

Abstract
Enteroviruses can induce human myocarditis, which can be modeled in mice inoculated with group B coxsackieviruses (CVB) and in which CVB evolve to produce defective, terminally deleted genomes. The 5' non-translated region (NTR) was enzymatically amplified from heart tissue of a fatal case of enterovirus-associated myocarditis in Japan in 2002. While no intact 5' viral genomic termini were detected, 5' terminal deletions ranged in size from 22−36 nucleotides. Sequence of the 5' third of this viral genome is of a modern strain, closely related to CVB2 strains isolated in Japan in 2002. A CVB3 chimera containing the 5’ NTR with a 22 nt deletion produced progeny virus upon transfection of HeLa cells. When the 5' 22 nucleotide deletion was repaired, the virus induced myocarditis in mice and replicated like wild-type virus in murine heart cells. This is the first report of these naturally occurring defective enteroviral genomes in human myocarditis.
Keywords: coxsackievirus, human myocarditis, defective enterovirus, 5’ terminal deletion
INTRODUCTION
The group B coxsackieviruses (CVB, serotypes 1−6) are typical human enteroviruses (HEV) classified within HEV species B (Stanway G., 2005). The genome is a single molecule of positive sense RNA, encoding 11 proteins from within a single open reading frame (ORF). The ORF is flanked on both the 5' and 3' ends by non-translated regions (NTR) (Racaniello, 2007) with both NTRs essential for maximal virus replication(Brown et al., 2004). Numerous studies in which either the 5' or 3' NTR was altered by experimental mutational or deletional analysis have shown that viral replication is deleteriously, usually lethally, affected (Barton, O'Donnell, and Flanegan, 2001; Brown et al., 2005; Murray et al., 2004; Trono, Andino, and Baltimore, 1988).
Myocarditis (inflammation of the heart muscle) has been frequently associated with an HEV etiology (Kim, 2002). Acute myocarditis is believed to be a precursor to dilated cardiomyopathy [DCM; (Mason, 2003; Spotnitz and Lesch, 2006)], a serious disease that can lead to death in lieu of transplantation. Human enteroviral RNA has also been detected in dilated cardiomyopathic hearts and in myocarditic hearts (Andreoletti et al., 2000; Archard et al., 1998; Bowles et al., 1989; Kammerer, Kunkel, and Korn, 1994; Rey et al., 2001; Satoh et al., 1994; Tracy et al., 1990). Despite the detection of HEV RNA in adult myocarditis and DCM, there has been a near complete inability to isolate infectious enterovirus in cell culture from these tissues (Rey et al., 2001) with only rare reports in the literature (Longson, Cole, and Davies, 1969; Monaldi, Benedetto, and Tentorimontalto, 1963; Soutar, 1971). This is quite different from HEV associated pediatric myocarditis in which infectious virus can usually be readily isolated (Fechner, Smith, and Middlekamp, 1963; Gear and Measroch, 1973; Piraino, Sedmak, and Raab, 1982; Porres et al., 1985). Similar observations have been made in mouse models of myocarditis (Gauntt and Huber, 2003; Kim et al., 2001; Woodruff, 1980), in which viral RNA can often be detected in heart tissue of mice experimentally inoculated with CVB3 long after lytic virus is not detectable using cell culture (Kim et al., 2005; Klingel et al., 1992; Reetoo et al., 2000).
The likely mechanism underlying the disparity between the detection of enteroviral RNA in the absence of culturable virus, was discovered by studying CVB infections in the mouse heart (Kim et al., 2005). In these studies, following inoculation of mice with CVB3, heart tissue was screened at various times post-infection for infectious virus. When these viral RNA populations were examined, no intact 5' genomic termini were detected and a variety of deletions existed with sizes ranging from 7 to 49 nt in length. These 5' terminal deletions (TD) had profound impacts upon the viral biology. As virus replication was so slowed that cytopathic effects (cpe) were not observed, virus titers had to be quantitated by RT-mediated qPCR analysis of RNA genomes in infected cells. Measurements of the positive to negative strand viral RNA ratio in infected cell cultures showed them to be close to unity rather than the high positive to negative ratios normally seen in wild-type virus infected cells (Kim et al., 2005). Interestingly, this finding extended into virion-encapsidated RNA as well, with CsCl-purified virion preparations showing the presence of negative as well as positive strand RNA. Clearly, packaging of negative strand RNA which cannot be translated upon infection of a cell, also served to enhance the attenuation of replication brought about by the TD state. Recently, we have demonstrated that similar TD mutations can occur in primary cell cultures (Kim, Chapman and Tracy, 2008). These findings have defined a novel and unsuspected aspect of HEV biology.
We were curious, therefore, to test the hypothesis that TD mutations occurring naturally during HEV replication in the human could similarly be associated with human heart disease in the absence of a detectable cytopathic virus population. Oka et al. published a case report on two patients with fulminant myocarditis (Oka et al., 2005), one of whom showed evidence of enterovirus involvement. We used formalin-fixed, paraffin-embedded heart tissue from this adult case of fatal, HEV-associated myocarditis in Japan in 2002, to demonstrate that TD genomes with deletions of up to 36 nt were detected in myocardium in the absence of detectable wild-type viral genomes. Sequence analysis of the 5' third of the genome identified the virus as a CVB2 with high identity to CVB2 strains circulating in Japan in 2002. To our knowledge, this is the first report to show that HEV TD genomes can also naturally occur in human beings. As persistent expression of CVB proteins and 2Aprotease alone are sufficient for induction of cardiomyopathy in the mouse (Wessely et al., 1998a) (Xiong et al., 2007) and as detection of CVB TD genomes in a diseased human heart disease is likely due to persistent, defective HEV replication (Chapman, 2008), there is now a mechanism to link of acute viral myocarditis with postviral DCM (Mason, 2003; Spotnitz and Lesch, 2006).
RESULTS
Extent of myocarditis and enterovirus infection in the heart muscle
In 2002, an adult male died of myocarditis in Mito, Japan; the details of this enterovirus-associated fatal adult myocarditis case have been described previously (Oka et al., 2005). Using formaldehyde-fixed, paraffin-embedded heart muscle samples from which to cut sections, we used immunohistochemistry to confirm and extend earlier work (Oka et al., 2005) that enteroviral capsid protein was detectable in the tissue. Diffuse lymphocytic infiltrates were observed throughout sections from all tissue samples used in this study together with localized myocarditic lesions and muscle damage (Figure 1A,B), consistent with the published diagnosis of fulminant myocarditis (Oka et al., 2005). Immunohistochemical staining for HEV capsid protein VP1 demonstrated widespread infection in the heart muscle (Figure 1D, F). Detection of viral protein was adjacent to but seldom overlapped the localized inflammatory infiltrates, a finding that has been observed previously by others in cases of HEV-associated myocarditis and DCM (Zhang et al., 2000) as well as in persistent viral infections in a murine model of CVB1-induced myositis (Tam et al., 1991). Immunohistochemical staining of sections from a clinically normal human heart sample (Figure 1G,H) for HEV capsid protein VP1 were negative.
Figure 1. Enterovirus protein in heart from case of fulminant myocarditis.
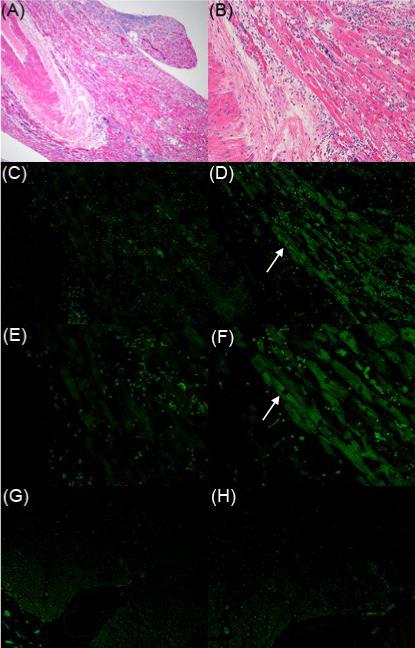
Serial sections of formalin-fixed, paraffin-embedded heart were stained with hematoxylin and eosin (A, B), or with an isotype negative control antibody (C, E), or with an antibody against a conserved epitope in HEV capsid protein VP1 (D, F). A non-infected human heart was also stained with the HEV capsid antibody as a negative control (G, H). Original magnification, ×40 (A), ×100 (B-D, G), ×200 (E, F, H). Arrows in D and F indicate the same position.
Detection of 5' terminal deletions in the viral RNA in the myocarditic heart muscle
We tested for the presence of deleted 5' termini in RNA samples derived from heart sections that had been initially assayed for the presence of HEV RNA using enterovirus B specific primers (Table 1) designed from conserved sequences [data not shown, (Chapman et al., 1990)]. Complementary DNA (cDNA) generated from total heart RNA using the conserved HEV primer E1 (Chapman et al., 1990) was used for PCR amplification with primers specific for 5’ terminal sequences of HEV-B cDNA and the primer E3Sub (Table 1). The orientation of these primers and location within the generic HEV 5' nontranslated region (5’NTR) is outlined in Fig. 2A. Each of the 5’ terminus primers amplified cDNA from the wild type CVB3/28 positive control RNA (Fig. 2B). However, only S3EntB (nt 33−58) and S4EntB (nt 50−79) efficiently amplified sequences in the heart cDNA (Fig. 2B), although a weak signal was obtained using S2ENTB (nt 21−46). The inability to amplify heart cDNA with primers S and S1ENTB (nt 1−25) and only weakly using a primer located further in from the 5' end (S2ENTB), recalled previous results obtained from mouse hearts (Kim et al., 2005) and cell cultures (Kim, 2008) that indicated 5’ terminal sequences were missing from the viral RNA.
Table 1.
|
Primera |
Genomic location,bStrandc |
Nucleotide sequence (5′ to 3′)d |
|---|---|---|
|
Primers for detection of cDNA | ||
| E1 |
644−627, − |
CACCGGATGGCCAATCCA |
| E2 |
450−464, + |
TCCGGCCCCTGAATG |
| E3 |
563−537, − |
ACACGGACACCCAAAGTAGTCGGTTCC |
| E3Sub |
549−535, − |
AGTAGTCGGTTCCGC |
| E3REV |
537−562, + |
GGAACCGACTACTTTGGGTGTCCGTG |
| E5 |
378−361, − |
GCAGGCCGCCAACGCAGC |
| S |
1−20, + |
TTAAAACAGCCTGTGGGTTG |
| S1ENTB |
1−25, + |
TTAAAACAGCCTGTGGGTTGTTCCC |
| S2ENTB |
21−46, + |
TTCCCACCCACAGGGCCCACTGGGCG |
| S3ENTB |
33−58, + |
GGGCCCACTGGGCGCTAGCACTCTGG |
| S4ENTB |
50−79, + |
GCACTCTGGTATCACGGTACCTTTGTGCGC |
|
Primers for generation of cDNA | ||
| With E1 | ||
| E8 |
65−82, + |
GGTACCTTTGTGCGCCTG |
| With E2 | ||
| DREVENTB |
766−745, − |
GCGTTGACACTTGAGCTCCC |
| With Mito5NTR | 644−673, + | GACCAATAGAGCGATCGTCTATCTATTTG |
| VP4REV |
1046−1025, − |
CGACCACATTGGCACACTCTTG |
| With MitoVP4 | 916−942, + | GGACATTATGATAAARTCTATG |
| VP2REVC |
1537−1514, − |
GGTGCGCAAGTTKATCCACTGRTG |
| With MitoVP4 | ||
| VP2REVA |
1582−1557, − |
GGCACACTGTTTATRTATGGCATBAC |
| With MitoVP2 | 1444−1468, + | GACCGATGTCCAAACTGCAGTGTGC |
| CVB2VP1REV |
2605−2582, − |
CTGGTTTGCATCGTRTCACTRGG |
|
Primers for cloning | ||
| 22RIBOZ1 |
22−44, + |
ATGAGGCCGAAAGGCCGAAAACCCGGTATCCCGGGTTCCCCACCCACAGGGCCCACTGGGC |
| 22RIBOZ2 |
|
CACTATAGGGCGCGGGTGGGTGGGCTGATGAGGCCGAAAGGCCGAAAACCCGGTATC |
| TD22RIBOZPCRT7 |
|
GACCGCGGCCGCGTAATACGACTCACTATAGGGCGCGGGTGGGTGGGCTGATGAGG |
| DREV |
766−745,− |
GCGTTGATACTTGAGCTCCC |
| ENTBCONSENSUS |
1−47,+ |
TTAAAACAGCCTGTGGGTTGTTCCCACCCACAGGGCCCACTG |
| 28RIBOZ1 |
1−20,+ |
TGAGGCCGAAAGGCCGAAAACCCGGTATCCCGGGTTCTTAAAACAGCCTGTGGGTTG |
| 28RIBOZ2 |
|
GACACTGATCCGCGGGTGTTTTAACTGATGAGGCCGAAAGGCCGAAAACCCGGTATC |
| 28RIBOZPCRT7 | GACCGCGGCCGCGTAATACGACTCACTATAGGGCGCGGGTGTTTTAACTGATGAGG | |
With the exception of DC-Tail, 5CMV, 3CMV, 28RIBOZPCR, sequences of primers are based on the CVB3/28 genome (GenBank Accession no. AY752944).
Numbering as in CVB3/28 (Genbank Accession no. AY752944)
Positive is same sense as genomic RNA, negative is antisense to genomic RNA.
Underlined sequence is not genomic cDNA in cloning primers.
Figure 2. cDNA from human myocarditic heart was not amplified with 5’ terminal enterovirus-specific primers.
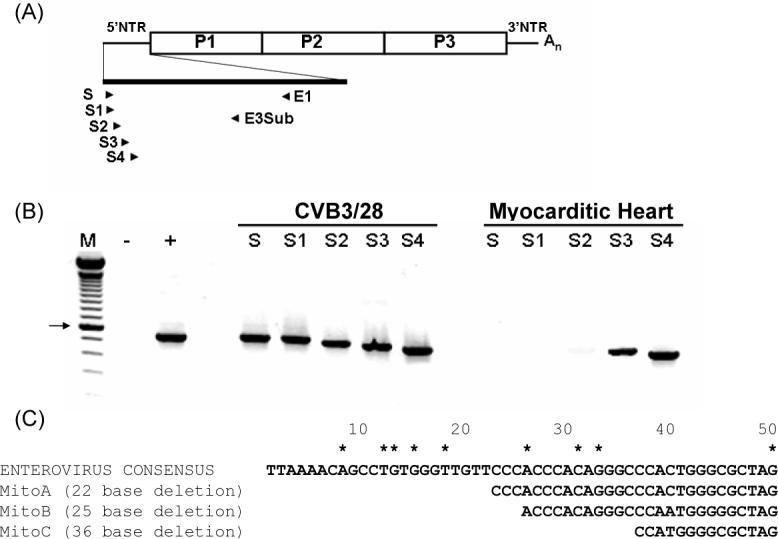
(A) Orientation of primers within the 5' NTR. (B) E1-primed cDNA of total RNA from human myocarditic heart or purified CVB3/28 was amplified with 5’ terminal primers S, S1ENTB-S4ENTB and E3Sub in separate PCRs and then electrophoresed in 1.5% agarose. M, 100 bp ladder (Invitrogen); -, PCR with S and E3Sub without template; +, PCR with S and E3Sub with pCVB3/28. Abbreviations for S1EntB-S4 EntB are S1-S4. Arrow indicates 600 bp band. (C) Alignment of the 5’ terminal sequences of CVB2/Mito with the enterovirus B consensus sequence. Three sizes of 5' terminal deletions were cloned and sequenced (22, 25 and 36 nt). Numbering begins from the 5' terminus. Asterisks indicate positions of previous terminal deletions detected in hearts of mice (Kim et al., 2005) and in cell culture passage (Kim, Chapman, and Tracy, 2008).
We then tested directly for 5' terminal deletions using sequence analysis. Heart cDNA primed with E1 was tailed with dG10, amplified with E5 and a primer specific for the oligo dG tail [DCTail which has a 3' oligo dC10; (Kim et al., 2005)], and cloned. Only four unique clones were obtained due to the low yield of tailed cDNA. Two clones showed deletions of the 5' nt 1−22, one had nt 1−24 deleted, and one had nt 1−35 deleted relative to consensus 5’ terminal sequence derived from 90 HEV-B 5’ terminal sequences in the Genbank database (Fig. 2C). Control experiments using this approach with cDNA from wild type CVB3/28 stocks generated clones with 10 C residues at the 5' terminus upstream of the authentic HEV-B consensus 5’ terminus (Kim et al., 2005). One of the 4 clones had a 5’ terminal C10 tract followed by sequence that aligned with nt 26−360 of the enterovirus B sequence (Fig. 2C, MitoB) while in two clones, the C10 tract was followed by sequence aligning to nt 23−360 (Fig. 2C, Mito A). Because nt 23−25 are CCC in the HEV-B consensus, it is possible that the deletions in these genomic termini might be 22−25 nt in length, although 25 and 22 nt are the most likely deletions based upon the C10 tract. Similarly, the final clone (Fig. 2C, MitoC) most likely had a deletion of 36 nt, although 35−38 nt deletions are similarly possible. Like the previously described CVB3-TD genomes isolated from mouse hearts (Kim et al., 2005) or cell cultures (Kim, 2008), the deletions disrupt or delete regions in the secondary structure termed domain I (Bailey and Tapprich, 2007): the loss of nt 1−22 and 1−25 delete stem a, while the greater deletion (nt 1−36) delete stems a and b of domain I. Together, these results demonstrated that the population of HEV RNA in the human heart contained 5' terminal genomic deletions.
The nucleotide and amino acid sequences of the heart virus identify it as a modern strain of CVB2
Sequence analysis using overlapping cloned cDNAs was carried out to determine the identity of the virus in the heart muscle. The sequence of the 5' 2,563 nt of the heart HEV RNA (GenBank Accession no. EU177671) was determined as outlined in Methods. A BLAST search of GenBank using this sequence revealed closest identity to the genomes of the six CVB serotypes, with the sequences showing highest identity (92−93%) being partial sequences of CVB2 isolates from 2002 in Kanagawa, Japan (GenBank Accession nos. AB162751, AB162752). Alignment of the 2,563 nt sequence to the prototype CVB genomes of serotypes 1−6 (GenBank Accession nos. AF081485, M16560, AY752944, X05690, AF114383, AF114384) revealed the heart virus sequence shared highest nt identity (84%) with that of CVB2, strain Ohio-1 (Polacek et al., 1999) with significantly lower identities shared by genomes of the other 5 CVB serotypes (74−75%). The relatively low identity with the prototype CVB2 sequence (and to other prototype CVB genomes) in comparison to the 2002 Kanagawa CVB2 isolates, was not surprising given that the prototype strains circulated more than 50 years ago (Melnick et al., 1950). However, a recently published study of an HEV in type I diabetes patients (Dotta et al., 2007) generated a sequence which shared >99% overall nt identity with the CVB4 prototype strain Benschoten originally isolated in 1951 (Sickles, Feorino, and Plager, 1955), with just one nt mismatch in the 5’NTR. As RT-PCR has a risk of contamination with laboratory strains [see, for example, (Giacca et al., 1994)], it can be concluded that the CVB4, “Tuscany” strain (Dotta et al., 2007), was derived from contamination with the 58 year old prototype strain. The 84% identity of the Mito sequence with the CVB2 prototype and higher identity with sequences of CVB2 isolates from the same time and geography demonstrates the Mito sequenceis not due to contamination.
Oberste and colleagues (Nix, Oberste, and Pallansch, 2006; Oberste et al., 1999a; Oberste et al., 1999b) have demonstrated that sequence analysis of the HEV VP1 genomic region correlates with HEV serologic identity; virus strains within a specific serotype share ≥75% nucleotide identity or 85% amino acid identity. The VP2 puff region [aa2129−2180 (Muckelbauer et al., 1995)] also has a similar level of intraserotype (but not interserotype) identity and can be used reliably to type HEV strains (Nasri et al., 2007). As we were unable to sequence further than 2,563 nt, we analyzed the sequence encoding the entire VP2 capsid protein (nt 950−1721), comparing it to those from other CVB genomes. The closest identity again occurred with CVB2 (84−85%; GenBank Accession nos. EF174468, EF174469, AF081485, AF085363), with greater divergence from other CVB serotypes (72−74%). We then examined the VP2 Puff sequence (aa197−250), a structure that contributes to the determination of serotype (He et al., 2001). When compared to those of modern and prototype CVB2 strains, the heart virus sequence revealed highest identity with other CVB2 strains (Table 2). Taken together, these results indicated that the heart virus was a modern (ca. 2002) strain of CVB2 and was subsequently termed CVB2/Mito.
Table 2.
Alignment of the predicted amino acid sequence of the VP2 Puff from the heart virus (Mito) with CVB serotype strains.
| Year | 197 250 | |
|---|---|---|
| MITO | 2002 | PEAEMGCTNKENTPLFEKLCGQDNAKEFSREGPTVSEGATDVQTAVCNAGMGVG |
| CVB2/SE-96−72087 | 1996 | ..................................I................... |
| CVB2/SE-05−90025 | 2005 | ...................................A.................. |
| CVB2/Ohio | (prototype) | .................N..........T.....I.K................. |
| CVB4/SE-04−85460 | 2004 | .........V..A TYGD...GET..Q.EQN---AAT.E.A............. |
| CVB4/SE-04−84828 | 2004 | .........V..A TYGD...GET..Q.EQN---AAT.E.A............. |
| CVB4/Benschoten | (prototype) | .........A..A AYGD...GET..S.EQN---AAT.K.A............. |
| CVB4/E2 | 1958 | .........A..A TYGD...GET..Q.EQN---AVT.E.A............. |
| CVB6/Schmitt | (prototype) | .......S.LN.A AAD.SAGEV.RQ.TV.P---AN.QNQ.....H..A...A |
| CVB1/SE-02−71582 | 2002 | .......S.LN...E AE.S.G.T..M.TDTKVG-ESN.KK.....W....... |
| CVB1/SE-99−94869 | 1999 | .......S.LN...E AE.S.G.T..M.TDTKVG-ESN.KK.....W....... |
| CVB1/Tucson | (prototype) | .......S.LD...E VE.S.G.S.RL.TDTQVG-ESNEKK.....W....... |
| CVB1/Conn-5 | (prototype) | .......S.LN...K AE.S.G...RM.TDTEVG-TSNDKK.....W....... |
| CVB5/SE-03−79895 | 2003 | .......ATLA.K.DLKS.SNGET.SM.ESQN---.T.Q.A..AN.I....... |
| CVB5/Peterborough | 1954 | .......ATLA.K.DQKS.SNGET.NT.DSQN---TT.Q.A..AN.I....... |
| CVB5/Faulkner | (prototype) | .......ATLA.K.DQKS.SNGET.NV.ESQ---N.S.Q.A..AN.I.....I. |
| CVB3/SE-94−51301 | 1994 | .......ATLD...SS.E.L.G.A....ADK-.VA.GSNKL..R..Y.....I. |
| CVB3/AS | 1977 | .......ATLD...SSAE.L.G.S....ADK-.VA.GSNKL..RV.Y....... |
| CVB3/CO | 1978 | .......ATLD...SS.E.L.G.A....ADK-.VA.GSNKL..RV.Y.....I. |
| CVB3/GA | 1956 | .......ATLD...SSAE.L.G.S.M..TDK-.VAAGSNKL..RV.H....... |
| CVB3/Nancy | (prototype) | .......ATLD...SSAE.L.GDS....ADK-.VA.GSNKL..RV.Y....... |
GenBank Accession nos: CVB2/SE-96−72087, ABI31471; CVB2/SE-05−90025, ABI31477; CVB2/Ohio, AAD46138; CVB4/SE-04−85460, ABI31513; CVB4/SE-04−84828, ABI31533; CVB4/Benschoten, P08292; CVB4/E2, Q86887; CVB6/Schmitt, AAF21972; CVB1/SE-99−94869, ABI31571; CVB1/SE-02−71582, ABI31508; CVB1/Tucson, AAO84299; CVB1/Conn-5, AAC00531; CVB5/SE-03−79895, ABI31514; CVB5/Peterborough, Q03053; CVB5/Faulkner, AAF21971; CVB3/SE-94−51301, ABI31532; CVB3/AS, AAD53727; CVB3/CO, AAD53726; CVB3/GA, AAT79531; CVB3/Nancy, AAA42931; CVB3/20, AAV34213; CVB3/31−1−93, AAG23918; CVB3/28, AAV34212. VP2 “Puff” sequence of strains selected based on alignment with CVB3 2129−2180. Dot indicates conserved sequence relative to Mito; - indicates deletion.
Terminally-deleted CVB2/Mito 5’ NTR in a CVB3 genome produces non-cytopathic virus but restoration of the 5’ terminus to the consensus HEV-B sequence produces a cytopathic virus
We replaced the 5' NTR of the infectious cDNA copy of the cardiovirulent strain CVB3/28 genome (Tracy et al., 2002) with the CVB2/Mito 5' NTR containing the 22 nt deletion to study how the CVB2/Mito 5' NTR would function in an intact CVB genome. The 5' NTRs of enteroviruses have been shown to be functionally interchangeable, producing infectious progeny viruses (Chapman et al., 2000a; Lukashev et al., 2003; Semler, Johnson, and Tracy, 1986). No cpe was observed following transfection of this cDNA, termed CVB3/5NTRMitoTD23 [the TD23 genome is named for the first nt of the genome relative to the enterovirus consensus; (Kim et al., 2005)], into HeLa cell cultures (Fig. 3A), although transfection of the control CVB3/28 cDNA caused widespread cpe within 48 hours (Fig. 3A). However, upon restoration of the 22 nt 5' terminal deletion (CVB3/5NTRMito), a lytic phenotype was restored (Fig. 3A).
Figure 3. CVB3/5NTRMitoTD23 replicates without cytopathic effect but restoration of 5’ terminal sequence to HEV-B consensus results in cytopathic replication.

(A) HeLa cell monolayers were transfected with T7 RNA polymerase transcripts of pCMVT7r5NTRMitoTD23 (CVB3/5NTRMitoTD23), pCMVT7r5Mito (CVB3/5NTRMito), pCVB3−28 (CVB3/28) or mock transfected, control. Monolayers were fixed at 72 hours and stained with crystal violet. (B) cDNA from RNA of 72 hour cultures of transfected HeLa monolayers was amplified with primer E1 and primers S1ENTB (lane 1), S2ENTB (lane 2), S3ENTB (lane 3), S4ENTB (lane 4) and electrophoresed in 1.5% agarose. Lane M, Hi-Lo DNA Marker (Minnesota Molecular, Minneapolis, MN); lane -, PCR with S4ENTB and E1 without template cDNA. Arrow indicates 750 bp.
To verify that the transfection which did not generate cpe was in fact productive and that transfected cDNA genomes produced viral RNA, this experiment was repeated with HeLa cultures transfected with RNA from CVB3/28, CVB3/5NTRMitoTD23, CVB3/5NTRMito, or mock transfected as described above. Total RNA was isolated from each culture, DNase treated, and assayed for the presence of viral RNA by RT-PCR using E1 and primers specific for the 5’ terminus. Despite the lack of cpe in CVB3/5NTRMitoTD23 infected cultures, viral RNA was readily detected (Fig. 3B) with 5' end primers outside of the deleted sequence. Both the wild type (positive control) CVB3/28 RNA and the RNA from the cultures infected with the CVB3/5NTRMito (repaired deletion) strain were detected with all of the primers as expected, (Fig. 3B); these strains both induced complete cpe (Fig. 3A).
When supernatants of the CVB3/5NTRMitoTD23 infected cultures were treated with ribonuclease, passed through 0.2 micron filters, and placed on fresh HeLa cell monolayers, no cpe resulted but viral RNA remained detectable (data not shown), indicating that infection was due to a normal infectious process by virions and not by adventitious contamination with free viral RNA. Measurement of the ratio of positive to negative strand viral RNA in a purified CVB3/5NTRMitoTD23 stock by real time RT-mediated quantitative PCR was carried out. This revealed that both negative and positive strand viral RNA was detected in RNA isolated from CsCl-purified virions with a positive/negative strand ratio of 1.6:1. This finding with the chimeric CVB3/Mito5NTRTD23 strain was consistent with known CVBTD biology established with murine viral isolates (Kim et al., 2005), confirming that CVBTD strains can encapsidate either negative or positive stranded RNA. By comparison, stocks of parental strain CVB3/28 showed no detectable negative strand RNA as expected from prior work (Kim et al., 2005; Novak and Kirkegaard, 1991). Together, these results showed that the CVB2/Mito 22 nt deletion incurred the same type of replication lesion in the chimeric CVB3/5'NTRMitoTD23 strain as did other TD strains described previously (Kim et al., 2005).
Analysis of CVB3/5NTRMito in cell culture
Stocks of the chimeric CVB3/5NTRMito strain were analyzed in single step growth curves together with control virus strains, cardiovirulent CVB3/28 and noncardiovirulent CVB3/GA (Lee et al., 2005), in HeLa cell and MHC cultures. The chimeric CVB3/5NTRMito strain replicated with similar initial kinetics in HeLa cultures as the two wild-type CVB3 strains, reaching a similar yield of infectious particles two hours later (Fig. 4A). HeLa cultures are a rich environment for the growth and replication of HEV, making it difficult to differentiate virus strains that differ by replication phenotype (Agol et al., 1989; Lee et al., 2005). However, when these virus strains were similarly analyzed in cultures of MHC, a cell culture system in which cardiovirulent CVB3 strains replicate to higher levels than non-cardiovirulent strains (Kim, 2008), CVB3/5NTRMito and CVB3/28 replicated similarly in rate and extent, while the non-cardiovirulent CVB3/GA replicated more slowly to 3 orders of magnitude lower titer (Fig. 4B). These results demonstrated that with an intact 5' terminal sequence, the 5’NTR of CVB2/Mito permitted the chimeric virus to replicate like a cardiovirulent strain.
Figure 4. CVB3/5NTRMito replicates in murine heart cells like virulent CVB3/28 and but not avirulent CVB3/GA.

Single step growth curves were carried out in HeLa (A) and MHC (B) monolayers using triplicate wells initially seeded with 100,000 cells per well. Monolayers were inoculated with CVB3/5NTRMito, CVB3/28, or CVB3/GA at an MOI of 25. Virus titers were determined on HeLa cell monolayers. Square, CVB3/5NTRMito; filled circle, CVB3/28; open circle, CVB3/GA.
Inoculation of mice with CVB3/5NTRMito induces myocarditis
To investigate whether the chimeric virus with the restored 22 nt would induce myocarditis, male A/J mice (4−5 weeks old) were inoculated with 1×105 TCID50 equivalents of the defective strain (CVB3/5NTRMitoTD23), the restored strain (CVB3/5NTRMito), or the control strain (CVB3/28). Hearts from control mice, inoculated with 100 mM saline (virus diluent) (Fig. 5A) were normal, and indistinguishable from the hearts from mice inoculated with the defective strain CVB3/5NTRMitoTD23 (Fig. 5C), despite the presence of viral RNA in the myocardium (as determined by RT-PCR; data not shown). Inoculation with CVB3/5NTRMito with the restored 5' genomic terminus, induced myocarditis (Fig. 5D) to an extent similar to that induced by the control, cardiovirulent strain, CVB3/28 (Fig. 5B). These results demonstrate that the CVB2/Mito 5' NTR is capable of working in synergy with the rest of a cardiovirulent CVB genome to generate myocarditis.
Figure 5. CVB3/5NTRMitoTD23 shows no pathogenic effects in mouse heart but CVB3/5NTRMito with a restored 5’ terminus causes myocarditis.

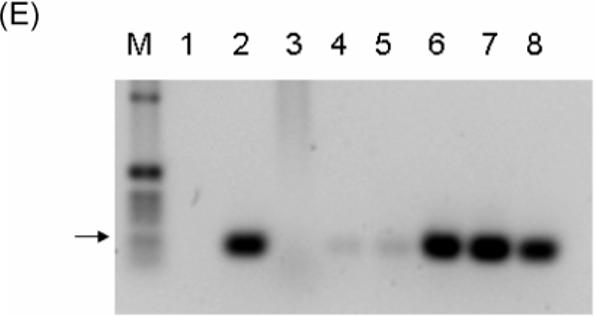
Male A/J mice were mock infected (saline) (A), or inoculated with 1×105 TCID50 units of CVB3/28 (B), or the equivalent of 1×105 TCID50 units of CVB3/5NTRMitoTD23 (C), or 1×105 TCID50 units of CVB3/5NTRMito (D). At day 14 post inoculation hearts were fixed in 10% buffered formalin and sections stained with hematoxylin and eosin. Typical lesions indicated by arrows. Original magnification, ×100. E. Heart homogenates of mock-infected, CVB3/5NTRMitoTD23, CVB3/5NTRMito, or CVB3/28 inoculated A/J mice were used to inoculate HeLa cell monolayers. cDNA from RNA of 72 hour cultures of transfected HeLa monolayers was amplified with primer E1 and E2 and electrophoresed in 1.5% agarose. Lane M, 1 kb ladder (Invitrogen); lane 1, PCR with E1 and E2 without template cDNA, lane 2 with CVB3/28 viral cDNA, lane 3 with cDNA of mock-infected heart homogenate culture, lanes 4, 5 with cDNA from CVB3/5NTRMitoTD23 heart homogenate cultures, lanes 6, 7 with cDNA from CVB3/5NTRMito heart homogenate cultures, lane 8 with cDNA from CVB3/28 heart homogenate culture. Arrow indicates 510 bp.
DISCUSSION
This report documents for the first time that group B coxsackieviruses can generate heretofore unsuspected 5' terminal genomic deletions during replication in the human host subsequent to a naturally-occurring infection. Using RNA isolated from formalin-fixed, paraffin-embedded heart muscle samples from a case of fulminant myocarditis in 2002 in Mito, Japan (Oka et al., 2005) in which HEV protein and RNA were detected, we have identified the virus as a CVB2 strain with close identity to the Kanagawa strain of CVB2 which circulated in Japan in 2002. Importantly, while we were unable to detect intact 5' viral genomic termini, deletions at the 5' termini of at least 22−36 nt were present. These data from a human sample closely mirror results first obtained from mouse heart tissue following experimental CVB3 inoculation (Kim et al., 2005) and more recently, from CVB3-inoculated cell cultures (Kim, 2008), thereby demonstrating that this newly-discovered aspect of HEV biology has clinical relevance in addition to the basic description of a new aspect of the enterovirus replication cycle.
The discovery that HEV can delete the 5' terminal genomic sequences during replication and thereby can persist in the host for long periods of time [as has been shown in the mouse; (Kim et al., 2005)], has led to the demonstration of dramatic changes in the replication cycle of such viruses. Terminally-deleted CVB3 strains isolated from mouse heart or tissue culture (Kim, 2008; Kim et al., 2005), similar to the CVB2/Mito genome population characterized here, replicate very slowly and to low titers. CVB3TD strains persisted for months in murine heart tissue but did not induce cytopathic effects when heart homogenates were inoculated onto HeLa cell monolayer cultures (Kim et al., 2005). Because progeny infectious virus can nonetheless be detected in such cultures, cell lysis undoubtedly occurs; however, due to the very slow replication rate of the CVBTD strains, cpe in cell cultures is not observable by light microscopy. No viral or genomic RNA replication.was noted in a recent study (Hunziker, Cornell, and Whitton, 2007) of a CVB3 genome with an engineered deletion of the 5' terminal 32nt. In that study, viral translation was assessed by labeling transfected cells for 30 minutes, while RNA replication was assayed by slot-blot. However, previous work which required accumulation of viral proteins for 24 hours for detection by western blot and enzymatic amplification for detection of viral RNA (Kim, Chapman, and Tracy, 2008), strongly suggests that these assays would not have been sufficiently sensitive to detect the low levels of virus found in cultures of CVB3TD.
The slow replication of CVBTD strains is linked to the deletions in the domain I RNA structure, a structure that is known to be crucial for HEV replication (Andino, Rieckhof, and Baltimore, 1990; Lyons et al., 2001; Sharma, O'Donnell, and Flanegan, 2005). The interesting observation that the normally high positive/negative viral RNA strand ratios in wild type infected cells (Novak and Kirkegaard, 1991) decrease to near unity with CVB3TD viruses, further indicates that 5' terminal deletions deleteriously affect positive strand viral RNA synthesis. The discovery of TD genomes has also provided a mechanistic explanation for the detection of nearly equimolar levels of positive and negative strand RNA that have been reported during persistence of CVB in mice (Klingel et al., 1992; Tam and Messner, 1999). Because CVBTD (but not wild type) virions can contain either negative or positive stranded viral RNA with nearly equal probability (Kim et al., 2005), CVBTD attenuation is further enhanced because negative strand viral RNA cannot be used as an mRNA upon infection. We have also shown that passage of wild type CVB3 in cardiac primary cell cultures can also generate TD strains (Kim, 2008), although this was not observed in immortal cell cultures such as HeLa. These latter findings indicate that the type of host cell and tissue plays a key role in determining whether TD genome populations can displace wild type virus. We have not yet explored whether other organs may harbor similar populations of CVBTD strains following experimental inoculation of mice.
Our discovery that TD genomes occur in human tissue raises the question of their impact upon human health. Following experimental inoculation of mice with CVB3, we (Kim et al., 2005) and others (Klingel et al., 1992; Reetoo et al., 2000) have shown that viral RNA can be detected in mouse heart muscle for weeks, well past the time that cytolytic virus can be isolated in susceptible reporter cell cultures. Long-term persistence of an HEV infection in the apparent absence of lytic virus, has similarly been observed in human hearts by analysis of sequential endomyocardial biopsies (Jin et al., 1990; Kuhl et al., 2005; Stille-Siegener M, 1993). Although the myocarditic heart used in this study was from an acute stage of enteroviral myocarditis, no non-deleted 5’ ends of viral genomes were detected by cloning or by amplification of 5’NTR sequence, indicating that in this tissue the process of selection of deleted genomes is rapid. Although the possibility of repeated HEV infections cannot be rigorously disproven in clinical studies, we propose here that HEV persistence which occurs via generation of a TD genome population, is the more likely mechanism. How could a slowly-replicating, persisting TD virus population damage the heart? A plausible mechanism was suggested by work in which a CVB3 genome was cloned in a transgenic mouse line under the cardiac myosin promoter (Wessely et al., 1998b). The VP4/VP2 autocatalytic cleavage site (Wessely et al., 1998a) was engineered to prevent correct cleavage of the VP0 protein, a lesion that prevented production of infectious virus particles. These transgenic mice developed enlarged hearts, reminiscent of dilated cardiomyopathy, a pathologic outcome that was subsequently demonstrated to be due solely to expression of 2Apro (Xiong et al., 2007), one of two CVB proteases, in the host. It is well established that the expression of HEV proteins can alter host cell protein expression and secretion (Belov and Ehrenfeld, 2007; Kundu et al., 2005; Kuyumcu-Martinez et al., 2004; Lloyd, Jense, and Ehrenfeld, 1987). CVB proteins can cleave and disrupt host cell proteins such as dystrophin (Badorff et al., 1999), a cytoskeletal protein altered in X-linked dilated cardiomyopathy (Towbin et al., 1993), and host translation factors such as poly A binding protein (Joachims, Van Breugel, and Lloyd, 1999; Kerekatte et al., 1999). Such changes can cause cell and tissue dysfunction in the infected host. In the current fatal case of fulminant myocarditis described by Oka and colleagues (Oka et al., 2005), the CVB2 population had already evolved to a TD form and viral proteins were expressed and accumulating in many intact cardiomyocytes at the time of death (Fig. 2). Had the patient survived such an extensive myocardial infection (which produced sufficient virus for verification of the 5’ terminal deletion), there would have been the potential for extensive alteration of myocardial function by expression of the viral proteins.
The development of DCM as a consequence of viral myocarditis has been discussed extensively in the medical literature (D'Ambrosio et al., 2001; Figulla, 2004; Kawai, 1999; Mason, 2003) and has been reviewed and summarized recently (Spotnitz and Lesch, 2006). A question frequently cited in these reviews has been the role that HEV infections play in the development of DCM; whether symptoms are due to direct viral-mediated effects on cell function during persistent infection as discussed above or due to inflammation and autoimmunity persisting post-infection (Fairweather, Frisancho-Kiss, and Rose, 2005; Huber, 2006). Although we have not yet documented the presence of TD forms in DCM, this study demonstrates that HEV in human heart infections can rapidly evolve to the TD form, a form capable of persisting far beyond the acute stage (Kim et al., 2005). The results presented here and elsewhere (Kim, 2008; Kim et al., 2005) have now documented a natural and previously unsuspected mechanism by which cardiotropic HEV strains can persist as a productive infection of the heart, providing an environment in which cleavage of and damage to host cell proteins by viral proteases expressed from the persistently infectious viral genome, can accumulate with time with deleterious outcomes.
How does a TD genome population persist in the face of an activated adaptive immune response? We theorize that TD genomes arise through RNA polymerase error; in support of this, while the HEV 3Dpol has been shown to be a processive enzyme, small amounts of prematurely terminated transcripts have been detected (Rodriguez-Wells, Plotch, and DeStefano, 2001) . We propose that TD genomes are present in any replicating HEV population and that this defective quasipecies population can rapidly become dominant if the environment changes to favor them over lytic wild type HEV replication (Domingo et al., 2006). At present, we cannot assay for the presence of TD genomes in the presence of intact wild-type genomes, and can therefore demonstrate TD RNA only when the TD RNA population has becomes dominant. In experimentally-inoculated mice (Kim et al., 2005), we have shown that TD genomes become detectable in the latter part of the acute viral replication period when the adaptive immune system has been activated and has largely cleared the wild type virus from the host. Activation of the serotype-specific adaptive immune response against the infecting virus is a prime candidate for the significant change in the environment that leads to clearing of fast-replicating wild type virus without eradication of the TD population. In support of this, inoculation of scid mice (which lack functional B and T cells) with poorly pathogenic CVB strains results in long-term persistence of infectious, cytopathic virus (S Tracy, unpubl. data).
But why aren't TD populations also eradicated in the immunocompetent host? We theorize that it is related to the TD virus population's slow replication rate, which may be linked with low antigen levels or expression. Following inoculation of immunocompetent mice with cloned CVB3TD strains (Kim, 2008; Kim et al., 2005), we have been unable to detect neutralizing antiviral antibodies (lowest dilution, 1:6), although antibodies that bind purified virions used as target antigens in ELISA are detectable, albeit at lower concentrations than in sera of mice inoculated with wild type CVB3 (N. Chapman, S. Tracy, unpublished data). Together with the profound defects in TD virus replication, these observations suggest that TD virus populations are poorly antigenic, most likely due to low virus antigen loads in the host. These results may also define why areas of extensive lymphocytic infiltration border but seldom overlap with, areas of viral protein expression found in this study (Figure 1) and in work of others (Tam et al., 1991; Zhang et al., 2000). Thus, even under the scrutiny of a thoroughly activated adaptive immune system, HEV persistence can occur via a significantly restrained lifestyle compared to the acute, high-titer, cell-damaging replication normally associated with HEV infections (Pallansch, 2001).
METHODS
Cell culture and viruses
CVB3/28 (Tracy et al., 2002) and CVB3/GA (Lee et al., 2005) have been cloned, sequenced and described previously. Virus stocks were prepared in HeLa cell monolayers as previously described (Kim et al., 2005) or propagated in cultures of primary murine heart cells (MHC). MHC cultures were derived from minced ventricular tissue of a male C3H/HeJ mouse at 6 weeks of age as described (Toraason et al., 1989) with minor modification. Briefly, mouse heart tissue was finely chopped, washed in saline, and trypsinized (50 μg/ml; Worthington Biochemical Corp., Lakewood, NJ) at 50 μg/ml in calcium and magnesium-free Hanks balanced salt solution with 30 mM 2,3 butanedione 2-monoxime for 20 hours at 6°C. This was then treated with collagenase (100 U/ml; Worthington Biochemical Corp.) for 1 hour at 37°C in Claycomb medium (SAFC Biosciences, Inc.; Lenexa, KS), and passed through a 70 micron cell strainer (BD Falcon, San Jose, CA). The filtrate was placed into culture in Claycomb medium supplemented with 10% fetal bovine serum, 1 U/ml penicillin, 100 μg/ml streptomycin, 0.1 mM norepinephrine and 2 mM L-glutamine at 37°C, 5% CO2. MHC cultures were used between passages 4 to 15.
Detection of viral capsid protein in human heart tissue by immunohistochemistry
Formalin-fixed, paraffin-embedded heart muscle samples (approximately 9 cm3) were used to cut sections for immunohistochemistry and RNA isolation (below). Immunohistochemical staining for viral capsid protein was performed essentially as described (Drescher et al., 2004; Tracy et al., 2002). Briefly, following deparaffinization, rehydration, and steaming in 10 mM sodium citrate (pH = 6.0) to expose epitopes, slides were blocked with normal goat serum (Sigma; St. Louis, MO). Detection was performed using a mouse monoclonal antibody against a common antigen in the enteroviral capsid protein VP1 (Clone 5-D8/1; DAKO Corp., Carpinteria, CA). Control slides received an isotype control sera (mouse IgG2a isotype control; eBioscience, San Diego, CA). Antibody was detected with an Alexa UO 488-labeled goat anti-mouse IgG (H+L) antibody (Invitrogen; San Diego, CA).
Isolation of human heart RNA
About 40−50 sections (6 micron) from a tissue block were cut for each RNA preparation. Sections were deparaffinized in xylene, washed successively with 100%, 90% and 70% ethanol, washed once in 10mM Tris-HCl, pH 8.0, then mixed with 6,000 units/ml proteinase K (Ambion; Austin, TX) in 10 mM Tris-HCl, pH 8.0, 100 mM NaCl, 25 mM EDTA, 1% w/v SDS, for 18 hours at 56°C. Total RNA was then isolated on silica-based matrix columns (ZR Viral RNA Kit; Zymo Research, Orange, CA). Over the course of this work, enteroviral RNA was isolated from three different samples of the myocarditic heart muscle. RT-PCR for enteroviral RNA (below) was performed prior to using the RNA for characterizing the viral genome.
Virus identification strategy
Because the serotype of the HEV strain in the myocarditic heart had not been previously identified (Oka et al., 2005), we proceeded to amplify the viral genome in overlapping fragments for sequence analysis. Purified heart RNA was short in length after extraction from the formalin fixed heart and it was necessary to use numerous reverse transcription and PCR steps to acquire overlapping amplimers for progressive sequence analysis of the HEV genome inward from the 5' end. We first amplified the viral 5' non-translated region (NTR) because 5' NTR sequences are well-conserved among the HEV (Racaniello, 2001), followed by amplimers within the capsid protein coding region of the genome. All primers used are listed in Table 1. HEV-conserved primers targeting the 5’ NTR [E8 (Kim et al., 2005) with E1 (Chapman et al., 1990) and E2 (Chapman et al., 1990) with DRevEntB (reverse complement of nt 745−766, Table 1)] were used to amplify and sequence 662 nt of the 5’ NTR. Heart virus-specific sense primers (Mito5NTR, MitoVP4, MitoVP2, Table 1) were designed using this 5’ NTR sequence as well as subsequently amplified cDNA sequences. cDNA was amplified using sense and antisense primers (VP4REV, VP2REVC, VP2REVA, CVB2VP1REV; Table 1) based on an human enterovirus B (HEV-B) consensus sequence from alignment of 90 complete HEV-B genomes in GenBank (November, 2005). We used an HEV-B consensus sequence because of the established association of the CVB (classified as HEV-B) with myocarditis (Kim et al., 2001).
cDNA was transcribed in 20 μL reactions containing 100−200 ng of heart RNA, 10 pmol of primer E1 (Chapman et al., 1990), DREVENTB, VP4Rev, VP2REVC, VP2REVA, or CVB2VP1REV (Table 1) and 1 unit Improm-II reverse transcriptase (Promega; Madison WI), 3mM MgCl2 and 0.5 mM each dNTP in Improm-II buffer. The cDNA was either diluted 10 fold with water prior to PCR or concentrated by ethanol precipitation. cDNA was amplified using Taq polymerase (Promega) (Kim et al., 2005). For the detection of the viral genome, amplification was carried out using primers E1 and E2 (Chapman et al., 1990). For characterization of 5' terminal sequences, the primer E3Sub (Table 1) was paired with S (Kim et al., 2005) or any of the SENTB primers (Table 1). For cloning of the viral 5' NTR, E1-primed cDNA was tailed with dG10 residues using terminal deoxynucleotidyl transferase (Promega), amplified with a primer annealing to the 3’ G10, DC-Tail (Kim et al., 2005) and E5 (Table 1), then ligated into pPCR-Script-Amp (Strataclone PCR; Stratagene, La Jolla, CA) as described previously (Kim et al., 2005). Colonies with insert-containing plasmids were identified by PCR using primers that flank the polylinker (M13forward and reverse; Stratagene). Plasmid DNA was purified and both strands sequenced using the same primers.
Generation of a chimeric infectious CVB3 cDNA with the Mito 5’ NTR
Overlap extension (Horton et al., 1989) was used to generate a complete 5’ NTR from smaller fragments. The cDNA of the 22 nucleotide deletion in the CVB2Mito genome was amplified with DC-Tail (Kim et al., 2005) and E5. A clone containing the E8-E1 fragment of the viral sequence (nt 65−644) was used as a template to amplify with primers E8 and E1. These amplimers were purified from an agarose gel, mixed in equimolar ratios in water, denatured at 94°C for 1 minute, cooled to room temperature, and amplified by PCR with terminal primers DC-Tail and E1. Similarly, cDNA amplified from cloned cDNA containing nt 644−1046 with Mito5NTR and VP4REV was denatured with the product of the first overlap PCR, cooled to room temperature, and amplified with DREV and 23RIBOZ1 (Table 1). Subsequent amplification with DREV and 23RIBOZ2 (Table 1) followed by amplification with DREV and TD23RIBOZPCRT7 (Table 1) was used to generate a 5’ NTR with an added NotI site, T7 RNA polymerase promoter, a ribozyme which cleaves RNA transcripts at nt 23(Herold and Andino, 2000) and a SacI site at nt 751. Using the NotI and SacI sites, the deleted 5’ NTR was substituted for the CVB3/28 5’ NTR in a subclone containing nt 1−2011 of cDNA of CVB3/28 to generate pBST7rTD235NTR. This recombinant Mito/CVB3 sequence was subsequently ligated into the pCVB3−28 plasmid which has a full length infectious cDNA (Tracy et al., 2002) using the NotI site and an XhoI site (at nt 2011) to generate pCMVT7r5NTRMitoTD23.
To generate a chimeric CVB3/28 genome with the CVB2/Mito 5' NTR and a restored 5’ terminal sequence (nt 1−22), cDNA was amplified from pBST7rTD235NTR with successive PCRs using DREV and ENTBCONSENSUS (Table 1), then with DREV and 28RIBOZ1 (Kim et al., 2005), with DREV and 28RIBOZ2 (Kim et al., 2005) and finally with DREV and 28RIBOZPCRT7 (Table 1) to generate a NotI site, a ribozyme and the consensus 5’ terminal 22 nucleotides generated by alignment of 90 5’ terminal containing enterovirus B sequences from GenBank. This cDNA was cloned into the pCMVT7r28 infectious cDNA as described above to generate pCMVT7r5NTRMIitoCVB3.
Generation of progeny virus from infectious cDNA clones
RNA polymerase T7 was used to transcribe RNA from pCMVT7r5NTRMitoCVB3, pCMVT7r5NTRMitoTD23, and pCMVT7r28 (Ribomax; Promega). HeLa cells (1×106 per well) growing as a monolayer in 6 well plates, were transfected with 10 μg T7 transcript RNA of pCMVT7r28 or pCMVT7r5NTRMitoCVB3 (Transmessenger; Qiagen, Valencia CA), then incubated at 37°C for 72 hours or until 90% cytopathic effect was observed. Plates were harvested by freezing and thawing three times, after which the lysate was centrifugally cleared of cellular debris. Virus stocks (CVB3/5NTRMito, CVB3/28) were prepared on HeLa cells and titered as described previously (Kim et al., 2005). Virus was generated from pCMVT7r5NTRMitoTD23 (termed CVB3/5NTRMitoTD23) by transfection of HeLa and concentration as described previously (Kim et al., 2005).
Determination of positive to negative strand viral RNA ratios
To determine positive to negative polarity ratios of viral RNA, RNA was purified from virus-infected cells (ZR Viral RNA Kit, Zymo Research, Promega). T7 (positive strand) and SP6 (negative strand) transcripts from a subclone of CVB3/28 (pBSCVB3NXh containing nt 1−2011 of the CVB3 genome) were prepared (Ribomax) for positive and negative strand controls. RNA was purified by annealing a biotinylated strand-specific primer (E3 for annealing to positive strand and E3REV for negative strand) to RNA and purified using streptavidin-labeled magnetic beads (Dynabeads M-280 streptavidin; Dynal Biotech, New York NY) as described previously (Kim et al., 2005). cDNA was reversed transcribed from purified positive strand using primer E1 and from negative strand with primer E2 (Chapman et al., 1990). For quantitative PCR, ten percent of cDNA was used with E1 and E2 at 0.125 OD260/ml in DyNAmo SYBR Green qPCR reaction mix (Finnzyme; Finland). Ten-fold dilutions of 2.16 × 1011 copies through 2.16 × 103 copies of T7 or SP6 transcripts of pBSCVB3NXh were used for the generation of a standard curve. Cycling times were 1 cycle at 95°C for 15 s, 45 cycles at 95°C for 20 s, 55°C for 20 s and 72°C for 20 s and finishing with a final extension at 72°C for 10 min. qPCRs were carried out using an Opticon 2 DNA Engine (MJ Research, Waltham, MA). The copy number of viral RNA samples was determined based on a standard curve of C(t) level versus copy number of the T7 (positive strand) or SP6 (negative strand) RNA polymerase transcripts.
Single-step growth curves
Single step growth curves were performed as previously described (Chapman et al., 2000b) with CVB3/5NTRMito, cardiovirulent CVB3/28 (Tracy et al., 2002) and nonvirulent CVB3/GA (Lee et al., 2005) using a multiplicity of infection (MOI) of 25 TCID50 per cell on HeLa and MHC monolayer cultures. Time points were taken by freezing cultures at −70°C, after which infectious titers were assayed on HeLa cell monolayers.
Inoculation of mice
Five 3−4 week old A/J male mice (Jackson Laboratory; Bar Harbor, ME) were inoculated intraperitoneally with 1×105 TCID50 units of CVB3/28 or CVB3/5NTRMito, or with 0.1 ml concentrated CVB3/5NTRMitoTD23 [the concentration of positive strand RNA by quantitative PCR was equivalent to 104 TCID50 (Kim et al., 2005)] . At day 14 post-inoculation, the pancreas and half of each heart from each mouse were formalin-fixed, embedded, sectioned and stained with hematoxylin and eosin for pathology. HeLa cell cultures (1×105 cells in 24 well plates) were inoculated with the homogenate made from the other heart half and incubated for 72 hours (Kim et al., 2005). Total RNA was then prepared from these cell cultures and viral cDNA amplified with E1 and E2 primers as described above.
Acknowledgements
Support for this work was provided in part by grants from the American Heart Association (N.M.C.), the Juvenile Diabetes Research Foundation International and the American Diabetes Association (S.T), and the USPHS and the Multiple Sclerosis Society (K.M.D.). We also thank F. Schiff of the ERACE Foundation, the Stein family, M. Guthrie, and E. Barnett for their generosity in supporting this work in memory of loved ones.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Bibliography
- Agol VI, Drozdov SG, Ivannikova TA, Kolesnikova MS, Korolev MB, Tolskaya EA. Restricted growth of attenuated poliovirus strains in cultured cells of a human neuroblastoma. J Virol. 1989;63(9):4034–8. doi: 10.1128/jvi.63.9.4034-4038.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andino R, Rieckhof GE, Baltimore D. A functional ribonucleoprotein complex forms around the 5' end of poliovirus RNA. Cell. 1990;63(2):369–80. doi: 10.1016/0092-8674(90)90170-j. [DOI] [PubMed] [Google Scholar]
- Andreoletti L, Bourlet T, Moukassa D, Rey L, Hot D, Li Y, Lambert V, Gosselin B, Mosnier JF, Stankowiak C, Wattre P. Enteroviruses can persist with or without active viral replication in cardiac tissue of patients with end-stage ischemic or dilated cardiomyopathy. J Infect Dis. 2000;182(4):1222–7. doi: 10.1086/315818. [DOI] [PubMed] [Google Scholar]
- Archard LC, Khan MA, Soteriou BA, Zhang H, Why HJ, Robinson NM, Richardson PJ. Characterization of Coxsackie B virus RNA in myocardium from patients with dilated cardiomyopathy by nucleotide sequencing of reverse transcription-nested polymerase chain reaction products. Hum Pathol. 1998;29(6):578–84. doi: 10.1016/s0046-8177(98)80006-3. [DOI] [PubMed] [Google Scholar]
- Badorff C, Lee GH, Lamphear BJ, Martone ME, Campbell KP, Rhoads RE, Knowlton KU. Enteroviral protease 2A cleaves dystrophin: evidence of cytoskeletal disruption in an acquired cardiomyopathy. Nat Med. 1999;5(3):320–6. doi: 10.1038/6543. [DOI] [PubMed] [Google Scholar]
- Bailey JM, Tapprich WE. Structure of the 5' nontranslated region of the coxsackievirus b3 genome: Chemical modification and comparative sequence analysis. J Virol. 2007;81(2):650–68. doi: 10.1128/JVI.01327-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barton DJ, O'Donnell BJ, Flanegan JB. 5' cloverleaf in poliovirus RNA is a cis-acting replication element required for negative-strand synthesis. Embo J. 2001;20(6):1439–48. doi: 10.1093/emboj/20.6.1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belov GA, Ehrenfeld E. Involvement of cellular membrane traffic proteins in poliovirus replication. Cell Cycle. 2007;6(1):36–8. doi: 10.4161/cc.6.1.3683. [DOI] [PubMed] [Google Scholar]
- Bowles NE, Rose ML, Taylor P, Banner NR, Morgan-Capner P, Cunningham L, Archard LC, Yacoub MH. End-stage dilated cardiomyopathy. Persistence of enterovirus RNA in myocardium at cardiac transplantation and lack of immune response. Circulation. 1989;80(5):1128–36. doi: 10.1161/01.cir.80.5.1128. [DOI] [PubMed] [Google Scholar]
- Brown DM, Cornell CT, Tran GP, Nguyen JH, Semler BL. An authentic 3' noncoding region is necessary for efficient poliovirus replication. J Virol. 2005;79(18):11962–73. doi: 10.1128/JVI.79.18.11962-11973.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown DM, Kauder SE, Cornell CT, Jang GM, Racaniello VR, Semler BL. Cell-dependent role for the poliovirus 3' noncoding region in positive-strand RNA synthesis. J Virol. 2004;78(3):1344–51. doi: 10.1128/JVI.78.3.1344-1351.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman N, Ragland A, Leser JS, Hofling K, Willian S, Semler B, Tracy S. A group B coxsackievirus/poliovirus 5' non-translated region chimera can act as an attenuated vaccine strain in mice. J Virol. 2000a;74:4047–4056. doi: 10.1128/jvi.74.9.4047-4056.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman NM, Kim K-S. Persistent coxsackievirus infection: enterovirus persistence in chronic myocarditis and dilated cardiomyopathy. Curr Top Microbiol Immunol. 2008;323 doi: 10.1007/978-3-540-75546-3_13. [DOI] [PubMed] [Google Scholar]
- Chapman NM, Ragland A, Leser JS, Hofling K, Willian S, Semler BL, Tracy S. A group B coxsackievirus/poliovirus 5' nontranslated region chimera can act as an attenuated vaccine strain in mice. J Virol. 2000b;74(9):4047–56. doi: 10.1128/jvi.74.9.4047-4056.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman NM, Tracy S, Gauntt CJ, Fortmueller U. Molecular detection and identification of enteroviruses using enzymatic amplification and nucleic acid hybridization. J Clin Microbiol. 1990;28(5):843–50. doi: 10.1128/jcm.28.5.843-850.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Ambrosio A, Patti G, Manzoli A, Sinagra G, Di Lenarda A, Silvestri F, Di Sciascio G. The fate of acute myocarditis between spontaneous improvement and evolution to dilated cardiomyopathy: a review. Heart. 2001;85(5):499–504. doi: 10.1136/heart.85.5.499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domingo E, Martin V, Perales C, Grande-Perez A, Garcia-Arriaza J, Arias A. Viruses as quasispecies: biological implications. Curr Topics Microbiol Immunol. 2006;299:51–82. doi: 10.1007/3-540-26397-7_3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dotta F, Censini S, van Halteren AG, Marselli L, Masini M, Dionisi S, Mosca F, Boggi U, Muda AO, Prato SD, Elliott JF, Covacci A, Rappuoli R, Roep BO, Marchetti P. Coxsackie B4 virus infection of beta cells and natural killer cell insulitis in recent-onset type 1 diabetic patients. Proc Natl Acad Sci U S A. 2007;104(12):5115–20. doi: 10.1073/pnas.0700442104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drescher KM, Kono K, Bopegamage S, Carson SD, Tracy S. Coxsackievirus B3 infection and type 1 diabetes development in NOD mice: insulitis determines susceptibility of pancreatic islets to virus infection. Virology. 2004;329(2):381–94. doi: 10.1016/j.virol.2004.06.049. [DOI] [PubMed] [Google Scholar]
- Fairweather D, Frisancho-Kiss S, Rose NR. Viruses as adjuvants for autoimmunity: evidence from Coxsackievirus-induced myocarditis. Rev Med Virol. 2005;15(1):17–27. doi: 10.1002/rmv.445. [DOI] [PubMed] [Google Scholar]
- Fechner RE, Smith MG, Middlekamp JN. Coxsackie B virus infection of the newborn. Am J Pathol. 1963;42:493–505. [PMC free article] [PubMed] [Google Scholar]
- Figulla HR. Transformation of myocarditis and inflammatory cardiomyopathy to idiopathic dilated cardiomyopathy: facts and fiction. Med Microbiol Immunol. 2004;193(2−3):61–4. doi: 10.1007/s00430-003-0205-y. [DOI] [PubMed] [Google Scholar]
- Gauntt C, Huber S. Coxsackievirus experimental heart diseases. Front Biosci. 2003;8:e23–35. doi: 10.2741/928. [DOI] [PubMed] [Google Scholar]
- Gear JH, Measroch V. Coxsackievirus infections of the newborn. Prog Med Virol. 1973;15:42–62. [PubMed] [Google Scholar]
- Giacca M, Severini GM, Mestroni L, Salvi A, Lardieri G, Falaschi A, Camerini F. Low frequency of detection by nested polymerase chain reaction of enterovirus ribonucleic acid in endomyocardial tissue of patients with idiopathic dilated cardiomyopathy. J Am Coll Cardiol. 1994;24(4):1033–40. doi: 10.1016/0735-1097(94)90866-4. [DOI] [PubMed] [Google Scholar]
- He Y, Chipman P, Howitt J, Bator C, Whitt M, Baker T, Kuhn RJ, Anderson C, Freimuth P, Rossmann MG. Interaction of coxsackievirus B3 with the full length coxsackievirus-adenovirus receptor. Nat Struct Biol. 2001;8:874–878. doi: 10.1038/nsb1001-874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herold J, Andino R. Poliovirus requires a precise 5' end for efficient positive-strand RNA synthesis. J Virol. 2000;74(14):6394–400. doi: 10.1128/jvi.74.14.6394-6400.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horton RM, Hunt HD, Ho SN, Pullen JK, Pease LR. Engineering hybrid genes without the use of restriction enzymes: gene splicing by overlap extension. Gene. 1989;77(1):61–8. doi: 10.1016/0378-1119(89)90359-4. [DOI] [PubMed] [Google Scholar]
- Huber SA. Autoimmunity in coxsackievirus B3 induced myocarditis. Autoimmunity. 2006;39(1):55–61. doi: 10.1080/08916930500484906. [DOI] [PubMed] [Google Scholar]
- Hunziker IP, Cornell CT, Whitton JL. Deletions within the 5'UTR of coxsackievirus B3: consequences for virus translation and replication. Virology. 2007;360(1):120–8. doi: 10.1016/j.virol.2006.09.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin O, Sole MJ, Butany JW, Chia WK, McLaughlin PR, Liu P, Liew CC. Detection of enterovirus RNA in myocardial biopsies from patients with myocarditis and cardiomyopathy using gene amplification by polymerase chain reaction. Circulation. 1990;82(1):8–16. doi: 10.1161/01.cir.82.1.8. [DOI] [PubMed] [Google Scholar]
- Joachims M, Van Breugel PC, Lloyd RE. Cleavage of poly(A)-binding protein by enterovirus proteases concurrent with inhibition of translation in vitro. J Virol. 1999;73(1):718–27. doi: 10.1128/jvi.73.1.718-727.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kammerer U, Kunkel B, Korn K. Nested PCR for specific detection and rapid identification of human picornaviruses. J Clin Microbiol. 1994;32(2):285–91. doi: 10.1128/jcm.32.2.285-291.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawai C. From myocarditis to cardiomyopathy: mechanisms of inflammation and cell death: learning from the past for the future. Circulation. 1999;99(8):1091–100. doi: 10.1161/01.cir.99.8.1091. [DOI] [PubMed] [Google Scholar]
- Kerekatte V, Keiper BD, Badorff C, Cai A, Knowlton KU, Rhoads RE. Cleavage of Poly(A)-binding protein by coxsackievirus 2A protease in vitro and in vivo: another mechanism for host protein synthesis shutoff? J Virol. 1999;73(1):709–17. doi: 10.1128/jvi.73.1.709-717.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim K-S, Chapman NM, Tracy S. Replication of coxsackievirus B3 in primary cell lines generates novel viral genomic deletions. J Virol. 2008 doi: 10.1128/JVI.01774-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim KS, Chapman NM, Tracy S. Replication of coxsackievirus B3 in primary cell cultures generates novel viral genome deletions. J Virol. 2008;82(4):2033–7. doi: 10.1128/JVI.01774-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim KS, Hofling K, Carson SD, Chapman NM, Tracy S. The primary viruses of myocarditis. In: Cooper L, Knowlton K, editors. Myocarditis: From Bench to Bedside. Humana Press; Totowa, NJ: 2002. pp. 23–54. [Google Scholar]
- Kim KS, Hufnagel G, Chapman NM, Tracy S. The group B coxsackieviruses and myocarditis. Rev Med Virol. 2001;11(6):355–68. doi: 10.1002/rmv.326. [DOI] [PubMed] [Google Scholar]
- Kim KS, Tracy S, Tapprich W, Bailey J, Lee CK, Kim K, Barry WH, Chapman NM. 5'-Terminal deletions occur in coxsackievirus B3 during replication in murine hearts and cardiac myocyte cultures and correlate with encapsidation of negative-strand viral RNA. J Virol. 2005;79(11):7024–41. doi: 10.1128/JVI.79.11.7024-7041.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klingel K, Hohenadl C, Canu A, Albrecht M, Seemann M, Mall G, Kandolf R. Ongoing enterovirus-induced myocarditis is associated with persistent heart muscle infection: quantitative analysis of virus replication, tissue damage, and inflammation. Proc Natl Acad Sci U S A. 1992;89(1):314–8. doi: 10.1073/pnas.89.1.314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhl U, Pauschinger M, Seeberg B, Lassner D, Noutsias M, Poller W, Schultheiss HP. Viral persistence in the myocardium is associated with progressive cardiac dysfunction. Circulation. 2005;112(13):1965–70. doi: 10.1161/CIRCULATIONAHA.105.548156. [DOI] [PubMed] [Google Scholar]
- Kundu P, Raychaudhuri S, Tsai W, Dasgupta A. Shutoff of RNA polymerase II transcription by poliovirus involves 3C protease-mediated cleavage of the TATA-binding protein at an alternative site: incomplete shutoff of transcription interferes with efficient viral replication. J Virol. 2005;79(15):9702–13. doi: 10.1128/JVI.79.15.9702-9713.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuyumcu-Martinez NM, Van Eden ME, Younan P, Lloyd RE. Cleavage of poly(A)-binding protein by poliovirus 3C protease inhibits host cell translation: a novel mechanism for host translation shutoff. Mol Cell Biol. 2004;24(4):1779–90. doi: 10.1128/MCB.24.4.1779-1790.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee CK, Kono K, Haas E, Kim KS, Drescher KM, Chapman NM, Tracy S. Characterization of an infectious cDNA copy of the genome of a naturally occurring, avirulent coxsackievirus B3 clinical isolate. J Gen Virol. 2005;86(Pt 1):197–210. doi: 10.1099/vir.0.80424-0. [DOI] [PubMed] [Google Scholar]
- Lloyd RE, Jense HG, Ehrenfeld E. Restriction of translation of capped mRNA in vitro as a model for poliovirus-induced inhibition of host cell protein synthesis: relationship to p220 cleavage. J Virol. 1987;61(8):2480–8. doi: 10.1128/jvi.61.8.2480-2488.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longson M, Cole FM, Davies D. Isolation of a Coxsackie virus group B, type 5, from the heart of a fatal case of myocarditis in an adult. J Clin Pathol. 1969;22(6):654–8. doi: 10.1136/jcp.22.6.654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukashev AN, Lashkevich VA, Ivanova OE, Koroleva GA, Hinkkanen AE, Ilonen J. Recombination in circulating enteroviruses. J Virol. 2003;77(19):10423–31. doi: 10.1128/JVI.77.19.10423-10431.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyons T, Murray KE, Roberts AW, Barton DJ. Poliovirus 5'-terminal cloverleaf RNA is required in cis for VPg uridylylation and the initiation of negative-strand RNA synthesis. J Virol. 2001;75(22):10696–708. doi: 10.1128/JVI.75.22.10696-10708.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mason JW. Myocarditis and dilated cardiomyopathy: an inflammatory link. Cardiovasc Res. 2003;60(1):5–10. doi: 10.1016/s0008-6363(03)00437-1. [DOI] [PubMed] [Google Scholar]
- Melnick JL, Ledinko N, Kaplan AS, Kraft LM. Ohio strains of a virus pathogenic for infant mice, Coxsackie group; simultaneous occurrence with poliomyelitis virus in patients with summer grippe. J Exp Med. 1950;91(2):185–95. doi: 10.1084/jem.91.2.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monaldi T, Benedetto A, Tentorimontalto T. Pericarditis Infection Due to Coxsackie Virus Group B, Type 2. Br Med J. 1963;2(5370):1451–2. doi: 10.1136/bmj.2.5370.1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muckelbauer JK, Kremer M, Minor I, Diana G, Dutko FJ, Groarke J, Pevear DC, Rossmann MG. The structure of coxsackievirus B3 at 3.5 A resolution. Structure. 1995;3(7):653–67. doi: 10.1016/s0969-2126(01)00201-5. [DOI] [PubMed] [Google Scholar]
- Murray KE, Steil BP, Roberts AW, Barton DJ. Replication of poliovirus RNA with complete internal ribosome entry site deletions. J Virol. 2004;78(3):1393–402. doi: 10.1128/JVI.78.3.1393-1402.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nasri D, Bouslama L, Omar S, Saoudin H, Bourlet T, Aouni M, Pozzetto B, Pillet S. Typing of Human Enterovirus by Partial Sequencing of VP2. J Clin Microbiol. 2007;45(8):2370–9. doi: 10.1128/JCM.00093-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nix WA, Oberste MS, Pallansch MA. Sensitive, seminested PCR amplification of VP1 sequences for direct identification of all enterovirus serotypes from original clinical specimens. J Clin Microbiol. 2006;44(8):2698–704. doi: 10.1128/JCM.00542-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novak JE, Kirkegaard K. Improved method for detecting poliovirus negative strands used to demonstrate specificity of positive-strand encapsidation and the ratio of positive to negative strands in infected cells. J Virol. 1991;65(6):3384–7. doi: 10.1128/jvi.65.6.3384-3387.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oberste MS, Maher K, Kilpatrick DR, Flemister MR, Brown BA, Pallansch MA. Typing of human enteroviruses by partial sequencing of VP1. J Clin Microbiol. 1999a;37(5):1288–93. doi: 10.1128/jcm.37.5.1288-1293.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oberste MS, Maher K, Kilpatrick DR, Pallansch MA. Molecular evolution of the human enteroviruses: correlation of serotype with VP1 sequence and application to picornavirus classification. J Virol. 1999b;73(3):1941–8. doi: 10.1128/jvi.73.3.1941-1948.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oka K, Oohira K, Yatabe Y, Tanaka T, Kurano K, Kosugi R, Murata M, Hakozaki H, Nishikawa T, Tsutsumi Y. Fulminant myocarditis demonstrating uncommon morphology--a report of two autopsy cases. Virchows Arch. 2005;446(3):259–64. doi: 10.1007/s00428-004-1173-3. [DOI] [PubMed] [Google Scholar]
- Pallansch MA, Roos RP. Enteroviruses: polioviruses, coxsackieviruses, echoviruses and newer enteroviruses. In: Knipe PMHDM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE, editors. Fields virology. Vol. 1. Lippincott Williams & Wilkins; Philadelphia, PA: 2001. pp. 723–775. 2. [Google Scholar]
- Piraino FF, Sedmak G, Raab K. Echovirus 11 infections of newborns with mortality during the 1979 enterovirus season in Milwaukee, Wis. Public Health Rep. 1982;97(4):346–53. [PMC free article] [PubMed] [Google Scholar]
- Polacek C, Lundgren A, Andersson A, Lindberg AM. Genomic and phylogenetic characterization of coxsackievirus B2 prototype strain Ohio-1. Virus Res. 1999;59(2):229–38. doi: 10.1016/s0168-1702(98)00140-3. [DOI] [PubMed] [Google Scholar]
- Porres ER, Werthammer J, Moss N, Bernstein JM, Belshe RB. Fatal coxsackievirus B4 infection in a neonate. South Med J. 1985;78(10):1254–6. doi: 10.1097/00007611-198510000-00028. [DOI] [PubMed] [Google Scholar]
- Racaniello VR. Picornaviridae: the viruses and their replication. In: Knipe HPM, D.M., editors. Fields virology. 4th ed. Vol. 1. Lippincott/The Williams & Wilkins Co.; Philadelphia, Pa: 2001. pp. 685–722. 2. [Google Scholar]
- Racaniello VR. Picornaviridae: the viruses and their replication. In: Howley P, Knipe D, editors. Fields virology. 1 Wolters Kluwer Lippincott/The Williams & Wilkins Co.; Philadelphia, PA: 2007. [Google Scholar]
- Reetoo KN, Osman SA, Illavia SJ, Cameron-Wilson CL, Banatvala JE, Muir P. Quantitative analysis of viral RNA kinetics in coxsackievirus B3-induced murine myocarditis: biphasic pattern of clearance following acute infection, with persistence of residual viral RNA throughout and beyond the inflammatory phase of disease. J Gen Virol. 2000;81(Pt 11):2755–62. doi: 10.1099/0022-1317-81-11-2755. [DOI] [PubMed] [Google Scholar]
- Rey L, Lambert V, Wattre P, Andreoletti L. Detection of enteroviruses ribonucleic acid sequences in endomyocardial tissue from adult patients with chronic dilated cardiomyopathy by a rapid RT-PCR and hybridization assay. J Med Virol. 2001;64(2):133–40. doi: 10.1002/jmv.1028. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Wells V, Plotch SJ, DeStefano JJ. Primer-dependent synthesis by poliovirus RNA-dependent RNA polymerase (3D(pol)). Nucleic Acids Res. 2001;29(13):2715–24. doi: 10.1093/nar/29.13.2715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satoh M, Tamura G, Segawa I, Hiramori K, Satodate R. Enteroviral RNA in dilated cardiomyopathy. Eur Heart J. 1994;15(7):934–9. doi: 10.1093/oxfordjournals.eurheartj.a060613. [DOI] [PubMed] [Google Scholar]
- Semler B, Johnson V, Tracy S. A molecular recombinant plasmid from cDNA clones of poliovirus and coxsackievirus produces an infectious virus that is temperature sensitive. Proc Natl Acad Sci USA. 1986;83:1777–1781. doi: 10.1073/pnas.83.6.1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma N, O'Donnell BJ, Flanegan JB. 3'-Terminal sequence in poliovirus negative-strand templates is the primary cis-acting element required for VPgpUpU-primed positive-strand initiation. J Virol. 2005;79(6):3565–77. doi: 10.1128/JVI.79.6.3565-3577.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sickles GM, Feorino P, Plager H. Isolation and type determination of Coxsackie virus, group B, in tissue culture. Proc Soc Exp Biol Med. 1955;88(1):22–4. doi: 10.3181/00379727-88-21481. [DOI] [PubMed] [Google Scholar]
- Soutar CA. Unusual case of viral pericarditis. Lancet. 1971;1(7697):498. doi: 10.1016/s0140-6736(71)91114-7. [DOI] [PubMed] [Google Scholar]
- Spotnitz MD, Lesch M. Idiopathic dilated cardiomyopathy as a late complication of healed viral (Coxsackie B virus) myocarditis: historical analysis, review of the literature, and a postulated unifying hypothesis. Prog Cardiovasc Dis. 2006;49(1):42–57. doi: 10.1016/j.pcad.2006.06.001. [DOI] [PubMed] [Google Scholar]
- Stanway G, B. F., Christian P, Hovi T, Hyypia T, King AMQ, Knowles NJ, Lemon SM, Minor PD, Pallansch MA, Palmenberg AC, Skern T. Picornaviridae. In: F. C., Mayo MA, Maniloff J, Desselberger U, Ball LA, editors. Virus taxonomy. Eighth report of the International Committee on Taxonomy of Viruses. Elsevier/Academic Press; London, UK: 2005. [Google Scholar]
- Stille-Siegener M, H. A., Klingel K, Kandolf R, Mall G, Kreuzer H, Figulla HR. Interferon therapy in enterovirus-associated idiopathic dilated cardiomyopathy. In: K. R., Figulla HR, McManus B, editors. Idiopathic dilated cardiomyopathy. Springer-Verlag; Berlin Heidelberg: 1993. [Google Scholar]
- Tam PE, Messner RP. Molecular mechanisms of coxsackievirus persistence in chronic inflammatory myopathy: viral RNA persists through formation of a double-stranded complex without associated genomic mutations or evolution. J Virol. 1999;73(12):10113–21. doi: 10.1128/jvi.73.12.10113-10121.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tam PE, Schmidt AM, Ytterberg SR, Messner RP. Viral persistence during the developmental phase of Coxsackievirus B1-induced murine polymyositis. J Virol. 1991;65(12):6654–60. doi: 10.1128/jvi.65.12.6654-6660.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toraason M, Luken ME, Breitenstein M, Krueger JA, Biagini RE. Comparative toxicity of allylamine and acrolein in cultured myocytes and fibroblasts from neonatal rat heart. Toxicology. 1989;56(1):107–17. doi: 10.1016/0300-483x(89)90216-3. [DOI] [PubMed] [Google Scholar]
- Towbin JA, Hejtmancik JF, Brink P, Gelb B, Zhu XM, Chamberlain JS, McCabe ER, Swift M. X-linked dilated cardiomyopathy. Molecular genetic evidence of linkage to the Duchenne muscular dystrophy (dystrophin) gene at the Xp21 locus. Circulation. 1993;87(6):1854–65. doi: 10.1161/01.cir.87.6.1854. [DOI] [PubMed] [Google Scholar]
- Tracy S, Chapman NM, McManus BM, Pallansch MA, Beck MA, Carstens J. A molecular and serologic evaluation of enteroviral involvement in human myocarditis. J Mol Cell Cardiol. 1990;22(4):403–14. doi: 10.1016/0022-2828(90)91476-n. [DOI] [PubMed] [Google Scholar]
- Tracy S, Drescher KM, Chapman NM, Kim KS, Carson SD, Pirruccello S, Lane PH, Romero JR, Leser JS. Toward testing the hypothesis that group B coxsackieviruses (CVB) trigger insulin-dependent diabetes: inoculating nonobese diabetic mice with CVB markedly lowers diabetes incidence. J Virol. 2002;76(23):12097–111. doi: 10.1128/JVI.76.23.12097-12111.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trono D, Andino R, Baltimore D. An RNA sequence of hundreds of nucleotides at the 5' end of poliovirus RNA is involved in allowing viral protein synthesis. J Virol. 1988;62(7):2291–9. doi: 10.1128/jvi.62.7.2291-2299.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wessely R, Henke A, Zell R, Kandolf R, Knowlton KU. Low-level expression of a mutant coxsackieviral cDNA induces a myocytopathic effect in culture: an approach to the study of enteroviral persistence in cardiac myocytes. Circulation. 1998a;98(5):450–7. doi: 10.1161/01.cir.98.5.450. [DOI] [PubMed] [Google Scholar]
- Wessely R, Klingel K, Santana LF, Dalton N, Hongo M, Jonathan Lederer W, Kandolf R, Knowlton KU. Transgenic expression of replication-restricted enteroviral genomes in heart muscle induces defective excitation-contraction coupling and dilated cardiomyopathy. J Clin Invest. 1998b;102(7):1444–53. doi: 10.1172/JCI1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodruff JF. Viral myocarditis. A review. Am J Pathol. 1980;101(2):425–84. [PMC free article] [PubMed] [Google Scholar]
- Xiong D, Yajima T, Lim BK, Stenbit A, Dublin A, Dalton ND, Summers-Torres D, Molkentin JD, Duplain H, Wessely R, Chen J, Knowlton KU. Inducible cardiac-restricted expression of enteroviral protease 2A is sufficient to induce dilated cardiomyopathy. Circulation. 2007;115(1):94–102. doi: 10.1161/CIRCULATIONAHA.106.631093. [DOI] [PubMed] [Google Scholar]
- Zhang H, Li Y, Peng T, Aasa M, Zhang L, Yang Y, Archard LC. Localization of enteroviral antigen in myocardium and other tissues from patients with heart muscle disease by an improved immunohistochemical technique. J Histochem Cytochem. 2000;48(5):579–84. doi: 10.1177/002215540004800501. [DOI] [PubMed] [Google Scholar]