

Abstract
Marubium vulgare (horehound) and Prunus serotina (wild cherry) have been traditionally used for the treatment of inflammatory-related symptoms such as cold, fever, and sore throat. In this report, we show that extracts of anti-inflammatory horehound leaves and wild cherry bark exhibit anti-proliferative activity in human colorectal cancer cells. Both horehound and wild cherry extracts cause suppression of cell growth as well as induction of apoptosis. We found that horehound extract up-regulates pro-apoptotic non-steroidal anti-inflammatory drug-activated gene (NAG-1) through trans-activation of the NAG-1 promoter. In contrast, wild cherry extract decreased cyclin D1 expression and increased NAG-1 expression in HCT-116 and SW480 cell lines. Treatment with wild cherry extract resulted in the suppression of β-catenin/T cell factor transcription, as assessed by TOP/FOP reporter constructs, suggesting that suppressed β-catenin signaling by wild cherry extract leads to the reduction of cyclin D1 expression. Our data suggest the mechanisms by which these extracts suppress cell growth and induce apoptosis involve enhanced NAG-1 expression and/or down-regulation of β-catenin signaling, followed by reduced cyclin D1 expression in human colorectal cancer cells. These findings may provide mechanisms for traditional anti-inflammatory products as cancer chemopreventive agents.
Keywords: horehound, NAG-1, wild cherry, cyclin D1, β-catenin signaling, human colorectal cancer cells
Introduction
Arid regions in the southwest United States are rich in highly specialized plants containing compounds with a high potential medical interest. Thus, the plants from this area have a long tradition of use by locals for various medicinal purposes such as analgesics, stimulants, sedatives, antibiotics, and aphrodisiacs. A number of medicinal plants have shown anti-inflammatory activities and their extracts are used for a wide range of ailments. Recently, the total antioxidant capacity of wild medicinal plants widely used in New Mexico has been investigated (1). In particular, the aqueous extracts from wild plants such as the leaves of Marubium vulgare elicit a high degree of antioxidant avtivity. Some of the beneficial effects of these plant extracts may be attributed to antioxidant activity. Marubium vulgare (horehound) and Prunus serotina (wild cherry) have been traditionally used for the treatment of inflammatory-related symptoms such as cold, fever, and sore throat. M. vulgare is also used for respiratory problems such as asthma and cough. Indeed, several siterpenoids including marrubiin and marrubenol were isolated from M. vulgare and may be responsible for its activity against respiratory problems. Several phenylpropanoid derivatives were also isolated as potential anti-inflammatory compounds. In addition, several flavonoids and antocyanidines have been found in P. serotina (2). Exploring commonly used natural products that can be collected or purchased locally will increase the chances of finding new therapeutic agents, because in most cases their traditional uses have already been well documented. However, the molecular mechanisms responsible for potential human health benefits derived from many plant extracts have not been elucidated and must be studied in vitro, followed by validation in pre-clinical studies.
Non-steroidal anti-inflammatory drug (NSAID)-activated gene (NAG-1), a member of the TGF-β gene superfamily, was identified from cyclooxygenase-deficient, indomethacin-induced human colorectal cancer HCT-116 cells and has been found to stimulate apoptosis (3). NAG-1 expression is increased independently of prostaglandin formation in human colorectal cancer cells by NSAIDs (4). It has been reported that treatment with purified NAG-1 induces apoptosis in prostate cancer cells (5). In addition, inhibition of phosphatidylinositol 3-kinase activity, which causes the induction of apoptosis and growth suppression, up-regulates NAG-1 in HCT-116 cells (6). These data support the hypothesis that NAG-1 is linked to apoptosis and that its reduction may promote tumorigenesis. NAG-1 expression is also up-regulated by several anti-tumorigenic polyphenol products such as resveratrol, genistein, catechins, and indol-3-compounds as well as known anti-inflammatory compounds (7–10). Thus, NAG-1 suppression plays a pivotal role in colorectal tumorigenesis and its expression may be involved in the chemo-preventive effects of several anti-tumorigenic compounds. Another important pathway in colon carcinogenesis involves β-catenin signaling. Dysregulation of the Wnt pathway including altered β-catenin status is attributed to the early stages of colorectal tumorigenesis in almost all cases of this disease. Loss of APC gene function or mutations in the β-catenin gene lead to aberrant nuclear migration of β-catenin and subsequent overexpression of a number of genes directly related to oncogenesis, such as c-Myc and cyclin D1 (11–13). Since activation of the Wnt/β-catenin pathway is thought to play a central role in colorectal carcinogenesis, attenuation of this signaling pathway presents a promising approach for the prevention of colon tumorigenesis. Recent studies demonstrated that β-catenin/T cell factor (TCF) signaling in several colorectal cancer cell lines are down-regulated by NSAIDs and phytochemicals such as aspirin, indomethacin, tea catechins and quercetin (14–16).
In this report, we explored the effects of M. vulgare leaves and P. serotina bark extracts on anti-tumorigenicity of human colorectal cancer cells. Here, we report that these extracts exhibit anti-proliferative effects in human colorectal cancer cells, and the mechanisms are involved in up-regulated NAG-1 expression and suppression of β-catenin signaling followed by the reduction of cyclin D1 expression.
Materials and methods
Cell lines and reagents
Cell lines were purchased from American Type Culture Collection (Manassas, VA). Human colorectal adenocarcinoma, HCT-116 cells were maintained in McCoy’s 5A medium supplemented with 10% fetal bovine serum (FBS) and gentamycin (10 μg/ml). Human colorectal adenocarcinoma, SW480 cells, were grown in RPMI-1640 medium supplemented with 10% FBS and gentamycin. Anti-p53 and actin antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Anti-cyclin D1 antibody was obtained from Cell Signaling Technology (Beverly, MA). Anti-human-NAG-1 antibody was described previously (3).
Plant materials and extraction procedures
Leaves of M. vulgare (horehound) and bark of P. serotina (wild cherry) were used. Plant materials (20 g) were ground and extracted with 200 ml methanol for 48 h in a 250 ml separatory funnel. Extracts were concentrated under vacuum using a rotary evaporator at 40°C. Extracts were transferred to 20 ml glass vials either by scrapping or dissolving into methanol, followed by sonication and drying under nitrogen.
Western blot analysis
Cells were washed with phosphate-buffered saline (PBS), lysed with RIPA buffer (1x PBS, 1% Nonidet P-40, 0.5% sodium deoxycholate, 0.1% SDS) supplemented with protease inhibitors (1 μg/ml aprotinin, 1 μg/ml leupeptin and 1 mM phenylmethylsulfonyl fluoride) and phosphatase inhibitors (1 mM Na3VO4 and 1 mM NaF). The soluble protein concentrations were determined with a BCA protein assay kit (Pierce Biotechnology, Rockford, IL). Total protein (30 μg) was separated by SDS-polyacrylamide gel electrophoresis and transferred for 1 h onto nitrocellulose membrane (Osmonics, Minnetonka, MN). The blots were blocked for 1 h with 5% skim milk in Tris-buffered saline/0.05% Tween-20 and probed with primary antibody at 4°C overnight. After washing, the blots were treated with horseradish peroxidase-conjugated secondary antibody for 1 h and washed several times. The signal was detected by enhanced chemiluminescence system (Amersham Biosciences, Piscataway, NJ) and quantified with Scion Image Software (Scion Corp., Frederick, MD).
β-catenin-TCF/LEF reporter gene (TOPFLASH) assay
TOPFLASH contains six copies of the TCF/lymphoid enhancer factor (TCF/LEF)-binding site upstream of a TK promoter. FOPFLASH contains a mutated TCF/LEF-binding site. Those constructs were kindly provided by Dr H.C. Crawford, State University of New York at Stony Brook. SW480 cells were plated in 12-well plates with 105 cells/well and grown for 16 h. Plasmid mixtures containing 0.5 μg of TOPFLASH or FOPFLASH were transfected with 0.05 μg of pRL-null (Promega, Madison, WI) by Lipofectamine (Invitrogen, Carlsbad, CA) according to the manufacturer’s protocol. After transfection, the medium was replaced with serum-free medium and the cells were treated with plant extracts for 24 h. Cells were harvested in luciferase lysis buffer, and luciferase activity was determined and normalized to the pRL-null luciferase activity using a dual luciferase assay kit (Promega, Madison, WI). Data were expressed as the ratio of TOPFLASH to FOPFLASH.
NAG-1 promoter and luciferase assay
The NAG-1 promoter clone linked to luciferase (pNAG−966/+41) has been previously reported (8). HCT-116 cells were plated in 12-well plates with 105 cells/well. Plasmid mixtures containing 0.5 μg of reporter vector and 0.05 μg of pRL-null were transfected with Lipofectamine. After transfection, indicated plant extracts were treated for 24 h. Luciferase activity was determined and normalized to the pRL-null luciferase activity using a dual luciferase assay kit.
Flow cytometric analysis
Cells were labeled with FITC-labeled Annexin V and propidium iodide (PI) using an Annexin V-FITC apoptosis detection kit (BD Biosciences PharMingen, San Diego, CA), according to the manufacturer’s instructions. Briefly, HCT-116 cells were plated with 3×105 cells/well in 6-well plates and grown until they reached 60% confluence. The cells were treated with indicated plant extracts (100 and 250 μg/ml) in the presence of serum for 24 h. The cells were collected, washed with PBS, and stained with Annexin V-FITC and PI. A total of 10,000 cells were examined by flow cytometry using a Beckman Coulter Epics XL equipped with ADC and ModFit LT software. Cells were gated on side scatter and forward scatter to exclude debris. Doublets were eliminated using peak versus integral analysis. Cells that stained Annexin V-FITC positive were determined as apoptotic cells from the total gated cells. Cells that stained PI positive/Annexin V-FITC negative were determined as necrotic cells.
Cell staining
HCT-116 cells were grown on the poly-L-lysine-coated coverslips and treated with plant extracts for 24 h. The cells were washed with PBS and fixed with methanol for 3 min. Morphological analysis was performed by staining with Giemsa solution (Acros Organics, Morris Plains, NJ). Cellular morphology was examined by light microscopy (×100).
Cell proliferation assay
The cell proliferation assay was performed using a CellTiter 96 AQueous One Solution Cell Proliferation Assay (Promega). The assay was carried out according to the manufacturer’s protocol. Briefly, cells were plated with 1,000 cells/well and 100 μl of media in 96-well plates. After 16 h, the cells were treated with the indicated compounds (100 and 250 μg/ml) in the presence of serum and incubated for 48 h. Methanethiosulfonate/phenazine methosulfate solution (20 μl/well) was added and the cells were incubated for 1 h at 37°C. Absorbance was read at 490 nm using a microplate reader (Universal Microplate Reader, ELX 800, Bio-Tek Instruments, Winooski, VT).
Results
Horehound leaves and wild cherry bark extracts cause apoptosis and cell growth suppression in human colorectal cancer cells
The cellular morphology was initially examined after treatment of HCT-116 cells with different concentration of either horehound leaves or wild cherry bark extract. After Wrights-Giemsa staining, the horehound treatment did not show any change in morphology, compared to vehicle-treated HCT-116 cells. Typical cellular morphologic changes, including cell membrane blebs, and the cytoplasm and nuclear chromatin condensation, were observed in 10 μg/ml of wild cherry-treated cells, whereas most of the cells showed necrotic features at the concentration of 100 μg/ml treatment (Fig. 1), indicating that wild cherry extracts exhibit cytotoxic effects in HCT-116 cells. We also investigated the effects of horehound and wild cherry extracts on cell growth of HCT-116 cells. The cells were treated with vehicle, horehound (100 and 250 μg/ml) or wild cherry extracts (100 and 250 μg/ml) in the presence of serum for 48 h, and then cell growth was measured. Both extracts significantly suppressed cell growth of HCT-116 at concentration of 250 μg/ml (Fig. 2A). Subsequently, flow cytometric analysis was performed to determine whether these extracts exhibit apoptotic effect in HCT-116 cells. As shown in Fig. 2B, both of these extracts induced apoptosis, although a significant number of necrotic cells were observed in the wild cherry extract-treated cells.
Figure 1.

Morphologic changes of HCT-116 cells treated with plant extracts. HCT-116 cells were treated with vehicle, horehound (10 and 100 μg/ml) or wild cherry extracts (10 and 100 μg/ml) in the absence of serum. After 24 h, the cells were stained with Giemsa solution and observed by light microscopy at x100 magnification.
Figure 2.
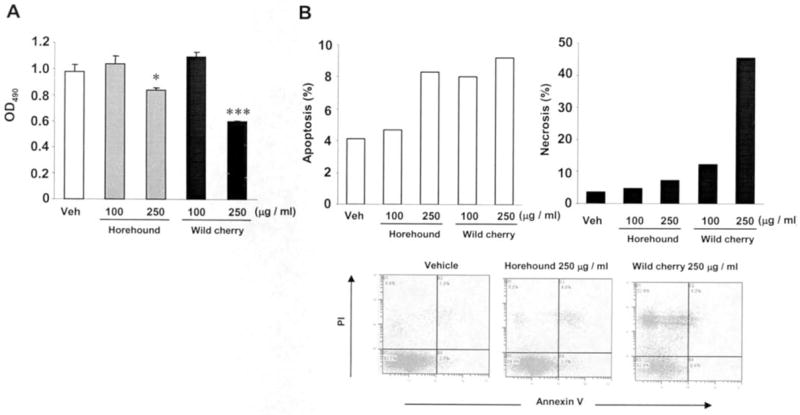
Horehound leaves and wild cherry bark extracts suppress HCT-116 cell growth and induce apoptosis in human colorectal cancer cells. (A) HCT-116 cells were plated at 1,000 cells/well in 96-well plates and treated with vehicle, horehound (100 and 250 μg/ml) or wild cherry extracts (100 and 250 μg/ml) in the presence of serum. After 48 h, cell growth was measured using the CellTiter 96 AQueous One Solution Cell Proliferation Assay. Values are expressed as mean ± SD of 6 replicates. Statistical significance is according to Student’s t-test. *P<0.05, ***P<0.001 versus vehicle-treated cells. (B) HCT-116 cells were plated at 3×105 cells/well in 6-well plates and treated with vehicle, horehound (100 and 250 μg/ml) or wild cherry extracts (100 and 250 μg/ml) in the presence of serum. After 24 h, the cells were stained with Annexin V and PI and analyzed by flow cytometry as described in Materials and methods. Values are expressed as mean of 2 replicates.
Effects of horehound leaves and wild cherry bark extracts on pro-apoptotic and cell cycle-regulatory gene expressions
To determine the molecular mechanism by which these extracts cause apoptosis and/or cell growth suppression, expression of apoptosis- and/or cell cycle regulation-related genes were assessed by Western blot analysis. HCT-116 cells were treated with vehicle, horehound (10 and 100 μg/ml) or wild cherry extracts (10 and 100 μg/ml) for 24 h. As shown in Fig. 3, the expression of pro-apoptotic protein NAG-1 was increased by both extracts at the concentration of 100 μg/ml. We have also examined other plant extracts, Anemopsis californica and Rumex hymenosepalus, from New Mexico; however, these extracts do not induce NAG-1 expression at the concentration of 100 μg/ml (data not shown). In addition to the positive effect on NAG-1 expression, treatment with wild cherry extract dramatically decreased cyclin D1 expression at the concentration of 100 μg/ml. Similarly, treatment with wild cherry extract at the concentration of 100 μg/ml reduced expression of p53, a tumor suppressor protein. Treatment with horehound had no effect on p53 expression (Fig. 3). These data suggest that horehound and cherry extracts may affect different mechanisms to induce cell growth arrest in HCT-116 cells.
Figure 3.
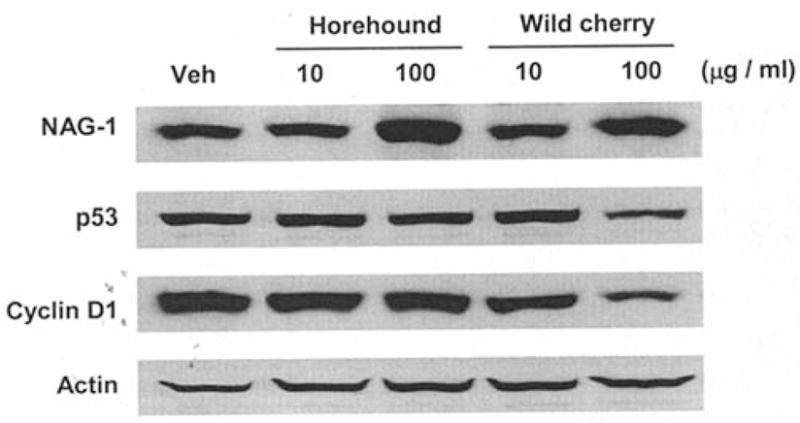
Effects of horehound leaves and wild cherry bark extracts treatment on NAG-1, p53 and cyclin D1 expressions in HCT-116 cells. HCT-116 cells were treated with vehicle, horehound (10 and 100 μg/ml) or wild cherry extracts (10 and 100 μg/ml) in the absence of serum for 24 h. The cell lysates were harvested to perform Western blot analysis. Equal loading was confirmed by determining actin immunoreactivity.
Wild cherry bark extract transactivates NAG-1 promoter
To investigate the underlying mechanism by which horehound and wild cherry extracts induce NAG-1 expression, the transcriptional activity of NAG-1 was assessed by luciferase activity. A NAG-1 promoter construct containing −966/+41 (pNAG−966/+41) was transfected into HCT-116 cells, and luciferase activity was measured. Consistent with Western blot analysis, wild cherry extract significantly transactivated the NAG-1 promoter at the concentration of 100 μg/ml. The marginal change in NAG-1 promoter activity was seen in horehound extract-treated cells only with lower concentration of horehound extract (10 μg/ml) (Fig. 4). These results suggest that wild cherry extract induces NAG-1 expression at the transcriptional level, whereas horehound extract did not affect the pNAG−966/+41 promoter to enhance NAG-1 expression.
Figure 4.
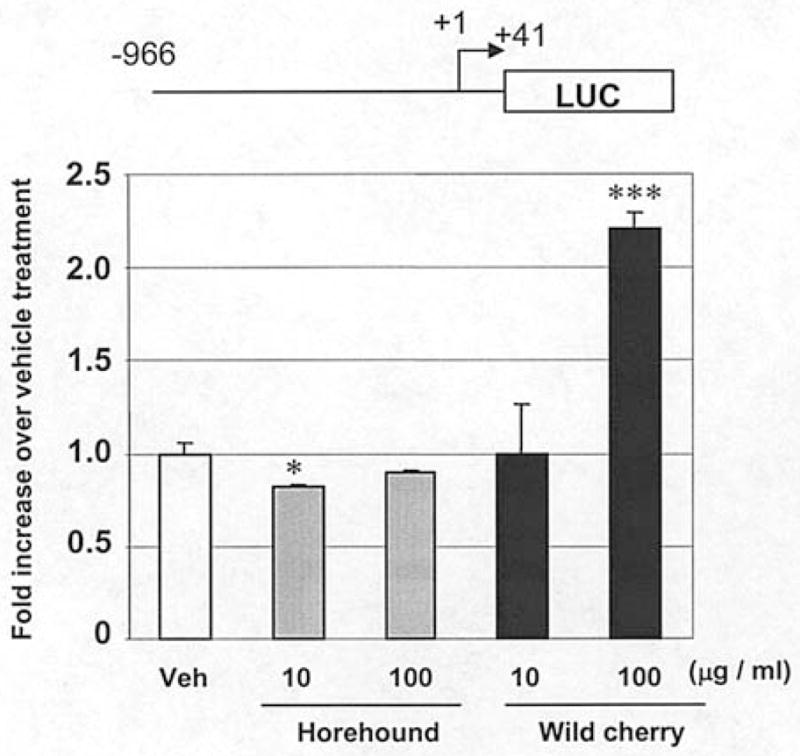
NAG-1 promoter activity on horehound leaves and wild cherry bark extracts treatment. pNAG−966/+41 (0.5 μg) was transiently transfected with 0.05 μg of pRL-null vector into HCT-116 cells. Subsequently, the cells were treated with vehicle, horehound (10 and 100 μg/ml) or wild cherry extracts (10 and 100 μg/ml) in the absence of serum for 24 h. The promoter activities were measured by luciferase activity. Transfection efficiency for luciferase activity was normalized to the Renilla luciferase (pRL-null vector) activity. The y axis shows fold increase of relative luciferase unit (RLU) compared to RLU of vehicle-treated cells. The date represents mean ± SD from three independent experiments. Statistical significance is according to Student’s t-test. *P<0.05, ***P<0.001 versus vehicle-treated cells.
Wild cherry bark extract suppresses β-catenin signaling
It has been established that association of β-catenin with the TCF/LEF family of transcription factors promotes expression of several genes such as cyclin D1, which plays an important role in the development and progression of colorectal tumorigenesis (13). Since wild cherry extract significantly decreased cyclin D1 expression, we investigated whether wild cherry extract modulates β-catenin signaling. The β-catenin-dependent TCF transcriptional activity was measured in human colorectal adenocarcinoma SW480 cells, which has a constitutively activated β-catenin/TCF pathway (17). First, we confirmed that cyclin D1 expression is reduced by the treatment with wild cherry extract in SW480 cells (Fig. 5A). Treatment with 100 μg/ml of wild cherry extract significantly suppressed β-catenin/TCF transcriptional activity in SW480 cells, compared to vehicle treatment; however horehound extract did not affect β-catenin signaling (Fig. 5B). This observation is consistent with Western blot analysis of cyclin D1 (Fig. 3) and further supports evidence for suppression of β-catenin signaling by wild cherry extract treatment resulting in reduced cyclin D1 expression.
Figure 5.
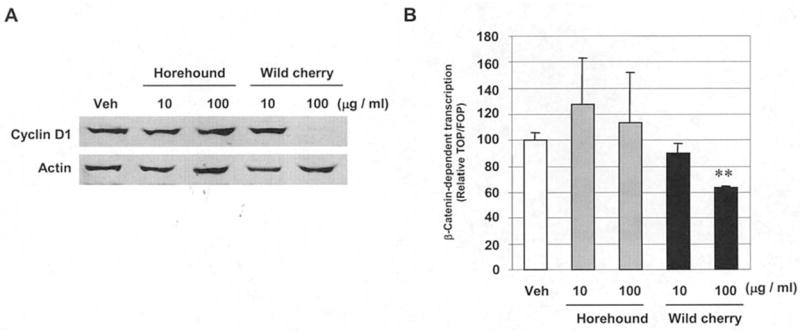
Wild cherry bark extract suppresses β-catenin signaling in SW480 cells. (A) SW480 cells were treated with vehicle, horehound (10 and 100 μg/ml) or wild cherry extract (10 and 100 μg/ml) in the absence of serum for 24 h. The cell lysates were harvested to perform Western blot analysis. (B) TOPFLASH or FOPFLASH β-catenin reporter construct (0.5 μg) was transiently transfected with 0.05 μg of pRL-null vector into HCT-116 cells, and then the cells were treated with vehicle, horehound (10 and 100 μg/ml) or wild cherry extract (10 and 100 μg/ml) in the absence of serum for 24 h. Transfection efficiency for luciferase activity was normalized to the Renilla luciferase activity. Data were expressed as the ratio of TOPFLASH to FOPFLASH. Each value represents mean ± SD from three independent transfections. The value obtained from vehicle-treated cells was defined as 100%. Statistical significance is according to Student’s t-test. **P<0.01 versus vehicle-treated cells.
Discussion
M. vulgare (horehound) and P. serotina (wild cherry) have been traditionally used for the treatment of inflammatory-related symptoms such as colds, fevers, and sore throats. In this report, we have shown that anti-inflammatory horehound leaves and wild cherry bark exhibit anti-proliferating activity in human colorectal cancer cells.
Anti-inflammatory drugs such as NSAIDs have also been shown to possess cancer chemopreventive properties. Similarly, many phytochemicals have potent anti-inflammatory as well as cancer chemopreventive properties. NAG-1, identified from an indomethacin-induced gene library, has anti-tumorigenic activity and stimulates apoptosis in colon cancer and other cell lines (3,5,18). These studies indicate that NAG-1 is a highly promising target gene for cancer chemoprevention. Thus, we investigated the effects of anti-inflammatory horehound leaves and wild cherry bark on cancer progression. Interestingly, both of these extracts increased NAG-1 expression (Fig. 3) as well as induced apoptosis in HCT-116 cells (Fig. 2B). We also found that treatment with these extracts suppressed cell growth (Fig. 2A). To determine the molecular mechanisms of NAG-1 induction, NAG-1 promoter activity was analyzed. Wild cherry extract clearly trans-activated NAG-1 promoter, resulting in increased protein expression (Fig. 4). Although horehound extract increased NAG-1 protein expression, it did not affect the promoter activity within −966 bp region. Our previous data suggest that NAG-1 induction is affected by several mechanisms including post-transcriptional regulation (19). Thus, horehound extracts may induce NAG-1 expression by either a different region of NAG-1 promoter or through post-transcriptional regulation.
Cyclin D1 protein is known to be overexpressed in colorectal adenocarcinoma (20). Amplification or overexpression of cyclin D1 plays a crucial role in the development of a subset of colon cancer through, in part, its cell cycle regulating function (21). Indeed, treatment of HCT-116 and SW480 cells with wild cherry extract dramatically decreases cyclin D1 expression, suggesting that reduction of cyclin D1 expression may be in part responsible for wild cherry extract-induced cell growth arrest. A number of oncogenic signals induce cyclin D1 expression through distinct DNA sequences in the promoter, including Ras (22), Src (23), ErbB2 (24), β-catenin (25), STAT5 (26), and simian virus 40 small tumor antigen (27). Colorectal carcinogenesis is closely linked to Wnt/β-catenin signaling pathway. Moreover, recent studies showed that anti-inflammatory drugs and anti-inflammatory phytochemicals such as NSAIDs and catechins can down-regulate β-catenin/TCF signaling in colorectal cancer cells (14–17,28). Therefore, whether wild cherry extract modulates β-catenin/TCF-mediated transcription was determined with TOP/FOP constructs. As expected, wild cherry extract significantly suppressed β-catenin/TCF signaling. These data suggest that suppression of β-catenin by wild cherry extract may result in cyclin D1 reduction.
NAG-1 promoter has been reported to contain two p53 binding sites. Interestingly, some dietary compounds such as resveratrol (8) and genistein (10) induce NAG-1 through a p53-dependent pathway. Thus, p53 expression was analyzed after treatment with these extracts. None of the extracts increased p53 expression, suggesting that p53 is not required for induction of NAG-1 expression. Tumor suppressor gene p53 mutations are frequently observed in human colon tumors (29–31), resulting in a loss in its tumor suppressor function and thereby diminishing the ability of such agents to prevent cancer from developing through p53-dependent mechanisms. Therefore, it can be beneficial to explore natural compounds that act through a p53-independent mechanism. On the other hand, p53 expression was reduced by treatment with wild cherry extract at the concentration of 100 μg/ml. At the same time, suppression of β-catenin signaling was observed in wild cherry extract (100 μg/ml)-treated cells. Reduction of p53 expression in wild cherry extract-treated cells may be explained in part by suppressed β-catenin signaling, because it has been reported that overexpression of β-catenin results in accumulation of p53 (32).
In summary, our data show that anti-inflammatory M. vulgare (horehound) leaves and P. serotina (wild cherry) bark extracts have anti-proliferative activity in human colorectal cancer cells and the mechanisms may involve enhanced NAG-1 expression and/or down-regulation of β-catenin signaling. It has been known that horehound leaves contain several siterpenoids, including marrubiin and marrubenol, and wild cherry bark contains flavonoids and antocyanidines; these compounds may be responsible for the chemopreventive effects of the extracts, but further detailed investigation is necessary. Our findings provide insight into a new implication for traditional anti-inflammatory natural plants found in southwest United States as potential novel cancer chemopreventive agents.
Acknowledgments
We thank Jada Huskey for her critical reading of manuscript, and Dr Seong-Ho Lee and Nichelle Whitlock for comments on the manuscript. We also thank Dr Howard C. Crawford (State University of New York) for providing TOPFLASH and FOPFLASH constructs. This work was supported by a grant from the National Institutes of Health (R21CA109423).
Abbreviations
- NSAID
non-steroidal anti-inflammatory drug
- NAG-1
NSAID-activated gene
- TCF
T cell factor
- PI
propidium iodide
References
- 1.Van der Jagt TJ, Ghattas R, Van der Jagt DJ, Crossey M, Glew RH. Comparison of the total antioxidant content of 30 widely used medicinal plants of New Mexico. Life Sci. 2002;70:1035–1040. doi: 10.1016/s0024-3205(01)01481-3. [DOI] [PubMed] [Google Scholar]
- 2.Buchalter L. Identification of monomeric and polymeric 5,7,3′4′-tetrahydroxyflavan-3,4-diol from tannin extract of wild cherry bark USP, Prunus serotina Erhart, family Rosaceae. J Pharm Sci. 1969;58:1272–1273. doi: 10.1002/jps.2600581026. [DOI] [PubMed] [Google Scholar]
- 3.Baek SJ, Kim KS, Nixon JB, Wilson LC, Eling TE. Cyclo-oxygenase inhibitors regulate the expression of a TGF-beta superfamily member that has proapoptotic and antitumorigenic activities. Mol Pharmacol. 2001;59:901–908. [PubMed] [Google Scholar]
- 4.Baek SJ, Wilson LC, Lee CH, Eling TE. Dual function of non-steroidal anti-inflammatory drugs (NSAIDs): inhibition of cyclooxygenase and induction of NSAID-activated gene. J Pharmacol Exp Ther. 2002;301:1126–1131. doi: 10.1124/jpet.301.3.1126. [DOI] [PubMed] [Google Scholar]
- 5.Liu T, Bauskin AR, Zaunders J, et al. Macrophage inhibitory cytokine 1 reduces cell adhesion and induces apoptosis in prostate cancer cells. Cancer Res. 2003;63:5034–5040. [PubMed] [Google Scholar]
- 6.Yamaguchi K, Lee SH, Eling TE, Baek SJ. Identification of non-steroidal anti-inflammatory drug-activated gene (NAG-1) as a novel downstream target of phosphatidylinositol 3-kinase/AKT/GSK-3beta pathway. J Biol Chem. 2004;279:49617–49623. doi: 10.1074/jbc.M408796200. [DOI] [PubMed] [Google Scholar]
- 7.Baek SJ, Kim JS, Jackson FR, Eling TE, McEntee MF, Lee SH. Epicatechin gallate-induced expression of NAG-1 is associated with growth inhibition and apoptosis in colon cancer cells. Carcinogenesis. 2004;25:2425–2432. doi: 10.1093/carcin/bgh255. [DOI] [PubMed] [Google Scholar]
- 8.Baek SJ, Wilson LC, Eling TE. Resveratrol enhances the expression of non-steroidal anti-inflammatory drug-activated gene (NAG-1) by increasing the expression of p53. Carcinogenesis. 2002;23:425–434. doi: 10.1093/carcin/23.3.425. [DOI] [PubMed] [Google Scholar]
- 9.Lee SH, Kim JS, Yamaguchi K, Eling TE, Baek SJ. Indole-3-carbinol and 3,3′-diindolylmethane induce expression of NAG-1 in a p53-independent manner. Biochem Biophys Res Commun. 2005;328:63–69. doi: 10.1016/j.bbrc.2004.12.138. [DOI] [PubMed] [Google Scholar]
- 10.Wilson LC, Baek SJ, Call A, Eling TE. Non-steroidal anti-inflammatory drug-activated gene (NAG-1) is induced by genistein through the expression of p53 in colorectal cancer cells. Int J Cancer. 2003;105:747–753. doi: 10.1002/ijc.11173. [DOI] [PubMed] [Google Scholar]
- 11.Bienz M, Clevers H. Linking colorectal cancer to Wnt signaling. Cell. 2000;103:311–320. doi: 10.1016/s0092-8674(00)00122-7. [DOI] [PubMed] [Google Scholar]
- 12.Polakis P. Wnt signaling and cancer. Genes Dev. 2000;14:1837–1851. [PubMed] [Google Scholar]
- 13.Wong NA, Pignatelli M. Beta-catenin - a linchpin in colorectal carcinogenesis? Am J Pathol. 2002;160:389–401. doi: 10.1016/s0002-9440(10)64856-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dihlmann S, Siermann A, von Knebel Doeberitz M. The non-steroidal anti-inflammatory drugs aspirin and indomethacin attenuate beta-catenin/TCF-4 signaling. Oncogene. 2001;20:645–653. doi: 10.1038/sj.onc.1204123. [DOI] [PubMed] [Google Scholar]
- 15.Orner GA, Dashwood WM, Blum CA, Diaz GD, Li Q, Dashwood RH. Suppression of tumorigenesis in the Apc(min) mouse: down-regulation of beta-catenin signaling by a combination of tea plus sulindac. Carcinogenesis. 2003;24:263–267. doi: 10.1093/carcin/24.2.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Park CH, Chang JY, Hahm ER, Park S, Kim HK, Yang CH. Quercetin, a potent inhibitor against beta-catenin/Tcf signaling in SW480 colon cancer cells. Biochem Biophys Res Commun. 2005;328:227–234. doi: 10.1016/j.bbrc.2004.12.151. [DOI] [PubMed] [Google Scholar]
- 17.Nath N, Kashfi K, Chen J, Rigas B. Nitric oxide-donating aspirin inhibits beta-catenin/T cell factor (TCF) signaling in SW480 colon cancer cells by disrupting the nuclear beta-catenin-TCF association. Proc Natl Acad Sci USA. 2003;100:12584–12589. doi: 10.1073/pnas.2134840100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li PX, Wong J, Ayed A, et al. Placental transforming growth factor-beta is a downstream mediator of the growth arrest and apoptotic response of tumor cells to DNA damage and p53 overexpression. J Biol Chem. 2000;275:20127–20135. doi: 10.1074/jbc.M909580199. [DOI] [PubMed] [Google Scholar]
- 19.Newman D, Sakaue M, Koo JS, et al. Differential regulation of non-steroidal anti-inflammatory drug-activated gene in normal human tracheobronchial epithelial and lung carcinoma cells by retinoids. Mol Pharmacol. 2003;63:557–564. doi: 10.1124/mol.63.3.557. [DOI] [PubMed] [Google Scholar]
- 20.Arber N, Hibshoosh H, Moss SF, et al. Increased expression of cyclin D1 is an early event in multistage colorectal carcinogenesis. Gastroenterology. 1996;110:669–674. doi: 10.1053/gast.1996.v110.pm8608874. [DOI] [PubMed] [Google Scholar]
- 21.Fu M, Wang C, Li Z, Sakamaki T, Pestell RG. Minireview: cyclin D1: normal and abnormal functions. Endocrinology. 2004;145:5439–5447. doi: 10.1210/en.2004-0959. [DOI] [PubMed] [Google Scholar]
- 22.Albanese C, Johnson J, Watanabe G, et al. Transforming p21ras mutants and c-Ets-2 activate the cyclin D1 promoter through distinguishable regions. J Biol Chem. 1995;270:23589–23597. doi: 10.1074/jbc.270.40.23589. [DOI] [PubMed] [Google Scholar]
- 23.Lee RJ, Albanese C, Stenger RJ, et al. pp60(v-src) induction of cyclin D1 requires collaborative interactions between the extra-cellular signal-regulated kinase, p38, and Jun kinase pathways. A role for cAMP response element-binding protein and activating transcription factor-2 in pp60(v-src) signaling in breast cancer cells. J Biol Chem. 1999;274:7341–7350. doi: 10.1074/jbc.274.11.7341. [DOI] [PubMed] [Google Scholar]
- 24.Lee RJ, Albanese C, Fu M, et al. Cyclin D1 is required for transformation by activated Neu and is induced through an E2F-dependent signaling pathway. Mol Cell Biol. 2000;20:672–683. doi: 10.1128/mcb.20.2.672-683.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shtutman M, Zhurinsky J, Simcha I, et al. The cyclin D1 gene is a target of the beta-catenin/LEF-1 pathway. Proc Natl Acad Sci USA. 1999;96:5522–5527. doi: 10.1073/pnas.96.10.5522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Matsumura I, Kitamura T, Wakao H, Tanaka H, Hashimoto K, Albanese C, Downward J, Pestell RG, Kanakura Y. Transcriptional regulation of the cyclin D1 promoter by STAT5: its involvement in cytokine-dependent growth of hematopoietic cells. EMBO J. 1999;18:1367–1377. doi: 10.1093/emboj/18.5.1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Watanabe G, Howe A, Lee RJ, et al. Induction of cyclin D1 by simian virus 40 small tumor antigen. Proc Natl Acad Sci USA. 1996;93:12861–12866. doi: 10.1073/pnas.93.23.12861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Williams JL, Nath N, Chen J, Hundley TR, Gao J, Kopelovich L, Kashfi K, Rigas B. Growth inhibition of human colon cancer cells by nitric oxide (NO)-donating aspirin is associated with cyclooxygenase-2 induction and beta-catenin/T-cell factor signaling, nuclear factor-kappaB, and NO synthase 2 inhibition: implications for chemoprevention. Cancer Res. 2003;63:7613–7618. [PubMed] [Google Scholar]
- 29.Elsaleh H, Powell B, Soontrapornchai P, et al. p53 gene mutation, microsatellite instability and adjuvant chemotherapy: impact on survival of 388 patients with Dukes’ C colon carcinoma. Oncology. 2000;58:52–59. doi: 10.1159/000012079. [DOI] [PubMed] [Google Scholar]
- 30.Pfeifer GP, Denissenko MF. Formation and repair of DNA lesions in the p53 gene: relation to cancer mutations? Environ Mol Mutagen. 1998;31:197–205. doi: 10.1002/(sici)1098-2280(1998)31:3<197::aid-em1>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]
- 31.Voskuil DW, Kampman E, van Kraats AA, et al. p53 over-expression and p53 mutations in colon carcinomas: relation to dietary risk factors. Int J Cancer. 1999;81:675–681. doi: 10.1002/(sici)1097-0215(19990531)81:5<675::aid-ijc1>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- 32.Damalas A, Ben-Ze’ev A, Simcha I, et al. Excess beta-catenin promotes accumulation of transcriptionally active p53. EMBO J. 1999;18:3054–3063. doi: 10.1093/emboj/18.11.3054. [DOI] [PMC free article] [PubMed] [Google Scholar]