

Abstract
Gap junctions establish direct intercellular conduits between adjacent cells and are formed by the hexameric organization of protein subunits called connexins (Cx). It is unknown whether the proinflammatory milieu that ensues during CNS infection with S. aureus, one of the main etiologic agents of brain abscess in humans, is capable of eliciting regional changes in astrocyte homocellular gap junction communication (GJC) and, by extension, influencing neuron homeostasis at sites distant from the primary focus of infection. Here we investigated the effects of S. aureus and its cell wall product peptidoglycan (PGN) on Cx43, Cx30, and Cx26 expression, the main Cx isoforms found in astrocytes. Both bacterial stimuli led to a time-dependent decrease in Cx43 and Cx30 expression; however, Cx26 levels were elevated following bacterial exposure. Functional examination of dye coupling, as revealed by single-cell microinjections of Lucifer yellow, demonstrated that both S. aureus and PGN inhibited astrocyte GJC. Inhibition of protein synthesis with cyclohexamide (CHX) revealed that S. aureus directly modulates, in part, Cx43 and Cx30 expression, whereas Cx26 levels appear to be regulated by a factor(s) that requires de novo protein production; however, CHX did not alter the inhibitory effects of S. aureus on astrocyte GJC. The p38 MAPK inhibitor SB202190 was capable of partially restoring the S. aureus-mediated decrease in astrocyte GJC to that of unstimulated cells, suggesting the involvement of p38 MAPK-dependent pathway(s). These findings could have important implications for limiting the long-term detrimental effects of abscess formation in the brain which may include seizures and cognitive deficits.
Keywords: connexin 43, connexin 26, connexin 30, astrocyte, gap junction, bacteria, neuroinflammation, central nervous system
INTRODUCTION
Many cell types within multi-cellular systems must communicate with each other to function properly. In the CNS, communication via gap junctions is critical for the homeostatic functions of non-excitable cells such as astrocytes. Gap junction channels are formed by the joining of two hemichannels (connexons) between adjacent cells each composed of hexameric protein subunits called connexins (Cx). Several connexins have been reported to be expressed by astrocytes including connexin 43 (Cx43), connexin 26 (Cx26), and connexin 30 (Cx30) (Altevogt and Paul, 2004; Dermietzel et al., 2000; Nagy et al., 2001, 2003).
It has been well established that astrocytes play an important role in maintaining CNS homeostasis by regulating extracellular pH, K+, and glutamate levels through their ion channels and membrane transporters (Anderson and Swanson, 2000; Ransom, 2000; Ransom and Orkand, 1996). Moreover, astrocytes conduct signaling by propagating Ca2+ waves over considerable distances from the focus of initiation (Scemes et al., 1998; Schipke et al., 2002). All of these functions have been shown to be influenced, in part, by gap junction communication (GJC) (Charles et al., 1992; Farahani et al., 2005; Froes and de Carvalho, 1998; Naus and Bani-Yaghoub, 1998; Naus et al., 1999; Rozental et al., 2000a; Spray, 1999; Stout et al., 2002; Ye et al., 2003). Astrocytes represent the largest gap junction-coupled cell population of the CNS which results in extensive syncytium formation. Functionally, the astrocyte syncytium serves to effectively dilute substances cleared from the extracellular environment such as glutamate, and facilitates the trafficking of substances such as glucose and its metabolites at the blood–brain barrier (BBB), forming a link between cerebral vascular endothelium and neurons over long distances (Froes and de Carvalho, 1998; Spray, 1999).
In addition to physiological maintenance, growing evidence suggests that the astrocyte syncytium plays an important role in various CNS pathological conditions such as brain ischemia and hemorrhage, epilepsy, and brain tumors (Nakase and Naus, 2004; Naus et al., 1999; Rouach et al., 2002). However, the functional consequences resulting from changes in the extent of astrocyte coupling remain an area of debate and are likely influenced by the context of CNS damage (Farahani et al., 2005; Perez Velazquez et al., 2003). Neuroinflammation is a hallmark of various CNS pathologies such as trauma, bacterial meningitis, brain abscess, Alzheimer’s disease, and multiple sclerosis, which share a general feature of reactive gliosis characterized, to varying degrees, by the proliferation and hypertrophy of activated astrocytes (Eikelenboom et al., 2002; Griffin and Mrak, 2002; Kielian, 2004; Koedel et al., 2002; McGeer and McGeer, 2002; Nau and Bruck, 2002; Scheld et al., 2002). When activated by an appropriate stimulus, astrocytes have the capacity to produce robust amounts of proinflammatory mediators which may have profound effects on GJC (Dong and Benveniste, 2001; Esen et al., 2004; Kim et al., 2005; Smits et al., 2001). Indeed, the gram-negative bacterial cell wall component lipopolysaccharide (LPS) has been shown to attenuate GJC in primary rat astrocytes, which was attributed, in part, to the autocrine/paracrine action of nitric oxide (NO), since the iNOS inhibitor NG-monoethyl-L-arginine (NMMA) was capable of restoring gap junction coupling (Bolanos and Medina, 1996). In addition, treatment of human fetal astrocytes with the proinflammatory cytokine interleukin-1β (IL-1β) resulted in reduced Cx43 expression concomitant with an attenuation in GJC (John et al., 1999). Taken together, these findings suggest that inflammatory stimuli and proinflammatory mediators can regulate the expression of gap junction proteins and subsequent communication in astrocytes.
Recently we have demonstrated that astrocytes are capable of recognizing the gram-positive bacterium S. aureus and its cell wall component peptidoglycan (PGN), and respond by elaborating numerous proinflammatory mediators including NO, IL-1β, and TNF-α (Esen et al., 2004). S. aureus is one of the main pathogens of parenchymal brain infection leading to the establishment of brain abscess (Townsend and Scheld, 1998). In the experimental brain abscess model which has been developed in our laboratory using S. aureus, we have shown that IL-1β and TNF-α play critical roles in CNS pathology (Kielian et al., 2004). Interestingly, these same proinflammatory cytokines have recently been reported by others to exert dramatic effects on glial homocellular GJC (Chanson et al., 2001; Duffy et al., 2000; Faustmann et al., 2003; John et al., 1999). Based upon this evidence, we propose that proinflammatory mediators released by S. aureus-activated astrocytes in developing brain abscesses may be capable of modulating the nature and extent of GJC, with changes initiated in the proximity of the lesion and consequently extending to affect the surrounding astrocytic syncytial network.
The current studies were designed to investigate the effects of S. aureus and its cell wall product PGN on Cx isoform expression and homocellular GJC in astrocytes. Both stimuli led to a time-dependent decrease in Cx43 and Cx30 levels with a concomitant increase in Cx26 expression. In addition, both S. aureus and PGN were found to significantly attenuate the extent of astrocyte gap junction coupling as evaluated by single-cell microinjections of Lucifer yellow. Inhibition of protein synthesis with cyclohexamide (CHX) revealed that S. aureus directly modulates, in part, Cx43 and Cx30 expression, whereas Cx26 levels appear to be regulated by factor(s) that require de novo protein production. However, despite these differences, CHX did not alter the inhibitory effects of S. aureus on astrocyte GJC. The p38 MAPK pathway appears to play an important role in regulating astrocyte GJC in response to S. aureus since the p38 MAPK inhibitor SB202190 was capable of partially restoring the S. aureus-mediated decrease in astrocyte GJC to that of unstimulated cells. These results suggest that gram-positive bacteria such as S. aureus can influence astrocyte Cx isoform expression and subsequent homocellular GJC either directly or indirectly through the release of proinflammatory mediators.
MATERIALS AND METHODS
Primary Astrocyte Culture and Reagents
Neonatal C57BL/6 mice (1–6 days of age) were used to prepare mixed glial cultures as previously described (Esen et al., 2004). Briefly, when cultures reached confluency (1–2 weeks), flasks were shaken at 200 rpm overnight at 37°C to recover microglia. The supernatant containing microglia was collected and fresh medium was added to the flasks and incubated another 5–7 days. This was repeated 3–4 times, whereupon flasks were trypsinized and cells resuspended in astrocyte medium supplemented with 0.1 mM of the microglial cytotoxic agent L-leucine methyl ester (L-LME; Sigma-Aldrich, St. Louis, MO). Astrocytes were treated with L-LME for at least 2 weeks prior to use in experiments. The purity of astrocyte cultures was determined by immuno-histochemical staining using antibodies for CD11b (BD PharMingen, San Diego, CA) and GFAP (DAKO Corp., Carpinteria, CA) to detect microglia and astrocytes, respectively. The purity of astrocyte cultures prepared in this manner was routinely ≥95%.
Throughout this study, astrocytes were seeded into 6-well plates or 35 mm dishes at 1 × 106 or 5 × 105, respectively, and incubated overnight. The following day, cells were stimulated with heat-inactivated S. aureus (107 cfu; strain RN6390, kindly provided by Dr. Ambrose Cheung, Dartmouth Medical School) or 10 μg/mL PGN (Fluka, St. Louis, MO) for 24 h. These doses were selected based on previous studies demonstrating optimal proinflammatory mediator induction in astrocytes without any evidence of cytotoxicity (Esen et al., 2004). In studies designed to examine the requirement of de novo protein synthesis on Cx mRNA expression and functional GJC, astrocytes were treated with the protein synthesis inhibitor CHX (Sigma). CHX treatment was initiated 1 h prior to S. aureus exposure and was maintained throughout the period of S. aureus stimulation (6, 12, or 24 h). The amount of CHX used for these experiments (10 μM) was optimized by dose-response studies and was not found to induce astrocyte toxicity as determined by a standard MTT assay (Esen et al., 2004) (data not shown).
All reagents and culture media were verified to have endotoxin levels <0.03 EU/mL as determined by Limulus amebocyte lysate assay (LAL; Associates of Cape Cod, Falmouth, MA).
Quantitative Real-Time RT-PCR
Total RNA from astrocytes was isolated using the TriZol reagent and treated with DNAse1 (both from Invitrogen, Carlsbad, CA) prior to use in quantitative real-time RT-PCR (qRT-PCR) studies. GAPDH primers and TAMRA TaqMan probes were designed as previously described (Esen et al., 2004) and synthesized by Applied Biosystems (ABI, Foster City, CA). ABI Assays-on-Demand™ Taqman kits were utilized to examine Cx26, Cx30, and Cx43 expression. The RT reaction was conducted using the iScript cDNA™ synthesis kit (Bio-Rad, Hercules, CA) according to the manufacturer’s instructions. Real-time PCR reactions were performed using the iCycler™ kit and analyzed using the iCycler IQ™ multicolor real-time PCR detection system (both from Bio-Rad). Cx expression levels in primary astrocytes were calculated after normalizing cycle thresholds against the housekeeping gene GAPDH and are presented as the fold-induction (2−ΔΔCt) value relative to unstimulated cells.
Protein Extraction and Western Blotting
The effects of heat-inactivated S. aureus and PGN on astrocytic Cx26, Cx30, and Cx43 protein expression were evaluated by Western blot analysis as previously described (Esen et al., 2004). Astrocytic protein extracts (40 μg of total protein) were electrophoresed on 10% Tris-HCl Ready Gels (Bio-Rad) and transferred to a PVDF membrane (Immobilon-P, Millipore, Bedford, MA) using a semidry transfer apparatus (Bio-Rad). Blots were probed using polyclonal rabbit anti-mouse Cx26, Cx30, and Cx43 antibodies (all from Zymed, San Francisco, CA; Cat nos. 51–2800, 71–2200, and 71–0700, respectively) (Penes et al., 2005; Rash et al., 2001) followed by a donkey anti-rabbit IgG-HRP conjugate (Jackson Immunoresearch, West Grove, PA). Blots were developed using the ChemiGlow West substrate (Alpha Innotech, San Leandro, CA) and visualized by exposure to X-ray film. Following Cx isoform detection, blots were stripped and re-probed with a rabbit anti-actin polyclonal antibody (Sigma) for normalization. For experiments designed to demonstrate the specificity of Cx30 immunoreactive bands, Cx30 pAb (1 μg) was pre-adsorbed with a 100-fold excess of Cx30 blocking peptide (generously provided by Dr. David Phan, Invitrogen) for 2 h at room temperature in PBS/0.1% Tween/1% milk prior to membrane blotting. For quantitation, non-saturated autoradiographs were scanned and the pixel intensity for each band was determined using the Image/J program (NIH Image) and normalized to the amount of actin. Results are expressed in arbitrary units as the ratio of Cx26, Cx30, or Cx43 to actin.
Immunofluorescence Staining of Cx Isoforms and Confocal Microscopy
Cx isoform expression in astrocytes was examined using two-color immunofluorescence staining and subsequent confocal microscopy. Primary astrocytes were seeded onto glass coverslips (105 cells/coverslip) and incubated in 24-well plates overnight. The following day, cells were treated with either 107 heat-inactivated S. aureus or 10 μg/mL PGN for 6 or 24 h (three replicates per time point). At the end of each incubation period, cells were washed extensively with PBS and immediately fixed in ice-cold methanol. Astrocytes were incubated with the rabbit polyclonal Cx26, Cx30, or Cx43 antibodies described above in conjunction with a polyclonal rat anti-mouse GFAP antibody (Zymed) overnight at 4°C in a humidified chamber. Following numerous rinses in PBS, cells were incubated with biotinylated donkey anti-rabbit IgG and donkey anti-rat-FITC antibodies (to detect Cx isoform and GFAP expression, respectively; both from Jackson Immunoresearch, West Grove, PA) for 1 h at room temperature. Cx isoform expression was visualized by the addition of a streptavidin-Alexa Fluor 568 conjugate (Molecular Probes, Eugene, OR) and Hoechst 33342 (2 μM; Molecular Probes) was included for identification of nuclei. All antibody dilutions were prepared in PBS supplemented with 10% donkey serum (Jackson Immunoresearch) to minimize non-specific binding. Importantly, the anti-rabbit and anti-goat IgG secondary Abs employed in this study were pre-adsorbed with normal goat and rabbit serum, respectively, to prevent non-specific cross-reactivity with primary antibodies. Upon completion of the staining protocol, coverslips were mounted onto glass slides using the Prolong anti-fade reagent (Molecular Probes) and sealed using nail polish. Coverslips were imaged using a Zeiss laser scanning confocal microscope (LSM 510, Carl Zeiss Microimaging Inc, Thornwood, NY). Samples were scanned using a multi-track mode and images corresponding to each fluorophore were collected sequentially using individual lasers to eliminate bleed-through. Hoechst 33342 for nuclear visualization was excited by a 405 nm diode laser, FITC to visualize GFAP immunoreactivity was excited with a 488 nm argon laser, and Alexa Fluor 568 to demonstrate Cx isoform expression was excited with a 561 nm DPSS laser, with images collected using the appropriate emissions. The confocal pinhole was set to obtain an optical section thickness of 1.6 μm. To demonstrate colocalization of GFAP, Cx isoforms, and Hoechst 33342, RGB merges of individual confocal images were performed using the ImageJ software program (NIH Image).
Single-Cell Microinjections of Lucifer Yellow
Astrocytes were plated for lucifer yellow (LY) injections in 35 mm dishes (5 × 105 cells/dish) and incubated for 24 h. Subsequently, cells were stimulated with either 107 heat-inactivated S. aureus or 10 μg/mL PGN and incubated for another 24 h. During LY injections, astrocyte cultures were continuously superfused with carbogen (95% O2 and 5% CO2)-saturated Ames medium (Sigma) at a rate of 3–4 mL/min. The perfusion solution was pre-warmed to 37°C using an inline solution heater regulated by a dual channel heater (both from Warner Instruments, Hamden, CT). Cells were allowed to equilibrate for 15 min prior to microinjections. Sharp micro-pipettes (resistance 100–200 MΩ) were prepared from borosilicate filament tubing (OD 1.0 mm, ID 0.5 mm) using a micropipette puller (P-97 Fleming/Brown, both from Sutter Instruments, Novato, CA). Pipettes were backfilled with the gap junction-permeable dye LY CH (Sigma, 4% (w/v) LY in 150 mM LiCl) with a 34-gauge microfill syringe needle (World Precision Instruments, Sarasota, FL). LY was injected intracellularly by applying brief hyperpolarizing pulses generated by a micro-iontophoresis current generator (World Precision Instruments) through a micropipette that was carefully inserted into the cell guided by a micromanipulator (MP-225, Sutter Instruments). In studies designed to confirm the gap junction-dependent nature of LY spread in astrocytes, cells were treated with the gap junction blocker 18-α-glycyrrhetinic acid (AGA, Sigma). Briefly, a minimum of three LY intracellular injections were performed in unstimulated astrocyte cultures prior to AGA treatment to confirm the establishment of gap junction-dependent coupling. Subsequently, the same cultures were continuously perfused for 20 min in Ames medium supplemented with 25 μM AGA to allow for sufficient levels of inhibitor to accumulate within the culture medium, whereupon gap junction-dependent coupling was assessed while still in the presence of AGA. A minimum of 10 individual cells were microinjected with LY in each experimental group (unstimulated, unstimulated + AGA, S. aureus, and PGN) with results replicated in five independent experiments. To evaluate the mechanism(s) influencing astrocyte GJC in response to S. aureus, cells were treated with either CHX (10 μM) or the p38 MAP kinase inhibitor SB202190 (SB; 1 μM, Calbiochem, San Diego, CA), both of which were initiated 1 h prior to S. aureus exposure and maintained throughout the period of bacterial stimulation (12 or 24 h). LY injected cells were visualized using a fixed stage upright epifluorescence microscope (BX51WI, Olympus) equipped with a 40× water immersion objective lens (NA 0.8), a 12-bit intensified monochrome CCD camera (CoolSnap ES, Photo-metrics, Tucson, AZ), a multiple excitation filter controlled through a filter wheel (Prior Scientific, Rockland, MA), and a quad emission filter (Chroma, Rockingham, VT). Images were acquired and processed using MetaMorph software (Universal Imaging Corporation, Downingtown, PA). Images depicting the extent of LY spread are presented after background subtraction for LY (excitation wave length 425 nm, emission wave length 540 nm), to correct for autofluorescence.
Statistics
Significant differences between experimental groups were determined by the Student’s t-test at the 95% confidence interval using Sigma Stat (SPSS Science, Chicago, IL).
RESULTS
S. aureus and PGN Modulate Cx mRNA and Protein Expression in Astrocytes
Previous studies have demonstrated that inflammatory mediators elicited in response to the gram-negative bacterial cell wall product LPS inhibit GJC in several cell types (De Maio et al., 2000; Eugenin et al., 2003; Fernandez-Cobo et al., 1999). In addition, infection of astrocytes with Toxoplasma gondii has been shown to result in the loss of GJC (Campos de Carvalho et al., 1998). However, the effects of gram-positive bacteria on astrocyte homocellular GJC remain unknown. Due to its prevalence as a CNS pathogen and the finding that activated astrocytes are a hallmark of brain abscess (Baldwin and Kielian, 2004; Kielian, 2004), we evaluated the effects of S. aureus and its cell wall component PGN on astrocytic Cx mRNA expression by qRT-PCR. Interestingly, both S. aureus and PGN were found to differentially modulate the expression of various Cx isoforms in astrocytes. Specifically, both stimuli led to a significant decrease in both Cx30 and Cx43 mRNA expression (Figs. 1B,C, respectively), whereas Cx26 levels were augmented within 6 h following S. aureus and PGN exposure (Fig. 1A).
Fig. 1.

Both S. aureus and PGN modulate Cx isoform mRNA expression differentially in astrocytes. Primary astrocytes were seeded at 106 cells per well in 6-well plates and incubated overnight. The following day, cells were stimulated with either 107 heat-inactivated S. aureus or 10 μg/mL PGN for 6, 12, or 24 h, whereupon total RNA was isolated and examined for Cx26, 30 and 43 expression by qRT-PCR as described in the Materials and Methods. Gene expression levels were calculated after normalizing Cx26 (A), Cx30 (B) and Cx43 (C) signals against the housekeeping gene GAPDH and are presented in relative mRNA expression units (mean ± SEM of three independent experiments). Significant differences between untreated versus S. aureus- or PGN-stimulated astrocytes are denoted with asterisks (*P < 0.05; **P < 0.001).
To examine whether the observed alterations in Cx isoform mRNA expression correlated with changes in protein levels, Western blots were performed. As shown in Fig. 2, Cx43 protein levels were significantly attenuated following exposure to either S. aureus or PGN, with inhibition most prominent at 24 h post-treatment. In our hands, a well-characterized Cx30 polyclonal antibody (71–2200) produced two prominent bands corresponding to the predicted molecular weight of Cx30 monomers and dimers (Fig. 3A), the identity of which was confirmed by peptide blocking studies (Supplemental Figure 1). Evaluation of astrocytic Cx30 expression revealed an intriguing relationship between the monomeric and dimeric forms of the protein following bacterial exposure. Specifically, within 6 h following S. aureus or PGN stimulation, a decrease in Cx30 monomers was observed, whereas the dimer form of Cx30 remained relatively unaffected (Fig. 3). Cx26 protein expression was elevated following either S. aureus or PGN stimulation (Fig. 4) in agreement with mRNA levels (Fig. 1A). Collectively, these findings suggest that gram-positive bacterial stimuli modulate astrocytic Cx isoforms differentially and with distinct kinetics.
Fig. 2.
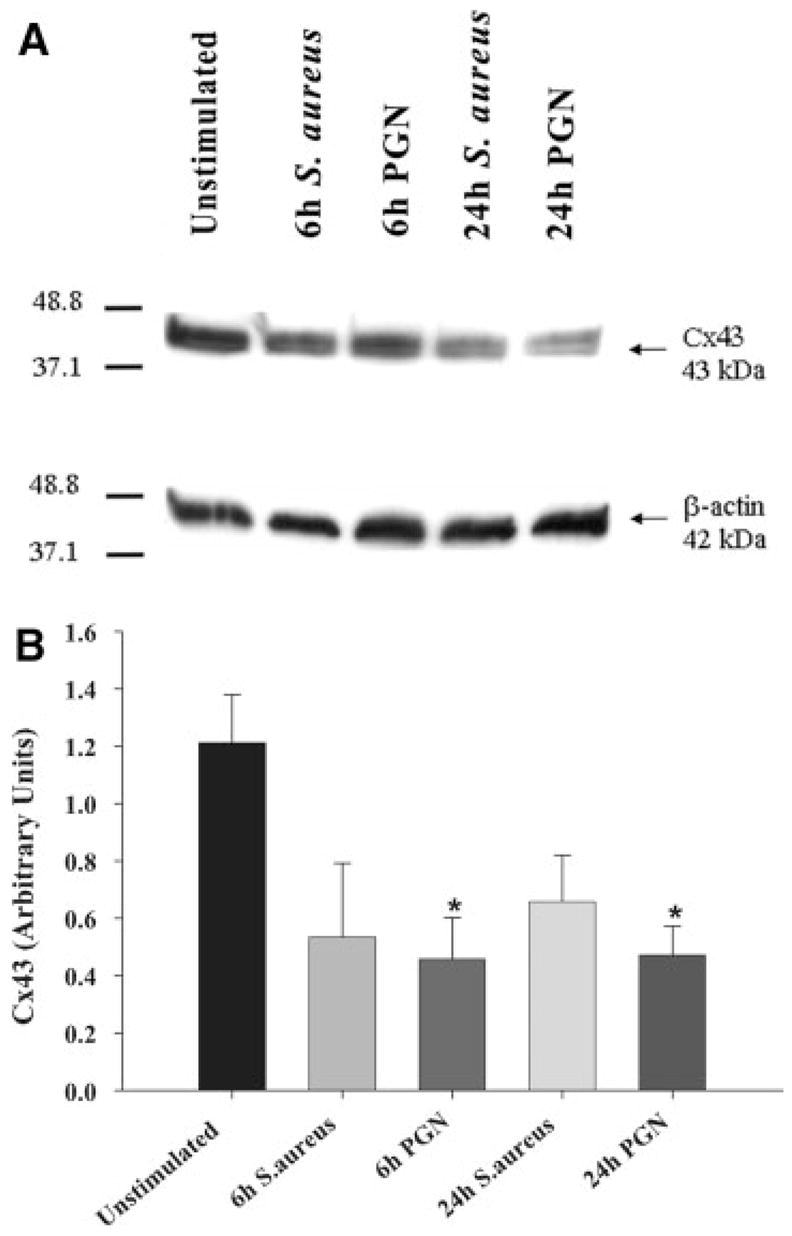
Cx43 protein levels are attenuated by S. aureus and PGN in astrocytes. Primary astrocytes were seeded at 106 cells per well in 6-well plates and incubated overnight. The following day, cells were stimulated with either 107 heat-inactivated S. aureus or 10 μg/mL PGN for 24 h, whereupon protein extracts from whole cell lysates (40 μg per sample) were evaluated for Cx43 expression by Western blotting as described in the Materials and Methods. Results are presented as the raw gel data (A) and quantitative analysis of Cx43 expression by densometric scanning (B). For quantitation in (B), the pixel intensity of each Cx43 band was normalized to the amount of actin included as a loading control. Results are expressed in arbitrary units as the ratio of Cx43 to actin and represent the mean ± SEM of three independent experiments. Significant differences are denoted with asterisks (*P < 0.05).
Fig. 3.
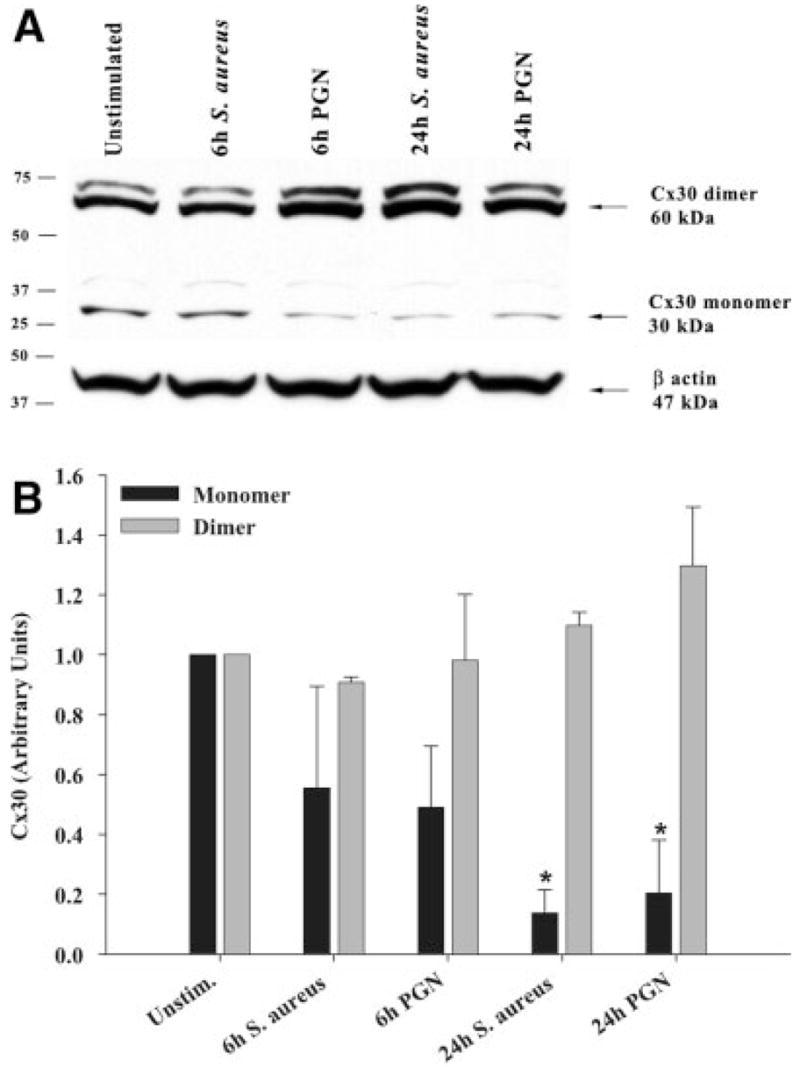
Effects of S. aureus and PGN on Cx30 protein expression. Primary astrocytes were seeded at 106 cells per well in 6-well plates and incubated overnight. The following day, cells were stimulated with either 107 heat-inactivated S. aureus or 10 μg/mL PGN for 24 h, whereupon protein extracts from whole cell lysates (40 μg per sample) were evaluated for Cx30 expression by Western blotting as described in the Materials and Methods. Results are presented as the raw gel data (A) and quantitative analysis of Cx30 expression by densometric scanning (B). For quantitation in (B), the pixel intensity of each Cx30 band was normalized to the amount of actin included as a loading control. Results are expressed in arbitrary units as the ratio of Cx30 to actin and represent the mean ± SEM of three independent experiments. Significant differences are denoted with asterisks (*P < 0.05).
Fig. 4.
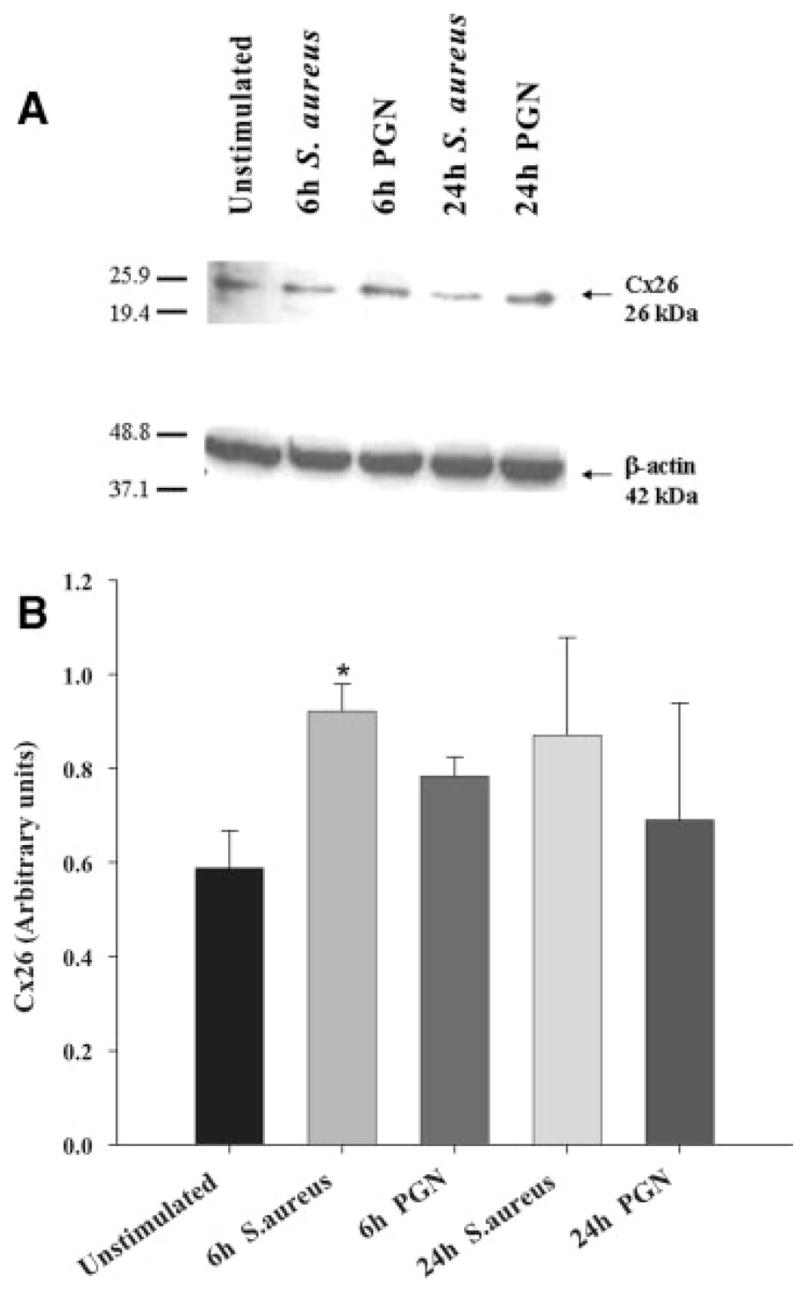
S. aureus and PGN augment astrocytic Cx26 protein expression. Primary astrocytes were seeded at 106 cells per well in 6-well plates and incubated overnight. The following day, cells were stimulated with either 107 heat-inactivated S. aureus or 10 μg/mL PGN for 24 h, whereupon protein extracts from whole cell lysates (40 μg per sample) were evaluated for Cx26 expression by Western blotting as described in the Materials and Methods. Results are presented as the raw gel data (A) and quantitative analysis of Cx26 expression by densometric scanning (B). For quantitation in (B), the pixel intensity of each Cx26 band was normalized to the amount of actin included as a loading control. Results are expressed in arbitrary units as the ratio of Cx26 to actin and represent the mean ± SEM of three independent experiments. Significant differences are denoted with asterisks (*P < 0.05).
S. aureus Influences Cx26, Cx30, and Cx43 Immunoreactivity
Although Western blots demonstrated that S. aureus and PGN differentially regulate Cx isoform expression in astrocytes, this approach does not allow for the direct visualization of Cx distribution in cells. Since previous studies have revealed that various treatments can lead to the re-distribution of Cx isoforms (Campos de Carvalho et al., 1998; Martinez and Saez, 2000), the expression of Cx26, Cx30 and Cx43 was examined in S. aureus- and PGN-stimulated astrocytes by immunofluorescence analysis. The expression of Cx43 was enhanced in unstimulated cells with increasing time in culture where it demonstrated a characteristic localization along junctional domains. Both S. aureus- and PGN-activated astrocytes consistently displayed lower Cx43 immunoreactivity at 24 h post-treatment compared with unstimulated cells (Fig. 5). Similar changes were observed with Cx30, where protein levels were drastically reduced in S. aureus- and PGN-treated cells (Fig. 6). Cx26 immunoreactivity was not detected in unstimulated astrocytes, but was found associated with cellular aggregates in response to both S. aureus and PGN (Fig. 7). Similar changes in Cx isoform expression were also observed at 6 h following S. aureus and PGN exposure (Supplemental Figures 2–4). Interestingly, unlike Cx43, we did not observe significant Cx30 and Cx26 localization along junctional domains (Figs. 6 and 7). Rather, these Cx isoforms appeared to be distributed throughout the cell, which suggests that S. aureus and PGN stimulation may elicit gap junction-independent functions for Cx30 and Cx26, although this possibility remains speculative at the present time. Collectively, the observed changes in Cx isoform immunoreactivity together with mRNA and protein expression suggest that the gram-positive bacterium S. aureus may be capable of influencing activation-dependent GJC.
Fig. 5.

S. aureus and PGN attenuate Cx43 immunoreactivity in astrocytes. Primary astrocytes were seeded on coverslips (105 cells/coverslip) and incubated overnight. The following day, astrocytes were stimulated with 107 heat-inactivated S. aureus or 10 μg/mL PGN for 24 h, whereupon cells were processed for immunofluorescence staining for Cx43 (red) and GFAP (green) as described in the Materials and Methods and imaged using a ×40 oil immersion objective lens by confocal microscopy. Nuclei were visualized using Hoechst 33342 (blue). Results are representative of five independent experiments.
Fig. 6.
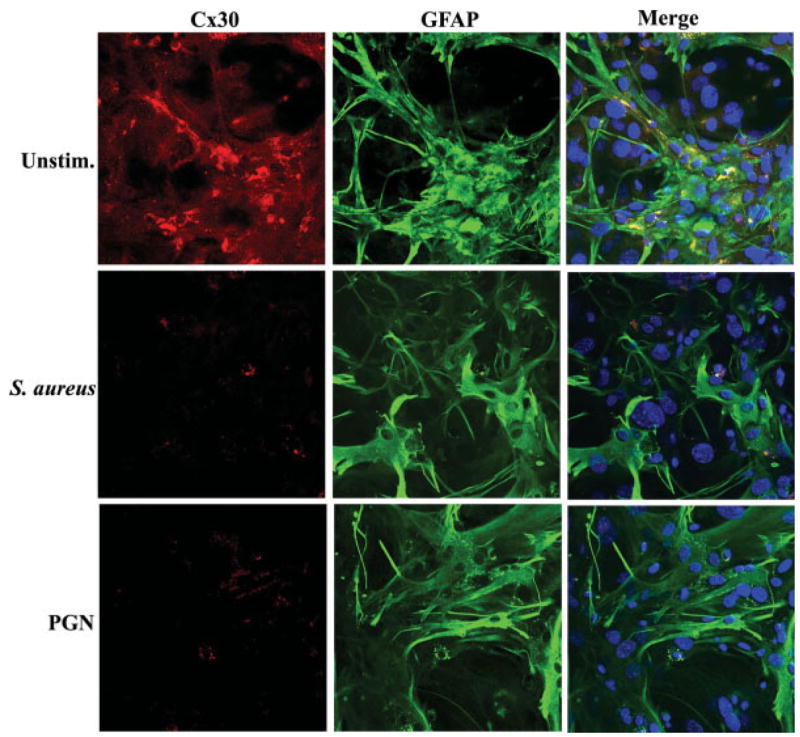
S. aureus and PGN reduce astrocytic Cx30 immunoreactivity. Primary astrocytes were seeded on glass coverslips (105 cells/coverslip) and incubated overnight. The following day, astrocytes were stimulated with 107 heat-inactivated S. aureus or 10 μg/mL PGN for 24 h, whereupon cells were processed for immunofluorescence staining for Cx30 (red) and GFAP (green) as described in the Materials and Methods and imaged using a ×40 oil immersion objective lens by confocal microscopy. Nuclei were visualized using Hoechst 33342 (blue). Results are representative of five independent experiments.
Fig. 7.
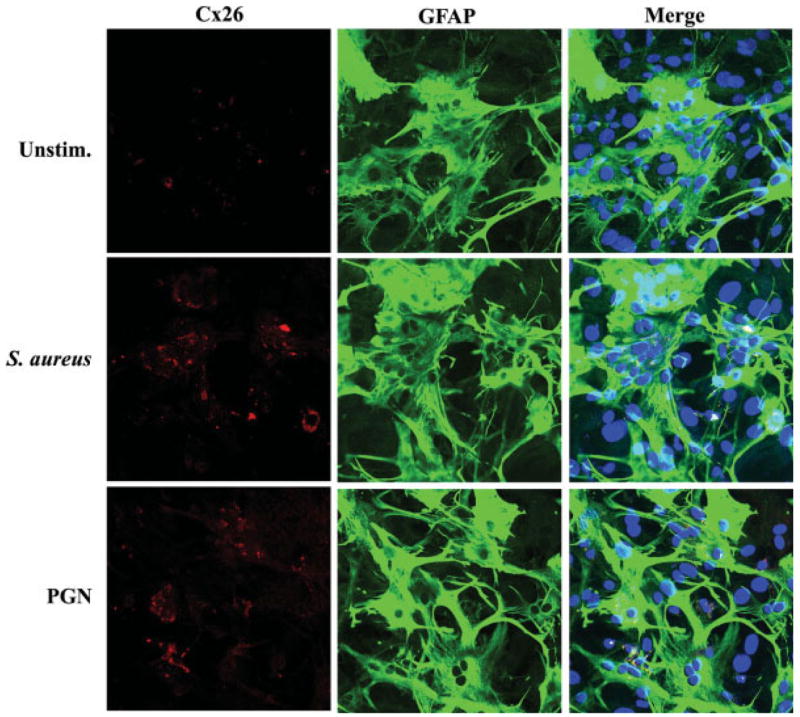
Astrocytic Cx26 immunoreactivity is enhanced following S. aureus and PGN stimulation. Primary astrocytes were seeded on glass coverslips (105 cells/coverslip) and incubated overnight. The following day, astrocytes were stimulated with 107 heat-inactivated S. aureus or 10 μg/mL PGN for 24 h, whereupon cells were processed for immunofluorescence staining for Cx26 (red) and GFAP (green) as described in the Materials and Methods and imaged using a ×40 oil immersion objective lens by confocal microscopy. Nuclei were visualized using Hoechst 33342 (blue). Results are representative of five independent experiments.
Astrocytic Homocellular GJC is Attenuated by Both S. aureus and PGN
To determine whether the observed changes in astrocytic Cx isoform expression following S. aureus and PGN stimulation translated into alterations in the activity of functional gap junction channels, GJC was evaluated in primary astrocytes using a single-cell microinjection technique with LY. This approach led to the finding that both S. aureus and PGN resulted in an activation-dependent decrease in GJC (Fig. 8). Quantitation of the number of dye coupled astrocytes revealed that an average of 7–10 cells were coupled under normal conditions; however both S. aureus and PGN significantly decreased the number of gap junction coupled astrocytes to an average of 2–3 cells per treatment (Fig. 8B). Importantly, the observed dye coupling in unstimulated astrocytes was completely inhibited by the gap junction blocker AGA (Rozental et al., 2000b), confirming the specificity of dye transfer through astrocyte gap junction channels (Figs. 8A,B). Collectively, the findings that S. aureus and PGN affect not only Cx isoform expression but also functional GJC indicate that gram-positive bacteria modulate both structural and functional aspects of astrocytic gap junctions.
Fig. 8.
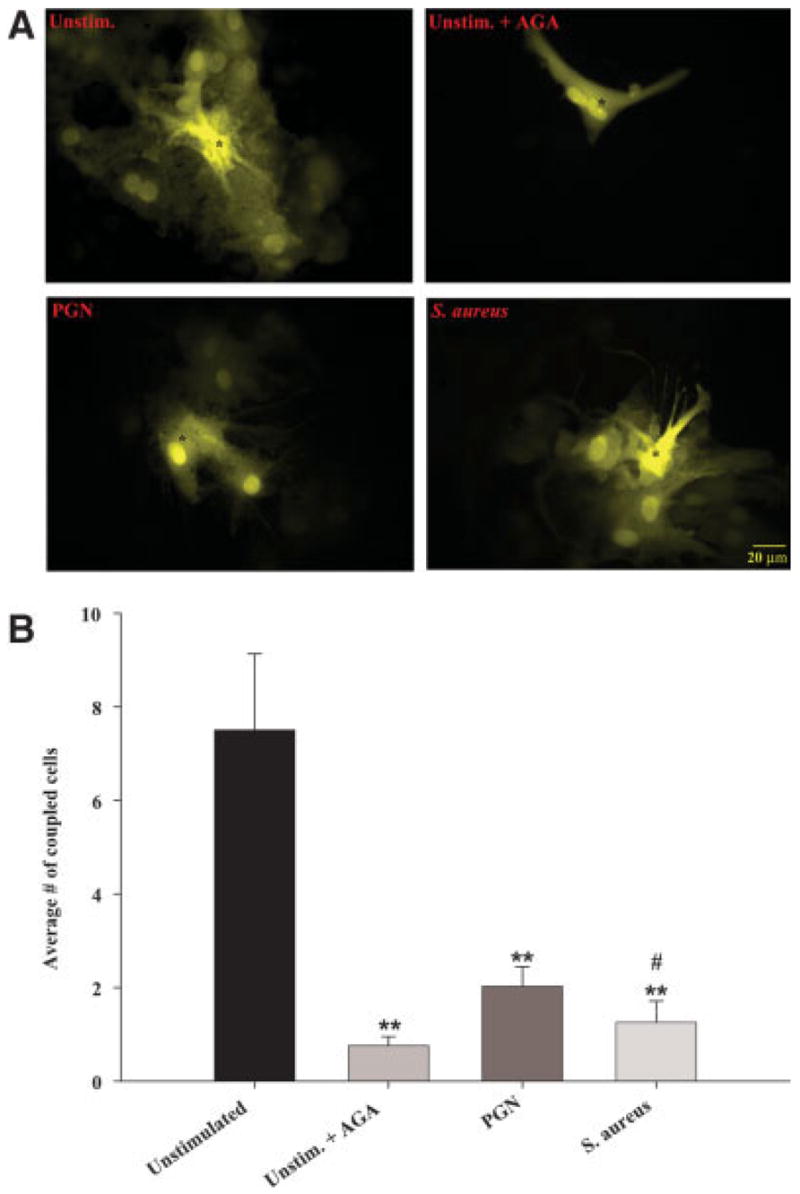
Both S. aureus and PGN attenuate homocellular gap junction communication in astrocytes. Primary astrocytes were seeded at 5 × 105 cells per well in 35-mm dishes and incubated overnight. The following day, cells were stimulated with either 107 heat-inactivated S. aureus or 10 μg/mL PGN for 24 h, whereupon functional GJC was evaluated by single-cell injections of LY (A) as described in the Materials and Methods. To confirm the specificity of dye transfer, unstimulated astrocytes were treated with the gap junction blocker AGA (25 μM) for 30 min prior to LY injection to inactivate gap junction channels. Microinjected cells are depicted by asterisks (*). In (B), the number of cells displaying dye transfer from each microinjected astrocyte is quantitated. Results are reported as the mean ± SEM from three independent experiments evaluating at least 10 injected cells per treatment group per experiment. Significant differences between unstimulated and AGA-, S. aureus- or PGN-treated cells are denoted with asterisks (**P < 0.001), whereas significant differences between S. aureus- and PGN-treated cells are denoted with a hatched sign (#P < 0.05). [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
S. aureus Exhibits Both Direct and Indirect Effects on Cx mRNA Expression
The modulation of Cx isoform expression by S. aureus could result from either a direct effect of signals initiated by bacteria binding to pattern recognition receptors such as Toll-like receptor 2 (TLR2) and/or indirectly as a consequence of proinflammatory mediators released by activated astrocytes (Esen et al., 2004). To begin to discriminate between these two possibilities, astrocytes were treated with the protein synthesis inhibitor cyclohexamide (CHX; 10 μM) prior to S. aureus stimulation. The ability of CHX to suppress proinflammatory mediator production in response to S. aureus and PGN stimulation was established prior to the initiation of these studies (data not shown). Inhibition of de novo protein synthesis with CHX revealed that the modulatory actions of S. aureus on Cx isoform mRNA expression are mediated by both direct and indirect effects. As shown in Fig. 9A, with CHX treatment the elevation in Cx26 expression normally seen after S. aureus stimulation was prevented, suggesting that the effect of S. aureus on Cx26 was indirect, possibly due to the autocrine/paracrine action of proinflammatory mediators known to be produced by S. aureus-stimulated astrocytes (Esen et al., 2004). In contrast, the suppression of Cx30 by S. aureus was found to be primarily a direct effect since CHX treatment did not alter the ability of S. aureus to attenuate Cx30 expression during acute time points following bacterial exposure (Fig. 9B). Interestingly, in the presence of cyclohexamide, Cx30 levels were significantly attenuated at 24 h following bacterial exposure compared with S. aureus treatment only (Fig. 9B), suggesting indirect effects due to mediator(s) released in response to bacterial stimulation. Interestingly, cyclohexamide did not alter the suppressive effect of S. aureus on Cx43 mRNA expression, which implies that S. aureus attenuates Cx43 mRNA levels directly (Fig. 9C). Taken together, these findings suggest that S. aureus modulates Cx isoform expression differently via either direct or indirect pathways, the latter of which may be mediated, in part, through the release of proinflammatory mediators by activated astrocytes.
Fig. 9.
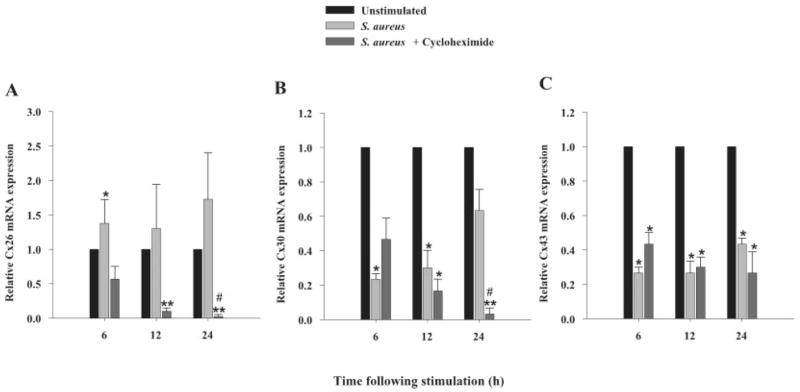
S. aureus exerts both direct and indirect effects on astrocytic Cx mRNA expression. Primary astrocytes were seeded at 106 cells per well in 6-well plates and incubated overnight. The following day, cells were pre-treated with the protein synthesis inhibitor CHX (10 μM) for 1 h prior to stimulation with 107 heat-inactivated S. aureus for 6, 12, or 24 h in the continued presence of CHX. At the indicated time points, total RNA was isolated and examined for Cx26, 30 and 43 expression by qRT-PCR as described in the Materials and Methods. Gene expression levels were calculated after normalizing Cx26 (A), Cx30 (B) and Cx43 (C) signals against the housekeeping gene GAPDH and are presented in relative mRNA expression units (mean ± SEM of three independent experiments). Significant differences between untreated versus S. aureus- or CHX treated S. aureus-stimulated astrocytes are denoted with asterisks (*P < 0.05, **P < 0.001), whereas significant differences between S. aureus- versus CHX treated S. aureus-stimulated astrocytes are denoted with a hatched sign (#P < 0.05).
De novo Protein Synthesis Is Not Essential for the Inhibitory Effects of S. aureus on Astrocyte GJC
Although our CHX results suggested that S. aureus modulates Cx isoform expression via both direct and indirect pathways, alterations in mRNA levels do not necessarily indicate that the activity of these proteins is affected. Therefore, to investigate the functional relevance of these Cx mRNA changes we have also examined the dynamic effects of CHX together with S. aureus on GJC. These studies were performed on astrocytes at 12 h following CHX and/or S. aureus exposure, since initial studies revealed that treatment with CHX alone for a 24 h period attenuated astrocyte GJC, emphasizing the dynamics of Cx protein turnover (data not shown). Inhibition of de novo protein synthesis by CHX had no effect on the ability of S. aureus to attenuate astrocyte GJC, suggesting that S. aureus modulates coupling via direct effects, at least at the early time interval examined in this study (Fig. 10). GJC in CHX-treated astrocytes remained similar to unstimulated cells at 12 h, indicating that the observed outcome was not due to confounding effects of CHX treatment.
Fig. 10.

The inhibitory effects of S. aureus on astrocyte GJC are direct. Primary astrocytes were seeded at 5 × 105 cells per well in 35-mm dishes and incubated overnight. The following day, cells were pre-treated with the protein synthesis inhibitor CHX (10 μM) for 1 h prior to stimulation with 107 heat-inactivated S. aureus for 12 h in the continued presence of CHX, whereupon functional GJC was evaluated by single-cell injections of LY (A) as described in the Materials and Methods. Microinjected cells are denoted by asterisks (*). In (B), the number of cells displaying dye transfer from each microinjected astrocyte is quantitated. Results are reported as the mean ± SEM from two independent experiments evaluating at least 10 injected cells per treatment group per experiment. Significant differences between unstimulated versus S. aureus alone or S. aureus + CHX are denoted with asterisks (*P < 0.05). [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
The Inhibitory Effects of S. aureus on Astrocyte GJC are Mediated, in Part, via a p38 MAPK-dependent Pathway
The direct effects of S. aureus on astrocyte gap junction coupling could be mediated either by the transcriptional modulation of Cx genes and/or activated kinases that influence the phosphorylation state of Cxs and subsequent GJC. Indeed, TLR2-dependent signaling, a receptor we have previously shown to be essential for mediating S. aureus recognition by astrocytes (Esen et al., 2004), leads to the activation of NF-κB and mitogen-activated protein kinase (MAPK) signaling cascades (Akira and Hemmi, 2003). To evaluate the importance of the MAPK pathway in mediating the S. aureus-dependent decrease in astrocyte GJC, cells were treated with the p38 MAPK inhibitor SB202190 (Ajizian et al., 1999; Singh et al., 1999). As shown in Fig. 11, p38 MAPK inhibition partially restored the level of GJC in S. aureus-treated astrocytes to that of unstimulated cells. In addition, SB202190 was also capable of partially reversing the S. aureus-dependent decrease in Cx43 protein expression (data not shown). Collectively, these findings indicate that S. aureus regulates astrocytic GJC, in part, via a p38 MAPK-dependent pathway. However, the finding that both Cx43 protein levels and functional GJC were not completely restored to baseline indicates that an additional factor (or factors) is also involved in mediating the S. aureus-induced reduction in astrocyte coupling.
Fig. 11.
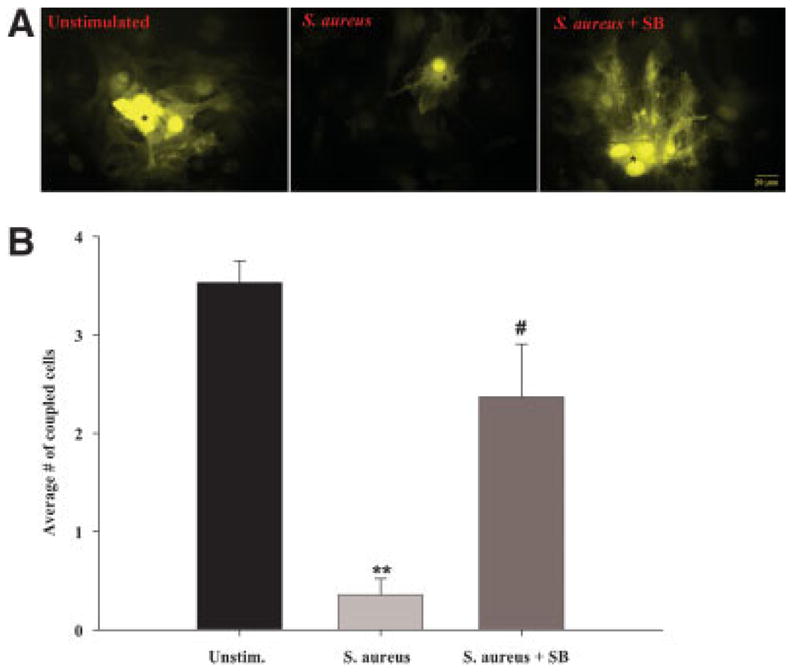
S. aureus attenuates astrocyte GJC, in part, via a p38 MAPK-dependent pathway. Primary astrocytes were seeded at 5 × 105 cells per well in 35-mm dishes and incubated overnight. The following day, cells were pre-treated with the p38 MAPK inhibitor SB202190 (SB, 1 μM) for 1 h prior to stimulation with 107 heat-inactivated S. aureus for 24 h in the continued presence of SB, whereupon functional GJC was evaluated by single-cell injections of LY (A) as described in the Materials and Methods. Microinjected cells are denoted by asterisks (*). In (B), the number of cells displaying dye transfer from each microinjected astrocyte is quantitated. Results represent the mean ± SEM of three independent experiments evaluating at least 10 injected cells per treatment group per experiment. Significant differences between unstimulated versus S. aureus treated cells are denoted with asterisks (**P < 0.001), whereas significant differences between S. aureus only versus S. aureus + SB treated astrocytes are denoted with a hatched sign (#P < 0.05). [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
DISCUSSION
Based on its prevalence as a CNS pathogen, we evaluated the effects of S. aureus and its cell wall component PGN on astrocytic gap junctional coupling. Here we demonstrate that both S. aureus and PGN differentially modulate Cx isoform expression, which appears to be regulated by a complex series of events. Specifically, both bacterial stimuli led to a decrease in astrocyte Cx43 and Cx30 expression concomitant with the inhibition of functional GJC. In contrast, Cx26 levels were enhanced in response to S. aureus and PGN. Our findings demonstrating the activation-dependent decrease in astrocyte Cx43 expression are in agreement with other reports demonstrating that Cx43 levels are downregulated in astrocytes in response to proinflammatory mediators such as NO and IL-1β (Bolanos and Medina, 1996; Duffy et al., 2000; John et al., 1999).
The observed inhibition of Cx43 and Cx30 expression in S. aureus- and PGN-stimulated astrocytes could be due to either direct and/or indirect effects, the latter of which may be mediated by the autocrine/paracrine action of proinflammatory cytokines released from activated astrocytes such as IL-1 and TNF-α. To discriminate between these two possibilities, astrocytes were treated with the protein synthesis inhibitor CHX. Inhibition of de novo protein synthesis with CHX did not alter the reduction in Cx43 expression that was consistently observed following bacterial exposure, suggesting that S. aureus directly regulates the expression of this Cx and potentially subsequent gap junction communication. Examination of dye coupling in CHX-treated cells revealed that S. aureus directly inhibits astrocytic GJC since CHX did not alter the inhibitory effects of bacteria on functional coupling at a relatively early time point following S. aureus exposure. This finding raises an important point, namely that alterations in Cx expression do not necessarily always translate into changes in functional coupling as evident by the ability of CHX to modulate the expression of certain Cx isoforms (i.e. Cx26). Alternatively, these results could be interpreted to mean that Cx43 represents the major Cx protein responsible for the observed alterations in astrocyte GJC following S. aureus stimulation since Cx43 levels remained unaffected by CHX, which mirrored the LY gap junction coupling data. However, we cannot exclude a potential indirect role for proinflammatory mediators such as TNF-α and IL-1β in modulating the S. aureus-dependent changes in astrocyte GJC, since many cell types harbor a pre-formed pool of these cytokines in an inactive state that would be CHX-insensitive. Therefore, examining gap junction coupling in IL-1 or TNF-α KO astrocytes would enable the direct assessment of these cytokines on astrocyte GJC in response to S. aureus.
Astrocyte activation by S. aureus occurs primarily via pattern recognition receptors such as Toll-like receptor 2 (TLR2), which we have recently shown to be pivotal for the recognition of S. aureus by astrocytes and subsequent induction of proinflammatory mediator release (Esen et al., 2004). As a consequence of TLR2-dependent signaling, NF-κB and MAPK signaling cascades are activated (Akira and Hemmi, 2003; Kaisho and Akira, 2002; Takeda and Akira, 2004). With regard to Cx regulation, these pathways could be envisioned to either directly influence the transcriptional activity of Cx genes and/or the expression of proinflammatory mediators that would act in an autocrine/paracrine fashion to modulate Cx levels. In addition to transcriptional regulation, activated kinases could influence the phosphorylation state of Cx proteins which in turn, would modulate GJC. Indeed, it has been reported that MAPK-dependent signal transduction pathways are capable of influencing the phosphorylation state of Cx43 (Warn-Cramer et al., 1996; Warn-Cramer and Lau, 2004) and inactivation of MAPKs with various inhibitors prevented the attenuation of GJC observed in response to pathophysiological stimuli (Cho et al., 2002; Lee et al., 2004). Similarly, our results with the p38 MAPK inhibitor SB202190 indicate that the MAPK pathway is responsible, in part, for the inhibitory effects of S. aureus on functional GJC in astrocytes. However, the participation of other kinases that have been shown to interact with Cx43, such as PKC (Martinez et al., 2002; Reynhout et al., 1992), casein kinase 1 (Cooper and Lampe, 2002), PKA (Burghardt et al., 1995; TenBroek et al., 2001), and c-Src (Li et al., 2005; Sorgen et al., 2004) cannot be excluded in modulating the S. aureus-induced decrease in Cx43 expression, and warrant further investigation.
Although to date, the majority of studies have focused on the functional implications of modulating Cx43 expression in astrocytes (Nakase et al., 2003; Naus et al., 1999; Spray et al., 1999), alterations in Cx30 levels in response to S. aureus may also impact astrocyte function and GJC. Indeed, it has been shown that Cx30 KO mice exhibit altered reactivity to novel environmental stimuli implying a role of Cx30 on neuronal activity (Dere et al., 2003). In this study, we found an interesting relationship between the monomeric and dimeric forms of Cx30 in activated astrocytes by Western blot analysis. Specifically, S. aureus and PGN treatment led to a decrease in Cx30 monomers, whereas the dimer form remained relatively unaffected. Interestingly, our immunofluorescence results using the same Cx30 polyclonal antibody mimicked the changes observed with the monomeric form of Cx30 in Western blot analysis, with both decreasing following bacterial stimulation. Although Western blots demonstrated that Cx30 dimer expression remained essentially unaltered following bacterial stimulation, it is intriguing why an overall decrease in Cx30 immunofluorescence staining was observed in both S. aureus- and PGN-treated cells at both 6 and 24 h after bacterial exposure. The reason for this finding is currently unknown.
In contrast to Cx43 and Cx30, the S. aureus-induced increase in Cx26 expression was reversed following inhibition of de novo protein synthesis, suggesting that Cx26 levels are regulated, in part, via an indirect manner. Recently, we have shown that astrocytes respond to S. aureus and PGN with the robust production of numerous proinflammatory mediators including IL-1β and TNF-α (Esen et al., 2004). An indirect effect of S. aureus on astrocyte Cx26 expression could be envisioned to occur through an autocrine/paracrine action since both TNF-α and IL-1β are capable of activating MAPK pathways upon binding to their cognate receptors (Hanada and Yoshimura, 2002). Since Cx26 is not phosphorylated it is more likely that any indirect effects of proinflammatory stimuli would be exerted at the level of transcription/translation (Kojima et al., 1999; Saez et al., 1998, 2005; Traub et al., 1989). Although Temme et al. (1998) have reported that IL-1 and TNF-α augment Cx26 while decreasing Cx32 mRNA expression in hepatocytes, to our knowledge, the role of proinflammatory mediators on modulating Cx26 levels in astrocytes has not yet been examined. Moreover, the functional consequences of augmented Cx26 expression in S. aureus- and PGN-stimulated astrocytes are unknown and warrant further investigation.
One intriguing finding that surfaced during the course of these studies related to the apparent disconnect between the degree of alterations in Cx43 and Cx30 expression detected by Western blots versus immunofluorescence staining. In particular, both Cx isoforms were detected at very low levels in S. aureus- or PGN-activated astrocytes when assessed by immunofluorescence staining and confocal microscopy, whereas the reduction in Cx expression observed in Western blots was not as dramatic. These results were highly reproducible and it is currently unclear why the activation-dependent changes in Cx43 and Cx30 expression were not more concordant between the two assays. One reason may relate to differences in the relative abundance of antigen between the two assays. For example, we load 40 μg of protein per lane for Western blot analysis to ensure strong signal detection. However, with immunofluorescence analysis, the amount of antigen present within individual cells is much more limited. In addition, our confocal analysis utilized very thin optical sections (1.6 μm), which may have also contributed to lower sensitivity since the images reported did not contain cumulative data of stacked confocal images. Therefore, what appears to be a modest decrease in Cx isoform expression in Western blot analysis could translate into a dramatic reduction in immunoreactivity due to decreased sensitivity from essentially a mass effect. In support of this possibility, it is important to note that although astrocytic Cx43 and Cx30 immuofluorescence staining is dramatically reduced in response to bacterial stimulation, these Cx isoforms are still detectable, albeit at very low levels.
The consequence(s) of S. aureus-dependent inhibition of astrocyte GJC is currently not known. Under physiological conditions, the astrocyte syncytium plays a supportive role through the transport of nutrients and waste products that are crucial for neuron homeostasis. However, under pathological conditions, this extended communication may also lead to the propagation of apoptotic and/or necrotic signals at distant sites within injured tissue leading to extended neuronal injury (Frantseva et al., 2002a,b; Lin et al., 1998). Thus, inhibition of GJC in response to injury could be envisioned to represent a protective defense mechanism of the CNS. However, in vivo studies from several groups have produced conflicting results regarding whether Cx expression is beneficial or detrimental in the context of disease pathogenesis (Farahani et al., 2005; Perez Velazquez et al., 2003). Since the brain is a complex organ with intricate relationships among its cellular constituents, it is not unexpected that different experimental paradigms such as tissue injury location, type of injury, and the resultant chemical and structural changes that occur in gap junctions may be key determinants of whether such changes are detrimental or beneficial for the outcome of various brain insults. The functional implications of changes in Cx isoform expression and GJC induced by S. aureus could be facilitated with studies of Cx43 heterozygous mice in the experimental brain abscess model that has been established in our laboratory (Kielian, 2004; Kielian et al., 2004), an area that we are actively pursuing.
In summary, S. aureus is capable of differentially modulating astrocyte Cx isoform expression that culminates in the inhibition of functional gap junction coupling. These activation-dependent changes were found to occur primarily via a direct manner, with the exception of Cx26 whose levels were influenced by de novo protein synthesis. The potential impact of these changes in the context of CNS infectious disease await testing the consequences of neuroinflammation on astrocytic GJC in acute cortical slices as well as in in vivo studies examining the relative importance of gap junction channels in the experimental brain abscess model.
Supplementary Material
This article contains supplementary material available via the Internet at http://www.interscience.wiley.com/jpages/0894-1491/suppmat.
Acknowledgments
The authors thank Dr. Gerry Dienel for equipment use during the initial phase of this study.
Grant sponsor: National Institute of Neurological Disorders and Stroke, NIH; Grant number: RO1 NS053487; Grant sponsor: Arkansas Biosciences Institute; Grant sponsor: Core Facility at UAMS supported by NINDS; Grant number: P30 NS047546; Grant sponsor: INBRE, NIH; Grant number: P20 RR6460; Grant sponsor: NCRR, NIH; Grant number: S10 RR19395.
References
- Ajizian SJ, English BK, Meals EA. Specific inhibitors of p38 and extracellular signal-regulated kinase mitogen-activated protein kinase pathways block inducible nitric oxide synthase and tumor necrosis factor accumulation in murine macrophages stimulated with lipopolysaccharide and interferon-gamma. J Infect Dis. 1999;179:939–944. doi: 10.1086/314659. [DOI] [PubMed] [Google Scholar]
- Akira S, Hemmi H. Recognition of pathogen-associated molecular patterns by TLR family. Immunol Lett. 2003;85:85–95. doi: 10.1016/s0165-2478(02)00228-6. [DOI] [PubMed] [Google Scholar]
- Altevogt BM, Paul DL. Four classes of intercellular channels between glial cells in the CNS. J Neurosci. 2004;24:4313–4323. doi: 10.1523/JNEUROSCI.3303-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson CM, Swanson RA. Astrocyte glutamate transport: Review of properties, regulation, and physiological functions. Glia. 2000;32:1–14. [PubMed] [Google Scholar]
- Baldwin AC, Kielian T. Persistent immune activation associated with a mouse model of Staphylococcus aureus-induced experimental brain abscess. J Neuroimmunol. 2004;151:24–32. doi: 10.1016/j.jneuroim.2004.02.002. [DOI] [PubMed] [Google Scholar]
- Bolanos JP, Medina JM. Induction of nitric oxide synthase inhibits gap junction permeability in cultured rat astrocytes. J Neurochem. 1996;66:2091–2099. doi: 10.1046/j.1471-4159.1996.66052091.x. [DOI] [PubMed] [Google Scholar]
- Burghardt RC, Barhoumi R, Sewall TC, Bowen JA. Cyclic AMP induces rapid increases in gap junction permeability and changes in the cellular distribution of connexin43. J Membr Biol. 1995;148:243–253. doi: 10.1007/BF00235042. [DOI] [PubMed] [Google Scholar]
- Campos de Carvalho AC, Roy C, Hertzberg EL, Tanowitz HB, Kessler JA, Weiss LM, Wittner M, Dermietzel R, Gao Y, Spray DC. Gap junction disappearance in astrocytes and leptomeningeal cells as a consequence of protozoan infection. Brain Res. 1998;790:304–314. doi: 10.1016/s0006-8993(97)01523-0. [DOI] [PubMed] [Google Scholar]
- Chanson M, Berclaz PY, Scerri I, Dudez T, Wernke-Dollries K, Pizurki L, Pavirani A, Fiedler MA, Suter S. Regulation of gap junctional communication by a proinflammatory cytokine in cystic fibrosis transmembrane conductance regulator-expressing but not cystic fibrosis airway cells. Am J Pathol. 2001;158:1775–1784. doi: 10.1016/S0002-9440(10)64133-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charles AC, Naus CC, Zhu D, Kidder GM, Dirksen ER, Sanderson MJ. Intercellular calcium signaling via gap junctions in glioma cells. J Cell Biol. 1992;118:195–201. doi: 10.1083/jcb.118.1.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho JH, Cho SD, Hu H, Kim SH, Lee SK, Lee YS, Kang KS. The roles of ERK1/2 and p38 MAP kinases in the preventive mechanisms of mushroom Phellinus linteus against the inhibition of gap junctional intercellular communication by hydrogen peroxide. Carcinogenesis. 2002;23:1163–1169. doi: 10.1093/carcin/23.7.1163. [DOI] [PubMed] [Google Scholar]
- Cooper CD, Lampe PD. Casein kinase 1 regulates connexin-43 gap junction assembly. J Biol Chem. 2002;277:44962–44968. doi: 10.1074/jbc.M209427200. [DOI] [PubMed] [Google Scholar]
- De Maio A, Gingalewski C, Theodorakis NG, Clemens MG. Interruption of hepatic gap junctional communication in the rat during inflammation induced by bacterial lipopolysaccharide. Shock. 2000;14:53–59. doi: 10.1097/00024382-200014010-00010. [DOI] [PubMed] [Google Scholar]
- Dere E, De Souza-Silva MA, Frisch C, Teubner B, Sohl G, Willecke K, Huston JP. Connexin30-deficient mice show increased emotionality and decreased rearing activity in the open-field along with neurochemical changes. Eur J Neurosci. 2003;18:629–638. doi: 10.1046/j.1460-9568.2003.02784.x. [DOI] [PubMed] [Google Scholar]
- Dermietzel R, Gao Y, Scemes E, Vieira D, Urban M, Kremer M, Bennett MV, Spray DC. Connexin43 null mice reveal that astrocytes express multiple connexins. Brain Res Brain Res Rev. 2000;32:45–56. doi: 10.1016/s0165-0173(99)00067-3. [DOI] [PubMed] [Google Scholar]
- Dong Y, Benveniste EN. Immune function of astrocytes. Glia. 2001;36:180–190. doi: 10.1002/glia.1107. [DOI] [PubMed] [Google Scholar]
- Duffy HS, John GR, Lee SC, Brosnan CF, Spray DC. Reciprocal regulation of the junctional proteins claudin-1 and connexin43 by interleukin-1β in primary human fetal astrocytes. J Neurosci. 2000;20:RC114. doi: 10.1523/JNEUROSCI.20-23-j0004.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eikelenboom P, Bate C, Van Gool WA, Hoozemans JJ, Rozemuller JM, Veerhuis R, Williams A. Neuroinflammation in Alzheimer’s disease and prion disease. Glia. 2002;40:232–239. doi: 10.1002/glia.10146. [DOI] [PubMed] [Google Scholar]
- Esen N, Tanga FY, DeLeo JA, Kielian T. Toll-like receptor 2 (TLR2) mediates astrocyte activation in response to the Gram-positive bacterium Staphylococcus aureus. J Neurochem. 2004;88:746–758. doi: 10.1046/j.1471-4159.2003.02202.x. [DOI] [PubMed] [Google Scholar]
- Eugenin EA, Branes MC, Berman JW, Saez JC. TNF-α plus IFN-γ induce connexin43 expression and formation of gap junctions between human monocytes/macrophages that enhance physiological responses. J Immunol. 2003;170:1320–1328. doi: 10.4049/jimmunol.170.3.1320. [DOI] [PubMed] [Google Scholar]
- Farahani R, Pina-Benabou MH, Kyrozis A, Siddiq A, Barradas PC, Chiu FC, Cavalcante LA, Lai JC, Stanton PK, Rozental R. Alterations in metabolism and gap junction expression may determine the role of astrocytes as ‘‘good samaritans’’ or executioners. Glia. 2005;50:351–361. doi: 10.1002/glia.20213. [DOI] [PubMed] [Google Scholar]
- Faustmann PM, Haase CG, Romberg S, Hinkerohe D, Szlachta D, Smikalla D, Krause D, Dermietzel R. Microglia activation influences dye coupling and Cx43 expression of the astrocytic network. Glia. 2003;42:101–108. doi: 10.1002/glia.10141. [DOI] [PubMed] [Google Scholar]
- Fernandez-Cobo M, Gingalewski C, Drujan D, De Maio A. Down-regulation of connexin 43 gene expression in rat heart during inflammation. The role of tumour necrosis factor. Cytokine. 1999;11:216–224. doi: 10.1006/cyto.1998.0422. [DOI] [PubMed] [Google Scholar]
- Frantseva MV, Kokarovtseva L, Naus CG, Carlen PL, MacFabe D, Perez Velazquez JL. Specific gap junctions enhance the neuronal vulnerability to brain traumatic injury. J Neurosci. 2002b;22:644–653. doi: 10.1523/JNEUROSCI.22-03-00644.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frantseva MV, Kokarovtseva L, Perez Velazquez JL. Ischemia-induced brain damage depends on specific gap-junctional coupling. J Cereb Blood Flow Metab. 2002a;22:453–462. doi: 10.1097/00004647-200204000-00009. [DOI] [PubMed] [Google Scholar]
- Froes MM, de Carvalho AC. Gap junction-mediated loops of neuronal-glial interactions. Glia. 1998;24:97–107. [PubMed] [Google Scholar]
- Griffin WS, Mrak RE. Interleukin-1 in the genesis and progression of and risk for development of neuronal degeneration in Alzheimer’s disease. J Leukoc Biol. 2002;72:233–238. [PMC free article] [PubMed] [Google Scholar]
- Hanada T, Yoshimura A. Regulation of cytokine signaling and inflammation. Cytokine Growth Factor Rev. 2002;13:413–421. doi: 10.1016/s1359-6101(02)00026-6. [DOI] [PubMed] [Google Scholar]
- John GR, Scemes E, Suadicani SO, Liu JS, Charles PC, Lee SC, Spray DC, Brosnan CF. IL-1β differentially regulates calcium wave propagation between primary human fetal astrocytes via pathways involving P2 receptors and gap junction channels. Proc Natl Acad Sci USA. 1999;96:11613–11618. doi: 10.1073/pnas.96.20.11613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaisho T, Akira S. Toll-like receptors as adjuvant receptors. Biochim Biophys Acta. 2002;1589:1–13. doi: 10.1016/s0167-4889(01)00182-3. [DOI] [PubMed] [Google Scholar]
- Kielian T. Immunopathogenesis of brain abscess. J Neuroinflam. 2004;1:16. doi: 10.1186/1742-2094-1-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kielian T, Bearden ED, Baldwin AC, Esen N. IL-1 and TNF-α play a pivotal role in the host immune response in a mouse model of Staphylococcus aureus-induced experimental brain abscess. J Neuropathol Exp Neurol. 2004;63:381–396. doi: 10.1093/jnen/63.4.381. [DOI] [PubMed] [Google Scholar]
- Kim JM, Oh YK, Lee JH, Im DY, Kim YJ, Youn J, Lee CH, Son H, Lee YS, Park JY, Choi IH. Induction of proinflammatory mediators requires activation of the TRAF, NIK, IKK and NF-κB signal transduction pathway in astrocytes infected with Escherichia coli. Clin Exp Immunol. 2005;140:450–460. doi: 10.1111/j.1365-2249.2005.02804.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koedel U, Scheld WM, Pfister HW. Pathogenesis and pathophysiology of pneumococcal meningitis. Lancet Infect Dis. 2002;2:721–736. doi: 10.1016/s1473-3099(02)00450-4. [DOI] [PubMed] [Google Scholar]
- Kojima T, Srinivas M, Fort A, Hopperstad M, Urban M, Hertzberg EL, Mochizuki Y, Spray DC. TPA induced expression and function of human connexin 26 by post-translational mechanisms in stably transfected neuroblastoma cells. Cell Struct Funct. 1999;24:435–441. doi: 10.1247/csf.24.435. [DOI] [PubMed] [Google Scholar]
- Lee KW, Jung JW, Kang KS, Lee HJ. p38 is a key signaling molecule for H-ras-induced inhibition of gap junction intercellular communication in rat liver epithelial cells. Ann N Y Acad Sci. 2004;1030:258–263. doi: 10.1196/annals.1329.032. [DOI] [PubMed] [Google Scholar]
- Li W, Hertzberg EL, Spray DC. Regulation of connexin43-protein binding in astrocytes in response to chemical ischemia/hypoxia. J Biol Chem. 2005;280:7941–7948. doi: 10.1074/jbc.M410548200. [DOI] [PubMed] [Google Scholar]
- Lin JH, Weigel H, Cotrina ML, Liu S, Bueno E, Hansen AJ, Hansen TW, Goldman S, Nedergaard M. Gap-junction-mediated propagation and amplification of cell injury. Nat Neurosci. 1998;1:494–500. doi: 10.1038/2210. [DOI] [PubMed] [Google Scholar]
- Martinez AD, Hayrapetyan V, Moreno AP, Beyer EC. Connexin43 and connexin45 form heteromeric gap junction channels in which individual components determine permeability and regulation. Circ Res. 2002;90:1100–1107. doi: 10.1161/01.res.0000019580.64013.31. [DOI] [PubMed] [Google Scholar]
- Martinez AD, Saez JC. Regulation of astrocyte gap junctions by hypoxia-reoxygenation. Brain Res Brain Res Rev. 2000;32:250–258. doi: 10.1016/s0165-0173(99)00086-7. [DOI] [PubMed] [Google Scholar]
- McGeer PL, McGeer EG. Local neuroinflammation and the progression of Alzheimer’s disease. J Neurovirol. 2002;8:529–538. doi: 10.1080/13550280290100969. [DOI] [PubMed] [Google Scholar]
- Nagy JI, Ionescu AV, Lynn BD, Rash JE. Coupling of astrocyte connexins Cx26, Cx30, Cx43 to oligodendrocyte Cx29, Cx32, Cx47: Implications from normal and connexin32 knockout mice. Glia. 2003;44:205–218. doi: 10.1002/glia.10278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagy JI, Li X, Rempel J, Stelmack G, Patel D, Staines WA, Yasumura T, Rash JE. Connexin26 in adult rodent central nervous system: Demonstration at astrocytic gap junctions and colocalization with connexin30 and connexin43. J Comp Neurol. 2001;441:302–323. doi: 10.1002/cne.1414. [DOI] [PubMed] [Google Scholar]
- Nakase T, Fushiki S, Naus CC. Astrocytic gap junctions composed of connexin 43 reduce apoptotic neuronal damage in cerebral ischemia. Stroke. 2003;34:1987–1993. doi: 10.1161/01.STR.0000079814.72027.34. [DOI] [PubMed] [Google Scholar]
- Nakase T, Naus CC. Gap junctions and neurological disorders of the central nervous system. Biochim Biophys Acta. 2004;1662:149–158. doi: 10.1016/j.bbamem.2004.01.009. [DOI] [PubMed] [Google Scholar]
- Nau R, Bruck W. Neuronal injury in bacterial meningitis: mechanisms and implications for therapy. Trends Neurosci. 2002;25:38–45. doi: 10.1016/s0166-2236(00)02024-5. [DOI] [PubMed] [Google Scholar]
- Naus CC, Bani-Yaghoub M. Gap junctional communication in the developing central nervous system. Cell Biol Int. 1998;22:751–763. doi: 10.1006/cbir.1998.0391. [DOI] [PubMed] [Google Scholar]
- Naus CC, Bani-Yaghoub M, Rushlow W, Bechberger JF. Consequences of impaired gap junctional communication in glial cells. Adv Exp Med Biol. 1999;468:373–381. doi: 10.1007/978-1-4615-4685-6_29. [DOI] [PubMed] [Google Scholar]
- Penes MC, Li X, Nagy JI. Expression of zonula occludens-1 (ZO-1) and the transcription factor ZO-1-associated nucleic acid-binding protein (ZONAB)-MsY3 in glial cells and colocalization at oligodendrocyte and astrocyte gap junctions in mouse brain. Eur J Neurosci. 2005;22:404–418. doi: 10.1111/j.1460-9568.2005.04225.x. [DOI] [PubMed] [Google Scholar]
- Perez Velazquez JL, Frantseva MV, Naus CC. Gap junctions and neuronal injury: protectants or executioners? Neuroscientist. 2003;9:5–9. doi: 10.1177/1073858402239586. [DOI] [PubMed] [Google Scholar]
- Ransom BR. Glial modulation of neural excitability mediated by extracellular pH: A hypothesis revisited. Prog Brain Res. 2000;125:217–228. doi: 10.1016/S0079-6123(00)25012-7. [DOI] [PubMed] [Google Scholar]
- Ransom BR, Orkand RK. Glial-neuronal interactions in non-synaptic areas of the brain: Studies in the optic nerve. Trends Neurosci. 1996;19:352–358. doi: 10.1016/0166-2236(96)10045-x. [DOI] [PubMed] [Google Scholar]
- Rash JE, Yasumura T, Dudek FE, Nagy JI. Cell-specific expression of connexins and evidence of restricted gap junctional coupling between glial cells and between neurons. J Neurosci. 2001;21:1983–2000. doi: 10.1523/JNEUROSCI.21-06-01983.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynhout JK, Lampe PD, Johnson RG. An activator of protein kinase C inhibits gap junction communication between cultured bovine lens cells. Exp Cell Res. 1992;198:337–342. doi: 10.1016/0014-4827(92)90388-o. [DOI] [PubMed] [Google Scholar]
- Rouach N, Avignone E, Meme W, Koulakoff A, Venance L, Blomstrand F, Giaume C. Gap junctions and connexin expression in the normal and pathological central nervous system. Biol Cell. 2002;94:457–475. doi: 10.1016/s0248-4900(02)00016-3. [DOI] [PubMed] [Google Scholar]
- Rozental R, Giaume C, Spray DC. Gap junctions in the nervous system. Brain Res Brain Res Rev. 2000a;32:11–15. doi: 10.1016/s0165-0173(99)00095-8. [DOI] [PubMed] [Google Scholar]
- Saez JC, Martinez AD, Branes MC, Gonzalez HE. Regulation of gap junctions by protein phosphorylation. Braz J Med Biol Res. 1998;31:593–600. doi: 10.1590/s0100-879x1998000500001. [DOI] [PubMed] [Google Scholar]
- Saez JC, Retamal MA, Basilio D, Bukauskas FF, Bennett MV. Connexin-based gap junction hemichannels: gating mechanisms. Biochim Biophys Acta. 2005;1711:215–224. doi: 10.1016/j.bbamem.2005.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scemes E, Dermietzel R, Spray DC. Calcium waves between astrocytes from Cx43 knockout mice. Glia. 1998;24:65–73. doi: 10.1002/(sici)1098-1136(199809)24:1<65::aid-glia7>3.0.co;2-#. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheld WM, Koedel U, Nathan B, Pfister HW. Pathophysiology of bacterial meningitis: Mechanism(s) of neuronal injury. J Infect Dis. 2002;186(Suppl 2):S225–S233. doi: 10.1086/344939. [DOI] [PubMed] [Google Scholar]
- Schipke CG, Boucsein C, Ohlemeyer C, Kirchhoff F, Kettenmann H. Astrocyte Ca2+ waves trigger responses in microglial cells in brain slices. FASEB J. 2002;16:255–257. doi: 10.1096/fj.01-0514fje. [DOI] [PubMed] [Google Scholar]
- Singh RP, Dhawan P, Golden C, Kapoor GS, Mehta KD. One-way !cross-talk between p38(MAPK) and p42/44(MAPK). Inhibition of p38 (MAPK) induces low density lipoprotein receptor expression through activation of the p42/44(MAPK) cascade. J Biol Chem. 1999;274:19593–19600. doi: 10.1074/jbc.274.28.19593. [DOI] [PubMed] [Google Scholar]
- Smits HA, van Beelen AJ, de Vos NM, Rijsmus A, van der Bruggen T, Verhoef J, van Muiswinkel FL, Nottet HS. Activation of human macrophages by amyloid-β is attenuated by astrocytes. J Immunol. 2001;166:6869–6876. doi: 10.4049/jimmunol.166.11.6869. [DOI] [PubMed] [Google Scholar]
- Sorgen PL, Duffy HS, Sahoo P, Coombs W, Delmar M, Spray DC. Structural changes in the carboxyl terminus of the gap junction protein connexin43 indicates signaling between binding domains for c-Src and zonula occludens-1. J Biol Chem. 2004;279:54695–54701. doi: 10.1074/jbc.M409552200. [DOI] [PubMed] [Google Scholar]
- Spray DC, Duffy HS, Scemes E. Gap junctions in glia: Types, roles, and plasticity. In: Matsas R, Tsacopoulos M, editors. The functional roles of glial cells in health and disease (Advances in experimental medicine and biology) New York: Plenum; 1999. pp. 339–359. [PubMed] [Google Scholar]
- Stout CE, Costantin JL, Naus CC, Charles AC. Intercellular calcium signaling in astrocytes via ATP release through connexin hemi-channels. J Biol Chem. 2002;277:10482–10488. doi: 10.1074/jbc.M109902200. [DOI] [PubMed] [Google Scholar]
- Takeda K, Akira S. Microbial recognition by Toll-like receptors. J Dermatol Sci. 2004;34:73–82. doi: 10.1016/j.jdermsci.2003.10.002. [DOI] [PubMed] [Google Scholar]
- Temme A, Traub O, Willecke K. Downregulation of connexin32 protein and gap-junctional intercellular communication by cytokine-mediated acute-phase response in immortalized mouse hepatocytes. Cell Tissue Res. 1998;294:345–350. doi: 10.1007/s004410051184. [DOI] [PubMed] [Google Scholar]
- TenBroek EM, Lampe PD, Solan JL, Reynhout JK, Johnson RG. Ser364 of connexin43 and the upregulation of gap junction assembly by cAMP. J Cell Biol. 2001;155:1307–1318. doi: 10.1083/jcb.200102017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Townsend GC, Scheld WM. Infections of the central nervous system. Adv Intern Med. 1998;43:403–447. [PubMed] [Google Scholar]
- Traub O, Look J, Dermietzel R, Brummer F, Hulser D, Willecke K. Comparative characterization of the 21-kD and 26-kD gap junction proteins in murine liver and cultured hepatocytes. J Cell Biol. 1989;108:1039–1051. doi: 10.1083/jcb.108.3.1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warn-Cramer BJ, Lampe PD, Kurata WE, Kanemitsu MY, Loo LW, Eckhart W, Lau AF. Characterization of the mitogen-activated protein kinase phosphorylation sites on the connexin-43 gap junction protein. J Biol Chem. 1996;271:3779–3786. doi: 10.1074/jbc.271.7.3779. [DOI] [PubMed] [Google Scholar]
- Warn-Cramer BJ, Lau AF. Regulation of gap junctions by tyro-sine protein kinases. Biochim Biophys Acta. 2004;1662:81–95. doi: 10.1016/j.bbamem.2003.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye ZC, Wyeth MS, Baltan-Tekkok S, Ransom BR. Functional hemichannels in astrocytes: A novel mechanism of glutamate release. J Neurosci. 2003;23:3588–3596. doi: 10.1523/JNEUROSCI.23-09-03588.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
This article contains supplementary material available via the Internet at http://www.interscience.wiley.com/jpages/0894-1491/suppmat.