

Abstract
Biochemical experiments have recently revealed that the p-S8 peptide, with an amino-acid sequence identical to the conserved fragment 83–93 (S8) of the HIV-1 protease, can inhibit catalytic activity of the enzyme by interfering with protease folding and dimerization. In this study, we introduce a hierarchical modeling approach for understanding the molecular basis of the HIV-1 protease folding inhibition. Coarse-grained molecular docking simulations of the flexible p-S8 peptide with the ensembles of HIV-1 protease monomers have revealed structurally different complexes of the p-S8 peptide, which can be formed by targeting the conserved segment 24–34 (S2) of the folding nucleus (folding inhibition) and by interacting with the antiparallel termini β-sheet region (dimerization inhibition). All-atom molecular dynamics simulations of the inhibitor complexes with the HIV-1 PR monomer have been independently carried out for the predicted folding and dimerization binding modes of the p-S8 peptide, confirming the thermodynamic stability of these complexes. Binding free-energy calculations of the p-S8 peptide and its active analogs are then performed using molecular dynamics trajectories of the peptide complexes with the HIV-1 PR monomers. The results of this study have provided a plausible molecular model for the inhibitor intervention with the HIV-1 PR folding and dimerization and have accurately reproduced the experimental inhibition profiles of the active folding inhibitors.
INTRODUCTION
Human immunodeficiency virus type 1 protease (HIV-1 PR) is a homodimeric enzyme which plays an important role in processing the viral polypeptide precursors (1–3). Crystal structures of the HIV-1 PR complexes have revealed that the protease flaps can exhibit a considerable mobility ranging from a closed form (in which the active site is occupied by a ligand), semiopen form (which is typically observed in the free HIV-1 PR), and a wide-open form (which allows the substrate and inhibitor access to the active site) (4–8). NMR experiments have shown that localized fluctuations of the HIV-1 PR flaps reflect a rapid dynamic equilibrium between the ensembles of semiopen and closed conformations, which are observed in the crystal structures of the HIV-1 PR. The ensemble of HIV-1 PR unbound structures can also include open-flap conformations as a rare event in a slow conformational exchange (9–12). Understanding HIV-1 PR flexibility and dynamics associated with the binding of HIV-1 PR substrates and inhibitors has been greatly enhanced using synergy of experiments and simulations. Mechanisms of HIV-1 PR flexibility and dynamics were studied at the atomic level, including flap openings in the activated dynamics studies (13,14), curling motions of the flaps observed in the long molecular dynamics (MD) simulations (15,16), high flexibility and opening of the flaps in the simulations of the HIV-1 PR and mutants (17–21), and role of solvation effects influencing flap collapse (22). The reduced complexity of a coarse-grained model of the HIV-1 PR dynamics has enabled us to capture statistically significant conformational transitions between open and closed forms of the protease and an accurate thermodynamic analysis of the HIV-1 PR motions occurring on the microsecond timescale (23–26). Recently, large-scale unconstrained all-atom MD simulations of HIV-1 PR have demonstrated the reversible opening of the flaps and evidence for multiple transitions between the closed, semiopen, and open conformations, in which rare opening events, when flaps separate and become less ordered, are accompanied by subsequent return to the thermodynamically stable semiopen state (27,28). The ability of certain mutants to alter the dynamic equilibrium between the open and closed enzyme forms of the HIV-1 PR may lead to an excessive stabilization of the semiopen conformation and drug-resistant forms of the enzyme (29–35).
The major areas that constitute the dimer interface are the active site region 24–29, forming the fireman's-grip hydrogen-bond network, and the four-stranded anti-parallel β-sheet, which is formed by interdigitation of the C-terminal and N-terminal residues of the HIV-1 PR. These interfaces are assembled from evolutionary and structurally conserved fragments 24–34 (S2) and 83–93 (S8) of the HIV-1 PR monomers, which constitute the critical components of the HIV-1 PR folding nucleus (Fig. 1 A). The protection patterns obtained for the HIV-1 PR units 24–34, 74–78, and 83–93 have shown that these conserved segments constitute the protease folding core (36). Although many crystal structures of HIV-PR and its complexes with various ligands have been solved, crystals of the natural HIV-PR monomer are difficult to obtain, due to the low dissociation constant of dimer and equilibrium concentration of monomer in solution. Solution NMR structures of the HIV-1 PR monomers have revealed similar tertiary folds of the HIV-1 PR monomer and a single subunit of the HIV-1 PR dimer, with some disorder in the terminal regions (37–40). Furthermore, destabilization of the interfacial network in the R87K, D29N, and T26A mutants can result in the formation of a stable monomeric structure of the HIV-1 PR single subunit. A comprehensive view of protein dynamics was recently inferred from NMR studies of the HIV-1 PR, suggesting that the flaps in the unliganded protease dimer may interact with each other in solution and that semiopen form may be a dominant conformation of the unliganded HIV-1 PR dimer and monomer (12,41). According to the computer simulations of the HIV-1 PR folding, the HIV-1 PR active dimer is likely formed by the association of folded HIV-1 PR monomers rather than by direct coupling between the monomer folding and binding (42). The dynamics of the HIV-PR monomer has been further explored using MD simulations of the HIV-PR monomer in water, which have revealed that stabilization of the termini in the β-sheet may be a dominant conformation of the solvated HIV-PR monomer (43).
FIGURE 1.

(A) Evolutionary and structurally conserved segments of the HIV-1 PR core are highlighted: the β-strand segment 24–34 and the 83–93 segment encompassing a single α-helix. (B) The folded units 83–93 and 24–34 of the HIV-1 PR core are shown in grayscale ribbons.
The diversity of the HIV-1 PR inhibition scenarios may include the conventional active site binding and alternative inhibitory mechanisms, based on blocking the assembly of the HIV-1 PR homodimer and disrupting the dimeric interface. Design of HIV- PR dimerization inhibitors is typically focused on disrupting four-stranded β-sheet and targeting flexible N- and C-termini that constitute most of the dimer interface (44–47). It was recently proposed that the peptide LDTGADDTVLE (p-S2) and the peptide NIIGRNLLTQI (p-S8), with the sequences identical to that of the conserved fragments 24–34 (S2) and 83–93 (S8) from the protease folding nucleus, may act as unconventional therapeutic agents intervening with the HIV-1 PR folding and dimerization (48–52). In particular, it was demonstrated that the p-S8 peptide (Fig. 1 A) can inhibit the enzyme folding and formation of the active dimer in the micromolar range (Ki = 2.58 μM) (48,49). The biochemical and circular dichroism spectroscopy experiments have further confirmed that the p-S8 peptide can inhibit catalytic activity of the enzyme, interfere with the assembly of the HIV-1 PR folding nucleus, and lead to partial destabilization of the active HIV-1 PR dimer (48–52). To improve pharmacological profiles of the folding inhibitor, a number of the p-S8 peptide analogs have been synthesized, including a shortened 83–92 derivative NIIGRNLLTQ and a N88D mutant NIIGRDLLTQI (Fig. 1 B). Both peptide analogs have exhibited a considerably better solubility than the wild-type folding inhibitor with the inhibition constants of Ki = 7.8 μM for a shortened 83–92 peptide and Ki = 15.8 μM for the N88D peptide mutant (48–52). Simulations of the p-S2 and p-S8 peptides in explicit solvent have provided the details of the docking mechanism of the two peptides (52). This study has shown that the p-S8 peptide can spontaneously fold into the structure of the 83–93 protease segment independently of the rest of the protein and retain the thermodynamic stability of this structure in isolation. In contrast, the peptide p-S2, corresponding to the fragment 24–34 of the protease-folding nucleus, is considerably more flexible. According to this study, binding of the p-S2 and p-S8 peptides may result in a stable complex, whereby a flexible p-S2 peptide can wrap around a folded structure of the p-S8 peptide by forming a network of specific hydrogen bonds, none of which involve residues from a single α-helix region (52).
The ultimate experiments probing the folding inhibition mechanism require a combination of biochemical and structural studies. However, x-ray crystallography is not readily applicable for these studies, since the necessary high concentrations of the enzyme would tend to shift the equilibrium toward the active dimer. NMR studies of the inhibitor intervention with the HIV-1PR dimer assembly are quite challenging, though may in principle assess the molecular basis of folding inhibition. We propose a hierarchical modeling strategy to dissect the molecular and energetic basis of the HIV-1 PR folding inhibition at atomic resolution. This approach includes the following stages:
Coarse-grained molecular docking simulations of the flexible p-S8 peptide with the ensembles of HIV-1 PR dimers and monomers (explicit flexibility of the inhibitor and implicit flexibility of the receptor) and subsequent free energy refinement of the low-energy docking solutions.
All-atom MD simulations of the flexible peptide—HIV-1 PR monomer complexes performed for the predicted folding and dimerization binding modes of the p-S8 peptide (explicit flexibility of both the inhibitor and the receptor).
Binding free energy calculations of the p-S8 peptide and its active analogs, which are performed using all-atom MD trajectories of the peptide complexes with the HIV-1 PR monomers in the folding and dimerization binding modes.
Using this hierarchical simulation approach, we demonstrate that the folding inhibitor may exhibit both folding and dimerization modes of inhibition by targeting conservative elements of the HIV-1 PR folding core and dimerization interface. This computational analysis presents a plausible molecular mechanism of the inhibitor intervention with the HIV-1 PR folding and dimerization, which is consistent with the available experimental data.
MATERIALS AND METHODS
HIV-1 PR conformational ensembles
The conformational ensemble of the HIV-1 PR crystal structures provides a convenient coarse-grained model of the enzyme mobility, reflecting a spectrum of the flap opening motions which are difficult to detect in the MD simulations. According to the recent structural analysis and classification of the HIV-1 PR apo structures (53,54), distinct families in HIV-1 PR may include semiopen, curled, and open conformations. The ensemble of presently available crystal structures of the apo HIV-1 PR was used in docking simulations to categorize structural landscape of the HIV-1 PR conformational states. This includes semiopen conformations with pdb entries 1hhp (55) and 3hvp (56). The curled-flaps conformations include pdb entries 3phv wild-type (57), 2g69 P53L mutant (58), 2hb4 2.15 Å wild-type, and 2hb2 2.3 Å sixfold L24I/M46I/F53L/L63P/V77I/V82A mutant) (53,54). The open-flaps conformations include pdb entries 2pc0 1.4 Å resolution wild-type (54), 1rpi ninefold mutant (59), and 1tw7 10-fold mutant (8). The HIV-1 PR conformations in complexes with the clinically approved HIV-1 inhibitors saquinavir, ritonavir, indinavir, nelfinavir, and amprenavir were used to represent a closed form of the enzyme (60–64). Overall, the ensemble of the HIV PR crystal structures can thereby represent a coarse-grained model of the flap-opening mechanism. The conformational ensemble of the HIV-1 monomeric conformations is initially obtained from the crystal structures of the unliganded HIV-1 PR and HIV-1 PR complexes with the inhibitors, followed by subsequent minimization and thermal equilibration procedures. The protonation state of Asp25 of every structure was adjusted as protonated according to experimental observations predicting that, under physiological conditions, only one of the two active site aspartates is protonated while the other remains in the carboxylated form. We have equilibrated the HIV-1 PR dimeric and monomeric structures at 300 K using MD simulations performed with the GROMACS package and OPLS/AMBER force field with 9428 water molecules and two chloride ions (65–67). The following steps were carried out: 1), a steepest descent energy minimization; 2), equilibration of water for 300 ps at 300 K keeping the heavy atoms of the protein constrained; and 3), a 300 ps dynamics at 300 K at constant volume to thermalize the system.
Coarse-grained molecular docking with the ensembles of the HIV-1 PR conformations
The knowledge-based simplified energetic model used in docking simulations includes intramolecular energy terms for the ligand, given by torsional and nonbonded functions, and intermolecular ligand-protein steric and hydrogen-bond interaction terms calculated from a piecewise linear potential summed over all protein and ligand heavy atoms (68). The piecewise linear potential parameters were refined to yield the experimental crystallographic structure of a set of ligand-protein complexes as the global energy minimum. Multiple docking simulations are performed using an evolutionary search algorithm based on the ideas of natural selection (68). For each docking simulation, the evolutionary search was performed for a total of 120 generations with a population size of 1200 members. During the course of 500 independent docking simulations peptide conformations and orientations are sampled in a parallelepiped that encompasses the binding site obtained from the crystallographic structure of the HIV-1 PR complexes with a large 20.0 Å cushion added to every side of this box surrounding the interface. The protein structure is held fixed in its minimized and equilibrated conformation, while rigid-body degrees of freedom and the peptide rotatable angles are treated as independent variables. Each member of the evolving population represents an encoded vector consisting of the rigid-body coordinates and the torsional angles about the peptide rotatable bonds. The initial peptide conformations are generated by randomizing the encoded vector, where the center of mass of the ligand is restricted to the rectangular parallelepiped that encompasses the HIV-1 PR crystal structures. The three rigid-body rotational degrees of freedom, as well as the torsional angles for all rotatable bonds are uniformly initialized between 0 and 360°. Bonds allowed to rotate include those linking sp3 hybridized atoms to either sp3 or sp2 hybridized atoms and single bonds linking two sp2 hybridized atoms. The employed coarse-grained modeling allows
An unbiased sampling of the large conformational space which entirely encompasses the ensemble of the HIV-1 PR conformations.
Effectively characterize the multitude of the binding modes for the folded inhibitor and determine putative binding interfaces of the inhibitors.
Molecular dynamics simulations
We have carried out two separate 10-ns simulations using the predicted folding and dimerization peptide binding modes with the HIV-1 PR monomers. MD simulations were done using NAMD 2.6 with the CHARMM22 force field (69). The respective systems were solvated in cubic boxes of TIP3P water, which extended at least 15 Å away from any given protein atom. The systems were then neutralized by adding the counterions Na+ and Cl− using Autoionize plug-in of VMD. The equilibration stage of simulations included:
Energy minimization of the protein for 5000 steps of steepest descent minimization converging to a value of 2000 kJ mol−1 nm−1.
Solvation and energy minimization of the system for 10,000 steps.
The entire system was subjected to a gradual temperature increase from 30 K to 300 K in intervals over 15 ps by increasing the temperature of Langevin damping and Langevin piston by 20 K in each step.
An NPT ensemble was used with periodic boundary conditions. Pressure was maintained at 1 atm using the Langevin piston method with a damping coefficient of 10 ps−1. The entire system was then equilibrated for 500 ps. Electrostatic interactions were computed using the particle-mesh Ewald algorithm (70). The list of nonbonded interactions was truncated at 10 Å and a switching cutoff distance of 8 Å is used for the Lennard-Jones interactions. All bonds involving hydrogen atoms were constrained using the SHAKE algorithm. A time-step of 2 fs was employed. During the productive phase, pressure and temperature were maintained at 1 atmosphere and 300 K respectively by using the Langevin piston method with a damping coefficient of 1 ps−1.
Binding free energy calculations
Binding free energies computations are done using the molecular mechanics AMBER force field (71) and the solvation energy term based on continuum generalized-Born and solvent-accessible surface area (GB/SA) solvation model (72,73). The details of the MM/GBSA model used in free energy simulations of biological systems have been extensively documented in our earlier studies (74–78). Here, for completeness of the presentation, we briefly outline the major ingredients of the computational model used in this study.
The binding free energy of the ligand-protein complex is computed as
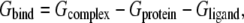 |
(1) |
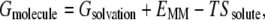 |
(2) |
where Gsolvation is the solvation free energy, EMM is the molecular mechanical energy of the molecule summing up the electrostatic Ees interactions, van der Waals contributions Evdw, and the internal strain energy Eint
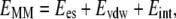 |
(3) |
Solvation free energy Gsolvation can be divided into nonpolar Gnonpol and polar Gpol components,
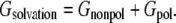 |
(4) |
The nonpolar contribution Gnonpol to the solvation free energy is computed as a function of the solvent accessible surface area (SA),
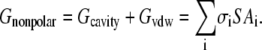 |
(5) |
In the GB/SA model, the Gcavity and Gvdw contributions are combined together via evaluating solvent-accessible surface areas.
The polar contribution Gpol to the solvation energy is computed as follows:
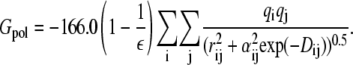 |
(6) |
The quasiharmonic approximation is typically used in the MM/GBSA calculations to assess the order of magnitude for the entropy loss during binding. Ssolute is the vibrational entropy of the molecule. The normal mode analysis is carried out and vibrational entropy is computed from classical statistical mechanics formula with the AMBER module nmode for the energy-minimized structures of the complex, the free protein, and free peptide without water molecules. A dielectric constant of 4rij, where rij is the distance between atoms i and j of the molecule, is used in the normal mode calculations.
Binding free energy evaluations can be performed using either separate trajectories of the complex, protein, and ligand (separate-trajectory protocol) or from a trajectory of the complex (single-trajectory protocol) (79). A single-trajectory protocol utilizes only the MD simulation of the complex, followed by separating a single trajectory of the complex into snapshots of the complex, the free protein, and the free ligand. In contrast, a separate-trajectory method involves analysis of three different independent MD simulations and uses the respective snapshots of the complex, the free protein, and the free ligand to compute the free energy of binding. These calculations are physically more meaningful and should, in principle, produce more accurate binding free energies, but at the potential expense of somewhat larger error bars. We have used both a single-trajectory protocol and a separate-trajectory method to compare the quality and consistency of the predictions using these approaches. Binding free energy evaluations are performed using MD trajectories of the p-S8 peptide complexes with the HIV-1 PR monomers in the folding and dimerization binding modes. In a single-trajectory protocol, the structures for the uncomplexed protease monomer and the peptide are generated by using the corresponding samples of the complex and separating the protein and peptide coordinates, followed by an additional minimization of the unbound protease monomer and unbound inhibitor. A single-trajectory protocol is less computationally intensive and may also be less sensitive because of cancellation of the intramolecular energies, caused by neglecting the effects of structural adaptation during binding. In a separate-trajectory protocol, we have performed three independent MD simulations of the complex, the peptide inhibitor, and the HIV-1 PR monomer. The obtained samples of the complex, the inhibitor, and the protease monomer have been used to compute the binding free energies. We assume that the predicted structures of the p-S8 peptide in complexes with the HIV-1 PR monomers are robust against minor chemical modifications of the truncated 83–92 analog NIIGRNLLTQ and the N88D mutant NIIGRDLLTQI. We have verified that this assumption may be indeed valid for our systems, because the equilibrium sampling of the active peptide analogs on a timescale of up to 2 ns follows closely the trajectory of the p-S8 folding inhibitor (data not shown). Consequently, the MM/GBSA binding free energy calculations of the peptide analogs have been performed combining the results of two independent long 10-ns MD trajectories of the wild-type p-S8 peptide in the folding and dimerization modes of inhibition. A respective mutation to the p-S8 peptide is introduced into each of the system snapshots and followed with the 10,000 steps of energy minimization.
RESULTS AND DISCUSSION
Coarse-grained peptide docking with the ensemble of the HIV-1 PR monomers
Molecular docking simulations of the p-S8 inhibitor have been conducted using a simplified, knowledge-based energy model with the ensembles of the HIV-1 PR dimers and monomers. Simulations were first carried out with the internally rigid peptide, kept in its native folded conformation and then with the fully flexible peptide, given the initial randomly distributed and fully extended inhibitor conformations. In this coarse-grained docking method, the explicit flexibility of the inhibitor is simulated given the implicit flexibility of the receptor, which is provided by structural differences within the ensemble of the HIV-1 PR conformations. This approach expands the realm of structural receptor models in simulations and assumes the presence of an ensemble of the multiple conformational states for both interacting molecules, which is similar to a conformational-selection model of biomolecular binding (80–83). Although there may exist certain differences in the timescale of the flap motions in the HIV-1 PR monomeric and dimeric states, the tertiary folds of the HIV-1 PR monomer, and a single subunit of the HIV-1 PR dimer are very similar, and can therefore exhibit a similar dynamic exchange between closed, open, and semiopen forms of the enzyme. On the other hand, global conformational fluctuations of the enzyme, such as those revealed by x-ray crystallographic studies, are difficult to directly probe in the flexible docking simulations. The proposed coarse-grained molecular docking of the flexible p-S8 peptide with the ensembles of HIV-1 PR structures and subsequent binding free energy refinement of the low-energy docking solutions presents a robust, yet an opportunistic approach for predicting binding modes of the inhibitor.
Structural analysis has revealed that the folded p-S8 peptide cannot be accommodated in the active site of the HIV-1 PR dimer, even with the flaps in their semiopen (pdb entry 1HHP) and wide-open (pdb entry 1tw7) forms. In contrast, we have discovered that low-energy complexes of the folded peptide can be preferably formed with the HIV-1 PR monomers, which may facilitate the inhibitor intervention with the HIV-1 PR folding and dimerization. Based on these preliminary insights and hypothesis, we have proceeded by simulating the p-S8 peptide folding and binding with the HIV-1 PR monomers. A series of 500 independent fully flexible docking simulations of the p-S8 peptide (with a total of 55 flexible bonds treated as independent degrees of freedom) have been performed with the ensemble of HIV-1 PR monomeric states. The binding free energy refinement of the coarse-grained docking solutions has identified two dominant classes of low-energy peptide conformations with the binding energetics in the range of −6 kcal/mol to −8 kcal/mol (Fig. 2). These structurally different, low-energy binding modes of the inhibitor are formed by either targeting the conserved segment 24–34 of the enzyme folding nucleus (folding inhibition mechanism) (Fig. 3, A and B) or interacting with the antiparallel β-sheet of the termini (dimerization inhibition mechanism) (Fig. 3, C and D). The low-energy peptide conformations correspond to a folded, α-helix-based structure of the 83–93 protease fragment and suggest a possibility of folding and binding coupling (80–83), according to which folding of the peptide may be required to ensure structural mimicry with the key elements of the HIV-1 PR dimer interface. The root mean-square deviations (RMSD) of the docked conformations are reported in the reference to the lowest energy structure of the p-S8 inhibitor corresponding to the folding mode of inhibition and reflect two structural clusters of low-energy docking solutions (Fig. 2 B). The docking results suggest that there may exist two dominant binding modes of the inhibitor: the first one corresponds to the folding mode of inhibition (conformations residing within RMSD = 2.0–2.5 Å from the reference conformation and binding energies in the range of −6 kcal/mol to −8 kcal/mol) and the second dimerization mode of binding (conformations residing within much larger RMSD = 6.0 Å from the reference conformation and similar energetics in the range of −6 kcal/mol to −8 kcal/mol).
FIGURE 2.

(A) The MM/GBSA binding free energies as a function of simulation run obtained from 500 flexible docking simulations. (B) The RMSD values of the docked conformations are reported in the reference to the lowest energy structure of the p-S8 inhibitor corresponding to the folding mode of inhibition and reflect two structural clusters of low-energy docking solutions.
FIGURE 3.

Structural characterization of the peptide-binding modes in complexes with the HIV-1 monomers. (A) The thermodynamically stable conformation of the rigid p-S8 peptide (in light blue) in the complex with the HIV-1 monomer (orange ribbons). The predicted folded conformation of the flexible p-S8 peptide (in yellow) binds to the HIV-1 PR monomer (green ribbons) in the same binding mode as determined in simulations with the rigid peptide. The bound peptide conformations overlayed with the conformation of the second HIV-1 PR monomer (violet ribbons) reveals structural mimicry with the fireman's-grip hydrogen-bond network present in the active dimer. (B) A closeup of the binding interface and specific interactions formed by the folded peptide in the folding mode of inhibition. (C) An alternative binding mode of the rigid p-S8 peptide (in light blue) and folded conformation of the flexible p-S8 peptide (in yellow) in the complex with the HIV-1 monomer (orange ribbons). The bound peptide conformations overlayed with the conformation of the second HIV-1 PR monomer (violet ribbons) mimics another important region of the dimerization interface. (D) A closeup of the binding interface and specific interactions formed by the folded peptide in the dimerization mode of inhibition. Atom-based representation of the peptide is shown in default colors and HIV-1 PR monomer conformations are shown in green ribbons.
In the first low-energy binding mode (referred to in the text as the folding binding mode), the folded p-S8 peptide is packed perpendicular to the conserved 24–34 fragment of the folding core (Fig. 3 A). The network of intermolecular hydrogen bonds which may be formed in the folding binding mode (Fig. 3 B) includes:
Interactions between carbonyl oxygen of Ile-84 from the peptide and backbone NH of Gly-27.
Interactions between the carboxylic acid group of the Asn-83 side chain from the peptide and the side-chain hydroxyl group of Thr-26.
Interactions between the side chains of the peptide Asp-88 and catalytic Asp-25.
Moreover, hydrogen bonds may be formed between the inhibitor and the backbone of the conserved Gly-49 and Ile-50 residues, which are unlikely to be mutated in the active protease variants (Fig. 3 B). The predicted hydrogen-bond interactions, which may be formed by the folded peptide with Asp-25, Thr-26, and Gly-27, mimic closely the fireman's-grip network of hydrogen bonds in the active HIV-1 PR dimer (Fig. 3, A and B). The predicted alternative mode of the peptide binding (referred in the text as the dimerization binding mode; see also Fig. 3, C and D) is prevalent in rationalizing dimerization inhibition mechanism and was recently confirmed by NMR experiments for a class of peptidic dimerization inhibitors (84). In this binding mode, the folded p-S8 peptide can interact with the residues from the anti-parallel β-sheet of the HIV-1 PR monomer and mimics a (Fig. 3 C). The specific interactions formed by the peptide in the dimerization binding mode include:
Hydrogen bonds formed by the side-chain amide group of the peptide Asn-83 with the Thr-96 and Ile-3 monomer residues.
Hydrogen-bonding formed between NH of Ile-85 and carbonyl oxygen of the protease Asn-98.
Interactions between the amide group of the Asn-88 carboxamide side chain and carbonyl oxygen of Leu-5 (Fig. 3 D).
The predicted binding modes of the p-S8 peptide with the HIV-1 PR monomers specify the low-energy structures, which may be formed by the folding inhibitor and provide a rationale for both folding and dimerization modes of inhibition. According to the proposed structural models of binding, the folding inhibitor may target the conserved HIV-1 PR regions mimicking specific interactions in the active HIV-1 PR dimer and thereby facilitate the inhibitor intervention with the HIV-1 PR folding and dimerization. In particular, folding mode of the p-S8 peptide inhibition may ensure structural mimicry of the binding interface with the fireman's-grip architecture of the monomer-monomer protease interactions (Fig. 4). The folding and binding coupling mechanism of the peptide inhibition is also consistent with a plausible scenario of the HIV-1 PR monomer destabilization through interfering with the formation of the HIV-1 PR folding core.
FIGURE 4.

A closeup of the dominant binding interface determined for the rigidly docked peptide NIIGRNLLTQI (shown in grayscale ribbons) in the complex with the HIV-1 PR monomer (A) and a closeup of the binding interface determined for the flexibly docked peptide (shown in grayscale ribbons)in the complex with the HIV-1 PR monomer (B). The predicted peptide structures reveal structural mimicry with the fireman's-grip hydrogen-bond network of the active dimer, which is depicted by the superposition with the segment 24–27 of the bound HIV-1 monomer.
All-atom molecular dynamics simulations of the peptide binding with the HIV-1 PR monomers
All-atom MD simulations of the inhibitor binding with the HIV-1 PR monomer are then performed to verify thermodynamic stability of the inhibitor complexes and suggest a possible mechanism of inhibiting HIV-1 PR folding and dimerization. In principle, a single long simulation of peptide binding with the HIV-1 PR monomers should ultimately result in conformational transitions between low-energy binding modes and allow to accurately evaluate the relative contribution of the folding and dimerization modes of inhibition to the thermodynamic equilibrium. In practice, however, it is unrealistic to expect MD simulations to adequately sample, structurally, very different binding interfaces of the peptide with the ensemble of HIV-1 PR monomers. Consequently, we have conducted separate 10 ns all-atom MD simulations, initiated from the predicted low-energy complexes corresponding respectively to the folding and dimerization binding modes of the p-S8 peptide. In these equilibrium MD simulations, which are subsequently used to calculate binding affinities of the folding inhibitors, flexibility of both the inhibitor and the receptor are explicitly considered. The atomic details of the peptide inhibition mechanism are determined by the equilibrium distribution of the inhibitor and protease monomer conformational states in the thermodynamically stable complexes.
Examination of the MD trajectories shows that after an ∼1-ns period of relaxation, the folding binding mode of the peptide complex with the HIV-1 PR monomer results in stable backbone fluctuations of RMSD = ∼1–1.5 Å for both the monomer and the peptide during the simulations (Fig. 5 A). The respective thermal fluctuations of the complex in the dimerization mode of inhibition can result in larger deviations throughout simulation period (Fig. 5 B). While the p-S8 peptide can experience equilibrium fluctuations in the range of 1.5 Å the monomer conformations can undergo larger fluctuations reaching 2.5–3.0 Å range (Fig. 5, A and B). To monitor the effect of the peptide binding on dynamics of the HIV-1 PR monomer residues, the root mean-square fluctuation (RMSF) values were also calculated, providing an estimate of the average residue fluctuations taken over simulation time (Fig. 5, C and D). In agreement with the existing body of structural and computational data, the termini, and the flap regions of the HIV-1 PR monomers retain a significant degree of flexibility in the course of simulations (Fig. 5 C). Furthermore, according to our results, the HIV-1 PR monomer termini tend to display considerably larger thermal fluctuations than the conserved protease segments 24–34 and 83–93 of the folding core (Fig. 5 C). This result is also consistent with the NMR structures of the HIV-1 PR monomers, suggesting a degree of disorder in the termini residues (37–40). However, the amplitude of the termini motions tends to decrease rather markedly after 7–8 ns of the simulation period, due to the progressive stabilization of the interactions between the inhibitor and the antiparallel region of the HIV-1 PR monomer (Fig. 5, A and B). On average, MD simulations of the peptide binding resulted in a more rigid form of the HIV-1 PR monomer in the folding mode of inhibition, whereas the dimerization mode produced increased fluctuations of the monomer residues (Fig. 5 C). Conversely, the thermal fluctuations of the p-S8 peptide residues are rather moderate in the dimerization mode of inhibition (Fig. 5 D). These results indicate that by stabilizing the secondary structure of the monomer termini and mimicking a considerable portion of the dimer interface, the folded p-S8 peptide may interfere with the dimerization of HIV-1 PR and therefore function as a disassociation inhibitor. These conclusions corroborate with the previous simulations of the HIV-PR monomer in water, which have demonstrated that the termini residues become assembled into a stable β-sheet on a longer timescale and become considerably less flexible (43). Consequently, a mechanism of structural mimicry and targeting hot spot residues may facilitate the inhibitor intervention with the assembly of the folding nucleus and HIV-1 PR dimerization.
FIGURE 5.

(A) The RMSD values for Cα atoms along the 10-ns trajectory corresponding to the folding inhibition mode. Time evolution of the peptide is shown in blue; time evolution of the HIV-1 PR monomer is shown in red. (B) The RMSD values for Cα atoms along the 10-ns trajectory corresponding to the dimerization inhibition mode. Time evolution of the peptide is shown in blue; time evolution of the HIV-1 PR monomer is shown in red. (C) The RMSF values of the HIV-1 PR monomer residues from 10-ns trajectories corresponding respectively to the folding inhibition mode (shown in blue) and dimerization mode (shown in red). (D) The RMSF values of the peptide residues (using numbering of the 83–93 protease monomer segment) from 10-ns trajectories corresponding respectively to the folding inhibition mode (shown in blue) and dimerization mode (shown in red).
According to many reported simulations (29,30), the opening process of the entire flap region may be triggered by curling of the flap tips. The ability of certain mutants to alter the equilibrium between the open and closed conformations, or to favor a semiopen form may lead to the drug resistant forms of the enzyme. In particular, 70-ns MS simulations in explicit solvent (30), which were performed on a multiple drug-resistant mutant and a wild-type HIV-1 PR, have confirmed that semiopen and open enzyme forms are the two stable forms found in the course of simulations. In our study, we have evaluated the extent of flap motions in simulations by measuring the distance between residue Ile-50 (Cα) located at the tip of the flap and the Cβ of the catalytic residue Asp-25 (Fig. 6, A and B). According to a previous definition proposed in Perryman et al. (17) and obtained from a crystal structure of a semiopen form of the HIV-1 PR, an I50 (Cα)-D25(Cβ) distance >15.8 Å would correspond to a ensemble of semiopen conformations. A comparison of the respective distances obtained from simulations with the folding inhibition mode (Fig. 6 A) and dimerization mode of binding (Fig. 6 B) suggests a similar dynamic behavior of the HIV-1 PR monomer flap in these inhibitor complexes. Conformational transitions between different flap conformations of the HIV-1 PR monomer have been monitored by measuring time evolution of the distances between Ile-50 (Cα) and the Cβ of the catalytic residue Asp-25 (Fig. 6) and have revealed equilibrium fluctuations between semiopen and open forms. Furthermore, blocking the binding interface and intervening with the consolidation of the HIV-1 PR folding core may lead to an additional stabilization of a semiopen flap conformation. The respective distances at the end of the simulation are in the range from 16 Å to 18 Å (Fig. 6, A and B), which is very similar to the distances observed in a previous 22-ns simulation of the V82F/I84V double mutant (17). The dynamics of the HIV-1 PR monomer observed in our simulations also closely resembles the conformational fluctuations observed for the HIV-1 PR monomers in 70-ns simulations of the wild-type HIV-1 PR, which predominantly sample a semiopen form of the enzyme and less frequently an open form, with average flap-tip to catalytic residue distances at 18 Å for the monomer A and 21 Å for the monomer B (30). Indeed, the results of our 10-ns simulations have shown a similar consistent emergence of the medium-range distances (15–22 Å) and a semiopen monomer ensemble in simulations with rare excursions into long-range distances (22–25 Å), corresponding to an open form. In addition, we have also estimated thermal fluctuations of the folding core 24–34 and 83–93 segments by monitoring time evolution of the distance between centers of mass of these segments (Fig. 6, C and D). A greater flexibility of the HIV-1 PR monomer folding core during simulations of the folding inhibition mode (Fig. 6 C) may be caused by the peptide interactions with the segment 24–34 of the monomer, partly interfering with the optimal packing of the folding core. The superposition of the protease monomer conformations corresponding to the MD snapshots after every 1 ns in the folding mode (Fig. 7 A) and dimerization mode of binding (Fig. 7 C) illustrates the extent of flexibility in the monomer flap during simulations. In agreement with the NMR studies of the protease monomer, the average structures of the peptide-monomer complex in the folding inhibition mode (Fig. 7 B) and dimerization binding mode (Fig. 7 D) do not produce noticeably wider flap fluctuations compared to that of the dimer. Overall, the results of MD simulations have confirmed thermodynamic stability of the inhibitor complexes with the HIV-1 PR monomers, which may have a functional role in inhibiting HIV-1 PR folding and dimerization.
FIGURE 6.

Time evolution of the distance between Ile-50 (Cα) and the Cβ of the Asp-25 in the course of 10-ns MD simulations corresponding to the folding inhibition mode (A) and dimerization inhibition mode (B). Time evolution of the distance between centers of mass of the protease monomer segments 24–34 and 83–93. The data gathered from 10-ns MD simulations corresponding to the folding inhibition mode (C) and dimerization inhibition mode (D).
FIGURE 7.

The superposition of the 10 HIV-1 PR monomer conformations corresponding to the MD snapshots after each 1 ns during 10-ns simulation of the folding-binding mode (A) and dimerization-binding mode (C). The average structure of the peptide complex with the HIV-1 PR monomer obtained from 10-ns MD trajectory in the folding mode of inhibition (B) and dimerization mode of inhibition (D).
The experimental data have indicated that the active peptide analogs can also inhibit the protease folding and formation of the active dimer (48–52). These peptide variants have demonstrated a similar activity profile to the original p-S8 inhibitor, with only a marginal twofold decrease in the binding affinity. We have performed binding free energy calculations of the p-S8 peptide and the active inhibitor analogs using MD trajectories of the p-S8 complexes with the HIV-1 PR monomers in both the folding and dimerization binding modes. Furthermore, to compare the quality and consistency of the energetic predictions, binding affinity evaluations have been carried out using different MM/GBSA protocols, which include analysis of the separate trajectories of the complex, protein, and the inhibitor (a separate-trajectory approach) and analysis of the trajectory of the complex (a single-trajectory approach) (Fig. 8). In agreement with the experiment, the results of MM/GBSA calculations have consistently revealed that truncation of the hydrophobic Ile-93 residue does not cause a considerable change in binding affinity, as this residue is only peripherally involved in the interaction network at the binding interface. A structurally predicted loss of a single hydrogen bond in the N88D peptide mutant results in a greater decrease of activity. Importantly, MD simulations of the p-S8 complex (a single trajectory approach) and an isolated p-S8 peptide (a separate trajectory approach) converge to a similar stable, α-helix-containing structure of the inhibitor. These data also agree with the earlier simulations of the p-S8 peptide in explicit solvent (52), which have shown that a stable, folded conformation of the p-S8 peptide in solution is similar to the p-S8 bound conformation in the complex with the pS2 peptide. Consequently, the binding free energy analysis derived from single-trajectory (Fig. 8 A) and separate-trajectory (Fig. 8 B) protocols is rather similar, as the effect of structural adaptation for the p-S8 peptide during binding is quite moderate. Interestingly, the binding free energies computed using a single-trajectory MM/GBSA protocol underestimate somewhat the absolute binding affinities (Fig. 8 A), whereas the binding free energies obtained from separate trajectories tend to slightly overestimate the binding affinities of the peptides (Fig. 8 A). Overall, the computed binding affinities are in agreement with the experimental values and provide an additional support to the proposed molecular basis of the inhibition mechanism. While we have been able to reproduce not only the overall trend in binding affinities, but also changes in the binding free energy values, it is worth stressing that the exact values of small binding free energies differences, which are on the scale of thermal fluctuations, should be taken with some caution as the results can be affected by numerous factors, including the force field and the length of MD simulations. Nevertheless, a comparative analysis of the binding free energies using both a single-trajectory and a separate-trajectory protocols provides a rather convincing evidence to
The consistency in predicting the correct trend of the binding affinity reduction in the active peptide analogs.
The effect of small deleterious changes in activity caused by minor chemical modifications in the inhibitor.
FIGURE 8.

Binding free energy analysis of the inhibitor binding using a single-trajectory protocol within the MM/GBSA approach (A) and a separate-trajectory protocol within the MM/GBSA approach (B). The experimental binding free energy values are displayed in solid bars and the computed binding free-energy values are displayed in shaded bars.
Structural mimicry in binding with the HIV-1 PR monomers
The results of our study suggest that folding and binding coupling during molecular recognition of the peptide may be necessary to ensure structural mimicry with the key elements of the dimer interface. According to the proposed mechanism, the folding inhibitor may target the conserved HIV-1 PR regions and thereby intervene with the HIV-1 PR folding and dimerization. Interestingly, a network of specific interactions formed by the folded p-S8 peptide with the HIV-1 PR monomers does not usually include contribution of highly structured residues from an α-helix region. Instead, the flexible portion of the peptide may mimic the fireman's-grip hydrogen-bond network and stabilize the antiparallel β-sheet of the termini. A more rigid, α-helix region of the folded peptide provides a considerable degree of structural mimicry with the second monomer at the binding interface. These results are consistent with a fly-casting mechanism of molecular recognition, which postulates that even in the presence of stable folded monomers, a flexible region of one of the binding partners may be utilized to bind weakly and speed up the reaction, followed by a folding transition to the final complex (85–87). Furthermore, in agreement with the principle of minimal frustration, the local interactions formed by the folding inhibitor in complexes in complexes with the HIV-1 PR monomers are similar to the respective interactions detected in simulations with an isolated 24–34 segment (52). Despite a diversity of the binding scenarios, the prevailing mechanism of folding inhibition may be determined by the unfrustrated inhibitor interactions with the conserved segment 24–34 of the HIV-1 PR folding nucleus. The proposed mechanism of targeting the assembly of the HIV-1 PR folding nucleus with the unfrustrated inhibitor may alleviate the emergence of drug resistant strains due a reduced probability to evolve mutants at the conserved sites.
Although binding affinity of the p-S8 peptide inhibitor is rather moderate compared to highly potent active site drugs, mechanistic details of the folding inhibition mechanism emerged from simulations may be utilized for improving binding potency and reducing peptidic nature of the folding inhibitor. We are currently pursuing structure-based design of peptido-mimetics of the p-S8 peptide by retaining the recognition component of the inhibitor, which is expected to mimic the fireman's-grip hydrogen-bond network or stabilize the antiparallel β-sheet of the termini. The peptidic nature of the inhibitor is reduced through modifications of the α-helix region. We suggest that in silico screening of the peptide analogs for elements of structural mimicry with the HIV-1 PR dimer interface may assist experimental techniques to probe alternative inhibitory mechanisms, based on blocking the assembly of the HIV-1 PR homodimer and potentially identifying new classes of dimerization HIV-1 PR inhibitors. Exploring binding specificities of the active peptide analogs to retroviral proteases may lead to more selective inhibitors, since the Gly-86/Arg-87/Asn-88 triad in the 83–93 segment is rather unique to this class of proteases and constitutes a crucial structural element involved in dimerization of the mature HIV-1 PR. The low dimer stability of the HIV-1 PR precursor relative to the mature protease, which is essential to allow initial recruitment of the polyproteins, may be arguably an apparent effect of the equilibrium between the partially folded and the folded, enzymatically active HIV-1 PR dimer (88). In a followup study, we are also planning to investigate whether the inhibitory function of the folding HIV-1 PR inhibitor could modulate equilibrium among HIV-1 PR partially unfolded, monomeric, and dimeric states. These studies may ultimately enable design of novel inhibitors that target the folded precursor monomer before the maturation of the HIV-1 protease, in contrast to inhibiting the dimerization of the mature protease.
CONCLUSIONS
The molecular basis of the HIV-1 protease inhibition through intervening with the protein folding and dimerization remains largely unknown, despite rapidly increasing experimental and theoretical efforts in this area. Considering experimental difficulties in probing the molecular basis of folding inhibition, we have proposed a modeling approach for understanding the molecular basis of the HIV-1 PR folding inhibition at atomic details. Using a combination of coarse-grained molecular docking, MD simulations, and binding free energy analysis, we have presented evidence that the folding inhibitor may exhibit both folding and dimerization modes of inhibition by targeting conservative elements of the HIV-1 PR. Coarse-grained molecular docking has shown that the p-S8 peptide can fold into an α-helix-based structure of the 83–93 fragment to ensure structural mimicry with the key elements of the dimer interface. All-atom MD simulations of the inhibitor folding and binding with the HIV-1 PR monomers provide atomic-level details of the putative binding mechanism, which may be fulfilled via the folding mode of inhibition (with the peptide targeting the conserved segment 24–34 of the enzyme-folding nucleus) and dimerization mode (with the peptide interacting with the antiparallel β-sheet of the termini). A mechanism of structural mimicry with the binding interface of the active dimer may reduce drug resistance due to binding to the conserved regions important for protease folding and stability. The performed microscopic analysis reconciles the experimental and computational results and rationalizes the molecular basis of folding inhibition for the active peptide analogs.
Editor: Ruth Nussinov.
References
- 1.Kohl, N. E., E. A. Emini, W. A. Schleif, L. J. Davis, J. C. Heimbach, R. A. Dixon, E. M. Scolnick, and I. S. Sigal. 1988. Active human immunodeficiency virus protease is required for viral infectivity. Proc. Natl. Acad. Sci. USA. 85:4686–4690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Seelmeier, S., H. Schmidt, V. Turk, and K. von der Helm. 1988. Human immunodeficiency virus has an aspartic-type protease that can be inhibited by pepstatin A. Proc. Natl. Acad. Sci. USA. 85:6612–6616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.McQuade, T. J., A. G. Tomasselli, L. Liu, V. Karacostas, B. Moss, T. K. Sawyer, R. L. Heinrikson, and W. G. Tarpley. 1990. A synthetic HIV-1 protease inhibitor with antiviral activity arrests HIV-like particle maturation. Science. 247:454–456. [DOI] [PubMed] [Google Scholar]
- 4.Wlodawer, A., and J. Vondrasek. 1998. Inhibitors of HIV-1 protease: a major success of structure-assisted drug design. Annu. Rev. Biophys. Biomol. Struct. 27:249–284. [DOI] [PubMed] [Google Scholar]
- 5.Kent, S., G. R. Marshall, and A. Wlodawer. 2000. Determining the 3D structure of HIV-1 protease. Science. 288:1590. [DOI] [PubMed] [Google Scholar]
- 6.Vondrasek, J., and A. Wlodawer. 2002. HIVdb: a database of the structures of human immunodeficiency virus protease. Proteins. 49:429–431. [DOI] [PubMed] [Google Scholar]
- 7.Gustchina, A., M. Jaskolski, and A. Wlodawer. 2006. Lessons learned fighting HIV can be applied to anti-cancer drug design. Cell Cycle. 5:463–464. [DOI] [PubMed] [Google Scholar]
- 8.Martin, P., J. F. Vickrey, G. Proteasa, Y. L. Jimenez, Z. Wawrzak, M. A. Winters, T. C. Merigan, and L. C. Kovari. 2005. “Wide-open” 1.3 Å structure of a multidrug-resistant HIV-1 protease as a drug target. Structure. 13:1887–1895. [DOI] [PubMed] [Google Scholar]
- 9.Ishima, R., D. I. Freedberg, Y. X. Wang, J. M. Louis, and D. A. Torchia. 1999. Flap opening and dimer-interface flexibility in the free and inhibitor-bound HIV protease, and their implications for function. Structure. 7:1047–1055. [DOI] [PubMed] [Google Scholar]
- 10.Freedberg, D. I., R. Ishima, J. Jacob, Y.-X. Wang, I. Kustanovich, J. M. Louis, and D. A. Torchia. 2002. Rapid structural fluctuations of the free HIV protease flaps in solution: relationship to crystal structures and comparison with predictions of dynamics calculations. Protein Sci. 11:221–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Katoh, E., J. M. Louis, T. Yamazaki, A. M. Gronenborn, D. A. Torchia, and R. Ishima. 2003. A solution NMR study of the binding kinetics and the internal dynamics of an HIV-1 protease-substrate complex. Protein Sci. 12:1376–1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Louis, J. M., R. Ishima, D. A. Torchia, and I. T. Weber. 2007. HIV-1 protease: structure, dynamics, and inhibition. Adv. Pharmacol. 55:261–298. [DOI] [PubMed] [Google Scholar]
- 13.Collins, J. R., S. K. Burt, and J. W. Erickson. 1995. Flap opening in HIV-1 protease simulated by “activated” molecular dynamics. Nat. Struct. Biol. 2:334–338. [DOI] [PubMed] [Google Scholar]
- 14.Hamelberg, D., and J. A. McCammon. 2005. Fast peptidyl cis-trans isomerization within the flexible Gly-rich flaps of HIV-1 protease. J. Am. Chem. Soc. 127:13778–13779. [DOI] [PubMed] [Google Scholar]
- 15.Kurt, N., W. R. P. Scott, C. A. Schiffer, and T. Haliloglu. 2003. Cooperative fluctuations of unliganded and substrate-bound HIV-1 protease: a structure-based analysis on a variety of conformations from crystallography and molecular dynamics simulations. Proteins. 51:409–422. [DOI] [PubMed] [Google Scholar]
- 16.Scott, W. R., and C. A. Schiffer. 2000. Curling of flap tips in HIV-1 protease as a mechanism for substrate entry and tolerance of drug resistance. Structure. 8:1259–1265. [DOI] [PubMed] [Google Scholar]
- 17.Perryman, A. L., J.-H. Lin, and J. A. McCammon. 2004. HIV-1 protease molecular dynamics of a wild-type and of the V82F/I84V mutant: possible contributions to drug resistance and a potential new target site for drugs. Protein Sci. 13:1108–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Perryman, A. L., J.-H. Lin, and J. A. McCammon. 2006. Restrained molecular dynamics simulations of HIV-1 protease: the first step in validating a new target for drug design. Biopolymers. 82:272–284. [DOI] [PubMed] [Google Scholar]
- 19.Perryman, A. L., J.-H. Lin, and J. A. McCammon. 2006. Optimization and computational evaluation of a series of potential active site inhibitors of the V82F/I84V drug-resistant mutant of HIV-1 protease: an application of the relaxed complex method of structure-based drug design. Chem. Biol. Drug Des. 67:336–345. [DOI] [PubMed] [Google Scholar]
- 20.Toth, G., and A. Borics. 2006. Closing of the flaps of HIV-1 protease induced by substrate binding: a model of a flap closing mechanism in retroviral aspartic proteases. Biochemistry. 45:6606–6614. [DOI] [PubMed] [Google Scholar]
- 21.Toth, G., and A. Borics. 2006. Flap opening mechanism of HIV-1 protease. J. Mol. Graph. Model. 24:465–474. [DOI] [PubMed] [Google Scholar]
- 22.Meagher, K. L., and H. A. Carlson. 2005. Solvation influences flap collapse in HIV-1 protease. Proteins. 58:119–125. [DOI] [PubMed] [Google Scholar]
- 23.Chang, C.-E., T. Shen, J. Trylska, V. Tozzini, and J. A. McCammon. 2006. Gated binding of ligands to HIV-1 protease: Brownian dynamics simulations in a coarse-grained model. Biophys. J. 90:3880–3885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Trylska, J., V. Tozzini, C.-e. A. Chang, and J. A. McCammon. 2007. HIV-1 protease substrate binding and product release pathways explored with coarse-grained molecular dynamics. Biophys. J. 92:4179–4187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chang, C.-E. A., J. Trylska, V. Tozzini, and J. A. McCammon. 2007. Binding pathways of ligands to HIV-1 protease: coarse-grained and atomistic simulations. Chem. Biol. Drug Des. 69:5–13. [DOI] [PubMed] [Google Scholar]
- 26.Tozzini, V., J. Trylska, C.-e. A. Chang, and J. A. McCammon. 2007. Flap opening dynamics in HIV-1 protease explored with a coarse-grained model. J. Struct. Biol. 157:606–615. [DOI] [PubMed] [Google Scholar]
- 27.Hornak, V., A. Okur, R. C. Rizzo, and C. Simmerling. 2006. HIV-1 protease flaps spontaneously close to the correct structure in simulations following manual placement of an inhibitor into the open state. J. Am. Chem. Soc. 128:2812–2813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hornak, V., A. Okur, R. C. Rizzo, and C. Simmerling. 2006. HIV-1 protease flaps spontaneously open and reclose in molecular dynamics simulations. Proc. Natl. Acad. Sci. USA. 103:915–920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lauria, A., M. Ippolito, and A. M. Almerico. 2007. Molecular dynamics studies on HIV-1 protease: a comparison of the flap motions between wild-type protease and the M46I/G51D double mutant. J. Mol. Model. 13:1151–1156. [DOI] [PubMed] [Google Scholar]
- 30.Seibold, S. A., and R. I. Cukier. 2007. A molecular dynamics study comparing a wild-type with a multiple drug resistant HIV protease: differences in flap and aspartate 25 cavity dimensions. Proteins. 69:551–565. [DOI] [PubMed] [Google Scholar]
- 31.Ode, H., S. Matsuyama, M. Hata, S. Neya, J. Kakizawa, W. Sugiura, and T. Hoshino. 2007. Computational characterization of structural role of the non-active site mutation M36I of human immunodeficiency virus type 1 protease. J. Mol. Biol. 370:598–607. [DOI] [PubMed] [Google Scholar]
- 32.Hou, T., and R. Yu. 2007. Molecular dynamics and free energy studies on the wild-type and double mutant HIV-1 protease complexed with amprenavir and two amprenavir-related inhibitors: mechanism for binding and drug resistance. J. Med. Chem. 50:1177–1188. [DOI] [PubMed] [Google Scholar]
- 33.Foulkes-Murzycki, J. E., W. R. P. Scott, and C. A. Schiffer. 2007. Hydrophobic sliding: a possible mechanism for drug resistance in human immunodeficiency virus type 1 protease. Structure. 15:225–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Young, T., R. Abel, B. Kim, B. J. Berne, and R. A. Friesner. 2007. Motifs for molecular recognition exploiting hydrophobic enclosure in protein-ligand binding. Proc. Natl. Acad. Sci. USA. 104:808–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jenwitheesuk, E., and R. Samudrala. 2005. Prediction of HIV-1 protease inhibitor resistance using a protein-inhibitor flexible docking approach. Antivir. Ther. 10:157–166. [PubMed] [Google Scholar]
- 36.Wallqvist, A., G. W. Smythers, and D. G. Covell. 1998. A cooperative folding unit in HIV-1 protease. Implications for protein stability and occurrence of drug-induced mutations. Protein Eng. 11:999–1005. [DOI] [PubMed] [Google Scholar]
- 37.Ishima, R., D. A. Torchia, S. M. Lynch, A. M. Gronenborn, and J. M. Louis. 2003. Solution structure of the mature HIV-1 protease monomer: insight into the tertiary fold and stability of a precursor. J. Biol. Chem. 278:43311–43319. [DOI] [PubMed] [Google Scholar]
- 38.Louis, J. M., R. Ishima, I. Nesheiwat, L. K. Pannell, S. M. Lynch, D. A. Torchia, and A. M. Gronenborn. 2003. Revisiting monomeric HIV-1 protease. Characterization and redesign for improved properties. J. Biol. Chem. 278:6085–6092. [DOI] [PubMed] [Google Scholar]
- 39.Ishima, R., R. Ghirlando, J. Tozser, A. M. Gronenborn, D. A. Torchia, and J. M. Louis. 2001. Folded monomer of HIV-1 protease. J. Biol. Chem. 276:49110–49116. [DOI] [PubMed] [Google Scholar]
- 40.Ishima, R., D. A. Torchia, and J. M. Louis. 2007. Mutational and structural studies aimed at characterizing the monomer of HIV-1 protease and its precursor. J. Biol. Chem. 282:17190–17199. [DOI] [PubMed] [Google Scholar]
- 41.Ishima, R., and J. M. Louis. 2007. A diverse view of protein dynamics from NMR studies of HIV-1 protease flaps. Proteins. 70:1408–1415. [DOI] [PubMed] [Google Scholar]
- 42.Levy, Y., A. Caflisch, J. N. Onuchic, and P. G. Wolynes. 2004. The folding and dimerization of HIV-1 protease: evidence for a stable monomer from simulations. J. Mol. Biol. 340:67–79. [DOI] [PubMed] [Google Scholar]
- 43.Yan, M.-C., J. W. Yu Sha, X.-Q. Xiong, J.-H. Ren, and M.-S. Cheng. 2007. Molecular dynamics simulations of HIV-1 protease monomer: assembly of N-terminus and C-terminus into β-sheet in water solution. Proteins. 70:731–738. [DOI] [PubMed] [Google Scholar]
- 44.Bannwarth, L., and M. Reboud-Ravaux. 2007. An alternative strategy for inhibiting multidrug-resistant mutants of the dimeric HIV-1 protease by targeting the subunit interface. Biochem. Soc. Trans. 35:551–554. [DOI] [PubMed] [Google Scholar]
- 45.Camarasa, M.-J., S. Velazquez, A. San-Felix, M.-J. Perez-Perez, and F. Gago. 2006. Dimerization inhibitors of HIV-1 reverse transcriptase, protease and integrase: a single mode of inhibition for the three HIV enzymes? Antiviral Res. 71:260–267. [DOI] [PubMed] [Google Scholar]
- 46.Bannwarth, L., A. Kessler, S. Pethe, B. Collinet, N. Merabet, N. Boggetto, S. Sicsic, M. Reboud-Ravaux, and S. Ongeri. 2006. Molecular tongs containing amino acid mimetic fragments: new inhibitors of wild-type and mutated HIV-1 protease dimerization. J. Med. Chem. 49:4657–4664. [DOI] [PubMed] [Google Scholar]
- 47.Lee, S.-G., and J. Chmielewski. 2006. Rapid synthesis and in situ screening of potent HIV-1 protease dimerization inhibitors. Chem. Biol. 13:421–426. [DOI] [PubMed] [Google Scholar]
- 48.Broglia, R. A., D. Provasi, F. Vasile, G. Ottolina, R. Longhi, and G. Tiana. 2006. A folding inhibitor of the HIV-1 protease. Proteins. 62:928–933. [DOI] [PubMed] [Google Scholar]
- 49.Broglia, R. A., G. Tiana, L. Sutto, D. Provasi, and F. Simona. 2005. Design of HIV-1-PR inhibitors that do not create resistance: blocking the folding of single monomers. Protein Sci. 14:2668–2681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Broglia, R. A., G. Tiana, L. Sutto, D. Provasi, and V. Perelli. 2007. Low-throughput model design of protein folding inhibitors. Proteins. 67:469–478. [DOI] [PubMed] [Google Scholar]
- 51.Broglia, R. A., G. Tiana, L. Sutto, D. Provasi, and F. Simona. 2006. The physics of protein folding and non-conventional drug design: attacking AIDS with its own weapons. La Rivista del Nuovo Cimento. 29:1–19. [Google Scholar]
- 52.Bonomi, M., F. L. Gervasio, G. Tiana, D. Provasi, R. A. Broglia, and M. Parrinello. 2007. Insight into the folding inhibition of the HIV-1 protease by a small peptide. Biophys. J. 93:2813–2821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Heaslet, H., V. Kutilek, G. M. Morris, Y.-C. Lin, J. H. Elder, B. E. Torbett, and C. D. Stout. 2006. Structural insights into the mechanisms of drug resistance in HIV-1 protease NL4-3. J. Mol. Biol. 356:967–981. [DOI] [PubMed] [Google Scholar]
- 54.Heaslet, H., R. Rosenfeld, M. Giffin, Y. C. Lin, K. Tam, B. E. Torbett, J. H. Elder, D. E. McRee, and C. D. Stout. 2007. Conformational flexibility in the flap domains of ligand-free HIV protease. Acta Crystallogr. D Biol. Crystallogr. 63:866–875. [DOI] [PubMed] [Google Scholar]
- 55.Spinelli, S., Q. Z. Liu, P. M. Alzari, P. H. Hirel, and R. J. Poljak. 1991. The three-dimensional structure of the aspartyl protease from the HIV-1 isolate BRU. Biochimie. 73:1391–1396. [DOI] [PubMed] [Google Scholar]
- 56.Wlodawer, A., M. Miller, M. Jaskolski, B. K. Sathyanarayana, E. Baldwin, I. T. Weber, L. M. Selk, L. Clawson, J. Schneider, and S. B. Kent. 1989. Conserved folding in retroviral proteases: crystal structure of a synthetic HIV-1 protease. Science. 245:616–621. [DOI] [PubMed] [Google Scholar]
- 57.Lapatto, R., T. Blundell, A. Hemmings, J. Overington, A. Wilderspin, S. Wood, J. R. Merson, P. J. Whittle, D. E. Danley, and K. F. Geoghegan. 1989. X-ray analysis of HIV-1 proteinase at 2.7 Å resolution confirms structural homology among retroviral enzymes. Nature. 342:299–302. [DOI] [PubMed] [Google Scholar]
- 58.Liu, F., A. Y. Kovalevsky, J. M. Louis, P. I. Boross, Y.-F. Wang, R. W. Harrison, and I. T. Weber. 2006. Mechanism of drug resistance revealed by the crystal structure of the unliganded HIV-1 protease with F53L mutation. J. Mol. Biol. 358:1191–1199. [DOI] [PubMed] [Google Scholar]
- 59.Logsdon, B. C., J. F. Vickrey, P. Martin, G. Proteasa, J. I. Koepke, S. R. Terlecky, Z. Wawrzak, M. A. Winters, T. C. Merigan, and L. C. Kovari. 2004. Crystal structures of a multidrug-resistant human immunodeficiency virus type 1 protease reveal an expanded active-site cavity. J. Virol. 78:3123–3132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Krohn, A., S. Redshaw, J. C. Ritchie, B. J. Graves, and M. H. Hatada. 1991. Novel binding mode of highly potent HIV-proteinase inhibitors incorporating the (R)-hydroxyethylamine isostere. J. Med. Chem. 34:3340–3342. [DOI] [PubMed] [Google Scholar]
- 61.Kempf, D. J., K. C. Marsh, J. F. Denissen, E. McDonald, S. Vasavanonda, C. A. Flentge, B. E. Green, L. Fino, C. H. Park, and X. P. Kong. 1995. ABT-538 is a potent inhibitor of human immunodeficiency virus protease and has high oral bioavailability in humans. Proc. Natl. Acad. Sci. USA. 92:2484–2488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chen, Z., Y. Li, E. Chen, D. L. Hall, P. L. Darke, C. Culberson, J. A. Shafer, and L. C. Kuo. 1994. Crystal structure at 1.9-Å resolution of human immunodeficiency virus (HIV) II protease complexed with L-735,524, an orally bioavailable inhibitor of the HIV proteases. J. Biol. Chem. 269:26344–26348. [PubMed] [Google Scholar]
- 63.Kaldor, S. W., V. J. Kalish, J. F. Davies II, B. V. Shetty, J. E. Fritz, K. Appelt, J. A. Burgess, K. M. Campanale, N. Y. Chirgadze, D. K. Clawson, B. A. Dressman, S. D. Hatch, D. A. Khalil, M. B. Kosa, P. P. Lubbehusen, M. A. Muesing, A. K. Patick, S. H. Reich, K. S. Su, and J. H. Tatlock. 1997. Viracept (nelfinavir mesylate, AG1343): a potent, orally bioavailable inhibitor of HIV-1 protease. J. Med. Chem. 40:3979–3985. [DOI] [PubMed] [Google Scholar]
- 64.Surleraux, D. L. N. G., A. Tahri, W. G. Verschueren, G. M. E. Pille, H. A. de Kock, T. H. M. Jonckers, A. Peeters, S. D. Meyer, H. Azijn, R. Pauwels, M.-P. de Bethune, N. M. King, M. Prabu-Jeyabalan, C. A. Schiffer, and P. B. T. P. Wigerinck. 2005. Discovery and selection of TMC114, a next generation HIV-1 protease inhibitor. J. Med. Chem. 48:1813–1822. [DOI] [PubMed] [Google Scholar]
- 65.Spoel, D. V. D., E. Lindahl, B. Hess, G. Groenhof, A. E. Mark, and H. J. C. Berendsen. 2005. GROMACS: fast, flexible, and free. J. Comput. Chem. 26:1701–1718. [DOI] [PubMed] [Google Scholar]
- 66.Christen, M., P. H. Hunenberger, D. Bakowies, R. Baron, R. Burgi, D. P. Geerke, T. N. Heinz, M. A. Kastenholz, V. Krautler, C. Oostenbrink, C. Peter, D. Trzesniak, and W. F. van Gunsteren. 2005. The GROMOS software for biomolecular simulation: GROMOS05. J. Comput. Chem. 26:1719–1751. [DOI] [PubMed] [Google Scholar]
- 67.Kutzner, C., D. van der Spoel, M. Fechner, E. Lindahl, U. W. Schmitt, B. L. de Groot, and H. Grubmuller. 2007. Speeding up parallel GROMACS on high-latency networks. J. Comput. Chem. 28:2075–2084. [DOI] [PubMed] [Google Scholar]
- 68.Verkhivker, G. M., D. Bouzida, D. K. Gehlhaar, P. A. Rejto, S. Arthurs, A. B. Colson, S. T. Freer, V. Larson, B. A. Luty, T. Marrone, and P. W. Rose. 2000. Deciphering common failures in molecular docking of ligand-protein complexes. J. Comput. Aided Mol. Des. 14:731–751. [DOI] [PubMed] [Google Scholar]
- 69.Phillips, J. C., R. Braun, W. Wang, J. Gumbart, E. Tajkhorshid, E. Villa, C. Chipot, R. D. Skeel, L. Kale, and K. Schulten. 2005. Scalable molecular dynamics with NAMD. J. Comput. Chem. 26:1781–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Essmann, U., L. Perera, M. L. Berkowitz, T. Darden, H. Lee, and L. G. Pedersen. 1995. A smooth particle mesh Ewald method. J. Chem. Phys. 103:8577–8593. [Google Scholar]
- 71.Wang, J., R. M. Wolf, J. W. Caldwell, P. A. Kollman, and D. A. Case. 2004. Development and testing of a general AMBER force field. J. Comput. Chem. 25:1157–1174. [DOI] [PubMed] [Google Scholar]
- 72.Srinivasan, J., J. Miller, P. A. Kollman, and D. A. Case. 1998. Continuum solvent studies of the stability of RNA hairpin loops and helices. J. Biomol. Struct. Dyn. 16:671–682. [DOI] [PubMed] [Google Scholar]
- 73.Kollman, P. A., I. Massova, C. Reyes, B. Kuhn, S. Huo, L. Chong, M. Lee, T. Lee, Y. Duan, W. Wang, O. Donini, P. Cieplak, J. Srinivasan, D. A. Case, and T. E. Cheatham III. 2000. Calculating structures and free energies of complex molecules: combining molecular mechanics and continuum models. Acc. Chem. Res. 33:889–897. [DOI] [PubMed] [Google Scholar]
- 74.Verkhivker, G. M., D. Bouzida, D. K. Gehlhaar, P. A. Rejto, S. T. Freer, and P. W. Rose. 2002. Monte Carlo simulations of the peptide recognition at the consensus binding site of the constant fragment of human immunoglobulin G: the energy landscape analysis of a hot spot at the intermolecular interface. Proteins. 48:539–557. [DOI] [PubMed] [Google Scholar]
- 75.Verkhivker, G. M., D. Bouzida, D. K. Gehlhaar, P. A. Rejto, S. T. Freer, and P. W. Rose. 2003. Computational detection of the binding-site hot spot at the remodeled human growth hormone-receptor interface. Proteins. 53:201–219. [DOI] [PubMed] [Google Scholar]
- 76.Verkhivker, G. M. 2004. Computational analysis of ligand binding dynamics at the intermolecular hot spots with the aid of simulated tempering and binding free energy calculations. J. Mol. Graph. Model. 22:335–348. [DOI] [PubMed] [Google Scholar]
- 77.Verkhivker, G. M. 2007. Computational proteomics of biomolecular interactions in the sequence and structure space of the tyrosine kinome: deciphering the molecular basis of the kinase inhibitors selectivity. Proteins. 66:912–929. [DOI] [PubMed] [Google Scholar]
- 78.Verkhivker, G. M. 2007. In silico profiling of tyrosine kinases binding specificity and drug resistance using Monte Carlo simulations with the ensembles of protein kinase crystal structures. Biopolymers. 85:333–348. [DOI] [PubMed] [Google Scholar]
- 79.Gilson, M. K., and H.-X. Zhou. 2007. Calculation of protein-ligand binding affinities. Annu. Rev. Biophys. Biomol. Struct. 36:21–42. [DOI] [PubMed] [Google Scholar]
- 80.Tsai, C. J., B. Ma, and R. Nussinov. 1999. Folding and binding cascades: shifts in energy landscapes. Proc. Natl. Acad. Sci. USA. 96:9970–9972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Tsai, C. J., S. Kumar, B. Ma, and R. Nussinov. 1999. Folding funnels, binding funnels, and protein function. Protein Sci. 8:1181–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kumar, S., B. Ma, C. J. Tsai, N. Sinha, and R. Nussinov. 2000. Folding and binding cascades: dynamic landscapes and population shifts. Protein Sci. 9:10–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Tsai, C. D., B. Ma, S. Kumar, H. Wolfson, and R. Nussinov. 2001. Protein folding: binding of conformationally fluctuating building blocks via population selection. Crit. Rev. Biochem. Mol. Biol. 36:399–433. [DOI] [PubMed] [Google Scholar]
- 84.Frutos, S., R. A. Rodriguez-Mias, S. Madurga, B. Collinet, M. Reboud-Ravaux, D. Ludevid, and E. Giralt. 2007. Disruption of the HIV-1 protease dimer with interface peptides: structural studies using NMR spectroscopy combined with [2-(13)C]-Trp selective labeling. Biopolymers. 88:164–173. [DOI] [PubMed] [Google Scholar]
- 85.Shoemaker, B. A., J. J. Portman, and P. G. Wolynes. 2000. Speeding molecular recognition by using the folding funnel: the fly-casting mechanism. Proc. Natl. Acad. Sci. USA. 97:8868–8873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Levy, Y., P. G. Wolynes, and J. N. Onuchic. 2004. Protein topology determines binding mechanism. Proc. Natl. Acad. Sci. USA. 101:511–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Levy, Y., J. N. Onuchic, and P. G. Wolynes. 2007. Fly-casting in protein-DNA binding: frustration between protein folding and electrostatics facilitates target recognition. J. Am. Chem. Soc. 129:738–739. [DOI] [PubMed] [Google Scholar]
- 88.Chatterjee, A., P. Mridula, R. K. Mishra, R. Mittal, and R. V. Hosur. 2005. Folding regulates autoprocessing of HIV-1 protease precursor. J. Biol. Chem. 280:11369–11378. [DOI] [PubMed] [Google Scholar]