

Abstract
We previously identified a cis-acting element within the 3′ untranslated region of CD40 ligand messenger RNA (mRNA) that is composed of three complex binding sites and acts to increase mRNA stability in both in vitro and in vivo systems. We now demonstrate the functional consequences of the three binding sites with respect to increasing both luciferase activity and mRNA stability in a heterologous transcript expressed in a T-cell line. The internal region B was shown to be a bona fide stability element because its presence increased luciferase activity fourfold over the unmodified transcript and its removal from the XbaI–HaeIII region resulted in rapid degradation of the transcript. Region A contained both a binding site for a polypyrimidine-tract-binding protein (PTB)-mediated complex (Complex I) and an upstream, adjacent sequence that was a negative regulator of mRNA stability. Region C bound Complex II, which contained both PTB and heterogeneous nuclear ribonucleoproteinL (hnRNPL), and was less effective as a stability element on its own compared to region B. Our findings demonstrate differential levels of activity for the three binding sites relative to the turnover of CD40 ligand mRNA, suggesting that the lack of binding of Complex I/II during the early stages of T-cell activation contributes to the rapid degradation of the CD40 ligand mRNA transcript.
Keywords: CD40 ligand, messenger RNA decay, polypyrimidine-tract-binding protein, RNA binding, T-cell activation
Introduction
The interaction of CD40 ligand (CD40L or CD154) expressed primarily on CD4+ T cells with CD40 expressed on antigen-presenting cells is critical for generating both innate and acquired immune responses (reviewed in reference 1). The requirement for CD40 signalling in acquired immunity has been demonstrated in animal models lacking either functional CD402 or CD40L3 and in humans suffering from X-linked hyper-immunoglobulin M syndrome (HIGMX-1), which involves an absence of functional CD40L activity.4–8 In these instances there is a clear absence of isotype switching in B cells, a lack of germinal centres, and deficient primary and secondary responses to thymus-dependent antigens (reviewed in reference 1).
The role of CD40L in establishing cell-mediated or innate immune responses is thought to occur primarily through the interaction of activated CD4+ T cells with CD40-expressing macrophage and dendritic cells (reviewed in references 9 and 10). Blocking antibodies or soluble CD40–immunoglobulin are known to be successful at abrogating the onset of numerous autoimmune disorders in mouse models of disease (reviewed in reference 11) and blocking CD40L and cytotoxic T-lymphocyte antigen interactions appear to be critical for preventing allograft rejection.12–14 Finally, inhibition of CD40L action directly interferes with atherogenesis in mice.15 Thus, CD40L is a key factor in the orchestration of an effective and comprehensive immune response. Furthermore, lack of expression, as well as inappropriate expression, leads to acute pathological consequences.
The complexity of CD40L expression is underscored by several studies showing that the time–course and extent of expression are dependent on the type of stimuli as well as costimulatory interactions provided by B cells or antigen-presenting cells.16–26 The expression of CD40L is tightly regulated within the context of T-cell activation and accordingly a number of signalling pathways that activate T cells also induce CD40L expression.27–29 Transcriptional regulation is dependent on nuclear factor of activated T cells, and transcription factors E3 and EB binding to promoter elements,30–35 as well as a nuclear factor-κB-dependent binding to an enhancer located 3′ of the coding region.33 In addition to transcriptional regulation, mounting evidence indicates that post-transcriptional mechanisms are utilized during T-cell activation to control CD40L gene expression.36–40 Specifically, at early times of T-cell receptor engagement, when the CD40L gene is highly transcribed, the messenger RNA (mRNA) is rapidly degraded with a half-life (t½) of less than 40 min. In contrast, at late times of activation the message becomes three- to fourfold more stable under conditions where the rate of transcription is decreased.41 Surprisingly, the stability of the CD40L transcript is only marginally increased by costimulatory signals,36,38,41 which is different from the regulation observed for both the interleukin-2 and tumour necrosis factor-α transcripts.42,43
In experiments to further analyse the post-transcriptional regulation of CD40L mRNA we identified a region in the 3′ untranslated region (UTR) that bound two ribonucleoprotein complexes (termed Complex I and Complex II) only at late times of T-cell activation (defined by XbaI and HaeIII). In defining the composition of the primary RNA-binding complex (Complex I), it has been found that the major protein is the 55 000 molecular weight polypyrimidine-tract-binding protein (PTB) or heterogeneous nuclear ribonucleoprotein I (hnRNP-I)44,45 whereas nucleolin is a second component of Complex I that interacts with both the RNA and PTB.46 Complex I binds two distinct regions within a highly pyrimidine-rich stretch of sequence in the 3′ UTR (regions A and B) and the 63-nucleotide CU-rich sequence within region B has been defined as the minimal binding site for this complex. Furthermore, Complex II binds to the 3′-most sequences defined by region C.44 We also demonstrated that a CU binding protein directly stabilized the CD40L mRNA by depleting complex binding in vitro and in transfection studies showed that a CD40L transcript containing sites A, B and C was more highly stable compared to a transcript lacking these sites.40,44
In our current work we extend our earlier findings by defining the sequence limits of Complex I and II binding within regions A and C and analyse the effect of inserting the Complex I/II binding region into a stable, heterologous transcript. Also, we identify hnRNPL as a component factor of Complex II. Specifically, we demonstrate that the CD40L stability element acts to inhibit the decay rate of the Renilla luciferase transcript and is therefore a bona fide stability element.
Materials and methods
Cell lines and antibodies
The human D1.1 T-cell line is a CD40L-expressing Jurkat subclone that has been previously described.17,47 Jurkat/D1.1 cells were cultured in RPMI-1640 medium supplemented with 10% fetal bovine serum, 2 mm glutamine, 100 μg/ml streptomycin and 100 U/ml of penicillin (RPMI-complete). To isolate cytoplasmic extracts, 1 × 107 to 2 × 107 cells were harvested, washed once with 1 ml ice-cold 1× phosphate-buffered saline and lysed in 200 μl cold extraction buffer [40 mm KCl, 10 mm HEPES (pH 7·9), 3 mm MgCl2, 1 mm dithiothreitol (DTT), 5% glycerol, 0·2% Nonidet P-40, 0·5 mm phenylmethylsulphonyl fluoride and 20 ml protease inhibitor cocktail (Sigma-Aldrich Chemical Co., St Louis, MO)]. Cells were incubated on ice for 5 min, centrifuged at 16 000 g for 10 min at 4° and the supernatant was collected. Protein concentrations of cell supernatants were determined by Bradford Assay (BioRad Corp., Hercules, CA) and aliquots were frozen for later use. The anti-PTB hybridoma CRL-2501 was obtained from American Type Culture Collection (ATCC; Manassas, VA). The anti-hnRNPL monoclonal antibody was purchased from Sigma-Aldrich.
RNA probes
The following probes were generated for use in RNA-binding studies using 5′ primers that contained a T7 promoter and unique 3′ primers: T7-1300-E1, 5′-cgtaatacgactcactatagggctagaacgtctaacacagtggaga-3′ (forward) and 5′tgaaagagagagatggagagagagagagagatt-3′ (reverse); T7-1349-E1, 5′-cgtaatacgactcactataggggccaccctctcggacagt-3′ (forward) and 5′-tgaaagagagagatggagagagagagagagatt-3′ (reverse); T7-E5-HaeIII and E5-HinfI templates were synthesized using primers: 5′-cgtaatacgactcactatagggagtctcttccctcccccagtctctctt-3′ (forward) and 5′-agagaactgactagcaacggc-ctga-3′ (reverse), followed by digestion with HaeIII and HinfI enzymes (Promega, Madison, WI) respectively; the T7-E5-1518, 5′-cgtaatacgactcactatagggagtctcttccctcccccagtctctctt-3′ (forward) and 5′ttagaaagggggattga-3′ (reverse); the T7-E5-HΔCa, 5′-cgtaatacgactcactatagggagtctcttccctcccccagtctctctt-3′ (forward) and 5′-cctgactcttagaaagggggattg-3′ (reverse); T7-1518-HaeIII probe, 5′-tccccctttccgtaatacgactcactatagggtaacacacacaca-3′ (forward), and 5′-agagaactgactagcaacggcctga-3′ (reverse), followed by digestion with HaeIII. Polymerase chain reaction (PCR) was carried out in 10 mm Tris–HCl (pH 9·0), 50 mm KCl, 0·1%Triton X-100, 1·5 mm MgCl2, 0·2 mm dNTPs, 200 ng of each primer, 50 ng of DNA template, and 2·5 U Taq polymerase (Promega). Cycling parameters were as follows: one cycle of 2 min at 94°, 30 cycles of 94° for 30 seconds, 62° for 20 seconds, and 72° for 45 seconds. The annealing temperature varied depending on the primer set that was used.
For the synthesis of 32P-labelled RNA probes 0·5 μg template DNA; 0·4 mm each of rATP, rGTP and rCTP; 0·04 mm rUTP; 30 mm DTT; 20 U of RNasin (Promega); 2·5 mm cap analogue (Amersham Biosciences, Piscataway, NJ); T7 transcription buffer (40 mm Tris–HCl 7·9; 6 mm MgCl2; 2 mm spermidine and 10 mm NaCl); 25–40 μCi [α-32P]rUTP and 5 U T7 RNA polymerase (Promega) were used at 37° for 1 hr, treated with RQ1 RNase-free DNase at 37° for 15 min and centrifuged through G25 columns (Amersham Biosciences) to remove the unincorporated nucleotides. For the Site C probes [α-32P]CTP was used as label and corresponding changes to unlabelled nucleotide concentrations were made.
RNA-electrophoretic mobility shift assays and antibody interference analysis
RNA electrophoretic mobility shift assays (R-EMSA) were performed as described previously.44 Typically 5·0 μg of protein extracts was incubated with 4 × 104 counts per minute (c.p.m.) of RNA probes synthesized in vitro in 20 μl RNA-binding buffer containing 270 ng Escherichia coli transfer RNA. In antibody interference experiments, 1–2 μl of test or control polyclonal antibodies (1 mg/ml) was added to the reaction for 1 hr before the addition of probe. Following a 30-min incubation at room temperature, RNase mix (40 U RNase T1, 10 ng RNase A and 0·015 U RNase V1; Ambion, Austin, TX) was added and the reactions were incubated at 37° for 30 min. Following the addition of 100 mg heparin, the reactions were incubated for 10 min on ice. Samples were resolved on a 7% native acrylamide gel in 0·25× Tris–borate–ethylenediaminetetraacetic acid buffer at 200 V for 2–4 hr. The gels were dried and visualized by autoradiography. In competition experiments, increasing amounts (50, 100, 200 and 400 ng) of unlabelled oligonucleotide was added to the reaction 1 hr before addition of the probe.
Ultraviolet cross-linking assay
Reactions (20 μl) containing 1 μg yeast transfer RNA, 40 mm KCl, 10 mm HEPES (pH 7·9), 3 mm MgCl2, 1 mm DTT, 5% glycerol, 20 U of RNasin, 20 μg Jurkat/D1.1 extract, and 2 × 104 c.p.m. in-vitro-synthesized RNA probe were incubated at room temperature for 30 min. RNase T1 (100 U) was added to the reaction and incubated at 37° for 20 min. Proteins were ultraviolet (UV) cross-linked to probes by exposing the reactions to 254 nm for 30 min at 0°. Ten nanograms RNaseA and 0·01 U RNaseV1 was added to the reaction and the mixture was incubated at 37° for 30 min. Samples were boiled for 5 min and underwent electrophoresis through a 4% stacking gel/10% separating gel for 5 hr at 30 mA. The gel was fixed in 10% acetic acid, and visualized by autoradiography. Competition experiments were carried out as described above.
Generation of luciferase-based constructs
DNA templates for the synthesis of 3′ UTR fragments were generated by PCR and fragments representing the different regions of the CD40L stability element were subcloned into the XbaI site of the pRLSV40 vector (Promega). DNA templates for the synthesis of 3′ UTR fragments were generated as previously described.40 The following sequences were generated with 200 ng specific primers as indicated: Sites A+B+C, 5′-cgtggctagaacgtctaacacagtggaga-3′ (forward), and 5′-agagaactgactagcaacggcctga-3′ (reverse); Sites A + B, 5′-cgtggctagaacgtctaacacagtggaga-3′ and 5′-agagaactgactagcaacggcctga-3′ (reverse); Sites B + C, 5′-cgtggctagaacgtctaacacagtggaga-3′ (forward) and 5′-tgaaagagagagatggagagagagagagagatt-3′ (reverse); Site A, 5′-cgtaatacgactcactatagggctagaacgtctaacacagtggag-3′ (forward) and 5′-tgaaagagagagatggagagagagagagagatt-3′ (reverse); Site B, 5′-cgtggtctctctctctcaacctcttt-3′ (forward) and 5′-agagaactgactagcaacggcctga-3′ (reverse); Site C, 5′-cgtggagtctcttccctcccccagtctctctt-3′ (forward) and 5′-agagaactgactagcaacggcctga-3′ (reverse); and Site D, 5′-ccgttgctagtcagttctctt-3′ (forward) and 5′cctcaccctcatctatagtgg-3′ (reverse). The pRL-D construct was made by ligating site A and site D PCR products for 2 hr at room temperature after treatment of the individual fragments with 10 U T4PNK (New England Brolabs, Ipswich, MA) for 30 min at 37°. Following PCR of the A-D fragment, the A-D and C fragments were treated with kinase and ligated together as described above. The pRL-A5′ insert was generated using forward primer; 5′-gctctagaacgtctaacaca-3′ (1315) and reverse primer; 5′-gctctagagcggtaactgtccgagagggtggc-3′ The 3′-site fragment was amplified with Sites A+B+C forward and reverse primers (see above) and cloned into the XbaI site of pRLSV40.
Transfection of Jurkat/D1.1 cells
Jurkat/D1.1 cells (1 × 107) were transiently transfected with 2·5 μg plasmid DNA together with 12·5 μg pGL2 DNA. 400 μl of this cell suspension was pulsed once at 250 mV, 960 μF using the Bio-Rad Gene Pulser (Bio-Rad Corp., Hercules, CA). Transfected cells were incubated at 37°, in 5% CO2 in an additional 4 ml complete RPMI-1640 for 36–48 hr. Cultures were harvested by centrifugation and used to measure luciferase activity using the Dual-Luciferase kit (Promega) together with the Glow Max 20/20 luminometer as described by the manufacturer (Promega). Three independent transfections were performed for each construct and three separate readings were performed for each sample. Results were averaged and the ratio of Renilla luciferase activity over firefly luciferase activity was calculated to determine relative luminescence. The normalized ratio for each construct was divided by the normalized ratio for the control construct pRLSV40 to give the fold induction of each. To generate stable clones of Jurkat/D1.1 cells expressing the pRLSV40 and pRLABC constructs, 107 cells were electroporated with one pulse at 250 mV 960 mF capacitance in 400 μl serum-free RPMI containing 20 μg plasmid DNA and 1 μg pCDNA3 (cotransfected for G418 resistance). Cells were selected 48 hr after transfection in RPMI containing 2 mg/ml G418. After drug selection, cells were subcloned by limiting dilution and independent clones were assayed for Renilla luciferase expression using the Dual Luciferase Assay kit. Relative light units were normalized to μg of total protein by Bradford assay.
Messenger RNA stability assays
Transiently transfected cells (2 × 107) were cultured for 40–44 hr post-transfection followed by treatment with 50 μg/ml 5,6-dichlorobenzimidizole 1-m-d-ribofuranoside (DRB) to inhibit transcriptional elongation over 4 hr. Total RNA was isolated using the High Pure RNA Isolation Kit (Roche Diagnostics, Indianapolis, IN) and complementary DNA (cDNA) was synthesized using the Transcriptor First-Strand cDNA Synthesis Kit (Roche Diagnostics). Semi-quantitative PCR was performed using 1 μl cDNA in a 25 μl reaction containing 50 mm KCl, 10 mm Tris–HCl (pH 9·0), 0·1% Triton X-100, 1·5 mm MgCl2, 0·2 mm dNTPs, 1·25 U Taq polymerase in Buffer B (Promega), with 100 ng of Renilla 5′ primer (5′-GATACATTGAT5GAGTTTGGAC-3′) and Renilla-3′ primer (5′-TTTTTTTTTTTTTTTTTTTGTTGTTA-3′). To amplify β-actin control, 1 μl of 1 : 100 dilution of cDNA in a 25-μl reaction was established with 50 ng of primers 5′β-actin 5′-GCATCCTCACCCTGAAGTA-3′ (forward) and 3′β-actin 5′-TGTGGTGCCAGATTTTGTCC-3′ (reverse). The reactions were prepared on ice, heated for 1 min at 94°, followed by 32 cycles of 94° for 30 seconds, 55° for 30 seconds, and 72° for 30 seconds. Ten microlitres was electrophoresed on a 2% agarose/TBE gel and bands were quantified using the Kodak 1D analysis software. To analyse mRNA decay in stable transfectants, 2 × 107 Jurkat/D1.1 cells were cultured in RPMI-complete and a total of 5 × 106 cells were removed at 1-hr intervals over an 8-hr time–course. The RNA and cDNA were isolated and prepared as described above. Quantitative PCR was performed using the following primers: Renilla luciferase forward, 5′-GAAGTTGGTCGTGAGGCACT-3′ and reverse, 5′-TCCGTTTCCTTTGTTCTGGA-3′; quantitative PCR was performed using both the Mx4000 Multiplex PCR System and FastStart SYBR Green Master Mix according to the manufacturer's directions (Stratagene, LaJolla, CA and Roche Diagnostics, respectively). After the amplification a melt curve was performed to ensure amplicon homogeneity. Decay of a specific RNA transcript was determined by the 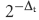 method comparing each time-point to the zero time-point to quantify the percentage of RNA remaining48,49 using the MxPro-Mx3000P software.
method comparing each time-point to the zero time-point to quantify the percentage of RNA remaining48,49 using the MxPro-Mx3000P software.
Cell fractionation of Jurkat/D1.1 cells
For each construct, 1 × 108 Jurkat/D1.1 cells were transiently transfected and incubated for 24 hr before polysome isolation. After transfection, 1 × 108 cells were incubated in 100 μg/ml cyclohexamide (Sigma-Aldrich) for 15 min at 37°. Following treatment, the cells were washed three times in cold plain RPMI-1640. Cells were resuspended in 1·5 ml ice-cold Buffer A [10 mm Tris–HCl (pH 7·6), 1 mm potassium acetate, 1·5 mm magnesium acetate, 2 mm DTT] supplemented with 500 U RNasin (Promega) and 1·6× Protease Inhibitor cocktail (Sigma-Aldrich). The cell suspension was passed thorough a 211/2 gauge needle 20 times and the crude lysates were centrifuged at 13 400 g for 10 min. The supernatants were layered over 1·5 ml 30% sucrose cushion and centrifuged at 130 000 g for 2·5 hr at 4° in SW55-Ti rotor. The S130 extract was carefully removed from the tube and stored in aliquots at − 80°. The remaining ribosome pellet was resuspended in 1·3 ml Buffer A and KCl was added to a final concentration of 0·3 m with stirring for 15 min at 4°. The polysomes were repelleted through the sucrose cushion and the supernatant (high-salt wash fraction) was divided into aliquots and stored at − 80°. The remaining polysome pellet was resuspended and all fractions were quantified for protein using the Bradford assay (BioRad). RNA was isolated from 500 μg polysomal fractions using Trizol LS reagent following the manufacturer's protocol (Invitrogen). RNA samples were subject to reverse transcription and amplified using quantitative PCR as described above. Primers used to amplify 28S ribosomal RNA were: forward primer 5′-TTCACCCACTAATAGGGAACG-3′; and reverse primer 5′-TGGCAACAACACATCATCAG-3′.
Results
Identification of the minimal Complex I binding site within region A
The stability region within the CD40L 3′ UTR is composed of three separate and adjacent binding regions (Fig. 1a) and extends from the XbaI site at nucleotide (nt) 1300 to the HaeIII site at nt 1589.40,44 Since only the minimal Complex I binding site for region B has been previously defined, our initial focus was to map the minimal complex binding sites for both regions A and C. The 110-nt XbaI-E1 region (region A, nt 1300–1410) consists of 31 random nt followed by 17 (C)s and 62 nt of CU-rich sequence. To establish whether the poly(C) tract and sequences 5′ to it were important for Complex I binding, a probe containing the complete A region and a probe containing only the CU-rich sequences (1348-E1) were synthesized in vitro, capped and used in R-EMSA with total cellular extract from Jurkat/D1.1 cells. As shown in Fig. 1(b), Complex I bound equally well to both the A region probe (lanes 1–3) and the 1348-E1 probe (lanes 4–6), indicating that sequences upstream of the CU-rich region, including the poly(C) tract, were not required for binding. To show that PTB was the major component of this complex anti-PTB and isotype control antibodies were added to reactions (lanes 7 and 8) and UV-cross-linking was carried out (lanes 9–11). Because the entire complex was supershifted with anti-PTB antibodies and there was a single UV-cross-linked band that ran at the size of PTB (50 000–55 000 molecular weight) we concluded that PTB was the major RNA-binding component of Complex I bound to the A site.
Figure 1.

The minimal A binding site is defined by a 62 nucleotide (nt) CU-rich region. (a) Schematic diagram of the CD40 ligand (CD40L) messenger RNA showing the three distinct Complex I/II binding sites in the 3′ untranslated region. The XbaI–HaeIII region, defined by nt 1300 to 1589, is divided into three individual sites: A (XbaI-E1, dashed underlined), B (E1-E5, underlined) and C (E5-HaeIII, boxed). (b) An RNA binding gel (RNA-electrophoretic mobility shift assay, lanes 1–8) and an ultraviolet-cross-linking gel using either a Site A (nt 1300-E1) probe (lanes 1–3 and 9–11) or a 3′ Site A probe defined by nt 1349–1410 (lanes 4–6). Reactions were set up with probe alone (lanes 1, 4 and 9), probe + RNase (lanes 2, 5 and 10) and probe + RNase + 20 μg Jurkat/D1.1 extract (lanes 3, 6 and 11). To analyse the presence of polypyrimidine-tract-binding protein (PTB), isotype control immunoglobulin (lane 7) or 1 μg anti-PTB monoclonal antibody (lane 8) was added to the reactions 1 hr before addition of the probe. All reactions were separated on a 7% polyacrylamide gel and analysed by exposure to film.
Multiple sequences within region C form the Complex II binding site
A similar analysis was carried out to define the minimal binding sequence in region C. We previously reported that this region, defined by nt 1473–1589 (Fig. 1a) bound Complex II, a PTB-containing complex that migrates with a slower mobility than the more abundant Complex I. Region C can be divided into a 43-nt CU-rich region directly adjacent to region B, a (CA)32 element downstream of the CU-rich region and 8 nt beyond the end of the CA repeat (Fig. 2a, top schematic). We first asked whether the sequences containing either the CU-rich stretch of sequence (E5-1518) or the CU-rich stretch plus the CA repeat (E5-1582) were sufficient for Complex II formation. In binding assays using extract from Jurkat/D1.1 cells and the E5-1518 or the E5-1582 probes (second and third schematics), we failed to observe binding to either of these probes, indicating that these sequences were insufficient to bind Complex II (Fig. 2b, lanes 6 and 9). Additionally, Complex II formation was restored when the 3′-most nucleotides (nt 1582–1589) were included in the probe, showing that these nucleotides were required for efficient Complex II formation. A question remained whether the CU-rich region plus the 8 nt downstream of the CA repeat were sufficient for binding or whether the CA repeat was also required. However, no Complex II formation was observed using an RNA probe that lacked the CA repeat but contained the 8 nt juxtaposed to the CU-rich region at nt 1518 (probe designated E5-HΔCA, Fig. 2a; Fig. 2b, lane 12). Finally, to establish whether the CA repeat plus the 8 nt between the HinfI and HaeIII sites were necessary and sufficient for Complex II binding a probe was engineered that contained only these nucleotides (probe 1518-HaeIII). We found that this probe formed a broad complex running just at and below Complex II (see lower arrow). Together these results identified the entire C region as being required for Complex II binding. However, in the absence of the upstream CU-rich sequence, a faster migrating complex bound to the CA-repeats in conjunction with the eight adjacent downstream nucleotides.
Figure 2.

The Complex II binding site is composed of all regions of Site C. (a) Schematic representation of probes derived from the CD40 ligand (CD40L) 3′ untranslated region Site C (E5-1589) that were used to map the Complex II binding site. (b) In vitro synthesized RNA probes spanning the C site, E5-HaeIII (lanes 1–3), E5-1518 (lanes 4–6), E5-HinfI (lanes 7–9), E5-HΔCA (lanes 10–12) and 1518-HaeIII (lanes 13–15) were used in RNA-electrophoretic mobility shift assay without (lanes 2, 5, 8, 11 and 14) or with (lanes 3, 6, 9, 12 and 15) 15 μg Jurkat/D1.1 cytoplasmic extract. Lanes 1, 4, 7, 10 and 13 show intact probes in the absence of RNase and extract.
PTB and hnRNPL bind to site C
Our binding studies to analyse Site C indicated that sequences outside the CU-rich core, which contained two canonical PTB binding sites (UCUU), were involved in complex formation. To identify proteins that bound to this region RNA-binding and competition experiments were carried out using unlabelled oligonucleotides. In R-EMSA, Complex II efficiently competed with the 100 ng oligo-dCT and a decrease in, but not a complete loss of, complex binding occurred when oligo-dCA was added as competitor (Fig. 3a, lanes 8–15). Oligo-U did not compete for Complex II binding at any concentration (lanes 4–7). The inclusion of anti-PTB but not control antibodies to the binding reaction completely blocked formation of Complex II (lanes 17 and 16, respectively). To extend our RNA-binding studies, UV cross-linking assays were carried out using the E5-HaeIII, Jurkat/D1.1 cytoplasmic extract and increasing concentrations of oligo-U, oligo-dCT and oligo-dCA to identify specific RNA-binding proteins. Six proteins with molecular weights between 30 000 and 100 000 bound to this probe and the addition of increasing amounts of oligo-U to remove non-specific binding resulted in the specific binding of four proteins in the range of 50 000–100 000 (Fig. 3b, lanes 3–8). As expected, increasing concentrations of oligo-dCT competed the doublet at approximately 55 000, a molecular weight that directly corresponded to the 55 000–57 000 forms of PTB (lanes 8–11). Notably, oligo-dCA competed with the 65 000 protein at the lowest concentration of oligo whereas the 75 000 and 100 000 proteins were competed with higher concentrations, suggesting that all three proteins were CA-binding proteins but with different binding affinities (lanes 12–15). Because hnRNPL is a well-characterized CA-binding protein with molecular weight 64 000–68 000, we included anti-hnRNPL or control antibodies in binding assays to identify proteins bound to Site C. As shown in Fig. 3(c), hnRNPL bound specifically to the 1518-HaeIII probe (lane 5) confirming that hnRNPL, in addition to PTB, is a component of Complex II.
Figure 3.

Polypyrimidine-tract-binding protein (PTB) and heterogeneous nuclear ribonucleoprotein L (hnRNPL) are components of Complex II. (a) Competition RNA electrophoretic mobility shift assay (R-EMSA) was performed using an in vitro transcribed, 32P-CTP-labelled 1518-HaeIII RNA probe in the absence (lane 3) or presence of 50 ng (lanes 4, 8 and 12), 100 ng (lanes 4, 9 and 13), 200 ng (lanes 6, 10 and 14) and 400 ng (lanes 7, 11 and 15) oligo-U (lanes 4–7), oligo-dCT (lanes 8–11) and oligo-dCA (lanes 12–15). To show the presence of PTB in Complex II, 1 μg isotype control immunoglobulin G (lane 16) or 1 μg anti-PTB monoclonal antibody (lane 17) was added to the binding reaction 1 hr before the addition of probe. Lanes 1 and 2 show the E5-HaeIII probe alone and with RNase mix in the absence of extract, respectively. (b) Ultraviolet cross-linking competition assay using an in vitro transcribed, 32P-CTP-labelled E5-HaeIII RNA probe in the presence of increasing amounts of unlabelled competitor RNA. The assay was conducted in the absence (lane 2) and presence (lanes 3–15) of 20 μg Jurkat/D1.1 cytoplasmic extract and RNase mix. Reactions were incubated with 25 ng (lanes 4, 8 and 12), 50 ng (lanes 5, 9 and 13), 100 ng (lanes 6, 10 and 14) and 200 ng (lanes 7, 11 and 15) unlabelled oligo-U (lanes 4–7), oligo-dCT (lanes 8–11) and oligo-dCA (lanes 12–15). Arrows indicate four specific RNA-binding proteins. Lane 1 shows the E5-HaeIII probe alone in the absence of RNase, extract, and competitor RNA. Asterisks indicate non-specific RNA binding proteins. (c) R-EMSA using an in vitro transcribed 1518-HaeIII probe in the presence of 20 μg cytoplasmic extract from Jurkat/D1.1 (lanes 3–5) and incubated with either no antibody (lane 3), 1 μg control immunoglobulin G (lane 4) or 1 μg anti-hnRNPL antibody (lane 5) before addition of the probe. Lane 2 shows reaction products from probe incubated in the presence of RNase mix in the absence of extract.
The CD40L stability element increases the expression of Renilla luciferase
To characterize the functional capacity of the CD40L stability element we established a luciferase-based expression system in which the binding sites were introduced individually or together into the 3′ UTR of the Renilla luciferase operon contained within the pRLSV40 vector (termed, pRLABC) (Fig. 4a). To control for transfection efficiency, an equal amount of the firefly luciferase-expressing vector, pGL2, was included in each transfection. The constructs were transiently transfected into Jurkat/D1.1 cells and luciferase activity was measured 48 hr later. As shown in Fig. 4(b), insertion of the three regions increased activity approximately threefold over the unmodified construct suggesting that the introduced sequence was having positive effects either on message stability or translation. Transfection of the pRL-ΔA and pRL-ΔC constructs increased luciferase activity to levels that were 3·9 times and 3·1 times that of the control plasmid, respectively, whereas activity of the pRL-ΔB construct consistently fell below control activity. Experiments using the single B region construct showed, on average, a 4·5-fold higher increase in activity and the single A and C regions had no stimulatory effect on Renilla activity. In contrast, the minimal A Site (1348-E1, defined above) increased luciferase activity approximately twofold over pRL-A, indicating that sequences upstream of the minimal A site (1300 and 1348) were negatively affecting luciferase activity at either the post-transcriptional or translational levels.
Figure 4.

Differential activity of the Complex I/II binding sites. (a) Schematic representations are shown of the different CD40 ligand (CD40L) 3′ untranslated regions that were cloned into the pRLSV40 plasmid between the XbaI and the simian virus 40 (SV40) late poly(A) site. (b) 2·5 μg of the different constructs were transiently transfected into 5 × 106 Jurkat/D1.1 cells with 15 μg pGL2 control plasmid and assayed for Renilla and firefly luciferase activity 48 hr post-transfection. The results represent the average of a minimum of three independent transfections and show the fold induction over the activity of the unmodified pRLSV40 plasmid.
The CD40L XbaI–HaeIII region stabilizes the Renilla luciferase mRNA
To determine if the XbaI–HaeIII region was affecting message turnover, stably transfected Jurkat/D1.1 cell lines were generated that contained relatively equal numbers of pRLABC and pRLSV40 integrations (data not shown). These cell lines were treated with DRB and RNA decay was measured by quantitative PCR over an 8-hr time–course. As shown in Fig. 5(a), there is a reproducible increase in the stability of the Renilla luciferase message in transcripts containing the CD40L stability region (solid line) compared to those lacking it (dotted line). This suggested that the increase in luciferase activity observed with the pRLABC construct was directly related to an increase in RNA levels mediated by enhanced stability of the transcript. Experiments were carried out to establish if the effect of Complex I binding was solely at the level of mRNA stability or whether there was an increase in transcript association with the ribosomes. Cytoplasmic extracts of stable Jurkat/D1.1 subclones expressing the Renilla luciferase transcript, either with or without the ABC regions, were fractionated by high-speed centrifugation into ribosome-associated and unbound fractions. A third fraction was collected that represented transcripts that were removed from the ribosome fraction with a high-salt wash. RNA was isolated and quantitative PCR was carried out to determine the percentage of Renilla luciferase transcript associated with each fraction. In three independent experiments there was no major change in distribution of Renilla mRNA, with or without the stability element, in the different subcellular fractions (Fig. 5b). The efficiency of cell fractionation was monitored by the distribution of 28S RNA into the ribosome-associated fraction. Thus, our results indicated that sequences within the XbaI–HaeIII region stabilize the CD40L mRNA but do not necessarily increase transcript association with translating ribosomes.
Figure 5.

The XbaI–HaeIII site acts as a stability element within a heterologous transcript. (a) Stable subclones of Jurkat/D1.1 cells expressing Renilla luciferase with (solid line) or without (broken line) the CD40 ligand (CD40L) XbaI–HaeIII stability region were analysed for messenger RNA stability by the addition of 50 μg/ml 5,6-dichlorobenzimidizole 1-m-d-ribofuranoside (DRB) over an 8-hr time course. Cells (5 × 106) were collected for each time-point, RNA was extracted and triplicate samples were quantified using quantitative polymerase chain reaction (qPCR). The fractions of total RNA remaining at each time-point are plotted. Results are the average of three independent experiments and curve-fitting was performed by non-linear regression. (b) Cytoplasmic extracts of stable subclones of Jurkat/D1.1 cells expressing Renilla luciferase with (dark rectangles, pRLABC) and without (light rectangles, pRLSV40) the CD40L XbaI–HaeIII region were fractionated into polysome-associated and non-associated fractions by centrifugation at 130 000 g. The pellet was further washed with high salt to release loosely associated proteins. Analysis of Renilla luciferase RNA (left graph) and 28S RNA (right graph) was carried out using qPCR.
The B site acts as a bona fide stability element
As shown in Fig. 4(b), removing Site B from the ABC region had a strong negative effect that resulted in a markedly lower level of luciferase activity compared to both the pRLABC and the pRLSV40 parent constructs. To explore whether the effect of this deletion was at the level of RNA decay, Jurkat/D1.1 cells were transiently transfected with either pRLABC or pRL-ΔB, treated with DRB for different time periods, and Renilla RNA decay was assayed by semi-quantitative and quantitative PCR (Fig. 6a,b). Notably, removal of the B binding site within the XbaI–HaeIII region resulted in the Renilla luciferase RNA being highly unstable. To rule out the possibility that removing the B site created a new endonuclease target site by bringing together two non-adjacent regions of the RNA, a construct was engineered that inserted a 65-base-pair sequence (nt 1589–1655) between the A and the C regions (termed pRL-D). Before cloning, this sequence was tested in R-EMSA and displayed no RNA-binding activity (data not shown). When the pRL-D construct was introduced into Jurkat/D1.1 cells a very low level of luciferase activity was observed that was similar to that of the pRL-ΔB construct (Fig. 6c). Also, the pRL-D transcript was less stable than the pRLABC transcript (data not shown). Because our transfection data suggested that there was a negative regulatory element in the A region (compare luciferase activity between pRL-A and 1348-E1, Fig. 4b) we engineered a construct (pRL-A5′) with only the sequences in the A region between nt 1300 and 1368 and tested its effect on RNA stability. In support of our prediction, insertion of these sequences resulted in a very low overall level of luciferase (Fig. 6c) and rapid degradation of the Renilla transcript (Fig. 6d). Therefore, our results support a model whereby both positive and negative regulatory elements are located within the XbaI–HaeIII region and that the B site specifically acts as a ‘stability’ element by obscuring an instability site in the A region.
Figure 6.

Site B is a bona fide stability element. (a) Jurkat/D1.1 cells were transiently transfected with either pRLABC (top panel) or pRL-ΔB (lower panel) and treated with 5,6-dichlorobenzimidizole 1-m-d-ribofuranoside (DRB) over a 4-hr time–course. Total RNA was isolated from cells treated with DRB for 8 hr and Renilla luciferase (lanes 1–5) or control β-actin (lanes 6–10) expression was analysed by semi-quantitative reverse transcription–polymerase chain reaction (RT-PCR). The RT-PCR was conducted with a concentration of complementary DNA that was previously determined to be in the linear range of amplification. (b) Time-points from three independent transfections were averaged and the SEM was calculated. Curve-fitting was performed by non-linear regression. (c) Schematic representation of the pRL-D construct and a graph of the fold induction of Renilla luciferase activity for each construct over the control pRLSV40 plasmid. Levels of activity for each construct were normalized to the firefly luciferase activity. Results represent the average of at least three independent experiments ± SEM. (d) Schematic representation of the pRL-A5′ construct and DRB assays using RNA from transiently transfected cells expressing pRL-1348-E1, pRLSV40 and pRL-A5′. RNA was isolated at four different time-points. The results represent the average ± SEM of two independent transfections.
Discussion
Our current findings extend previous work by identifying the minimal binding sites for Complexes I and II in the 3′ UTR of CD40L and showing that hnRNPL also binds to sequences within the third binding site. Furthermore, the B region, which had been previously identified as the minimum Complex I binding site,40 conferred the highest level of transcript stability. Importantly, our work begins to uncover a mechanism for CD40L stability by demonstrating that the central B region protects the transcript from the rapid degradation that is initiated by the A region.
In this study the minimum Complex I binding site in region A was identified as 62 nt of highly CU-rich sequence at the 3′ end adjacent to region B. In fact, a core region of 36 nt beginning at 1369 and extending to 1409 displayed 86% identity with a 36-residue CU-rich core in the B region located between nt 1429 and 1464. We hypothesize that the twofold difference in luciferase response of the pRL-A and pRL-1348-E1 constructs (1·0 and 2·3, respectively) reflects the delineation of a bona fide PTB complex binding site within the CU-rich core. Also, our findings with region A support a model whereby the nt 1300–1348 region, upstream of the CU-rich region contains sequences that destabilize the transcript. Among the different transcripts analysed by luciferase assay, the pRLΔB and pRL-D transcripts gave unusually low levels of activity. In fact the presence of the XbaI–HaeIII region lacking the region B PTB binding site (pRLΔB), markedly decreased the stability of the Renilla luciferase transcript, implying that other sequences within the XbaI–HaeIII region contributed to the rapid decay of the pRLΔB transcript. Reconciling these data with the decreased activity conferred by the pRL-A construct and the fourfold increase in activity provided by the A+B regions strongly indicated that deleting the PTB binding site within the B region exposed a putative ‘instability’ element in the A region. Our results with the pRL-A5′ construct, showing both very reduced Renilla activity and message stability, support this model. This type of scenario is similar to that of an identified endonuclease site in the α-globin 3′ UTR. Erythroid cell-enriched endoribonuclease activity is regulated by the α-globin poly(C)binding protein (αCP) complex that binds to a stability element in the 3′ UTR of the α-globin mRNA.50 Sequestration of either the poly(A)-binding protein or αCP results in the exposure of the endonuclease site and rapid degradation of the mRNA.50,51
Analysis of region C revealed that hnRNPL was binding to the CA-repeat and that both PTB and hnRNPL were components of Complex II. Unlike Complex I, where super-shifting was observed with antibodies to PTB, antibodies to either PTB or hRNPL resulted in the complete loss of Complex II formation. However, UV cross-linking studies using the entire C region probe and competition oligos suggest that under conditions where either PTB or hnRNPL is competed from the RNA, the other protein can still efficiently bind to the same transcript. Together these results suggested that PTB and hnRNPL are capable of binding to region C independently; however, both proteins are required for Complex II formation. Only when the CU-rich core is deleted does a second, stronger complex bind, which we would predict based on our super-shift assays is composed of hnRNPL alone. This interaction is similar to what has been previously reported showing that hnRNPL and PTB heterodimerize52,53 and bind to the murine inducible nitric oxide synthase 3′ UTR upon glucocorticoid activation.54
In conclusion, we have shown that the CD40L A+B+C region functions on its own in vivo as a stability element to redirect the turnover of a heterologous transcript. We have also independently analysed both the first and third stability regions and characterized proteins that bind to specific sites. Establishing a better understanding of the mechanisms that regulate CD40L gene expression throughout a time–course of T-cell activation will help elucidate the role that post-transcriptional mechanisms have in regulating global gene expression during an ongoing immune response.
Acknowledgments
We thank all members of the Covey laboratory for helpful discussions and critical comments on the manuscript. This work was supported by a grant from The National Institutes of Health (PO1 AI-57596) and a Grant-in-Aid from The American Heart Association to L.R.C. as well as a postdoctoral fellowship from the New Jersey Commission on Cancer Research to J.F.P.
References
- 1.Banchereau J, Bazan F, Blanchard D, et al. The CD40 antigen and its ligand. Annu Rev Immunol. 1994;12:881–922. doi: 10.1146/annurev.iy.12.040194.004313. [DOI] [PubMed] [Google Scholar]
- 2.Kawabe T, Naka T, Yishida K, et al. The immune response in CD40-deficient mice: impaired immunoglobulin class switching and germinal center formation. Immunity. 1994;1:167–78. doi: 10.1016/1074-7613(94)90095-7. [DOI] [PubMed] [Google Scholar]
- 3.Xu J, Foy TM, Laman JD, et al. Mice deficient for the CD40 ligand. Immunity. 1994;1:423–31. doi: 10.1016/1074-7613(94)90073-6. [DOI] [PubMed] [Google Scholar]
- 4.Aruffo A, Farrington M, Hollenbaugh D, et al. The CD40 ligand, gp39, is defective in activated T cells from patients with X-linked immunodeficiency with hyper-IgM. Cell. 1993;72:291–300. doi: 10.1016/0092-8674(93)90668-g. [DOI] [PubMed] [Google Scholar]
- 5.Korthauer U, Graf D, Mages HW, et al. Defective expression of T-cell CD40 ligand causes X-linked immunodeficiency with hyper-IgM. Nature. 1993;361:539–41. doi: 10.1038/361539a0. [DOI] [PubMed] [Google Scholar]
- 6.Allen RC, Armitage RJ, Conley ME, et al. CD40 ligand gene defects responsible for X-linked hyper-IgM syndrome. Science. 1993;259:990–3. doi: 10.1126/science.7679801. [DOI] [PubMed] [Google Scholar]
- 7.Fuleihan R, Ramesh N, Loh R, et al. Defective expression of the CD40 ligand in X-chromosome-linked immunoglobulin deficiency with normal or elevated IgM. Proc Nat Acad Sci USA. 1993;90:2170–3. doi: 10.1073/pnas.90.6.2170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Durandy A, Schiff C, Bonnefoy JY, Forveille M, Rousset F, Mazzei G, Milili M, Fischer A. Induction by anti-CD40 antibody or soluble CD40 ligand and cytokines of IgG, IgA, and IgE production by B cells from patients with X-linked hyper-IgM syndrome. Eur J Immunol. 1993;23:2294–9. doi: 10.1002/eji.1830230936. [DOI] [PubMed] [Google Scholar]
- 9.Foy TM, Aruffo A, Bajorath J, Buhlmann JE, Noelle RJ. Immune regulation by CD40 and its ligand GP39. Annu Rev Immunol. 1996;14:591–617. doi: 10.1146/annurev.immunol.14.1.591. [DOI] [PubMed] [Google Scholar]
- 10.Noelle RJ. CD40 and its ligand in host defense. Immunity. 1996;4:415–9. doi: 10.1016/s1074-7613(00)80408-2. [DOI] [PubMed] [Google Scholar]
- 11.Grewal IS, Flavell RA. CD40 and CD154 in cell-mediated immunity. Annu Rev Immunol. 1998;16:111–35. doi: 10.1146/annurev.immunol.16.1.111. [DOI] [PubMed] [Google Scholar]
- 12.Larsen CP, Elwood ET, Alexander DZ, et al. Long-term acceptance of skin and cardiac allografts after blocking CD40 and CD28 pathways. Nature. 1996;381:434–8. doi: 10.1038/381434a0. [DOI] [PubMed] [Google Scholar]
- 13.Hancock WW, Sayegh MH, Zheng XG, Peach R, Linsley PS, Turka LA. Costimulatory function and expression of CD40 ligand, CD80, and CD86 in vascularized murine cardiac allograft rejection. Proc Nat Acad Sci USA. 1996;93:13967–72. doi: 10.1073/pnas.93.24.13967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kirk AD, Harlan DM, Armstrong NN, et al. CTLA4-Ig and anti-CD40 ligand prevent renal allograft rejection in primates. Proc Nat Acad Sci USA. 1997;94:8789–94. doi: 10.1073/pnas.94.16.8789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mach F, Schonbeck U, Sukhova GK, Atkinson E, Libby P. Reduction of atherosclerosis in mice by inhibition of CD40 signaling. Nature. 1998;394:200–3. doi: 10.1038/28204. [DOI] [PubMed] [Google Scholar]
- 16.Lane P, Traunecker A, Inui S, Lanzavecchia A, Gray D. Activated human T cells express a ligand for the human B cell-associated antigen CD40 which participates in T cell-dependent activation of B lymphocytes. Eur J Immunol. 1992;22:2573–8. doi: 10.1002/eji.1830221016. [DOI] [PubMed] [Google Scholar]
- 17.Lederman S, Yellin MJ, Krichevsky A, Belko J, Lee JJ, Chess L. Identification of a novel surface protein on activated CD4+ T cells that induces contact-dependent B cell differentiation (Help) J Exp Med. 1992;175:1092–101. doi: 10.1084/jem.175.4.1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Castle BE, Kishimoto K, Stearns C, Brown ML, Kehry MR. Regulation of expression of the ligand for CD40 on T helper lymphocytes. J Immunol. 1993;151:1777–88. [PubMed] [Google Scholar]
- 19.de Boer M, Kasran A, Kwekkeboom J, Walter H, Vandenberghe P, Ceupens JL. Ligation of B7 with CD28/CTLA-4 on T cells results in CD40 ligand expression, interleukin-4 secretion and efficient help for antibody production by B cells. Eur J Immunol. 1993;23:3120–5. doi: 10.1002/eji.1830231212. [DOI] [PubMed] [Google Scholar]
- 20.Hermann P, Blanchard D, de Saint-Vis B, et al. Expression of a 32-kDa ligand for the CD40 antigen on activated human T lymphocytes. Eur J Immunol. 1993;23:961–4. doi: 10.1002/eji.1830230430. [DOI] [PubMed] [Google Scholar]
- 21.Klaus SJ, Pinchuk LM, Ochs HD, Law C-L, Fanslow WC, Armitage RJ, Clark EA. Costimulation through CD28 enhances T cell-dependent B cell activation via CD40-CD40L interaction. J Immunol. 1994;152:5643–52. [PubMed] [Google Scholar]
- 22.Yellin MJ, Sippel K, Inghirami G, et al. CD40 molecules induce down-modulation and endocytosis of T cell surface T cell-B cell activating molecule/CD40-L. J Immunol. 1994;152:598–608. [PubMed] [Google Scholar]
- 23.van Kooten C, Gaillard C, Galizzi J-P, Hermann P, Fossiez F, Banchereau J, Blanchard D. B cells regulate expression of CD40 ligand on activated T cells by lowering the mRNA level and through the release of soluble CD40. Eur J Immunol. 1994;24:787–92. doi: 10.1002/eji.1830240402. [DOI] [PubMed] [Google Scholar]
- 24.Nusslein HG, Frosch K-H, Woith W, Lane P, Kalden JR, Manger B. Increase of intracellular calcium is the essential signal for the expression of CD40 ligand. Eur J Immunol. 1996;26:846–50. doi: 10.1002/eji.1830260418. [DOI] [PubMed] [Google Scholar]
- 25.Ludewig B, Henn V, Schroder JM, Graf D, Kroczek RA. Induction, regulation, and function of soluble TRAP (CD40 ligand) during interaction of primary CD4+ CD45RA+ T cells with dendritic cells. Eur J Immunol. 1996;26:3137–43. doi: 10.1002/eji.1830261246. [DOI] [PubMed] [Google Scholar]
- 26.Jaiswal AI, Dubey C, Swain SL, Croft M. Regulation of CD40 ligand expression on naive CD4 T cells: a role for TCR but not co-stimulatory signals. Int Immunol. 1996;8:275–85. doi: 10.1093/intimm/8.2.275. [DOI] [PubMed] [Google Scholar]
- 27.Kwekkeboom J, de Rijk D, Kasran A, Barcy S, de Groot C, de Boer M. Helper effector function of human T cells stimulated by anti-CD3 mAb can be enhanced by co-stimulatory signals and is partially dependent on CD40-CD40 ligand interaction. Eur J Immunol. 1994;24:508–17. doi: 10.1002/eji.1830240303. [DOI] [PubMed] [Google Scholar]
- 28.Lindgren H, Axcrona K, Leanderson T. Regulation of transcriptional activity of the murine CD40 ligand promoter in response to signals through TCR and the costimulatory molecules CD28 and CD2. J Immunol. 2001;166:4578–85. doi: 10.4049/jimmunol.166.7.4578. [DOI] [PubMed] [Google Scholar]
- 29.Parra E, Mustelin T, Dohlsten M, Mercola D. Identification of a CD28 response element in the CD40 ligand promoter. J Immunol. 2001;166:2437–43. doi: 10.4049/jimmunol.166.4.2437. [DOI] [PubMed] [Google Scholar]
- 30.Fuleihan R, Ramesh N, Horner A, et al. Cyclosporin A inhibits CD40 ligand expression in T lymphocytes. J Clin Invest. 1994;93:1315–20. doi: 10.1172/JCI117089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schubert LA, King G, Cron RQ, Lewis DB, Aruffo A, Hollenbaugh D. The human gp39 promoter. J Biol Chem. 1995;270:29624–7. doi: 10.1074/jbc.270.50.29624. [DOI] [PubMed] [Google Scholar]
- 32.Tsytsykova AV, Tsitsikov EN, Geha RS. The CD40L promoter contains nuclear factor of activated T cells-binding motifs which require AP-1 binding for activation of transcription. J Biol Chem. 1996;271:3763–70. doi: 10.1074/jbc.271.7.3763. [DOI] [PubMed] [Google Scholar]
- 33.Schubert LA, Cron RQ, Cleary AM, et al. A T cell-specific enhancer of the human CD40 ligand gene. J Biol Chem. 2002;277:7386–95. doi: 10.1074/jbc.M110350200. [DOI] [PubMed] [Google Scholar]
- 34.Srahna M, Remacle JE, Annamalai K, Pype S, Huylebroeck D, Boogaerts MA, Vandenberghe P. NF-kappaB is involved in the regulation of CD154 (CD40 ligand) expression in primary human T cells. Clin Exp Immunol. 2001;125:229–36. doi: 10.1046/j.1365-2249.2001.01601.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Huan C, Kelly ML, Steele R, Shapira I, Gottesman SR, Roman CA. Transcription factors TFE3 and TFEB are critical for CD40 ligand expression and thymus-dependent humoral immunity. Nat Immunol. 2006;7:1082–91. doi: 10.1038/ni1378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Suarez A, Mozo L, Gayo A, Zamorano J, Gutierrez C. Requirement of a second signal via protein kinase C or protein kinase A for maximal expression of CD40 ligand. Involvement of transcriptional and posttranscriptional mechanisms. Eur J Immunol. 1997;27:2822–9. doi: 10.1002/eji.1830271112. [DOI] [PubMed] [Google Scholar]
- 37.Ford GS, Yin CH, Barnhart B, Sztam K, Covey LR. CD40 ligand exerts differential effects on the expression of Iγ transcripts in subclones of an IgM+ human B cell lymphoma line. J Immunol. 1998;160:595–605. [PubMed] [Google Scholar]
- 38.Rigby WF, Waugh MG, Hamilton BJ. Characterization of RNA binding proteins associated with CD40 ligand (CD154) mRNA turnover in human T lymphocytes. J Immunol. 1999;163:4199–206. [PubMed] [Google Scholar]
- 39.Murakami K, Ma W, Fuleihan R, Pober JS. Human endothelial cells augment early CD40 ligand expression in activated CD4+ T cells through LFA-3-mediated stabilization of mRNA. J Immunol. 1999;163:2667–73. [PubMed] [Google Scholar]
- 40.Barnhart B, Kosinski PA, Wang Z, Ford GS, Kiledjian M, Covey LR. Identification of a complex that binds to the CD154 3′ untranslated region: implications for a role in message stability during T cell activation. J Immunol. 2000;165:4478–86. doi: 10.4049/jimmunol.165.8.4478. [DOI] [PubMed] [Google Scholar]
- 41.Ford GS, Barnhart B, Shone S, Covey LR. Regulation of CD154 (CD40 ligand) mRNA stability during T cell activation. J Immunol. 1999;162:4037–44. [PubMed] [Google Scholar]
- 42.Lindsten T, June CH, Ledbetter JA, Stella G, Thompson CB. Regulation of lymphokine messenger RNA stability by a surface-mediated T cell activation pathway. Science. 1989;244:339–43. doi: 10.1126/science.2540528. [DOI] [PubMed] [Google Scholar]
- 43.Chen C-Y, Gatto-Konczak FD, Wu Z, Karin M. Stabilization of interleukin-2 mRNA by the c-Jun NH2-terminal kinase pathway. Science. 1998;280:1945–9. doi: 10.1126/science.280.5371.1945. [DOI] [PubMed] [Google Scholar]
- 44.Kosinski PA, Laughlin J, Singh K, Covey LR. A complex containing polypyrimidine tract-binding protein is involved in regulating the stability of CD40 ligand (CD154) mRNA. J Immunol. 2003;170:979–88. doi: 10.4049/jimmunol.170.2.979. [DOI] [PubMed] [Google Scholar]
- 45.Hamilton BJ, Genin A, Cron RQ, Rigby WF. Delineation of a novel pathway that regulates CD154 (CD40 ligand) expression. Mol Cell Biol. 2003;23:510–25. doi: 10.1128/MCB.23.2.510-525.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Singh K, Laughlin J, Kosinski PA, Covey LR. Nucleolin is a second component of the CD154 mRNA stability complex that regulates mRNA turnover in activated T cells. J Immunol. 2004;173:976–85. doi: 10.4049/jimmunol.173.2.976. [DOI] [PubMed] [Google Scholar]
- 47.Yellin MJ, Lee JJ, Chess L, Lederman S. A human CD4-leukemic subclone with contact-dependent helper function. J Immunol. 1991;147:3389–95. [PubMed] [Google Scholar]
- 48.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-delta delta C(T)) method. Methods. 2001;25:402–8. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 49.Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001;29:e45. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wang Z, Kiledjian M. Identification of an erythroid-enriched endoribonuclease activity involved in specific mRNA cleavage. EMBO J. 2000;19:295–305. doi: 10.1093/emboj/19.2.295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wang Z, Kiledjian M. The poly(A)-binding protein and an mRNA stability protein jointly regulate an endoribonuclease activity. Mol Cell Biol. 2000;20:6334–41. doi: 10.1128/mcb.20.17.6334-6341.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hahm B, Cho OH, Kim JE, Kim YK, Kim JH, Oh YL, Jang SK. Polypyrimidine tract-binding protein interacts with HnRNP L. FEBS Lett. 1998;425:401–6. doi: 10.1016/s0014-5793(98)00269-5. [DOI] [PubMed] [Google Scholar]
- 53.Dumitru CD, Ceci JD, Tsatsanis C, et al. TNF-alpha induction by LPS is regulated posttranscriptionally via a Tpl2/ERK-dependent pathway. Cell. 2000;103:1071–83. doi: 10.1016/s0092-8674(00)00210-5. [DOI] [PubMed] [Google Scholar]
- 54.Soderberg M, Raffalli-Mathieu F, Lang MA. Regulation of the murine inducible nitric oxide synthase gene by dexamethasone involves a heterogeneous nuclear ribonucleoprotein I (hnRNPI) dependent pathway. Mol Immunol. 2007;44:3204–10. doi: 10.1016/j.molimm.2007.01.029. [DOI] [PubMed] [Google Scholar]