, Elson CE. Isoprenoids suppress the growth of murine B16 melanomas in vitro and in vivo. Journal of Nutrition. 1997;127(5):668–74. doi: 10.1093/jn/127.5.668. [DOI] [PubMed] [Google Scholar]
- Hensley K, Benaksas EJ, Bolli R, Comp P, Grammas P, Hamdheydari L, Mou S, Pye QN, Stoddard MF, Wallis G, et al. New perspectives on vitamin E: gamma-tocopherol and carboxyelthylhydroxychroman metabolites in biology and medicine. Free Radic Biol Med. 2004;36(1):1–15. doi: 10.1016/j.freeradbiomed.2003.10.009. [DOI] [PubMed] [Google Scholar]
- Ho D, Yuen KH, Yap SP. Drug delivery system:formulation for fat-soluble drugs. (US patent 6,596,306). United States patent 6,596,306 2003
- Hong JY, Kim JK, Song YK, Park JS, Kim CK. A new self-emulsifying formulation of itraconazole with improved dissolution and oral absorption. J Control Release. 2006;110(2):332–8. doi: 10.1016/j.jconrel.2005.10.002. [DOI] [PubMed] [Google Scholar]
- Horvath G, Wessjohann L, Bigirimana J, Jansen M, Guisez Y, Caubergs R, Horemans N. Differential distribution of tocopherols and tocotrienols in photosynthetic and non-photosynthetic tissues. Phytochemistry. 2006;67(12):1185–95. doi: 10.1016/j.phytochem.2006.04.004. [DOI] [PubMed] [Google Scholar]
- Hosomi A, Arita M, Sato Y, Kiyose C, Ueda T, Igarashi O, Arai H, Inoue K. Affinity for alpha-tocopherol transfer protein as a determinant of the biological activities of vitamin E analogs. FEBS Letters. 1997;409(1):105–8. doi: 10.1016/s0014-5793(97)00499-7. [DOI] [PubMed] [Google Scholar]
- Hsu CY, Persons PE, Spada AP, Bednar RA, Levitzki A, Zilberstein A. Kinetic analysis of the inhibition of the epidermal growth factor receptor tyrosine kinase by Lavendustin-A and its analogue. J Biol Chem. 1991;266(31):21105–12. [PubMed] [Google Scholar]
- Ingraham CA, Cox ME, Ward DC, Fults DW, Maness PF. c-src and other proto-oncogenes implicated in neuronal differentiation. Mol Chem Neuropathol. 1989;10(1):1–14. doi: 10.1007/BF02969481. [DOI] [PubMed] [Google Scholar]
- Inokuchi H, Hirokane H, Tsuzuki T, Nakagawa K, Igarashi M, Miyazawa T. Anti-angiogenic activity of tocotrienol. Biosci Biotechnol Biochem. 2003;67(7):1623–7. doi: 10.1271/bbb.67.1623. [DOI] [PubMed] [Google Scholar]
- Iqbal J, Minhajuddin M, Beg ZH. Suppression of 7,12-dimethylbenz[alpha]anthracene-induced carcinogenesis and hypercholesterolaemia in rats by tocotrienol-rich fraction isolated from rice bran oil. Eur J Cancer Prev. 2003;12(6):447–53. doi: 10.1097/00008469-200312000-00002. [DOI] [PubMed] [Google Scholar]
- Iqbal J, Minhajuddin M, Beg ZH. Suppression of diethylnitrosamine and 2-acetylaminofluorene-induced hepatocarcinogenesis in rats by tocotrienol-rich fraction isolated from rice bran oil. Eur J Cancer Prev. 2004;13(6):515–20. doi: 10.1097/00008469-200412000-00009. [DOI] [PubMed] [Google Scholar]
- Ishizaki Y, Murota S. Arachidonic acid metabolism in cultured astrocytes: presence of 12-lipoxygenase activity in the intact cells. Neurosci Lett. 1991;131(2):149–52. doi: 10.1016/0304-3940(91)90600-x. [DOI] [PubMed] [Google Scholar]
- Ishizawar R, Parsons SJ. c-Src and cooperating partners in human cancer. Cancer Cell. 2004;6(3):209–14. doi: 10.1016/j.ccr.2004.09.001. [DOI] [PubMed] [Google Scholar]
- Jishage K, Arita M, Igarashi K, Iwata T, Watanabe M, Ogawa M, Ueda O, Kamada N, Inoue K, Arai H, et al. Alpha-tocopherol transfer protein is important for the normal development of placental labyrinthine trophoblasts in mice. Journal of Biological Chemistry. 2001;276(3):1669–72. doi: 10.1074/jbc.C000676200. [DOI] [PubMed] [Google Scholar]
- Johnson NL, Gardner AM, Diener KM, Lange-Carter CA, Gleavy J, Jarpe MB, Minden A, Karin M, Zon LI, Johnson GL. Signal transduction pathways regulated by mitogen-activated/extracellular response kinase kinase kinase induce cell death. J Biol Chem. 1996;271(6):3229–37. doi: 10.1074/jbc.271.6.3229. [DOI] [PubMed] [Google Scholar]
- Kaempf-Rotzoll DE, Traber MG, Arai H. Vitamin E and transfer proteins. Current Opinion in Lipidology. 2003;14(3):249–54. doi: 10.1097/00041433-200306000-00004. [DOI] [PubMed] [Google Scholar]
- Khanna S, Patel V, Rink C, Roy S, Sen CK. Delivery of orally supplemented alpha-tocotrienol to vital organs of rats and tocopherol-transport protein deficient mice. Free Radic Biol Med. 2005a;39(10):1310–9. doi: 10.1016/j.freeradbiomed.2005.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khanna S, Roy S, Parinandi NL, Maurer M, Sen CK. Characterization of the potent neuroprotective properties of the natural vitamin E alpha-tocotrienol. J Neurochem. 2006 doi: 10.1111/j.1471-4159.2006.04000.x. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khanna S, Roy S, Ryu H, Bahadduri P, Swaan PW, Ratan RR, Sen CK. Molecular basis of vitamin E action: tocotrienol modulates 12-lipoxygenase, a key mediator of glutamate-induced neurodegeneration. J Biol Chem. 2003;278(44):43508–15. doi: 10.1074/jbc.M307075200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khanna S, Roy S, Slivka A, Craft TK, Chaki S, Rink C, Notestine MA, DeVries AC, Parinandi NL, Sen CK. Neuroprotective properties of the natural vitamin E alpha-tocotrienol. Stroke. 2005b;36(10):2258–64. doi: 10.1161/01.STR.0000181082.70763.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khanna S, Venojarvi M, Roy S, Sen CK. Glutamate-induced c-Src activation in neuronal cells. Methods Enzymol. 2002;352:191–8. doi: 10.1016/s0076-6879(02)52019-x. [DOI] [PubMed] [Google Scholar]
- Khosla P, Patel V, Whinter JM, Khanna S, Rakhkovskaya M, Roy S, Sen CK. Postprandial levels of the natural vitamin e tocotrienol in human circulation. Antioxid Redox Signal. 2006;8(5–6):1059–68. doi: 10.1089/ars.2006.8.1059. [DOI] [PubMed] [Google Scholar]
- Kitagawa K, Matsumoto M, Hori M. Cerebral ischemia in 5-lipoxygenase knockout mice. Brain Res. 2004;1004(1–2):198–202. doi: 10.1016/j.brainres.2004.01.018. [DOI] [PubMed] [Google Scholar]
- Komiyama K, Iizuka K, Yamaoka M, Watanabe H, Tsuchiya N, Umezawa I. Studies on the biological activity of tocotrienols. Chemical & Pharmaceutical Bulletin. 1989;37(5):1369–71. doi: 10.1248/cpb.37.1369. [DOI] [PubMed] [Google Scholar]
- Kumar KS, Raghavan M, Hieber K, Ege C, Mog S, Parra N, Hildabrand A, Singh V, Srinivasan V, Toles R, et al. Preferential radiation sensitization of prostate cancer in nude mice by nutraceutical antioxidant gamma-tocotrienol. Life Sci. 2006;78(18):2099–104. doi: 10.1016/j.lfs.2005.12.005. [DOI] [PubMed] [Google Scholar]
- Kwon KJ, Jung YS, Lee SH, Moon CH, Baik EJ. Arachidonic acid induces neuronal death through lipoxygenase and cytochrome P450 rather than cyclooxygenase. J Neurosci Res. 2005;81(1):73–84. doi: 10.1002/jnr.20520. [DOI] [PubMed] [Google Scholar]
- Lau AF. c-Src: bridging the gap between phosphorylation- and acidification-induced gap junction channel closure. Sci STKE 2005. 2005;(291):pe33. doi: 10.1126/stke.2912005pe33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebeau A, Esclaire F, Rostene W, Pelaprat D. Baicalein protects cortical neurons from beta-amyloid (25–35) induced toxicity. Neuroreport. 2001;12(10):2199–202. doi: 10.1097/00001756-200107200-00031. [DOI] [PubMed] [Google Scholar]
- Lee JR, Koretzky GA. Extracellular signal-regulated kinase-2, but not c-Jun NH2-terminal kinase, activation correlates with surface IgM-mediated apoptosis in the WEHI 231 B cell line. J Immunol. 1998;161(4):1637–44. [PubMed] [Google Scholar]
- Lennmyr F, Ericsson A, Gerwins P, Akterin S, Ahlstrom H, Terent A. Src family kinase-inhibitor PP2 reduces focal ischemic brain injury. Acta Neurol Scand. 2004;110(3):175–9. doi: 10.1111/j.1600-0404.2004.00306.x. [DOI] [PubMed] [Google Scholar]
- Leonard SW, Paterson E, Atkinson JK, Ramakrishnan R, Cross CE, Traber MG. Studies in humans using deuterium-labeled alpha- and gamma-tocopherols demonstrate faster plasma gamma-tocopherol disappearance and greater gamma-metabolite production. Free Radic Biol Med. 2005;38(7):857–66. doi: 10.1016/j.freeradbiomed.2004.12.001. [DOI] [PubMed] [Google Scholar]
- Lepley RA, Muskardin DT, Fitzpatrick FA. Tyrosine kinase activity modulates catalysis and translocation of cellular 5-lipoxygenase. Journal of Biological Chemistry. 1996;271(11):6179–84. doi: 10.1074/jbc.271.11.6179. [DOI] [PubMed] [Google Scholar]
- Li Y, Maher P, Schubert D. A role for 12-lipoxygenase in nerve cell death caused by glutathione depletion. Neuron. 1997;19(2):453–63. doi: 10.1016/s0896-6273(00)80953-8. [DOI] [PubMed] [Google Scholar]
- Lodge JK, Hall WL, Jeanes YM, Proteggente AR. Physiological factors influencing vitamin e biokinetics. Ann N Y Acad Sci. 2004;1031:60–73. doi: 10.1196/annals.1331.006. [DOI] [PubMed] [Google Scholar]
- Maness PF, Aubry M, Shores CG, Frame L, Pfenninger KH. c-src gene product in developing rat brain is enriched in nerve growth cone membranes. Proc Natl Acad Sci U S A. 1988;85(14):5001–5. doi: 10.1073/pnas.85.14.5001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martirosyan DM, Miroshnichenko LA, Kulakova SN, Pogojeva AV, Zoloedov VI. Amaranth Oil Application for Coronary Heart Disease and Hypertension. Lipids Health Dis. 2007;6(1):1. doi: 10.1186/1476-511X-6-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIntyre BS, Briski KP, Gapor A, Sylvester PW. Antiproliferative and apoptotic effects of tocopherols and tocotrienols on preneoplastic and neoplastic mouse mammary epithelial cells. Proc Soc Exp Biol Med. 2000a;224(4):292–301. doi: 10.1046/j.1525-1373.2000.22434.x. [DOI] [PubMed] [Google Scholar]
- McIntyre BS, Briski KP, Tirmenstein MA, Fariss MW, Gapor A, Sylvester PW. Antiproliferative and apoptotic effects of tocopherols and tocotrienols on normal mouse mammary epithelial cells. Lipids. 2000b;35(2):171–80. doi: 10.1007/BF02664767. [DOI] [PubMed] [Google Scholar]
- Miyamoto T, Lindgren JA, Hokfelt T, Samuelsson B. Formation of lipoxygenase products in the rat brain. Adv Prostaglandin Thromboxane Leukot Res. 1987a;17B:929–33. [PubMed] [Google Scholar]
- Miyamoto T, Lindgren JA, Hokfelt T, Samuelsson B. Regional distribution of leukotriene and mono-hydroxyeicosatetraenoic acid production in the rat brain. Highest leukotriene C4 formation in the hypothalamus. FEBS Lett. 1987b;216(1):123–7. doi: 10.1016/0014-5793(87)80769-x. [DOI] [PubMed] [Google Scholar]
- Miyazawa T, Inokuchi H, Hirokane H, Tsuzuki T, Nakagawa K, Igarashi M. Anti-angiogenic potential of tocotrienol in vitro. Biochemistry (Mosc) 2004;69(1):67–9. doi: 10.1023/b:biry.0000016353.18007.39. [DOI] [PubMed] [Google Scholar]
- Mizushina Y, Nakagawa K, Shibata A, Awata Y, Kuriyama I, Shimazaki N, Koiwai O, Uchiyama Y, Sakaguchi K, Miyazawa T, et al. Inhibitory effect of tocotrienol on eukaryotic DNA polymerase lambda and angiogenesis. Biochem Biophys Res Commun. 2006;339(3):949–55. doi: 10.1016/j.bbrc.2005.11.085. [DOI] [PubMed] [Google Scholar]
- Mo H, Elson CE. Apoptosis and cell-cycle arrest in human and murine tumor cells are initiated by isoprenoids. Journal of Nutrition. 1999;129(4):804–13. doi: 10.1093/jn/129.4.804. [DOI] [PubMed] [Google Scholar]
- Musiek ES, Breeding RS, Milne GL, Zanoni G, Morrow JD, McLaughlin B. Cyclopentenone isoprostanes are novel bioactive products of lipid oxidation which enhance neurodegeneration. J Neurochem. 2006;97(5):1301–13. doi: 10.1111/j.1471-4159.2006.03797.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa K, Eitsuka T, Inokuchi H, Miyazawa T. DNA chip analysis of comprehensive food function: Inhibition of angiogenesis and telomerase activity with unsaturated vitamin E, tocotrienol. Biofactors. 2004;21(1–4):5–10. doi: 10.1002/biof.552210102. [DOI] [PubMed] [Google Scholar]
- Nesaretnam K, Ambra R, Selvaduray KR, Radhakrishnan A, Reimann K, Razak G, Virgili F. Tocotrienol-rich fraction from palm oil affects gene expression in tumors resulting from MCF-7 cell inoculation in athymic mice. Lipids. 2004;39(5):459–67. doi: 10.1007/s11745-004-1251-1. [DOI] [PubMed] [Google Scholar]
- Nesaretnam K, Dorasamy S, Darbre PD. Tocotrienols inhibit growth of ZR-75-1 breast cancer cells. Int J Food Sci Nutr. 2000;51(Suppl):S95–103. [PubMed] [Google Scholar]
- Nesaretnam K, Guthrie N, Chambers AF, Carroll KK. Effect of tocotrienols on the growth of a human breast cancer cell line in culture. Lipids. 1995;30(12):1139–43. doi: 10.1007/BF02536615. [DOI] [PubMed] [Google Scholar]
- Nesaretnam K, Stephen R, Dils R, Darbre P. Tocotrienols inhibit the growth of human breast cancer cells irrespective of estrogen receptor status. Lipids. 1998;33(5):461–9. doi: 10.1007/s11745-998-0229-3. [DOI] [PubMed] [Google Scholar]
- Ngah WZ, Jarien Z, San MM, Marzuki A, Top GM, Shamaan NA, Kadir KA. Effect of tocotrienols on hepatocarcinogenesis induced by 2-acetylaminofluorene in rats. American Journal of Clinical Nutrition. 1991;53(4 Suppl):1076S–1081S. doi: 10.1093/ajcn/53.4.1076S. [DOI] [PubMed] [Google Scholar]
- Nishiyama M, Okamoto H, Watanabe T, Hori T, Hada T, Ueda N, Yamamoto S, Tsukamoto H, Watanabe K, Kirino T. Localization of arachidonate 12-lipoxygenase in canine brain tissues. Journal of Neurochemistry. 1992;58(4):1395–400. doi: 10.1111/j.1471-4159.1992.tb11355.x. [DOI] [PubMed] [Google Scholar]
- Nishiyama M, Watanabe T, Ueda N, Tsukamoto H, Watanabe K. Arachidonate 12-lipoxygenase is localized in neurons, glial cells, and endothelial cells of the canine brain. Journal of Histochemistry & Cytochemistry. 1993;41(1):111–7. doi: 10.1177/41.1.8417106. [DOI] [PubMed] [Google Scholar]
- O’Byrne D, Grundy S, Packer L, Devaraj S, Baldenius K, Hoppe PP, Kraemer K, Jialal I, Traber MG. Studies of LDL oxidation following alpha-, gamma-, or delta-tocotrienyl acetate supplementation of hypercholesterolemic humans. Free Radical Biology & Medicine. 2000;29(9):834–845. doi: 10.1016/s0891-5849(00)00371-3. [DOI] [PubMed] [Google Scholar]
- Panfili G, Fratianni A, Irano M. Normal phase high-performance liquid chromatography method for the determination of tocopherols and tocotrienols in cereals. J Agric Food Chem. 2003;51(14):3940–4. doi: 10.1021/jf030009v. [DOI] [PubMed] [Google Scholar]
- Parker RA, Pearce BC, Clark RW, Gordon DA, Wright JJ. Tocotrienols regulate cholesterol production in mammalian cells by post-transcriptional suppression of 3-hydroxy-3-methylglutaryl-coenzyme A reductase. Journal of Biological Chemistry. 1993;268(15):11230–8. [PubMed] [Google Scholar]
- Paul R, Zhang ZG, Eliceiri BP, Jiang Q, Boccia AD, Zhang RL, Chopp M, Cheresh DA. Src deficiency or blockade of Src activity in mice provides cerebral protection following stroke. Nat Med. 2001;7(2):222–7. doi: 10.1038/84675. [DOI] [PubMed] [Google Scholar]
- Pearce BC, Parker RA, Deason ME, Dischino DD, Gillespie E, Qureshi AA, Volk K, Wright JJ. Inhibitors of cholesterol biosynthesis. 2. Hypocholesterolemic and antioxidant activities of benzopyran and tetrahydronaphthalene analogues of the tocotrienols. Journal of Medicinal Chemistry. 1994;37(4):526–41. doi: 10.1021/jm00030a012. [DOI] [PubMed] [Google Scholar]
- Pearce BC, Parker RA, Deason ME, Qureshi AA, Wright JJ. Hypocholesterolemic activity of synthetic and natural tocotrienols. Journal of Medicinal Chemistry. 1992;35(20):3595–606. doi: 10.1021/jm00098a002. [DOI] [PubMed] [Google Scholar]
- Porfirova S, Bergmuller E, Tropf S, Lemke R, Dormann P. Isolation of an Arabidopsis mutant lacking vitamin E and identification of a cyclase essential for all tocopherol biosynthesis. Proc Natl Acad Sci U S A. 2002;99(19):12495–500. doi: 10.1073/pnas.182330899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pyper JM, Bolen JB. Neuron-specific splicing of C-SRC RNA in human brain. J Neurosci Res. 1989;24(1):89–96. doi: 10.1002/jnr.490240113. [DOI] [PubMed] [Google Scholar]
- Qureshi AA, Bradlow BA, Brace L, Manganello J, Peterson DM, Pearce BC, Wright JJ, Gapor A, Elson CE. Response of hypercholesterolemic subjects to administration of tocotrienols. Lipids. 1995;30(12):1171–7. doi: 10.1007/BF02536620. [DOI] [PubMed] [Google Scholar]
- Qureshi AA, Burger WC, Peterson DM, Elson CE. The structure of an inhibitor of cholesterol biosynthesis isolated from barley. Journal of Biological Chemistry. 1986;261(23):10544–50. [PubMed] [Google Scholar]
- Qureshi AA, Mo H, Packer L, Peterson DM. Isolation and identification of novel tocotrienols from rice bran with hypocholesterolemic, antioxidant, and antitumor properties. Journal of Agricultural & Food Chemistry. 2000;48(8):3130–40. doi: 10.1021/jf000099t. [DOI] [PubMed] [Google Scholar]
- Qureshi AA, Peterson DM, Hasler-Rapacz JO, Rapacz J. Novel tocotrienols of rice bran suppress cholesterogenesis in hereditary hypercholesterolemic swine. J Nutr. 2001a;131(2):223–30. doi: 10.1093/jn/131.2.223. [DOI] [PubMed] [Google Scholar]
- Qureshi AA, Qureshi N, Hasler-Rapacz JO, Weber FE, Chaudhary V, Crenshaw TD, Gapor A, Ong AS, Chong YH, Peterson D. Dietary tocotrienols reduce concentrations of plasma cholesterol, apolipoprotein B, thromboxane B2, and platelet factor 4 in pigs with inherited hyperlipidemias. American Journal of Clinical Nutrition. 1991a;53(4 Suppl):1042S–1046S. doi: 10.1093/ajcn/53.4.1042S. [DOI] [PubMed] [Google Scholar]
- Qureshi AA, Qureshi N, Wright JJ, Shen Z, Kramer G, Gapor A, Chong YH, DeWitt G, Ong A, Peterson DM. Lowering of serum cholesterol in hypercholesterolemic humans by tocotrienols (palmvitee) American Journal of Clinical Nutrition. 1991b;53(4 Suppl):1021S–1026S. doi: 10.1093/ajcn/53.4.1021S. [DOI] [PubMed] [Google Scholar]
- Qureshi AA, Sami SA, Salser WA, Khan FA. Synergistic effect of tocotrienol-rich fraction (TRF(25)) of rice bran and lovastatin on lipid parameters in hypercholesterolemic humans. J Nutr Biochem. 2001b;12(6):318–329. doi: 10.1016/s0955-2863(01)00144-9. [DOI] [PubMed] [Google Scholar]
- Qureshi AA, Sami SA, Salser WA, Khan FA. Dose-dependent suppression of serum cholesterol by tocotrienol-rich fraction (TRF25) of rice bran in hypercholesterolemic humans. Atherosclerosis. 2002;161(1):199–207. doi: 10.1016/s0021-9150(01)00619-0. [DOI] [PubMed] [Google Scholar]
- Raederstorff D, Elste V, Aebischer C, Weber P. Effect of either gamma-tocotrienol or a tocotrienol mixture on the plasma lipid profile in hamsters. Ann Nutr Metab. 2002;46(1):17–23. doi: 10.1159/000046748. [DOI] [PubMed] [Google Scholar]
- Rahmat A, Ngah WZ, Shamaan NA, Gapor A, Abdul Kadir K. Long-term administration of tocotrienols and tumor-marker enzyme activities during hepatocarcinogenesis in rats. Nutrition. 1993;9(3):229–32. [PubMed] [Google Scholar]
- Reddanna P, Rao MK, Reddy CC. Inhibition of 5-lipoxygenase by vitamin E. FEBS Letters. 1985;193(1):39–43. doi: 10.1016/0014-5793(85)80075-2. [DOI] [PubMed] [Google Scholar]
- Rippert P, Scimemi C, Dubald M, Matringe M. Engineering plant shikimate pathway for production of tocotrienol and improving herbicide resistance. Plant Physiol. 2004;134(1):92–100. doi: 10.1104/pp.103.032441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy S, Lado BH, Khanna S, Sen CK. Vitamin E sensitive genes in the developing rat fetal brain: a high-density oligonucleotide microarray analysis. FEBS Lett. 2002;530(1–3):17–23. doi: 10.1016/s0014-5793(02)03309-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakai M, Okabe M, Tachibana H, Yamada K. Apoptosis induction by gamma-tocotrienol in human hepatoma Hep3B cells. J Nutr Biochem. 2005 doi: 10.1016/j.jnutbio.2005.11.001. [DOI] [PubMed] [Google Scholar]
- Sakai M, Okabe M, Yamasaki M, Tachibana H, Yamada K. Induction of apoptosis by tocotrienol in rat hepatoma dRLh-84 cells. Anticancer Res. 2004;24(3a):1683–8. [PubMed] [Google Scholar]
- Schubert D, Piasecki D. Oxidative glutamate toxicity can be a component of the excitotoxicity cascade. Journal of Neuroscience. 2001;21(19):7455–62. doi: 10.1523/JNEUROSCI.21-19-07455.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz JB, Lindenau J, Seyfried J, Dichgans J. Glutathione, oxidative stress and neurodegeneration. European Journal of Biochemistry. 2000;267(16):4904–11. doi: 10.1046/j.1432-1327.2000.01595.x. [DOI] [PubMed] [Google Scholar]
- Sen CK, Khanna S, Roy S. Tocotrienol: the natural vitamin E to defend the nervous system? Ann N Y Acad Sci. 2004;1031:127–42. doi: 10.1196/annals.1331.013. [DOI] [PubMed] [Google Scholar]
- Sen CK, Khanna S, Roy S. Tocotrienols: Vitamin E beyond tocopherols. Life Sci. 2006;78(18):2088–98. doi: 10.1016/j.lfs.2005.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen CK, Khanna S, Roy S, Packer L. Molecular basis of vitamin E action. Tocotrienol potently inhibits glutamate-induced pp60(c-Src) kinase activation and death of HT4 neuronal cells. Journal of Biological Chemistry. 2000;275(17):13049–55. doi: 10.1074/jbc.275.17.13049. [DOI] [PubMed] [Google Scholar]
- Serbinova E, Kagan V, Han D, Packer L. Free radical recycling and intramembrane mobility in the antioxidant properties of alpha-tocopherol and alpha-tocotrienol. Free Radical Biology & Medicine. 1991;10(5):263–75. doi: 10.1016/0891-5849(91)90033-y. [DOI] [PubMed] [Google Scholar]
- Serbinova EA, Packer L. Antioxidant properties of alpha-tocopherol and alpha-tocotrienol. Methods in Enzymology. 1994;234:354–66. doi: 10.1016/0076-6879(94)34105-2. [DOI] [PubMed] [Google Scholar]
- Sever N, Song BL, Yabe D, Goldstein JL, Brown MS, DeBose-Boyd RA. Insig-dependent ubiquitination and degradation of mammalian 3-hydroxy-3-methylglutaryl-CoA reductase stimulated by sterols and geranylgeraniol. J Biol Chem. 2003;278(52):52479–90. doi: 10.1074/jbc.M310053200. [DOI] [PubMed] [Google Scholar]
- Shah S, Gapor A, Sylvester PW. Role of caspase-8 activation in mediating vitamin E-induced apoptosis in murine mammary cancer cells. Nutr Cancer. 2003;45(2):236–46. doi: 10.1207/S15327914NC4502_14. [DOI] [PubMed] [Google Scholar]
- Shah S, Sylvester PW. Tocotrienol-induced caspase-8 activation is unrelated to death receptor apoptotic signaling in neoplastic mammary epithelial cells. Exp Biol Med (Maywood) 2004;229(8):745–55. doi: 10.1177/153537020422900806. [DOI] [PubMed] [Google Scholar]
- Shah SJ, Sylvester PW. Gamma-tocotrienol inhibits neoplastic mammary epithelial cell proliferation by decreasing Akt and nuclear factor kappaB activity. Exp Biol Med (Maywood) 2005a;230(4):235–41. doi: 10.1177/153537020523000402. [DOI] [PubMed] [Google Scholar]
- Shah SJ, Sylvester PW. Tocotrienol-induced cytotoxicity is unrelated to mitochondrial stress apoptotic signaling in neoplastic mammary epithelial cells. Biochem Cell Biol. 2005b;83(1):86–95. doi: 10.1139/o04-127. [DOI] [PubMed] [Google Scholar]
- Shay JW, Wright WE. Telomerase therapeutics for cancer: challenges and new directions. Nat Rev Drug Discov. 2006;5(7):577–84. doi: 10.1038/nrd2081. [DOI] [PubMed] [Google Scholar]
- Shohami E, Glantz L, Nates J, Feuerstein G. The mixed lipoxygenase/cyclooxygenase inhibitor SK&F 105809 reduces cerebral edema after closed head injury in rat. J Basic Clin Physiol Pharmacol. 1992;3(2):99–107. doi: 10.1515/jbcpp.1992.3.2.99. [DOI] [PubMed] [Google Scholar]
- Shun MC, Yu W, Gapor A, Parsons R, Atkinson J, Sanders BG, Kline K. Pro-apoptotic mechanisms of action of a novel vitamin E analog (alpha-TEA) and a naturally occurring form of vitamin E (delta-tocotrienol) in MDA-MB-435 human breast cancer cells. Nutr Cancer. 2004;48(1):95–105. doi: 10.1207/s15327914nc4801_13. [DOI] [PubMed] [Google Scholar]
- Shupnik MA. Crosstalk between steroid receptors and the c-Src-receptor tyrosine kinase pathways: implications for cell proliferation. Oncogene. 2004;23(48):7979–89. doi: 10.1038/sj.onc.1208076. [DOI] [PubMed] [Google Scholar]
- Solomons NW, Orozco M. Alleviation of vitamin A deficiency with palm fruit and its products. Asia Pac J Clin Nutr. 2003;12(3):373–84. [PubMed] [Google Scholar]
- Song BL, Debose-Boyd RA. Insig-dependent ubiquitination and degradation of 3-hydroxy-3-methylglutaryl coenzyme A reductase stimulated by delta - and gamma -tocotrienols. J Biol Chem. 2006 doi: 10.1074/jbc.M605575200. [DOI] [PubMed] [Google Scholar]
- Soriano P, Montgomery C, Geske R, Bradley A. Targeted disruption of the c-src proto-oncogene leads to osteopetrosis in mice. Cell. 1991;64(4):693–702. doi: 10.1016/0092-8674(91)90499-o. [DOI] [PubMed] [Google Scholar]
- Srivastava JK, Gupta S. Tocotrienol-rich fraction of palm oil induces cell cycle arrest and apoptosis selectively in human prostate cancer cells. Biochem Biophys Res Commun. 2006;346(2):447–53. doi: 10.1016/j.bbrc.2006.05.147. [DOI] [PubMed] [Google Scholar]
- Steiner M. Vitamin E: more than an antioxidant. Clin Cardiol. 1993;16(4 Suppl 1):I16–8. doi: 10.1002/clc.4960161306. [DOI] [PubMed] [Google Scholar]
- Sugano M, Koba K, Tsuji E. Health benefits of rice bran oil. Anticancer Research. 1999;19(5A):3651–7. [PubMed] [Google Scholar]
- Sun X, Shih AY, Johannssen HC, Erb H, Li P, Murphy TH. Two-photon Imaging of Glutathione Levels in Intact Brain Indicates Enhanced Redox Buffering in Developing Neurons and Cells at the Cerebrospinal Fluid and Blood-Brain Interface. J Biol Chem. 2006;281(25):17420–31. doi: 10.1074/jbc.M601567200. [DOI] [PubMed] [Google Scholar]
- Sundram K, Khor HT, Ong AS, Pathmanathan R. Effect of dietary palm oils on mammary carcinogenesis in female rats induced by 7,12-dimethylbenz(a)anthracene. Cancer Research. 1989;49(6):1447–51. [PubMed] [Google Scholar]
- Sundram K, Sambanthamurthi R, Tan YA. Palm fruit chemistry and nutrition. Asia Pac J Clin Nutr. 2003;12(3):355–62. [PubMed] [Google Scholar]
- Suzuki YJ, Tsuchiya M, Wassall SR, Choo YM, Govil G, Kagan VE, Packer L. Structural and dynamic membrane properties of alpha-tocopherol and alpha-tocotrienol: implication to the molecular mechanism of their antioxidant potency. Biochemistry. 1993;32(40):10692–9. doi: 10.1021/bi00091a020. [DOI] [PubMed] [Google Scholar]
- Sylvester PW, Nachnani A, Shah S, Briski KP. Role of GTP-binding proteins in reversing the antiproliferative effects of tocotrienols in preneoplastic mammary epithelial cells. Asia Pac J Clin Nutr. 2002;11(Suppl 7):S452–9. doi: 10.1046/j.1440-6047.11.s.7.9.x. [DOI] [PubMed] [Google Scholar]
- Sylvester PW, Shah S. Intracellular mechanisms mediating tocotrienol-induced apoptosis in neoplastic mammary epithelial cells. Asia Pac J Clin Nutr. 2005;14(4):366–73. [PubMed] [Google Scholar]
- Takahashi K, Loo G. Disruption of mitochondria during tocotrienol-induced apoptosis in MDA-MB-231 human breast cancer cells. Biochem Pharmacol. 2004;67(2):315–24. doi: 10.1016/j.bcp.2003.07.015. [DOI] [PubMed] [Google Scholar]
- Tan DT, Khor HT, Low WH, Ali A, Gapor A. Effect of a palm-oil-vitamin E concentrate on the serum and lipoprotein lipids in humans. American Journal of Clinical Nutrition. 1991;53(4 Suppl):1027S–1030S. doi: 10.1093/ajcn/53.4.1027S. [DOI] [PubMed] [Google Scholar]
- Tan S, Schubert D, Maher P. Oxytosis: A novel form of programmed cell death. Current Topics in Medicinal Chemistry. 2001;1(6):497–506. doi: 10.2174/1568026013394741. [DOI] [PubMed] [Google Scholar]
- Tarrago-Trani MT, Phillips KM, Lemar LE, Holden JM. New and existing oils and fats used in products with reduced trans-fatty acid content. J Am Diet Assoc. 2006;106(6):867–80. doi: 10.1016/j.jada.2006.03.010. [DOI] [PubMed] [Google Scholar]
- Terasawa Y, Ladha Z, Leonard SW, Morrow JD, Newland D, Sanan D, Packer L, Traber MG, Farese RV., Jr Increased atherosclerosis in hyperlipidemic mice deficient in alpha -tocopherol transfer protein and vitamin E. Proceedings of the National Academy of Sciences of the United States of America. 2000;97(25):13830–4. doi: 10.1073/pnas.240462697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas SM, Brugge JS. Cellular functions regulated by Src family kinases. Annual Review of Cell & Developmental Biology. 1997;13:513–609. doi: 10.1146/annurev.cellbio.13.1.513. [DOI] [PubMed] [Google Scholar]
- Traber MG, Arai H. Molecular mechanisms of vitamin E transport. Annual Review of Nutrition. 1999;19:343–55. doi: 10.1146/annurev.nutr.19.1.343. [DOI] [PubMed] [Google Scholar]
- Traber MG, Burton GW, Hamilton RL. Vitamin E trafficking. Annals of the New York Academy of Sciences. 2004;1031:1–12. doi: 10.1196/annals.1331.001. [DOI] [PubMed] [Google Scholar]
- van der Worp HB, Bar PR, Kappelle LJ, de Wildt DJ. Dietary vitamin E levels affect outcome of permanent focal cerebral ischemia in rats. Stroke. 1998;29(5):1002–5. doi: 10.1161/01.str.29.5.1002. discussion 1005–6. [DOI] [PubMed] [Google Scholar]
- Venkatesh TV, Karunanandaa B, Free DL, Rottnek JM, Baszis SR, Valentin HE. Identification and characterization of an Arabidopsis homogentisate phytyltransferase paralog. Planta. 2006;223(6):1134–44. doi: 10.1007/s00425-005-0180-1. [DOI] [PubMed] [Google Scholar]
- Wada S, Satomi Y, Murakoshi M, Noguchi N, Yoshikawa T, Nishino H. Tumor suppressive effects of tocotrienol in vivo and in vitro. Cancer Lett. 2005;229(2):181–91. doi: 10.1016/j.canlet.2005.06.036. [DOI] [PubMed] [Google Scholar]
- Wagner KH, Kamal-Eldin A, Elmadfa I. Gamma-tocopherol--an underestimated vitamin? Ann Nutr Metab. 2004;48(3):169–88. doi: 10.1159/000079555. [DOI] [PubMed] [Google Scholar]
- Whittle KJ, Dunphy PJ, Pennock JF. The isolation and properties of delta-tocotrienol from Hevea latex. Biochemical Journal. 1966;100(1):138–45. doi: 10.1042/bj1000138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wie MB, Koh JY, Won MH, Lee JC, Shin TK, Moon CJ, Ha HJ, Park SM, Kim HC. BAPTA/AM, an intracellular calcium chelator, induces delayed necrosis by lipoxygenase-mediated free radicals in mouse cortical cultures. Prog Neuropsychopharmacol Biol Psychiatry. 2001;25(8):1641–59. doi: 10.1016/s0278-5846(01)00202-0. [DOI] [PubMed] [Google Scholar]
- Yamamoto S. Mammalian lipoxygenases: molecular structures and functions. Biochimica et Biophysica Acta. 1992;1128(2–3):117–31. doi: 10.1016/0005-2760(92)90297-9. [DOI] [PubMed] [Google Scholar]
- Yano Y, Satoh H, Fukumoto K, Kumadaki I, Ichikawa T, Yamada K, Hagiwara K, Yano T. Induction of cytotoxicity in human lung adenocarcinoma cells by 6-O-carboxypropyl-alpha-tocotrienol, a redox-silent derivative of alpha-tocotrienol. Int J Cancer. 2005;115(5):839–46. doi: 10.1002/ijc.20809. [DOI] [PubMed] [Google Scholar]
- Yao Y, Clark CM, Trojanowski JQ, Lee VM, Pratico D. Elevation of 12/15 lipoxygenase products in AD and mild cognitive impairment. Ann Neurol. 2005;58(4):623–6. doi: 10.1002/ana.20558. [DOI] [PubMed] [Google Scholar]
- Yap SP, Yuen KH, Wong JW. Pharmacokinetics and bioavailability of alpha-, gamma- and delta-tocotrienols under different food status. J Pharm Pharmacol. 2001;53(1):67–71. doi: 10.1211/0022357011775208. [DOI] [PubMed] [Google Scholar]
- Yoneda T, Lowe C, Lee CH, Gutierrez G, Niewolna M, Williams PJ, Izbicka E, Uehara Y, Mundy GR. Herbimycin A, a pp60c-src tyrosine kinase inhibitor, inhibits osteoclastic bone resorption in vitro and hypercalcemia in vivo. J Clin Invest. 1993;91(6):2791–5. doi: 10.1172/JCI116521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu FL, Gapor A, Bender W. Evidence for the preventive effect of the polyunsaturated phytol side chain in tocotrienols on 17beta-estradiol epoxidation. Cancer Detect Prev. 2005;29(4):383–8. doi: 10.1016/j.cdp.2005.03.003. [DOI] [PubMed] [Google Scholar]
- Yu SG, Thomas AM, Gapor A, Tan B, Qureshi N, Qureshi AA. Dose-response impact of various tocotrienols on serum lipid parameters in 5-week-old female chickens. Lipids. 2006;41(5):453–61. doi: 10.1007/s11745-006-5119-1. [DOI] [PubMed] [Google Scholar]
- Yu W, Simmons-Menchaca M, Gapor A, Sanders BG, Kline K. Induction of apoptosis in human breast cancer cells by tocopherols and tocotrienols. Nutrition & Cancer. 1999;33(1):26–32. doi: 10.1080/01635589909514744. [DOI] [PubMed] [Google Scholar]
- Zingg JM, Azzi A. Non-antioxidant activities of vitamin E. Curr Med Chem. 2004;11(9):1113–33. doi: 10.2174/0929867043365332. [DOI] [PubMed] [Google Scholar]