

Abstract
Polymicrobial sepsis is characterized by an early, hyperdynamic phase followed by a late hypodynamic phase. Adrenomedullin (AM), a vasodilatory peptide, inhibits this transition from the early phase to the late phase. Adrenomedullin binding protein-1 (AMBP-1) enhances AM-mediated activities. The decrease of AMBP-1 levels in late sepsis reduces the vascular response to AM and produces the hypodynamic phase. Studies have indicated that the administration of LPS downregulates AMBP-1 production in the liver. Since hepatocytes are the primary source of AMBP-1 biosynthesis in the liver, we employed a co-culture strategy using hepatocyte and Kupffer cells to determine whether LPS directly or by increasing pro-inflammatory cytokines from Kupffer cells downregulates AMBP-1 production. Hepatocytes and Kupffer cells isolated from rats were co-cultured and treated with LPS for 24 hrs. LPS significantly attenuated AMBP-1 protein expression in a dose dependent manner. Since AMBP-1 is basically a secretory protein, cell supernatants from co-culture cells treated with LPS were examined for AMBP-1 protein levels. LPS treatment caused a dose related decrease in AMBP-1 protein secretion. Similarly, LPS treatment produced a significant decrease in AMBP-1 protein expression in hepatocytes and Kupffer cells cultured using transwell inserts. LPS had no direct effect on AMBP-1 levels in cultured hepatocytes or Kupffer cells alone. To confirm that the observed effects in co-culture were due to the cytokines released from Kupffer cells, hepatocytes were treated with IL-1β or TNF-α for 24 hrs and AMBP-1 expression was examined. The results indicated that both cytokines significantly inhibited AMBP-1 protein levels. Thus, pro-inflammatory cytokines released from Kupffer cells are responsible for downregulation of AMBP-1.
Keywords: adrenomedullin binding protein-1, LPS, sepsis, TNF-α, IL-1β
1. Introduction
Sepsis, septic shock and the ensuing multiple organ failure continue to be the most common cause of mortality in surgical intensive care units [1–3]. Enormous efforts have been devoted to the understanding of sepsis and septic shock to develop new therapeutic agents which can either reduce or prevent this ever increasing problem in surgical critical care. Many investigators have directed their focus to animal models to understand the physiological consequences of sepsis and septic shock. Cecal ligation and puncture (CLP) in rodents has been developed as a suitable animal model for sepsis since it can produces pathophysiology (i.e. appendicitis and peritonitis) similar to that of patients undergoing sepsis [4]. The cardiovascular response to polymicrobial sepsis induced by CLP is characterized by an early, hyperdynamic phase followed by a late, hypodynamic phase [5]. In addition to the decrease in hemodynamic parameters, pro-inflammatory cytokines play a pertinent role in the initiation and progression of sepsis. Among those, TNF-α, IL-1β and IL-6 became important mediators during sepsis [6–8]. Recent studies have indicated that adrenomedullin (AM), a novel vasodilatory peptide, plays an important role in initiating the hyperdynamic response during the early phase of polymicrobial sepsis [9–11]. The reduced vascular responsiveness to AM appears to be responsible for the transition of hyperdynamic phase to the hypodynamic phase in late sepsis [12]. Adrenomedullin binding protein-1 (AMBP-1), a specific AM binding protein shown to be identical to complement factor H, augments AM mediated activities [13–15]. The administration of AM in combination with AMBP-1 (AM/AMBP-1) prevented the decrease in systemic and regional hemodynamic parameters and prevented the transition from a hyperdynamic phase to the hypodynamic phase [16].
The liver is a major source of AMBP-1 [17, 18] and our previous studies indicated that both gene and protein expression of AMBP-1 are markedly reduced in hepatic tissues during the late phase of sepsis and endotoxemia [19]. However, it is unclear whether LPS directly or by increasing pro-inflammatory cytokines downregulates AMBP-1 in sepsis. To answer this question, a co-culture strategy of hepatocytes and Kupffer cells were employed in the current study. Hepatocytes and Kupffer cells were isolated individually, co-cultured or cultured in transwell inserts and AMBP-1 protein expression was examined. The effect of pro-inflammatory cytokines, TNF-α and IL-1β, on AMBP-1 protein expression in hepatocytes was also analyzed.
2. Materials and methods
2.1. Cell culture
Hepatocytes and Kupffer cells were isolated separately from 6–8 week old normal Sprague-Dawley rats by collagenase perfusion of the liver followed by Percoll gradient centrifugation as described previously by us [20, 21]. Hepatocytes were plated at a density of 1 × 106 cells/well in 6 well culture plates with Williams medium E containing 10% heat inactivated fetal bovine serum (FBS), and incubated for 4 hrs at 37°C in 5% CO2. Kupffer cells were plated at a similar density in 6 well culture plates with DMEM containing 10% FBS and incubated overnight at 37°C. All media were supplemented with 10 mM HEPES, pH 7.4, 2 mM L-glutamine, 100 units/ml penicillin and 100 μg/ml streptomycin. For co-culture experiments, Kupffer cells were isolated, plated at 0.5 × 106 cells/well in 6 well culture plates and incubated overnight in DMEM and washed twice with phosphate buffered saline (PBS) and once with Williams medium E. Hepatocytes were plated at a similar density and plated in direct contact with the attached Kupffer cells in Williams medium E and incubated at 37°C. For experiments using transwell inserts, Kupffer cells were isolated, plated at 1 × 106 cells/well in 12 well culture plates and incubated overnight in DMEM and washed twice with PBS and once with Williams medium E and incubated at 37°C. Hepatocytes were plated at a similar density in transwell inserts containing 0.4 μm pore size membrane and incubated at 37°C.
2.2. Treatment with LPS or cytokines
After 4 hrs in co-culture or culture in transwell inserts LPS (1.0–100 ng/ml) were added and the cells were incubated at 37°C for 24 hrs. In experiments with hepatocytes alone, 4 h after plating, cells were treated with LPS, IL-1β or TNF-α (1.0, 10 ng/ml) and incubated for 24 hrs. In experiments with Kupffer cells alone, cells were plated overnight, washed and treated with LPS.
2.3. Western blotting of AMBP-1 and β-actin
Following treatment, cells were washed in PBS and lysed in 50 mM HEPES pH 7.5, 250 mM sucrose, 2 mM EDTA, 1.0 % Triton-X-100 and a mixture of protease inhibitors. Cell lysates were prepared by sonication and centrifugation at 5,000 g at 4°C for 5 min. The resulting cell lysates (25 μg) were electrophoresed on NuPAGE 3–8% Tris-Acetate gels under non-reducing conditions and transferred to 0.45 μM nitrocellulose membrane (In Vitrogen, Carlsbad, CA). The membranes were blocked in TBS-T (10 mM Tris-HCl, 150 mM NaCl, 0.1% Tween-20) containing 5% non-fat milk for 1 hr at room temperature. Blots were incubated overnight at 4°C with 1:5000 dilution of goat anti-human complement factor H serum (Quidel, San Diego, CA), reacted with 1:10,000 dilution of HRP labeled anti-goat IgG for 1hr at room temperature and detected with chemiluminescence (ECL; Amersham Biosciences, Inc.). The same blot was stripped and reprobed with anti-β-actin antibody. Since the Western blot on AMBP-1 was electrophoresed under non-reducing conditions, as reported by other authors [15] the antibody to β-actin did not recognize the protein on the same blot. Therefore, duplicate aliquots of cell lysates (25 μg) were fractionated on NuPAGE 4–12% Bis-Tris gels under reducing conditions and transferred to nitrocellulose membrane and Western blotted using anti-β-actin antibody (Sigma-Aldrich, St. Louis, MO). All membranes were exposed to X-ray film and the band densities were quantitated using the Bio-Rad image system (Hercules, CA). The data are represented as the ratio between AMBP-1 and actin protein expression.
The cell supernatants from control, 10 and 100 ng/ml LPS treated samples were concentrated ten fold using Centricon-50 (Millipore Corp, Bedford, MA) and an aliquot (2 μl) from each sample was run on SDS-PAGE under non-reducing conditions and Western blotted using anti-AMBP-1 antibody as described above.
2.4. Statistical analysis
All data are expressed as mean + SE and analyzed by one-way analysis of variance (ANOVA) and Student-Newman-Keuls method. The differences in values were considered significant if P <0.05.
3. Results
3.1. LPS treatment in co-culture of hepatocytes and Kupffer cells downregulated AMBP-1 protein expression
We have previously shown that the administration of LPS to normal Sprague-Dawley rats downregulates AMBP-1 production in the liver [19]. Since hepatocytes are the primary source of AMBP-1 biosynthesis in the liver, a co-culture strategy using hepatocyte and Kupffer cells were employed to determine whether LPS directly or by increasing pro-inflammatory cytokines from Kupffer cells downregulates AMBP-1 production. Hepatocytes and Kupffer cells were isolated from normal Sprague-Dawley rats and cells were co-cultured and treated with LPS for 24 hrs. Protein extracts were subjected to Western analysis using anti-AMBP-1 antibody and anti-β-actin antibody was used as loading control. Treatment with 1.0, 10 and 100 ng/ml LPS in a co-culture of hepatocytes and Kupffer cells produced 35%, 40% and 50% inhibition of AMBP-1 protein expression, respectively (Fig. 1). A representative Western blot is shown in Figure 1A. Please note, β-actin levels were relatively similar among different experimental groups. The calculated ratios between AMBP-1 and actin protein expression (AMBP-1/β-actin) from a number of experiments are shown in Figure 1B. Since AMBP-1 is basically a secretory protein, we collected culture supernatants from the co-culture experiments after LPS treatment and AMBP-1 protein secretions were measured. LPS treatment at both 10 and 100 ng/ml produced 27% and 46% decrease of AMBP-1 proteins compared to untreated controls, respectively. However, no statistical differences between control and treatment groups were found (Fig. 2). This suggests that the decrease in the protein levels in cell lysates of co-culture samples reflects a decrease in active production rather than an increase in secretion. To further demonstrate the cross-feeding between these two cell types, hepatocytes and Kupffer cells were plated in transwell chambers separated by a 0.4 μm membrane, treated with LPS and cell lysates were examined for AMBP-1 protein expression. The transwell chamber with the 0.4 μm membrane allows free exchange of molecules such as proteins and cytokines while preventing direct contact between the two cell populations. LPS treatment at both 10 and 100 ng/ml exhibited 29% and 38%, decrease in AMBP-1 protein expression compared to control samples, respectively (Fig. 3). These results are similar to what was observed in the co-culture experiments (Figure 1). To determine whether LPS directly inhibits AMBP-1, hepatocytes were treated with LPS for 24 hrs and AMBP-1 expression was examined. The results showed that LPS treatment had no significant effect in AMBP-1 levels in cultured hepatocytes alone (Fig. 4). To rule out the possibility that the LPS effects on AMBP-1 levels in co-culture were due to the downregulation of AMBP-1 levels in Kupffer cells, cultured Kupffer cells were treated with LPS for 24 hrs and AMBP-1 expression was examined. AMBP-1 levels were extremely low in Kupffer cell culture and LPS had no further effect over the control levels (Fig. 5). These data suggest that the downregulation of AMBP-1 in LPS treated co-culture could be due to the increase of pro-inflammatory cytokines from Kupffer cells.
Figure 1.
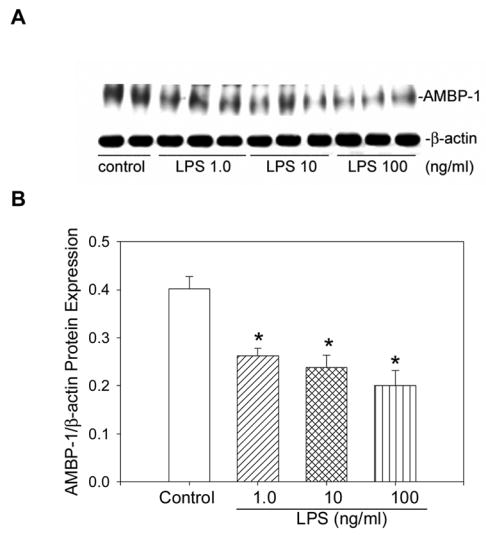
LPS treatment downregulates AMBP-1 in a co-culture of hepatocytes and Kupffer cells. A. Cells in co-culture for 4 hrs were treated with LPS (1.0, 10 and 100 ng/ml). 24 hr post treatment, cells were lysed in HEPES buffer containing protease inhibitors and protein levels were measured. Equal amounts of protein (25 μg) were electrophoresed on 3–8% NuPAGE Tris-Acetate gels under non-reducing conditions and Western blotted with anti-human complement factor H antibody (AMBP-1). Duplicate aliquots of protein (25 μg) were electrophoresed on 4–12% NuPAGE Bis-Tris gels under reducing conditions and Western blotted with anti-β-actin antibody (β-actin). B. The ratios between AMBP-1 and β-actin protein expressions (calculated as arbitrary densitometric values obtained from AMBP-1 protein expression divided by β-actin values in corresponding lanes) are shown. Data are presented as mean ± SE (n=6) and compared by one-way ANOVA and Student-Newman-Keuls method. *P<0.05 versus control.
Figure 2.
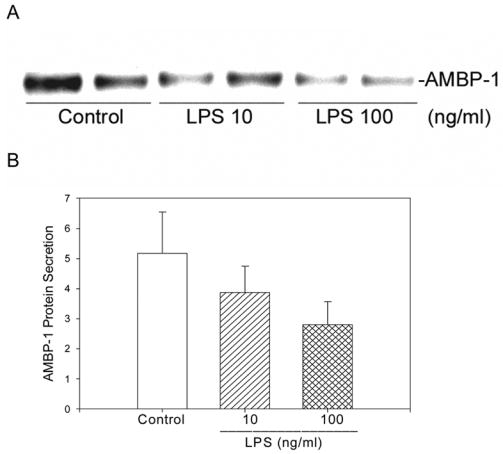
LPS treatment produced no increase in AMBP-1 secretion in a co-culture of hepatocytes and Kupffer cells. A. Cell supernatants from co-culture experiments treated with LPS were concentrated 10 fold with Centricon-50 and a 2 μl aliquot was analyzed by Western blot under non-reducing conditions using anti-AMBP-1 antibody. B. Arbitrary densitometric values obtained from the blot are shown. Data are presented as mean ± SE (n=3).
Figure 3.
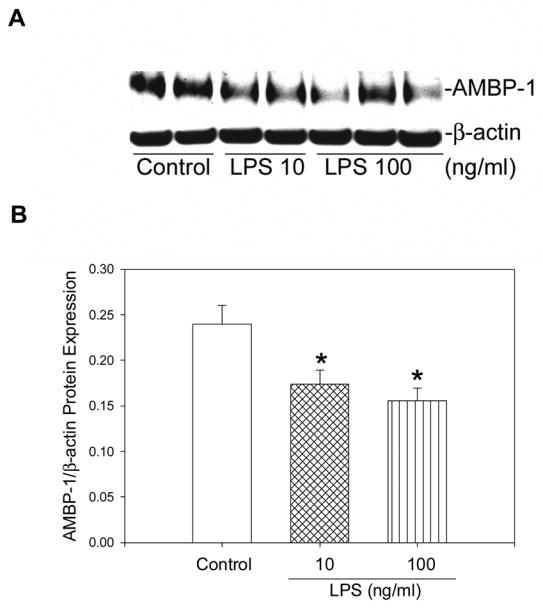
LPS treatment attenuates AMBP-1 expression in culture of hepatocytes and Kupffer separated by the use of 0.4 μm transwell inserts. A. Equal amount of proteins (25μg) from cell lysates were run on 3–8% NuPAGE gels under non-reducing conditions and Western blotted using anti-AMBP-1 antibody. Duplicate aliquots of protein (25 μg) were electrophoresed on 4–12% NuPAGE Bis-Tris gels under reducing conditions and Western blotted with anti-β-actin antibody (β-actin). B. The ratios between AMBP-1 and β-actin protein expressions are shown. Data are presented as mean ± SE (n=5) and compared by one-way ANOVA and Student-Newman-Keuls method. *P<0.05 versus control.
Figure 4.
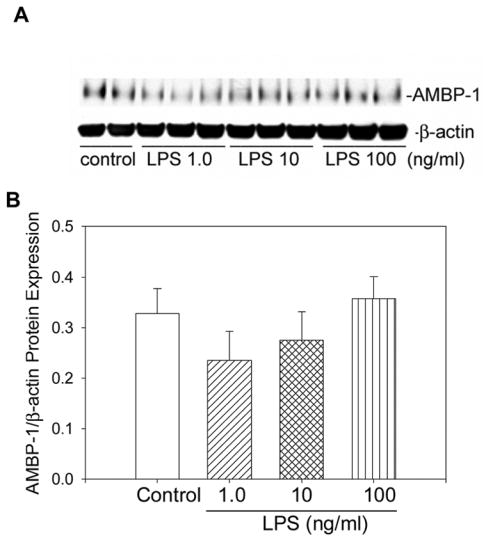
LPS treatment has no direct effect on AMBP-1 expression in hepatocytes. A. Hepatocytes in culture for 4 hrs were treated with LPS (1.0, 10 and 100 ng/ml) and incubated for 24 hrs at 37°C. Cells were lysed and equal amount of proteins (25 μg) were electrophoresed and Western blotted with either AMBP-1 or anti-β-actin antibody as described in the legend to Figure 1. B. The ratios between AMBP-1 and β-actin protein expression are shown. Data are presented as mean ± SE (n=6) and compared by ANOVA and Student-Newman-Keuls method.
Figure 5.
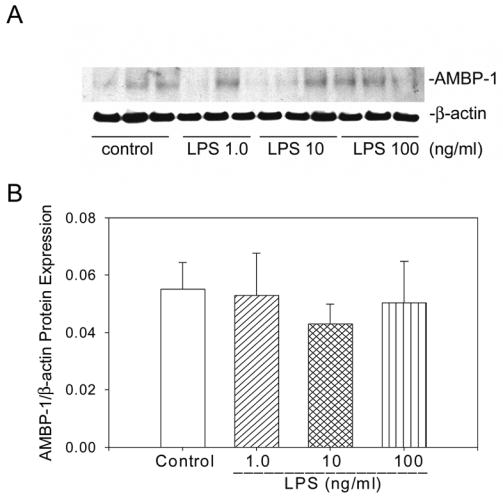
AMBP-1 protein expression is extremely low in Kupffer cells and LPS had no further effect on its expression. A. Kupffer cells in culture for 24 hrs were treated with LPS (1.0, 10 and 100 ng/ml) and incubated for 24 hrs at 37°C. Cells were lysed and equal amount of proteins (15 μg) were electrophoresed and Western blotted with either AMBP-1 or anti-β-actin antibody. B. The ratios between AMBP-1 and β-actin are shown. Data are presented as mean ± SE (n=6) and compared by ANOVA and Student-Newman-Keuls method.
3.2. Protein Expression of AMBP-1 is downregulated in Hepatocytes after IL-1β or TNF-α treatment
To test whether the observed downregulation of AMBP-1 in LPS treated co-culture was related to an increase in cytokine release, TNF-α and IL-1β levels in the co-culture cell supernatant were measured. LPS treatment at 100 ng/ml produced a significant increase in both TNF-α levels compared to untreated samples. In the case of IL-1β, hepatocytes alone produced a significant increase in IL-1β in the presence of 100 ng/ml LPS. These levels were not further increased in LPS treated co-culture [21]. To confirm that IL-1β and TNF-α indeed downregulate hepatocyte expression of AMBP-1 protein, hepatocytes were treated with either IL-1β or TNF-α for 24 hrs. The cell lysates were subjected to Western blotting and examined for AMBP-1 protein expression. Treatment with IL-1β at 1.0 and 10 ng/ml produced 53 and 61% inhibition of AMBP-1 protein levels, respectively (Fig. 6). Similarly, TNF-α at 1.0 and 10 ng/ml exhibited 53 and 50% inhibition of AMBP-1 protein levels, respectively (Fig. 6). No significant differences were observed in β-actin expression among different treatment groups and control.
Figure 6.
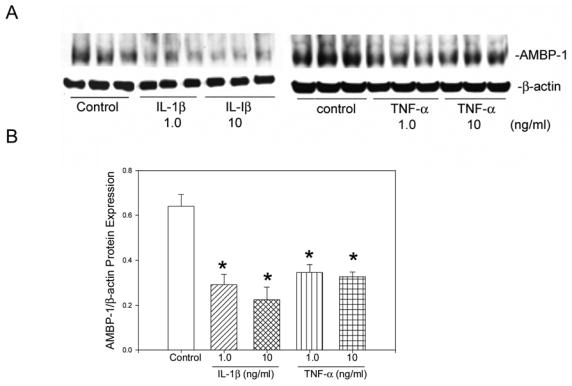
AMBP-1 protein expression in hepatocytes is significantly attenuated by IL-1β or TNF-α treatment. A. Hepatocytes in culture for 4 hrs were treated with IL-1β or TNF-α at 1.0 and 10 ng/ml and incubated for 24 hrs at 37°C. Cells were lysed and equal amount of proteins (25 μg) were electrophoresed and Western blotted with either AMBP-1 or anti-β-actin antibody. B. The ratios between AMBP-1 and β-actin protein expression are shown. Data are presented as mean ± SE (n=6) and compared by ANOVA and Student-Newman-Keuls method. *P<0.05 versus control.
4. Discussion
AMBP-1, identical to complement factor H, specifically binds AM and regulates AM’s bioactivities [13–15]. AMBP-1 is a 120 kDa molecular weight protein composed of 20 repetitive domains of short consensus repeats with approximately 60 amino acids in length. The main function of complement factor H known so far is to act as a cofactor of complement factor I for C3b cleaving and promotes the dissociation of C3bBb complex [22, 23]. Structure-functional analysis studies mapped two binding sites for AM in the complement factor H molecule which were distinct from the C3b binding sites [24]. The binding between AM and complement factor H has important physiological consequences. The presence of a binding protein can alter the biological function of a potent factor and determines its inhibitory or stimulatory capabilities. A recent study suggested that AM is degraded by metalloproteinase-2 and that the protein-protein binding interaction of AM with AMBP-1 prevents this degradation [25]. However, the mechanism of action is still unknown. In light of this report, it is plausible that AMBP-1 exerts its effects on AM by preventing the cleavage of AM by metalloproteinases. Recently we have shown that in a rat model of sepsis or endotoxemia, AMBP-1 gene and protein expression of hepatic tissue was significantly downregulated in the late phase of sepsis [19]. Even though AM is upregulated during sepsis primarily due to elevated levels of LPS [26], the vascular response to AM is significantly reduced in late sepsis [12]. This reduction in vascular response is attributed to the decrease in AMBP-1 levels. Thus, the hypodynamic response generally observed in late sepsis characterized by reduced vascular response to AM is presumably due to the decrease in AMBP-1 levels.
We have previously shown that administration of AM/AMBP-1 maintains cardiovascular response, decreases pro-inflammatory cytokine levels and reduces mortality in a rat model of sepsis [12, 16]. The combined treatment effectively suppressed LPS-induced TNF-α expression and release from Kupffer cells [20]. We have also shown that AMBP-1 is downregulated in the late phase of sepsis and LPS plays a critical role in the reduction of AMBP-1 [19]. In this study, we demonstrated that the downregulation of AMBP-1 in late sepsis or endotoxemia is due to the release of increased levels of pro-inflammatory cytokines from Kupffer cells rather than a direct effect of LPS. Our conclusion is based on the fact that [1] LPS treatment in a co-culture of hepatocytes and Kupffer cells significantly attenuated AMBP-1 protein expression, [2] no increase of AMBP-1 protein secretion was observed with LPS treatment in co-culture, [3] LPS treatment in transwell cultures of hepatocytes and Kupffer cells significantly downregulated AMBP-1 protein expression, [4] LPS at concentrations up to 100 ng/ml had no significant effect on AMBP-1 expression in cultured hepatocytes, [5] AMBP-1 expression is extremely low in Kupffer cells and LPS treatment did not show any further increase from the control levels. Thus, it was conceivable that LPS had no direct effect on AMBP-1 biosynthesis in the hepatocytes.
Our results with LPS treatment in rat hepatocytes is in good agreement with previous report that LPS at 1.0 and 10 ng/ml did not affect AMBP-1 protein expression [27]. A recent report using quantitative PCR suggests that hepatocytes contain mRNA for CD14 and TLR-4, the two receptors involved in LPS regulation, and that LPS at 100 μg/ml significantly upregulated the CD14 mRNA levels but not the TLR-4 levels [28]. However, our experiments were conducted with markedly lower concentrations of LPS (1–100 ng/ml) and that there were no significant changes in AMBP-1 levels in LPS treated hepatocyte cultures. Therefore, it is possible that the upregulation of CD14 expression and subsequent signaling via the CD14/TLR-4 receptors in hepatocytes occurs only at high concentrations of LPS. However, further investigation is warranted for such a conclusion.
In our study, constitutive level of AMBP-1 protein expression in Kupffer cells was at least ten fold lower than that seen in rat hepatocytes and no significant increase was evident with LPS treatment. However, it was reported using RT-PCR analysis, complement factor H mRNA was increased up to eight fold in LPS treated Kupffer cells [27]. Whether this increase in mRNA levels corresponds to an increase in secreted protein was not ascertained from that study. Our data clearly indicated that the source of complement factor H in the liver is the hepatocytes and that LPS up to 100 ng/ml concentrations, which will be specific for receptor-ligand interaction, had no direct effect on either the hepatocyte or Kupffer cell derived AMBP-1 production.
We have previously shown that co-culture with hepatocytes and Kupffer cells treated with 100 ng/ml LPS produced a 5-fold increase in TNF-α levels compared to untreated controls [21]. In addition, Hoebe KH et al. [29] and Scott MJ et al. [30] demonstrated that LPS treatment caused a significant increase in TNF-α and IL-6 release from co-culture of hepatocytes and Kupffer cells. Therefore, we hypothesized that the release of pro-inflammatory cytokines from Kupffer cells are responsible for the downregulation of AMBP-1 in LPS induced co-culture. To test this, we treated cultured hepatocytes with either TNF-α or IL-1β and examined AMBP-1 protein expression. Our results clearly indicated that both cytokines significantly downregulated AMBP-1 protein expression. Our study is in agreement with previous findings that IL-1β inhibits AMBP-1 secretion of cultured human umbilical vein endothelial cells [31–33]. It has been reported that interferon-γ also regulates AMBP-1 in primary hepatocytes [34]. Though our data clearly indicated that TNF-α and IL-1β significantly downregulated AMBP-1 protein expression, other pro-inflammatory cytokines such as IL-6 and interferon-γ could also regulate AMBP-1 expression in hepatocytes. Further studies are warranted to examine the role of either cytokine in AMBP-1 production in the co-culture system.
How do pro-inflammatory cytokines downregulate AMBP-1 protein expression? Cytokines are produced in response to cellular stress such as heat shock, ultraviolet light or adherence to a foreign surface. These stimuli often activate the mitogen activated protein kinases which phosphorylate transcription factors for gene expression. Infection and inflammation also use the same pathway for initiating cytokine gene expression. Most of the biological effects of IL-1β take place in cells following nuclear translocation of NFκB and AP-1, two known factors common to the IL-1β induced genes. Although the receptors for TNF-α and IL-1β are clearly different, the post receptor events could be similar. Unlike the IL-1β receptor, the TNF-α receptor contains “death domain” that recruits intracellular molecules involved in initiating programmed cell death or apoptosis. The pro-inflammatory cytokines such as TNF-α and IL-1β appears to trigger several events at the same time resulting in inflammation, tissue destruction and loss of function of the normal cellular activities [35]. It is plausible that these cytokines per se has no direct effect on the biosynthesis of AMBP-1 but rather they cause ultimate shut down of the cellular machinery. In that regard, it is highly unlikely that the cytokines caused necrosis of the hepatocytes within the 24 hr period studied due to the fact that we were able to detect similar levels of β-actin expression in cells treated and untreated with LPS. Our experiments did not address the role of apoptosis of hepatocytes in downregulation of AMBP-1. Therefore, further studies are needed to determine the mechanism by which pro-inflammatory cytokines downregulate AMBP-1.
Another possibility is proteolytic degradation of protein in general due to endotoxemia or sepsis. Since AMBP-1 is known to have a long half life, it is unlikely that specific degradation plays any role in LPS regulated AMBP-1 expression in hepatocytes. Future experiments using protease blockers could provide insights into this notion.
In summary, previously we have demonstrated that LPS administration downregulated AMBP-1 production in the liver. The present study was conducted to determine whether LPS directly or by increasing pro-inflammatory cytokines from Kupffer cells downregulate AMBP-1 biosynthesis. Our current data showed that pro-inflammatory cytokines, IL-1β and TNF-α released from Kupffer cells during sepsis are responsible for downregulation of AMBP-1.
Acknowledgments
This investigation was supported by NIH grants, R01GM057468 and R01HL076179 (P. Wang). The authors thank Mr. Weifeng Dong and Dr. Youxin Ji for their excellent technical assistances.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Baue AE. Sepsis research: what did we do wrong? What would Semmelweis do today? Shock. 2001;16:1–8. doi: 10.1097/00024382-200116010-00001. [DOI] [PubMed] [Google Scholar]
- 2.Cohen J, Guyatt G, Bernard GR, Calandra T, Cook D, Elbourne D, Marshall J, Nunn A, Opal S. New strategies for clinical trials in patients with sepsis and septic shock. Crit Care Med. 2001;29:880–886. doi: 10.1097/00003246-200104000-00039. [DOI] [PubMed] [Google Scholar]
- 3.Bernard GR, Vincent JL, Laterre PF, LaRosa SP, Dhainaut JF, Lopez-Rodriguez A, Steingrub JS, Garber GE, Helterbrand JD, Ely EW, Fisher CJ., Jr Efficacy and safety of recombinant human activated protein C for severe sepsis. N Engl J Med. 2001;344:699–709. doi: 10.1056/NEJM200103083441001. [DOI] [PubMed] [Google Scholar]
- 4.Chaudry IH, Wichterman KA, Baue AE. Effect of sepsis on tissue adenine nucleotide levels. Surgery. 1979;85:205–211. [PubMed] [Google Scholar]
- 5.Wang P, Chaudry IH. Mechanism of hepatocellular dysfunction during hyperdynamic sepsis. Am J Physiol. 1996;270:R927–938. doi: 10.1152/ajpregu.1996.270.5.R927. [DOI] [PubMed] [Google Scholar]
- 6.Ertel W, Morrison MH, Wang P, Ba ZF, Ayala A, Chaudry IH. The complex pattern of cytokines in sepsis. Association between prostaglandins, cachectin, and interleukins. Ann Surg. 1991;214:141–148. doi: 10.1097/00000658-199108000-00008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Waage A, Halstensen A, Espevik T. Association between tumour necrosis factor in serum and fatal outcome in patients with meningococcal disease. Lancet. 1987;1:355–357. doi: 10.1016/s0140-6736(87)91728-4. [DOI] [PubMed] [Google Scholar]
- 8.Debets JM, Kampmeijer R, van der Linden MP, Buurman WA, van der Linden CJ. Plasma tumor necrosis factor and mortality in critically ill septic patients. Crit Care Med. 1989;17:489–494. doi: 10.1097/00003246-198906000-00001. [DOI] [PubMed] [Google Scholar]
- 9.Wang P, Ba ZF, Cioffi WG, Bland KI, Chaudry IH. The pivotal role of adrenomedullin in producing hyperdynamic circulation during the early stage of sepsis. Arch Surg. 1998;133:1298–1304. doi: 10.1001/archsurg.133.12.1298. [DOI] [PubMed] [Google Scholar]
- 10.Koo DJ, Chaudry IH, Wang P. Mechanism of hepatocellular dysfunction during sepsis: the role of gut-derived norepinephrine (review) Int J Mol Med. 2000;5:457–465. doi: 10.3892/ijmm.5.5.457. [DOI] [PubMed] [Google Scholar]
- 11.Wang P. Adrenomedullin in sepsis and septic shock. Shock. 1998;10:383–384. doi: 10.1097/00024382-199811000-00013. [DOI] [PubMed] [Google Scholar]
- 12.Zhou M, Ba ZF, Chaudry IH, Wang P. Adrenomedullin binding protein-1 modulates vascular responsiveness to adrenomedullin in late sepsis. Am J Physiol Regul Integr Comp Physiol. 2002;283:R553–560. doi: 10.1152/ajpregu.00544.2001. [DOI] [PubMed] [Google Scholar]
- 13.Elsasser TH, Kahl S, Martinez A, Montuenga LM, Pio R, Cuttitta F. Adrenomedullin binding protein in the plasma of multiple species: characterization by radioligand blotting. Endocrinology. 1999;140:4908–4911. doi: 10.1210/endo.140.10.7157. [DOI] [PubMed] [Google Scholar]
- 14.Pio R, Elsasser TH, Martinez A, Cuttitta F. Identification, characterization, and physiological actions of factor H as an adrenomedullin binding protein present in human plasma. Microsc Res Tech. 2002;57:23–27. doi: 10.1002/jemt.10047. [DOI] [PubMed] [Google Scholar]
- 15.Pio R, Martinez A, Unsworth EJ, Kowalak JA, Bengoechea JA, Zipfel PF, Elsasser TH, Cuttitta F. Complement factor H is a serum-binding protein for adrenomedullin, and the resulting complex modulates the bioactivities of both partners. J Biol Chem. 2001;276:12292–12300. doi: 10.1074/jbc.M007822200. [DOI] [PubMed] [Google Scholar]
- 16.Yang S, Zhou M, Chaudry IH, Wang P. Novel approach to prevent the transition from the hyperdynamic phase to the hypodynamic phase of sepsis: role of adrenomedullin and adrenomedullin binding protein-1. Ann Surg. 2002;236:625–633. doi: 10.1097/00000658-200211000-00013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fowler DE, Yang S, Zhou M, Chaudry IH, Simms HH, Wang P. Adrenomedullin and adrenomedullin binding protein-1: their role in the septic response. J Surg Res. 2003;109:175–181. doi: 10.1016/s0022-4804(02)00086-0. [DOI] [PubMed] [Google Scholar]
- 18.Zipfel PF, Skerka C. Complement factor H and related proteins: an expanding family of complement-regulatory proteins? Immunol Today. 1994;15:121–126. doi: 10.1016/0167-5699(94)90155-4. [DOI] [PubMed] [Google Scholar]
- 19.Cui Y, Ji Y, Wu R, Zhou M, Wang P. Adrenomedullin binding protein-1 is downregulated during polymicrobial sepsis in the rat. Int J Mol Med. 2006;17:925–929. [PubMed] [Google Scholar]
- 20.Wu R, Zhou M, Wang P. Adrenomedullin and adrenomedullin binding protein-1 downregulate TNF-alpha in macrophage cell line and rat Kupffer cells. Regul Pept. 2003;112:19–26. doi: 10.1016/s0167-0115(03)00018-1. [DOI] [PubMed] [Google Scholar]
- 21.Wu R, Cui X, Dong W, Zhou M, Simms HH, Wang P. Suppression of hepatocyte CYP1A2 expression by Kupffer cells via AhR pathway: the central role of proinflammatory cytokines. Int J Mol Med. 2006;18:339–346. [PubMed] [Google Scholar]
- 22.Kuhn S, Skerka C, Zipfel PF. Mapping of the complement regulatory domains in the human factor H-like protein 1 and in factor H1. J Immunol. 1995;155:5663–5670. [PubMed] [Google Scholar]
- 23.Kuhn S, Zipfel PF. Mapping of the domains required for decay acceleration activity of the human factor H-like protein 1 and factor H. Eur J Immunol. 1996;26:2383–2387. doi: 10.1002/eji.1830261017. [DOI] [PubMed] [Google Scholar]
- 24.Martinez A, Pio R, Zipfel PF, Cuttitta F. Mapping of the adrenomedullin-binding domains in human complement factor H. Hypertens Res. 2003;26(Suppl):S55–59. doi: 10.1291/hypres.26.s55. [DOI] [PubMed] [Google Scholar]
- 25.Martinez A, Oh HR, Unsworth EJ, Bregonzio C, Saavedra JM, Stetler-Stevenson WG, Cuttitta F. Matrix metalloproteinase-2 cleavage of adrenomedullin produces a vasoconstrictor out of a vasodilator. Biochem J. 2004;383:413–418. doi: 10.1042/BJ20040920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yang S, Zhou M, Chaudry IH, Wang P. The role of lipopolysaccharide in stimulating adrenomedullin production during polymicrobial sepsis. Biochim Biophys Acta. 2001;1537:167–174. doi: 10.1016/s0925-4439(01)00069-2. [DOI] [PubMed] [Google Scholar]
- 27.Schlaf G, Demberg T, Beisel N, Schieferdecker HL, Gotze O. Expression and regulation of complement factors H and I in rat and human cells: some critical notes. Mol Immunol. 2001;38:231–239. doi: 10.1016/s0161-5890(01)00045-1. [DOI] [PubMed] [Google Scholar]
- 28.Hayashi T, Kishiwada M, Fujii K, Yuasa H, Nishioka J, Ido M, Gabazza EC, Suzuki K. Lipopolysaccharide-induced decreased protein S expression in liver cells is mediated by MEK/ERK signaling and NFkappaB activation: involvement of membrane-bound CD14 and toll-like receptor-4. J Thromb Haemost. 2006;4:1763–1773. doi: 10.1111/j.1538-7836.2006.02042.x. [DOI] [PubMed] [Google Scholar]
- 29.Hoebe KH, Witkamp RF, Fink-Gremmels J, Van Miert AS, Monshouwer M. Direct cell-to-cell contact between Kupffer cells and hepatocytes augments endotoxin-induced hepatic injury. Am J Physiol Gastrointest Liver Physiol. 2001;280:G720–728. doi: 10.1152/ajpgi.2001.280.4.G720. [DOI] [PubMed] [Google Scholar]
- 30.Scott MJ, Liu S, Su GL, Vodovotz Y, Billiar TR. Hepatocytes enhance effects of lipopolysaccharide on liver nonparenchymal cells through close cell interactions. Shock. 2005;23:453–458. doi: 10.1097/01.shk.0000160939.08385.f1. [DOI] [PubMed] [Google Scholar]
- 31.Dauchel H, Julen N, Lemercier C, Daveau M, Ozanne D, Fontaine M, Ripoche J. Expression of complement alternative pathway proteins by endothelial cells. Differential regulation by interleukin 1 and glucocorticoids. Eur J Immunol. 1990;20:1669–1675. doi: 10.1002/eji.1830200808. [DOI] [PubMed] [Google Scholar]
- 32.Brooimans RA, van der Ark AA, Buurman WA, van Es LA, Daha MR. Differential regulation of complement factor H and C3 production in human umbilical vein endothelial cells by IFN-gamma and IL-1. J Immunol. 1990;144:3835–3840. [PubMed] [Google Scholar]
- 33.Coulpier M, Andreev S, Lemercier C, Dauchel H, Lees O, Fontaine M, Ripoche J. Activation of the endothelium by IL-1 alpha and glucocorticoids results in major increase of complement C3 and factor B production and generation of C3a. Clin Exp Immunol. 1995;101:142–149. doi: 10.1111/j.1365-2249.1995.tb02290.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schlaf G, Beisel N, Pollok-Kopp B, Schieferdecker H, Demberg T, Gotze O. Constitutive expression and regulation of rat complement factor H in primary cultures of hepatocytes, Kupffer cells, and two hepatoma cell lines. Lab Invest. 2002;82:183–192. doi: 10.1038/labinvest.3780410. [DOI] [PubMed] [Google Scholar]
- 35.Dinarello CA. Proinflammatory cytokines. Chest. 2000;118:503–508. doi: 10.1378/chest.118.2.503. [DOI] [PubMed] [Google Scholar]