

Abstract
Electrostatic properties of cowpea chlorotic mottle virus (CCMV) and cucumber mosaic virus (CMV) were investigated using numerical solutions to the Poisson-Boltzmann equation. Experimentally, it has been shown that CCMV particles swell in the absence of divalent cations when the pH is raised from 5 to 7. CMV, although structurally homologous, does not undergo this transition. An analysis of the calculated electrostatic potential confirms that a strong electrostatic repulsion at the calcium binding sites in the CCMV capsid is most likely the driving force for the capsid swelling process during the release of calcium. The binding interaction between the encapsulated genome material (RNA) inside of the capsid and the inner capsid shell is weakened during the swelling transition. This probably aids in the RNA release process, but it is unlikely that the RNA is released through capsid openings due to unfavorable electrostatic interaction between the RNA and capsid inner shell residues at these openings. Calculations of the calcium binding energies show that Ca2+ can bind both to the native and swollen forms of the CCMV virion. Favorable binding to the swollen form suggests that Ca2+ ions can induce the capsid contraction and stabilize the native form.
Keywords: Computational Modeling, Supramolecular Assembly, Poisson-Boltzmann Equation, Electrostatic Potential, Simulation, Normal Mode Analysis, Swelling, Electrostatic Binding Energy
Introduction
Assembly and disassembly of a viral capsid, or a protein shell, are essential steps in the life cycle of a virus. Understanding of viral assembly/disassembly can provide additional insights to other oligomerization processes as in protein association in a signal transduction pathway, which occurs through a similar mechanism.1 Additionally, understanding the properties and function of the capsid shell alone is important for inferring all steps involved in host cell entry and release of the enveloped genetic material and can aid in proposing efficient ways of interfering with capsid assembly and stability via structure-based design of antiviral therapeutics.
One of the most intensely studied viruses has been the cowpea chlorotic mottle virus (CCMV), a positive strand RNA plant-infecting virus which belongs to the Bromoviridae family (Figure 1a and b). It is composed of a protein capsid and viral RNA which is packed inside. It has been widely used as a model system for understanding of biological assembly because of its icosahedral structure and the limited number of gene products required for its assembly. CCMV offers an accessible model system for examining the processes that regulate viral assembly, disassembly, and stability, both for theoretical and experimental studies.
Figure 1.
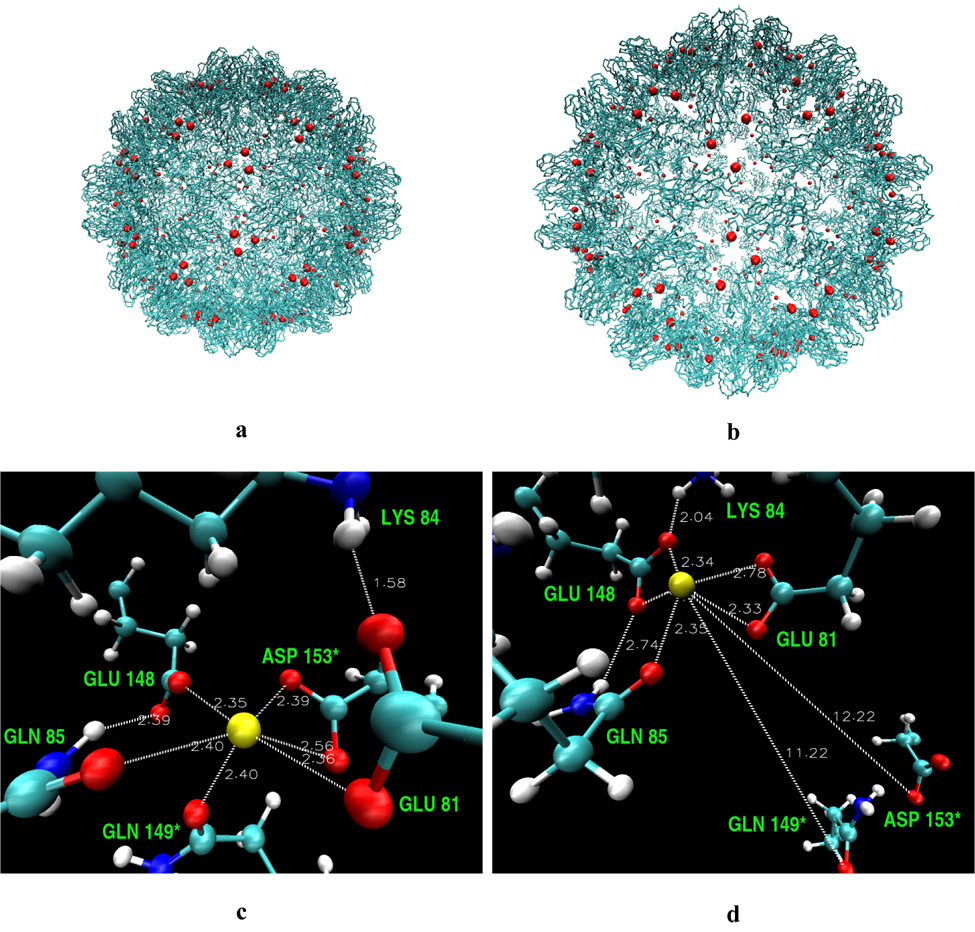
Backbone model of the CCMV capsid in the native (a) and swollen (b) state with calcium ions shown in red. Modeled calcium cavity residues in the native (c) and swollen (d) form of the CCMV virus. The most important distances from calcium ions (yellow spheres) are shown (in Å).
The crystal structure of CCMV has been solved to 3.2 Å resolution.2 The CCMV virion is comprised of 180 copies of the coat protein subunits arranged with T = 3 quasi-symmetry and organized in 20 hexameric and 12 pentameric capsomers (Figure 2). Each coat protein subunit consists of a β-barrel fold (residues 52–176) from which long N-terminal (residues 1–51) and C-terminal arms (residues 176–190) extend in opposite directions (Figure 2a). The individual pentameric and hexameric capsomers are linked into a comprehensive network by the coat protein C-termini. Additionally, each extended C-terminus is anchored by being clamped between the β-barrel core and the N-proximal loop of its partner subunit. The N-terminal arm of the coat protein participates in three different types of interactions. The first 26 amino acids are very basic and interact with negatively charged viral RNA. Amino acids 27–35 form the β-hexamer, which stabilizes hexameric capsomers, and amino acids 44–51 form the clamp for the extended C-terminal arm. This β-hexamer is located at the icosahedral threefold axes and is variant of the β-annulus observed in many plant virus capsids.3
Figure 2.
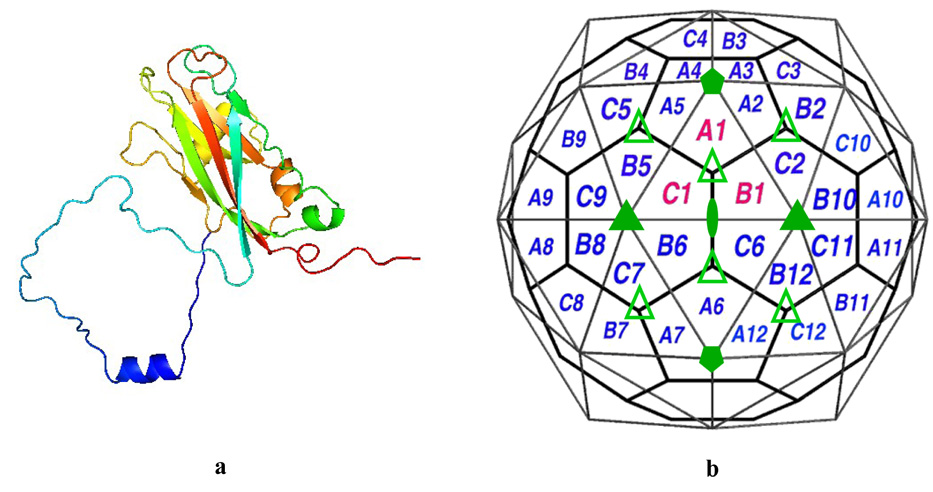
(a) A single CCMV capsid protein subunit (N-terminus arm blue, C-terminus is red). (b) Schematic representation of quasi-equivalent lattice model for CCMV. The 2-fold, 3-fold, 5-fold axes are identified by filled oval, triangles and pentagons respectively. The pseudo threefold axes are identified by empty triangles. The central triangle (labeled A1, B1 and C1) defines the icosahedral asymmetric unit. The polygons represent chemically identical protein subunits which represent the three slightly different geometrical environments of the coat protein.51
Inside the capsid, the single-stranded viral RNA is arranged as an interior shell, leaving the virion center hollow, and adopts an ordered conformation at each of the quasi threefold axis. The N-terminal 26 amino acids of the coat protein, which are highly basic and required for RNA packaging,4 extend into the interior of the capsid and interact with the RNA to neutralize phosphate charges. Additionally, the RNA interacts with 13 of the amino acids on the interior capsid surface. These include ionic interactions with number of lysines and arginines, and a stacking interaction between the base of two consecutive RNA nucleotides and a tryptophan side chain.
The capsid protein is multifunctional; in addition to having a role in encapsulation, it affects the viral movement in plants, transmission, symptom expression, and host range.
Native CCMV is stable at pH 5.0 but at pH 7.0 and in the absence of Ca2+ or Mg2+ the capsid diameter swells by approximately 10%. In this process the ionic strength remains low < 0.2M.5,6 This swelling can also be induced just by increasing pH of the solution while ionic strength is kept constant.2,7 Swelling is the result of an expansion of the virion capsid at the pseudo threefold axis while the contacts within the hexameric and pentameric morphological units are conserved (Figure 1a and b).2 Although the swelling creates large openings in the capsid at the pseudo-threefold axis, the viral RNA is not spontaneously released, rendering the swelling reversible.8,9 The capsid dimer subunit interactions are retained, but shifted in their geometry.10,11 The expanded state is stabilized by dimer and β-hexamer intersubunit contacts and, predominantly, by RNA-protein interactions. Empty capsids cannot swell without dissociating due to the absence of RNA-capsid stabilizing interaction. The biological significance of the swelling phenomenon is not well understood but it is believed that this mechanism is related to RNA release during infection.
The assembly process is thought to be initiated by the association of two protein subunits forming dimers and, consequently, pentamer of dimers.12 These form, with addition of other dimers, pentameric and hexameric capsomers. The overall assembly rate is high; infectious particles assemble in vitro in seconds.4
The first eight N-terminal amino acid residues are not essential for viral assembly in vitro; both empty and RNA-containing particles are assembled even when these residues are deleted.4 However, deletion of the first 25 N-terminal residues eliminates the ability of the virus to assemble RNA-containing particles, although empty particles are still produced in vitro. This experimental result is also supported by our recent simulation study.13 The deletion of the complete N-terminal arm prevents assembly of either RNA-containing or empty particles, presumably due to the importance of the β-hexamer structure for CCMV assembly.4
Based on several experimental mutation studies5–7 Albert et al. proposed that swelling may not be required for the RNA release from the capsid, and that the pentameric capsomer can undergo a different structural transition by forming a channel through which the viral RNA passes and becomes available to the cell’s replicating machinery.7 This has been supported by a recent computational study of RNA diffusion through a pseudo threefold channel in the capsid shell.14 However, an alternative interpretation of the experimental observation has been put forward which suggests that a fraction of the virions swell at neutral pH.15
The calcium electron density is not observed in the X-ray structure of the virus, but based on the experimental conditions of crystal growth and theoretical modeling, the calcium positions have been indicated.2,10 Three binding sites for the calcium ions in one unit of the CCMV virus have been proposed. They are situated at the interfaces of the subunits A-B, B-C, and C-A (Figure 2b). The calcium cavity is composed mainly of glutamic and aspartic acids (Figure 1c and d). These acidic residues interacting with calcium have been also seen in other calcium binding systems. Such a negatively charged surrounding is likely to bind a positively charged ion. It was proposed that binding of Ca2+ in the capsid cavities adds to the stability of the native virion.2
In a recent study,16 the calcium ions have been shown to be essential for the viral stability and for the assembly of the capsid in another virus, simian virus 40. Experiments were performed in which the negatively charged residues in the calcium cavity were mutated into a positively charged ones. The work has shown that the calcium ions mediate not only the virion assembly but also contribute to the initial process of cell entry. The studies were performed in vivo and provided the evidence for the controlled manner of calcium binding and release during the infection process. The study confirmed also that the calcium affects the structural flexibility of the virion.
In this study we explore the electrostatic properties of the CCMV viral capsid and also of cucumber mosaic virus (CMV). The CMV virus shows a remarkable similarity in overall architecture to CCMV but does not undergo the swelling phenomenon observed in CCMV.3,10 While in CCMV, a cluster of acidic residues is found at the pseudo threefold center which is likely to be responsible for the swelling, no such clusters exist in CMV.10,17 Analysis and comparison of electrostatic properties and in particular the electrostatic potential at the protein-protein interface at the pseudo threefold centers between CCMV and CMV should offer insights into functional differences between these two viral capsids. We also investigated the energetics of calcium binding to CCMV capsid cavities. Because the process of swelling is believed to require the removal of Ca2+ and, on the other hand, is reversible, calcium has to show favorable binding to both of the forms: contracted and expanded. The relative binding free energies to the native and swollen CCMV of the site-bound Ca2+ were calculated in this study based on the solution of the Poisson-Boltzmann equation in continuum solvent.
Materials and Methods
Theory
Poisson-Boltzmann Equation
The electrostatic calculations were based on Poisson-Boltzmann theory. For many years, this model has been successfully applied to biological molecules and the details of its implementation and application to such systems may be found in the following papers.18–20
The differential Poisson equation describes the electrostatic potential Φ(r) in a medium with a dielectric scalar field ε(r) and with a charge density ρ(r)
| (1) |
In a solvated molecular system, the charge density is divided into the fixed interior charge distribution of the molecule, ρint(r), and a mobile exterior charge density of the solvent and ions. The exterior is modeled as a dielectric continuum with the mobile ion density approximated by a Boltzmann distribution at temperature T. Equation 1, therefore, takes the form of a nonlinear Poisson-Boltzmann (PB) equation
| (2) |
where ε(r) is the dielectric constant of the solute or the solvent, k is the Boltzmann constant, λ equals 1 for ion-accessible regions and 0 elsewhere. The is the modified Debye-Hückel parameter defined as
| (3) |
where NA is the Avogadro constant, e the charge of an electron, and I the ionic strength. The nonlinear PB equation is often approximated by its linear form which is easier to solve,
| (4) |
Normal Mode Analysis
Normal mode analysis is a powerful technique to study large conformational rearrangements in biological systems. This approach generally entails the diagonalization of the matrix of second derivatives of the potential energy for the calculation of the independent collective modes of displacement. The direct calculation of atomic-level normal modes for viruses is computationally prohibitive for use in routine application, and some type of multi-scale approach is necessary. One such approach, the elastic network model,21 provides a simplified representation of the underlying energy surface for large systems that enables the study of the dynamics of these systems.22,23 The elastic network models can be constructed and normal mode analysis can be performed based on Cα’s, residue blocks, or even coarser granularities to yield good descriptions of large-scale conformational distortions in macromolecular assemblies.24 Further computational efficiency can be gained without significant loss in information through the addition of the rotation-translation block (RTB) method for the diagonalization of the matrix of second derivatives.25 In the present calculations we have used a Cα–only representation in constructing the elastic network models of the virus capsids and the RTB approach with individual blocks corresponding the each protein in the viral capsid for the normal mode calculations. Details of the elastic network models and associated normal mode methods can be found in the recent review by Tama and Brooks.26
It was established in our earlier studies of CCMV swelling that one of the lowest frequency normal modes well represents most of the conformational change observed between native and swollen CCMV.10 Using the normal mode directions from these calculations, intermediate structures along the swelling pathway were proposed and examined to investigate the origins of the observed pH triggered swelling in CCMV, and the key role of calcium binding and pKa shifts of acidic residues during expansion of the capsid were identified as critical components in the regulating the swelling process.10 In the present work we have extended the analysis of the electrostatic properties associated with the swelling processes by combining the normal mode calculations with whole-capsid electrostatic calculations. Since the NMA was performed using only the Cα atoms, fully atomic structures were obtained by superimposing the atomic detailed structure onto the Cα framework for each intermediate. These intermediate structures were used for the electrostatic calculations presented here.
Ligand Binding Energy Calculations
The free energy of ligand-protein association may be approximated in the form:27,28
| (5) |
where ΔGelec is the electrostatic contribution, ΔGnp the non-polar or hydrophobic term, and −TΔS describes the loss of entropy (conformational, translational and rotational) upon complexation. The electrostatic term is often calculated using the continuum Poisson-Boltzmann model,28,29 and the non-polar term is estimated based on the amount of the solvent accessible surface area (SASA) buried upon binding. We apply the above equation to calculate the relative strength of binding of site-bound Ca2+ between the native and expanded forms of the viral capsid.
In case of CCMV the volume of the Ca2+ is very small in comparison to the volume of ligands usually binding to proteins. Moreover, calcium is bound in a tight cavity and is inaccessible to solvent. The difference in the solvent accessible area upon binding of one Ca2+ is 129 Å2 and this difference is similar in the native and swollen forms of the virus. Estimating an upper limit of ΔGnp by multiplying the change in SASA by one of the highest proportionality constants applied in literature (60 cal/mol Å2)27,30 gives a value for ΔGnp less than 2 kcal/mol, which is negligible in comparison to the electrostatic contribution. Additionally, because we are interested in the relative binding energies between the native and expanded CCMV configurations, the nonpolar term cancels out because the contribution is similar in those two forms.
For similar reasons, the difference in entropy loss −TΔS should be roughly equal in both forms of CCMV, and significantly smaller than the difference in ΔGelec. Based on a rigid structure it is impossible to calculate −TΔS of calcium and residues upon binding but one may estimate this quantity based on literature and calculations for other systems.
We do not know how flexible are the residues without the presence of calcium. One of the glutamates Glu81 may be protonated without Ca2+ and stabilize the cavity residues;10 presence of water molecules may not be excluded. The difference in the SASA between the native structure with and without calcium is 111 Å2 suggesting that if we assume that the whole difference comes from the cavity it is 18.5 Å2 per residue. This is not a big number per residue and comes only from side-chains, main chains are shielded even prior Ca2+ binding, so we do not account for their entropy change. If we assume that the side-chains are ’free’ before and completely ’restricted’ to one rotamer upon calcium binding, we can estimate an upper limit of entropy loss for the cavity residues. The work of Pickett and Sternberg31,32 predicted an empirical scale of entropy loss per side-chain upon folding depending on the residue type. Following this scale the loss for the native cavity would be at most 11 kcal/mol and for the swollen 7.6 kcal/mol.
Translational and rotational entropy loss of calcium upon binding is also difficult to estimate. It was shown that Ca2+ forms stable clusters with up to eight water molecules.33 Therefore, its motion is already heavily constrained prior binding by interactions with aqueous environment. The interactions in the cavity stabilize calcium as well to override favorable interactions in water. The calculated entropy loss for ligands is typically between 4–7 kcal/mol [34,35, and references therein]. We suppose, that calcium is probably more restricted than an uncharged ligand binding to protein and the difference between translational and rotational entropy in water and in cavity will be closer to the lower value.
Overall, we expect the electrostatic term to be dominant in calcium binding;33 the electrostatic contribution to binding free energy may be written as:29
| (6) |
where the terms on the right are the electrostatic solvation (dehydration of ion upon binding) and coulombic pairwise charge-charge interaction, respectively. is the difference in energy of creating a dielectric boundary while immersing the complex, and the protein and calcium separately, in a high-dielectric medium. is the work of assembling of the atomic charges in a dielectric medium characterizing the protein.
We would like to note that there have been attempts to treat divalent cation binding more accurately within the PBE theoretical framework than presented here,36–38 but to rigorously account for ion desolvation and correlation as well as standard state issues divalent cations should be modeled explicitly. However, the present work only intends to focus on the changes in the electrostatic potential with viral capsid swelling, in a simple univalent ionic strength picture, and to note qualitatively the dramatic change in potential at the putative calcium binding site.
Computational Details
Electrostatic Calculations
The electrostatic calculations were performed using the Adaptive Poisson-Boltzmann Solver (APBS) package. The solution to the PBE is obtained as described by Baker et al.39 Specifically, the problem domain is divided into overlapping subdomains which are then used to assemble the final solution. The solution consists of a series of “focusing” calculations29 from the global domain to the smaller overlapping subdomains. During this focusing algorithm, energy integrals are calculated over subsets of the domain that are (a) unique to the processor of interest and (b) not part of a finer level of calculation. By performing the energy calculations in this manner, we ensure that energy contributions from all regions of the domain are calculated without any contributions being double-counted.
Since the first 26 N-terminal amino acid residues of the coat protein are unresolved in the X-ray crystal structure, the CCMV native structure (PDB ID 1CWP)2 with the added and optimized network of missing residues (1–26) was taken from our previous study.13 Normal mode analysis (NMA) was used to generate the CCMV geometry of the swollen particle. The CMV geometry was obtained from the Protein Data Bank40 (PDB ID 1F15).3 However, in this case we did not add any residues which were missing from the X-ray crystal structure (residues 1–28). In all of the electrostatic calculations the Cα geometries were used except for those in which ligand binding energies were computed, where the all-atom structures were employed. We performed several exploratory all-atom calculations and the resulting electrostatic potential was very similar to the potential calculated with Cα–only models. A similar approach was used in our previous study.41
In all of the APBS calculations the solute and solvent were assigned a dielectric constant of 2 and 78.5, respectively. The ionic strength of monovalent ions was set to 0.150 M with an ion exclusion radius of 2 Å. The dielectric boundary between solute and solvent was based on the molecular surface definition calculated with a probe sphere radius of 1.4 Å. For the boundary conditions the focusing method was applied.29,42
Considering the extremely large size of the system, the calculations were performed on the Blue Horizon and Data Star supercomputers using 1000 processors for each run. The dimensions of the CCMV capsid are 271 × 271 × 271 Å3 and 319 × 319 × 319 Å3 for the native and swollen forms (Cα model), respectively. The total charge of the system (modeled at pH 7) not including 180 calcium ions, with and without the missing 26 amino acid long N-terminal fragment, is +900 and −660e, respectively. The PB equation was solved on 543 × 543 × 543 Å3 numerical grid (the same grid size and position was used for the native and swollen structures for consistency) to a resolution of 0.4 Å (more than 2.5 billion grid points) both for the linear and nonlinear forms of the PB equation. The CMV capsid is slightly larger compared to native CCMV (285 × 285 × 285 Å3, Cα model), and the total charge is +720e. The CMV electrostatic calculation was performed on a 484 × 484 × 484 Å3 grid and the PB equation was solved to 0.4 Å resolution (> 1.7 billion grid points).
Ligand Binding Calculations
The calcium electron density is not observed in the X-ray structure of the virus, but based on the experimental conditions of crystal growth and theoretical modeling the calcium positions have been indicated.2,10 For each structure Ca2+ ions were added, with the position of calcium being given by the average of the coordinates of the six ligands.10 The hydrogens were added to each structure with CHARMM’s HBUILD command,43,44 according to the protonation states of the residues in solution at pH 7 (Asp, Glu, Lys, and Arg residues were assigned a net charge of −1 or +1, respectively; N and C-termini were kept charged and all other residues had a net charge of 0), and their positions were energy-minimized to avoid bad contacts. The positions of the calcium ions were also minimized.
First, the APBS calculations were performed for each calcium cavity and one protein unit composed of three subunits A, B, and C. The calcium cavities are not exactly symmetric and the rearrangement of the residues in each cavity differs slightly. Therefore, we chose to perform calculations for each cavity A, B and C and averaged the electrostatic contribution. For one protein unit three calciums were added at a time and the contribution was also averaged. We checked that adding one calcium separately to one protein unit and averaging over three ΔGelec gives similar result (within 2 kcal/mol) if adding three calciums at a time to the unit and dividing the output electrostatic energy by three. The latter approach is less time consuming therefore it was later used.
Three force field models were applied: a united atom model (charmm19), an all-atom model (charmm22),43,44 and a mixed charmm22/ESP model. The latter included a combination of the ESP charges assigned to the calcium cavity atoms (details below) and the charmm22 parameters assigned to the rest of the protein. In case of calculations for the calcium cavity only, the third model included just the ESP charges.
To derive the ESP charges for the calcium cavity residues, quantum mechanical calculations using the Gaussian’9845 program were performed. The modeled system was composed of six protein residues and the calcium ion (see Figure 1c and d). The Cα terminal atoms were modified into CH3 groups. The configuration was taken exactly as in the energy minimized protein. The density functional single point energy calculations with the B3LYP exchange and correlation functional46,47 and 6-31+G(d,p) basis set were carried out. The Breneman ESP derived charges (ChelpG),48 which gave a reduced partial charge on calcium of +1.6e, were used to calculate the APBS electrostatic energies with a mixed charmm22/ESP parameter model.
For the single calcium cavities, both the linear and nonlinear solutions of the PB equation converged up to a grid resolution of 0.15Å; for the one protein unit the linear PBE to 0.18 Å and nonlinear PBE to 0.35 Å; for the whole CCMV structure both PBE to 0.4 Å. The overall charge of subunits A, B and C including three Ca2+ ions is −5e; for one cavity is −2e. The same grid positioning and spacing were applied, both for the complex and binding systems. The binding energies were calculated with two methods and compared. In the direct method we calculated the difference between the PB energies for the complex and its constituents, and in the additive method, we calculated the differences in the solvation and coulombic contributions separately (see Eq. 6). The Coulomb interaction energy was evaluated as a pairwise charge-charge interaction.
Results and Discussion
Cowpea Chlorotic Mottle Virus (CCMV)
The calculated electrostatic potential for the native and swollen CCMV particles is shown in Figure 3 (presented as isosurface at +/− 1.0 kT/e). The outer electrostatic potential surface (Figure 3a and b) is predominantly negative with patches of positive electrostatic potential concentrated at the top of the hexameric and pentameric subunits, retaining symmetrical elements of the capsid. The electrostatic potential of the inner capsid shell (Figure 3c and d) is mostly positive as expected, since the inner shell is formed primarily by basic amino acid residues. These residues facilitate binding to the negatively charged RNA.
Figure 3.
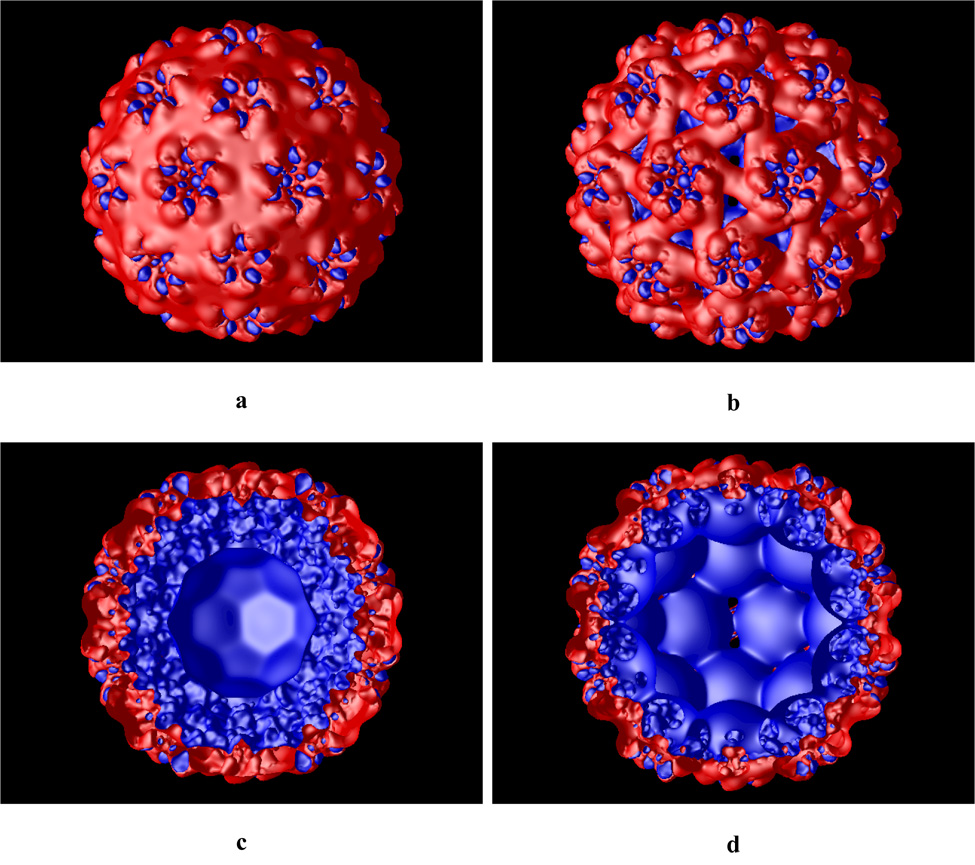
Calculated electrostatics isosurface (at +/− 1.0 kT/e) of the native (a and c) and swollen (b and d) CCMV viral particle. Panels c and d show cut through the capsid. Blue and red colors represent positive and negative potential, respectively.
Further insights can be gained when the calculated electrostatic potential is projected on the solvent accessible surface of the virion particle. This is shown in Figure 4 for both native and swollen structures. Again, the overall electrostatic potential at the outer capsid solvent accessible surface is negative (Figure 4a) and the highest concentration of the negative potential is at the pseudo threefold axis sites. These sites are mainly composed of a cluster of negatively charged aspartic acid residues, which are proposed to bind metal ions in pH-dependent manner. It is very likely that when the metal ion is removed from this site (for example by chelating to EDTA) the closely positioned negatively charged residues will experience strong electrostatic repulsion. One way for the capsid to relieve this strain is to increase inter-residue distance at this site. This is exactly what happens when the capsid swells as demonstrated by the earlier study of Tama and Brooks.10 The largest geometrical change in terms of inter atomic distances occurs at the pseudo threefold axis sites. While the hexameric and pentameric subunits rotate in the plane of the capsid surface during the capsid expansion the aspartic acid residues at the proposed metal binding site move laterally from each other minimizing electrostatic repulsion (Figure 4c and d).
Figure 4.
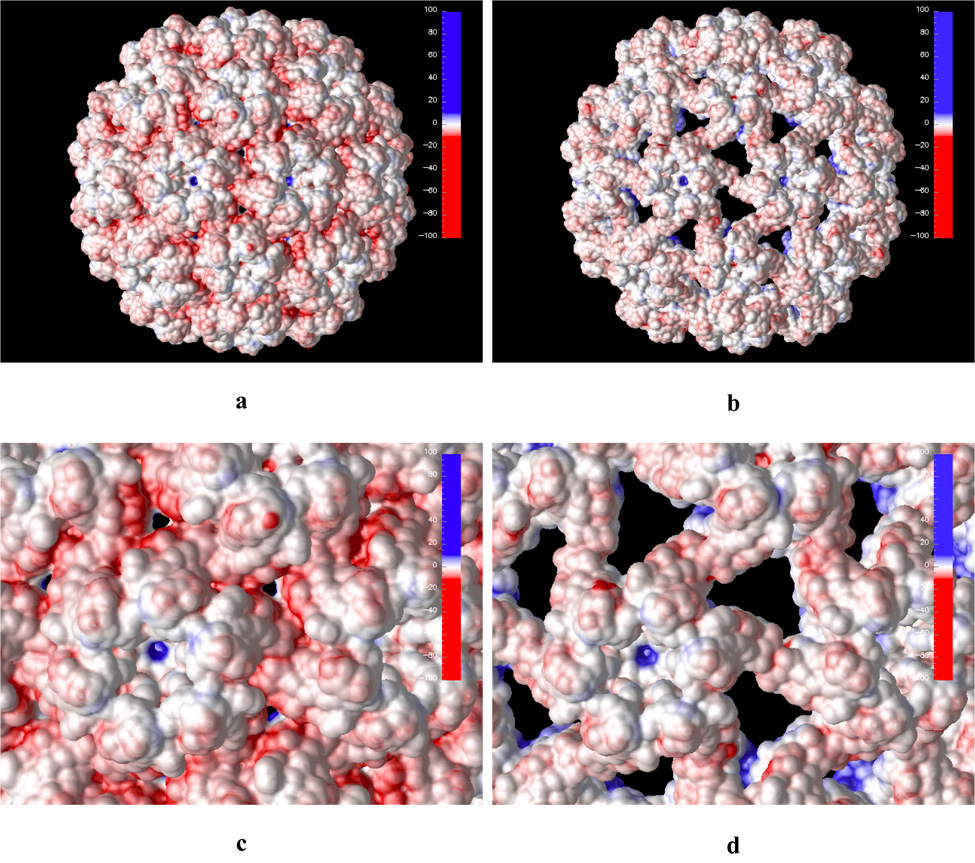
Calculated electrostatic potential projected on the solvent accessible surface of the native (a and c) and swollen (b and d) CCMV capsid. Subfigures c and d show closeups of the outer capsid shell. Blue and red colors represent positive and negative electrostatic potential, respectively. Hue intensity depicts strength of the potential (in kT/e units) as shown on the scale in the top right corner of each picture.
Positive electrostatic potential dominates at the inner CCMV surface shell as seen in Figure 5. A major source of this positive potential is the basic residues located among the first 26 amino acid N-terminal residues. These residues are also responsible for the binding interaction with negatively charged RNA. Experimentally, it was shown that deletion of the first 26 N-terminal amino acid residues suppresses assembly of RNA-containing particles, but not empty particles in vitro.4 We have calculated the electrostatic potential for the capsid without the first 26 amino acids in the N-terminal arm (Figure 6). The electrostatic potential on the inner capsid surface is still mostly positive but it is significantly weaker compared to the native structure. This indicates that the capsid shell with the first 26 N-terminal residues deleted can still favorably interact with negatively charged RNA but the interaction is not strong enough for in vitro assembly of RNA containing particle.4
Figure 5.
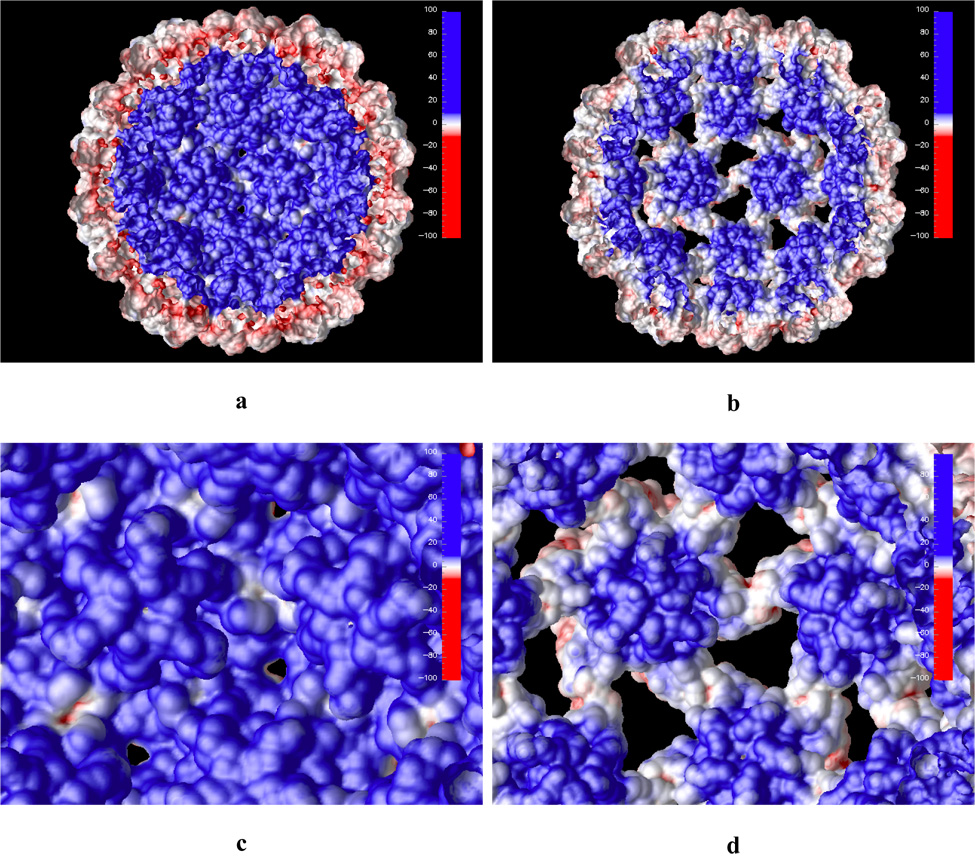
Electrostatic potential projected on the solvent accessible surface of the CCMV particle, inner shell surface view. Both the native (a and c) and swollen (b and d) structures are shown. Panels c and d show closeup views of the inner shell.
Figure 6.
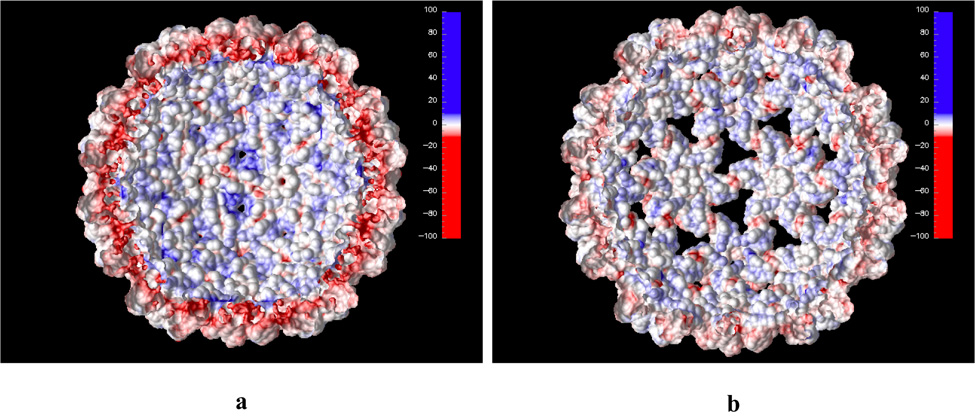
Calculated electrostatic potential projected on the solvent accessible surface of the inner shell of the CCMV capsid. The first 26 N-terminal amino acid residues were deleted. Panels a and b show the native and swollen structures, respectively.
As the capsid swells it is interesting to note that surface areas of negative electrostatic potential on the outer capsid shell are exposed (Figure 4b and d). These areas are located at the capsid shell openings which are created by the capsid expansion. Similarly, the inner swollen capsid surface exposes patches of negative electrostatic potential (Figure 5b and d). This may aid in releasing the enveloped genetic material since the exposed inner surface patches of negative potential will most likely decrease electrostatic binding energy to the negatively charged RNA inside of the capsid. However, since these exposed patches are located at the capsid shell openings at the pseudo threefold axis they will likely hinder spontaneous RNA release through this openings. We also note that counterion adsorption may stabilize this RNA/capsid protein interaction somewhat.
Cucumber Mosaic Virus (CMV)
The calculated electrostatic potential (visualized as an isosurface at +/− 1.0 kT/e) for the CMV capsid is shown in Figure 7. Although CCMV and CMV are structurally remarkably homologous, CMV has only 19% capsid protein sequence identity (34% similarity) to CCMV.3 This is reflected in a marked difference between their electrostatic properties. The electrostatic potential at the CMV outer capsid surface is far more neutral, with a similar distribution of negative and positive potential on the surface (Fig. 7). While the potential at the pentameric and hexameric subunits is negative, the rest of the capsid surface has a positive electrostatic potential. The negative potential at the hexameric and pentameric subunits is mainly due to charged amino acid residues in the βH-βI surface loop, which has been shown to play an important role in the CMV virion transmission.49 This βH-βI loop has a high degree of amino acid conservation among cucumoviruses and has the same length in CCMV but fewer acidic residues (two versus five).
Figure 7.
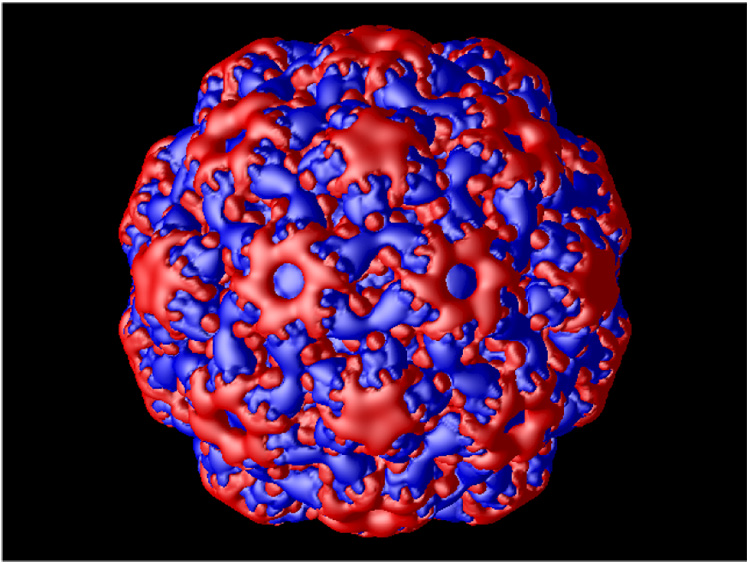
Calculated electrostatic potential presented as isosurface (at +/− 1.0 kT/e) of the CMV viral particle. Blue and red colors represent positive and negative potential, respectively.
A similar electrostatic potential distribution is observed when the potential is projected on the solvent accessible surface (Figure 8). A closeup at the outer capsid surface (Fig. 8b) reveals that there is no high electrostatic potential concentration between the hexameric and pentameric subunits as compared to CCMV. In fact, in CMV the cluster of aspartic residues at the pseudo threefold axis is replaced by complementing acids and bases, which could explain the insensitivity of the capsid to changes in pH or metal concentration.10,17
Figure 8.
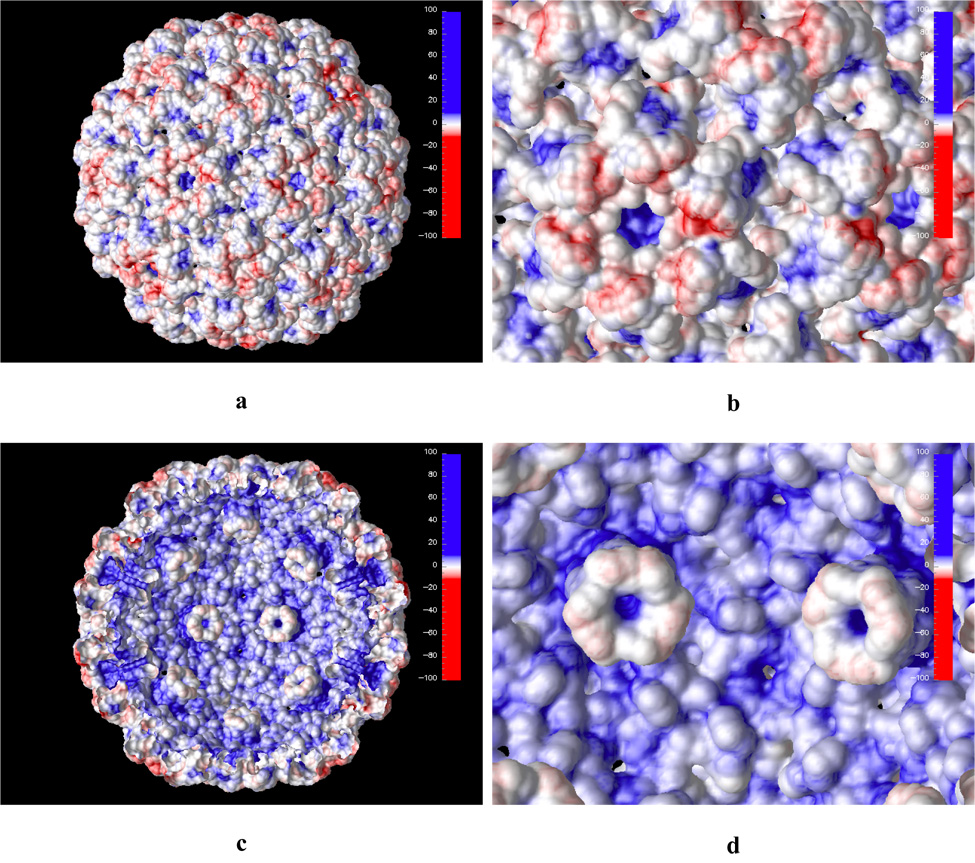
CMV calculated electrostatic potential projected on the solvent accessible surface. Both outer (a and b) and inner (c and d) surface of the capsid shell are shown. Subfigures b and d show a magnified view of the shell surface.
The relatively weak electrostatic potential on the CMV inner capsid surface (Fig. 8c and d), compared to the inner surface potential of the CCMV particle with first 26 N-terminal residues deleted (Figure 6) suggests that CMV without the first 28 N-terminal residues is also not likely to undergo assembly of RNA containing particles. This prediction is yet to be tested.
Calcium Binding to CCMV
Three binding sites for the calcium ions in one unit of the CCMV virus have been proposed.2,10 They are situated at the interface of the subunits A-B, B-C, and C-A. The calcium cavity residues are shown in Figure 1c and d. In the native form the cavity is composed of residues Glu81, Lys84, Gln85, Glu148, and Gln149* and Asp153* from the adjacent subunit. Two carbonyl and four carboxyl oxygens contribute to bonding of the Ca2 ion. Binding energies from gas-phase equilibria confirm that there is preference of Ca for six-ligand interactions.50 After the swelling, Gln149* and Asp153*, which lie at the border between subunits, escape from the cavity, the number of bonds drops to five, and, therefore, calcium binds weaker; however, electrostatic interactions are still favorable. The tendency to form a six coordinated calcium might be another reason for conformational change from the swollen to the native state. The affinity for the calcium in the cavity must be strong enough to prevent its escape into the surrounding aqueous solution. The net charge of the residues within 20 Å from the calcium ion changes from −5e in the native form to 0e in the swollen form, indicating that the environment in the swollen form is less electrostatically favorable to bind calcium.
The electrostatic binding energies for one calcium cavity and one native protein unit, comparing the direct and additive calculation method, and the linear and nonlinear solution to the PB equation, are presented in Table 1. The electrostatic contribution to binding calculated for one cavity is less negative than the average electrostatic binding energy of one calcium in one protein unit. This suggests that the local environment is not the only contribution to the binding. Indeed, the net charge of the cavity without calcium is −2e and within 20 Å of the ion is −5e, suggesting that there might be more favorable binding to the whole unit. The differences between the ΔGelec calculated with the direct and additive approach for one cavity and for one protein unit are less than 1 kcal/mol. The additive method requires twice as many PB calculations and additionally the calculation of the coulombic term. This method should be, in principle, more accurate because it does not depend on the placing of molecules on the grid. The direct method needs only three calculations, but with the same placing on the grid of both the complex, and the ligand and the protein, separately. The agreement of the two methods is very good, indicating that for this grid resolution one may use the simpler and faster approach. For one cavity, the energies determined with the linear solution to the PBE are in excellent agreement. For one unit the values cannot be compared because different convergence was obtained with the linear and nonlinear solutions to the PBE (see Methods Section).
Table 1.
Electrostatic contribution to Ca2+ binding free energy for fragments of the CCMV virion (kcal/mol). Results obtained directly and by splitting into solvation and coulombic contributions are compared. The energy terms are shown per one calcium, and for one cavity were averaged over cavities A, B and C; for one protein unit were averaged over three Ca2+.
| Force field | one calcium cavity | one protein unit | ||
|---|---|---|---|---|
| linear PBE | ||||
| charmm19 | −58.7 | −57.7 | −79.6 | −78.6 |
| charmm22 | −39.1 | −38.6 | −70.5 | −69.3 |
| charmm22/ESP | −43.2 | −43.1 | −67.9 | −66.9 |
| nonlinear PBE | ||||
| charmm19 | −58.9 | −57.3 | −74.8 | −73.6 |
| charmm22 | −39.3 | −38.9 | −65.0 | −64.8 |
| charmm22/ESP | −43.3 | −43.3 | −64.3 | −64.1 |
In case of one protein unit, we investigated if there is any cooperativity in the electrostatic contribution for the calcium binding. We compared ΔGelec of binding of one calcium alone with binding of the same calcium if another one was already present in the system. Calcium ions are between 11–13 Å apart in the native form. It turns out that the difference in ΔGelec is less than 3 kcal/mol, therefore, cooperativity is not detected with the PB approach. On the other hand we do not have evidence that it exists for this system.
Table 2 shows the electrostatic contributions to calcium binding energies for the whole native and swollen CCMV, and the corresponding differences in binding energies between the two forms. The relative calcium binding energies are substantial; the electrostatic contribution to the native form of the virus is at least three times more favorable, depending on the force field parameters used. If one compares the appropriate energy values of Table 1 and Table 2 for the native form of the virus, one may notice that for all the force fields ΔGelec calculated with the nonlinear PB equation is more favorable for the whole CCMV in comparison to the value determined for one protein unit, but the differences do not exceed 10%. This suggests that the one protein unit environment provides most of the favorable contribution for calcium binding.
Table 2.
Electrostatic contribution to Ca2+ binding energy for the whole CCMV virion (kcal/mol) obtained directly and by splitting into solvation and coulombic contributions. The energy terms are per one calcium ion and were calculated for the whole native and swollen CCMV virus with different force field parameters and averaged over 180 ions. and are the differences in the calculated electrostatic contribution per one calcium between the native and swollen CCMV structures.
| Native form | Swollen form | |||||
|---|---|---|---|---|---|---|
| Force field | ||||||
| linear PBE | ||||||
| charmm19 | −78.4 | −77.9 | −8.6 | −7.9 | −69.8 | −70.0 |
| charmm22 | −73.4 | −69.1 | −21.7 | −21.0 | −51.7 | −48.1 |
| charmm22/ESP | −70.3 | −66.9 | −20.3 | −19.8 | −50.0 | −47.1 |
| nonlinear PBE | ||||||
| charmm19 | −78.9 | −78.4 | −8.8 | −8.1 | −70.1 | −70.3 |
| charmm22 | −71.9 | −67.6 | −22.7 | −22.0 | −49.2 | −45.6 |
| charmm22/ESP | −70.0 | −66.4 | −21.0 | −20.6 | −49.0 | −45.8 |
The electrostatic contribution to binding energies of calcium to the swollen form is reduced but is still negative even with the united atom model force field of charmm19 (Table 2). This is expected because the surrounding overall charge (within 20 Å of Ca) is not as negative as in the native form. Moreover, in the native form the calcium ion is coordinated by six oxygens and in the swollen form only by five. An upper estimate of the −TΔS term for the swollen form does not exceed 12 kcal/mol (see Methods Section). Even without including the small (less than 2 kcal/mol) but favorable nonpolar contribution, calcium shows favorable binding for charmm22 and charmm22/ESP force fields. In charmm19 there would be no binding but it is an united atom model and it might not work well with such a complicated problem as binding of metal to a protein and may be too crude an approximation. Moreover, we estimate only an upper limit of the entropy loss and not the absolute value. With an all-atom parameter set, calcium is capable to bind to the swollen form of CCMV, which suggests that it facilitates the stabilization of the native form, and most probably induces the viral contraction. With a high concentration of Ca2+, the CCMV virus may not swell because its native form is highly stabilized. Therefore, the binding energy calculations show that calcium affects the structural flexibility of the virion, but when the virus is in its expanded form the electrostatic contribution is still favorable.
The results depend upon the parameters used in the PB equation, but we were interested in the differences of calcium affinity to the native and swollen states and not in the absolute binding energy values. We tested a few sets of charges and radii and also two approaches for calculating the binding energies, direct and by splitting into coulombic and desolvation contributions. Both methods gave comparable results. The main drawback of this kind of binding energy calculation is the single-conformation approximation, but nevertheless on average we see noticeable differences in the binding of calcium to the native and swollen forms of CCMV.
Conclusions
Assembly and disassembly and RNA release mechanisms of the cowpea chlorotic mottle viral capsid are still poorly understood, despite the capsid’s relative simplicity and available detailed experimental information. We have examined the role electrostatics plays in these processes. The calculated electrostatic potentials for the native and swollen CCMV structures confirm that electrostatic repulsion at the pseudo threefold axis sites is most likely the driving force for the capsid swelling. Strong positive electrostatic potential on the inside, inner capsid shell suggests that electrostatic interactions between enveloped, negatively charged RNA and the inner capsid shell are dominant. As the capsid swells, patches of negative electrostatic potential on the capsid surface are exposed, which destabilizes RNA-capsid interaction and may aid in the RNA release process. It is unlikely that the RNA could escape from the inside of the capsid (at neutral pH) through chanels formed by the swelling due to electrostatic repulsion.
Cucumber mosaic virus, which is highly homologous to CCMV, has complimentary distribution of negative and positive electrostatic potential at the pseudo threefold axis sites rendering the virion incapable of swelling.
The electrostatic contributions to binding free energy of site-bound Ca2+ to the whole CCMV were estimated by means of the continuum solvent PB approach. The size of the grid in the extremely large calculations exceeded 500 × 500 × 500 Å3 but we were able to reach a grid resolution of 0.4 Å (more than 2.5 billion grid points). The calculations show the differences in the electrostatic binding energies between the swollen and native CCMV, from 46 to 70 kcal/mol, depending on the force field parameters. The average electrostatic contribution in the native form is huge but the native form is already the most stable one, therefore, estimated binding energies to the native state are exaggerated. On the other hand, the energies must be strong relative to that with water to provide stability of the ion in protein relative to that in aqueous solution. Our calculations are restricted to two rigid conformations, with the swollen state being a model, that is why we can get only the qualitative picture and look at the relative energies in case of the native state.
Since binding of calcium is reversible, ions most probably start binding to the more expanded form and induce viral contraction. CCMV expansion takes away two cavity residues from the adjacent subunit and the number of coordinating oxygens drops from six to five, yet electrostatic contribution to calcium binding is still favorable. By estimating an upper limit of the nonpolar and entropic contributions, we see overall favorable calcium binding even in the swollen state. Small but favorable binding affinity to the expanded state may help in stabilizing the virus in its native form.
The average energies for one calcium ion binding to one protein unit and for the whole CCMV structure differ by less than 10%. However, the electrostatic binding energy to one calcium cavity is less negative, implicating the importance of the surrounding on binding. The linear and nonlinear PB equation give comparable values. The direct and additive method of calculating the binding energy gave similar results, suggesting that the effect of placing molecules on the grid does not contribute much in case of our system.
Acknowledgments
We are very grateful to Wayne Pfeiffer, Giri Chukkapalli and the San Diego Supercomputer Center for providing access to Blue Horizon and Data Star supercomputers and for their generous technical assistance. We would also like to thank the W. M. Keck Foundation and the NSF Center for Theoretical Biological Physics (CTBP) for providing additional computational resources. This work was supported in part by NIH, NSF, the National Biomedical Computing Resource, CTBP and Accelrys, Inc. NAB was supported by a Research Fellowship from the Alfred P. Sloan Foundation and grant GM069702 from the NIH. CLB and FT acknowledge the support of the NSF (CTBP) and NIH through the grant RR12255 (MMTSB). JT would like to acknowledge support by Polish Scientific Committee (115/E-343/BST-993/ICM/2004).
References
- 1.Harrison SC. Cell. 1996;86:341–343. doi: 10.1016/s0092-8674(00)80105-1. [DOI] [PubMed] [Google Scholar]
- 2.Speir JA, Munshi S, Wang GJ, Baker TS, Johnson JE. Structure. 1995;3:63–78. doi: 10.1016/s0969-2126(01)00135-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Smith TJ, Chase E, Schmidt T, Perry KL. J Virol. 2000;74:7578–7586. doi: 10.1128/jvi.74.16.7578-7586.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhao X, Fox JM, Olson NH, Baker TS, Young MJ. Virology. 1995;207:486–494. doi: 10.1006/viro.1995.1108. [DOI] [PubMed] [Google Scholar]
- 5.Fox JM, Zhao X, Speir JA, Young MJ. Virology. 1996;222:115–122. doi: 10.1006/viro.1996.0402. [DOI] [PubMed] [Google Scholar]
- 6.Fox JM, Albert FG, Speir JA, Young MJ. Virology. 1997;227:229–233. doi: 10.1006/viro.1996.8292. [DOI] [PubMed] [Google Scholar]
- 7.Albert FG, Fox JM, Young MJ. J Virol. 1997;71:4296–4299. doi: 10.1128/jvi.71.6.4296-4299.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bancroft JB. Adv Virus Res. 1970;16:99–134. doi: 10.1016/s0065-3527(08)60022-6. [DOI] [PubMed] [Google Scholar]
- 9.Jacrot B. Mol Biol. 1975;95:433–446. doi: 10.1016/0022-2836(75)90201-6. [DOI] [PubMed] [Google Scholar]
- 10.Tama F, Brooks CL., III J Mol Biol. 2002;318:733–747. doi: 10.1016/S0022-2836(02)00135-3. [DOI] [PubMed] [Google Scholar]
- 11.Liu H, Qu C, Johnson JE, Case DA. J Struct Biol. 2003;142:356–363. doi: 10.1016/s1047-8477(03)00028-5. [DOI] [PubMed] [Google Scholar]
- 12.Zlotnick A, Aldrich R, Johnson JM, Ceres P, Young MJ. Virology. 2000;277:450–456. doi: 10.1006/viro.2000.0619. [DOI] [PubMed] [Google Scholar]
- 13.Zhang D, Konecny R, Baker NA, McCammon JA. Biopolymers. 2004;75:325–337. doi: 10.1002/bip.20120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Isea R, Aponte C, Cipriani R. Biophys Chem. 2004;107:101–106. doi: 10.1016/S0301-4622(03)00193-5. [DOI] [PubMed] [Google Scholar]
- 15.Witz J, Brown F. Arch Virol. 2001;146:2263–2274. doi: 10.1007/s007050170001. [DOI] [PubMed] [Google Scholar]
- 16.Li PP, Naknanishi A, Tran MA, Ishizu KI, Kawano M, Phillips M, Handa H, Liddington RC, Kasamatsu H. J Virol. 2003;77:7527–7538. doi: 10.1128/JVI.77.13.7527-7538.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wikoff WR, Tsai CJ, Wang G, Baker TS, Johnson JE. Virology. 1997;232:91–97. doi: 10.1006/viro.1997.8543. [DOI] [PubMed] [Google Scholar]
- 18.Garrett AJM, Poladian L. Ann Phys. 1988;188:386–435. [Google Scholar]
- 19.Sharp KA, Honig B. Ann Rev Biophys Chem. 1990;19:301–332. doi: 10.1146/annurev.bb.19.060190.001505. [DOI] [PubMed] [Google Scholar]
- 20.Baker NA, McCammon JA. Electrostatic Interactions. In: Weissig H, Bourne PE, editors. Structural Bioinformatics. New York: John Wiley & Sons; 2003. [Google Scholar]
- 21.Tirion MM. Phys Rev Lett. 1996;77:1905–1908. doi: 10.1103/PhysRevLett.77.1905. [DOI] [PubMed] [Google Scholar]
- 22.Bahar I, Atilgan AR, Erman B. Fold Des. 1997;2:173–181. doi: 10.1016/S1359-0278(97)00024-2. [DOI] [PubMed] [Google Scholar]
- 23.Tama F, Sanejouand YH. Protein Eng. 2001;14:1–6. doi: 10.1093/protein/14.1.1. [DOI] [PubMed] [Google Scholar]
- 24.Tama F, Wriggers W, Brooks CL., III J Mol Biol. 2002;321:297–305. doi: 10.1016/s0022-2836(02)00627-7. [DOI] [PubMed] [Google Scholar]
- 25.Tama F, Gadea FX, Marques O, Sanejouand YH. Proteins. 2000;41:1–7. doi: 10.1002/1097-0134(20001001)41:1<1::aid-prot10>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- 26.Tama F, Brooks CL., III . Unveiling molecular mechanisms of biological functions in large macromolecular assemblies using elastic network normal mode analysis. In: Cui Q, Bahar I, editors. Normal Mode Analysis: Theory and Applications to Biological and Chemical Systems. CRC Press; 2005. [Google Scholar]
- 27.Froloff N, Windemuth A, Honig B. Prot Sci. 1997;6:1293–1301. doi: 10.1002/pro.5560060617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lazaridis T. Curr Org Chem. 2002;6:1319–1332. [Google Scholar]
- 29.Gilson MK, Sharp KA, Honig BH. J Comput Chem. 1988;9:327–335. [Google Scholar]
- 30.Nicholls A, Sharp KA, Honig B. Proteins. 1991;11:281–296. doi: 10.1002/prot.340110407. [DOI] [PubMed] [Google Scholar]
- 31.Pickett SD, Sternberg MJE. J Mol Biol. 1993;231:825–839. doi: 10.1006/jmbi.1993.1329. [DOI] [PubMed] [Google Scholar]
- 32.Doig AJ, Sternberg MJE. Prot Sci. 1995;4:2247–2251. doi: 10.1002/pro.5560041101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pavlov M, Siegbahn PEM, Sandstrom M. J Phys Chem A. 1998;102:219–228. [Google Scholar]
- 34.Lazaridis T, Masunov A, Gandolfo F. Proteins. 2002;47:194–208. doi: 10.1002/prot.10086. [DOI] [PubMed] [Google Scholar]
- 35.Swanson JMJ, Henchman RH, McCammon JA. Biophys J. 2004;86:67–74. doi: 10.1016/S0006-3495(04)74084-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Misra VK, Draper DE. J Mol Biol. 1999;294:1135–1147. doi: 10.1006/jmbi.1999.3334. [DOI] [PubMed] [Google Scholar]
- 37.Misra VK, Draper DE. J Mol Biol. 2000;299:813–825. doi: 10.1006/jmbi.2000.3769. [DOI] [PubMed] [Google Scholar]
- 38.Misra VK, Draper DE. Proc Natl Acad Sci USA. 2001;98:12456–12461. doi: 10.1073/pnas.221234598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Baker NA, Sept D, Joseph S, Holst MJ, McCammon JA. Proc Natl Acad USA. 2001;98:10037–10041. doi: 10.1073/pnas.181342398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Trylska J, Konecny R, Tama F, Brooks CL, III, McCammon JA. Biopolymers. 2004;74:423–431. doi: 10.1002/bip.20093. [DOI] [PubMed] [Google Scholar]
- 42.Yang AS, Gunner MR, Sampogna R, Sharp K, Honig B. Proteins Struct Func Gen. 1993;15:252–265. doi: 10.1002/prot.340150304. [DOI] [PubMed] [Google Scholar]
- 43.Brooks BR, Bruccoleri RE, Olafson BD, States DJ, Swaminathan S, Karplus M. J Comp Chem. 1983;4:187–217. [Google Scholar]
- 44.MacKerell AD, Jr, Brooks B, Brooks CL, III, Nilsson L, Roux B, Won Y, Karplus M. CHARMM: The Energy Function and Its Parameterization with an Overview of the Program. In: vR Schleyer P, et al., editors. The Encyclopedia of Computational Chemistry. Vol. 1. Chichester: John Wiley & Sons; 1998. [Google Scholar]
- 45.Frisch MJ, et al. Gaussian 98, Revision A.11.4. Pittsburgh, PA: Gaussian, Inc.; 2002. [Google Scholar]
- 46.Becke AD. J Chem Phys. 1993;98:5648–5652. [Google Scholar]
- 47.Lee C, Yang W, Parr RG. Phys Rev B. 1988;37:785–789. doi: 10.1103/physrevb.37.785. [DOI] [PubMed] [Google Scholar]
- 48.Chirlian LE, Franc MM. J Comp Chem. 1987;8:894–905. [Google Scholar]
- 49.Liu S, He X, Park G, Josefsson C, Perry KL. J Virol. 2002;76:9756–9762. doi: 10.1128/JVI.76.19.9756-9762.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Peschke M, Blades AT, Kebarle P. J Am Chem Soc. 2000;122:10440–10449. [Google Scholar]
- 51.Reddy VS, Natarajan P, Okerberg B, Li K, Damodaran KV, Morton RT, Brooks CL, III, Johnson JE. J Virol. 2001;75:11943–11947. doi: 10.1128/JVI.75.24.11943-11947.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]