

Summary
To intoxicate host cells, cholera toxin (CT) holotoxin must be activated. This process requires the intracellular dissociation of the enzymatic CTA1 domain from the holotoxin components CTA2 and B5, followed by subsequent interaction with the host factor ARF6-GTP. We report the first NMR-based solution structural data for the Cholera Toxin enzymatic domain (CTA1). We show that this free enzymatic domain partially unfolds at the C-terminus, and binds its protein partners at both the beginning and end of this activation process. Deviations from random coil chemical shifts (Δδcoil) indicate helix formation in the activation loop, which is essential to open the toxin’s active site, occurs prior to its association with human protein ARF6. We performed NMR titrations of both free CTA1 and an active CTA1:ARF6-GTP complex with NAD+, which revealed that the formation of the complex does not significantly enhance NAD+ binding. Partial unfolding of CTA1 is further illustrated by using bis-ANS fluorescence as an indicator of the exposed hydrophobic character of the free enzyme, which is substantially reduced when bound to ARF6-GTP. We propose that the primary role of ARF6’s allostery is to induce refolding of the C-terminus of CTA1. Thus, as a folded globular toxin complex, CTA1 escapes the chaperone and proteasomal degradation machinery in the cytosol and then proceeds to ADP-ribosylate its target Gsα, triggering the downstream events associated with the pathophysiology of cholera.
Keywords: NMR, cholera toxin, ARF6, partial unfolding, allostery
Introduction
Cholera Toxin (CT) is an intracellular-acting toxin responsible for much of the pathophysiology associated with cholera, a pandemic disease caused by Vibrio cholerae (1;2). The massive rice-water diarrhea, which is the hallmark of cholera, occurs only after CT enters intestinal epithelial cells and ADP-ribosylates the -subunit of the adenylate cyclase regulatory protein, Gs (3–6). Analogous to other intracellular-acting toxins (7), the αpotency of CT and other members of the AB5 toxin family is dependent on the activation of the toxin’s enzymatic domain during the cellular entry process (8;9).
The CT holotoxin crystal structure (10) is a non-covalent AB5 complex assembled from two distinct polypeptides, an A subunit (CTA1A2) and homo-pentameric B subunits (B5). CTA1A2 is a 240 amino acid (27.2 kDa) polypeptide comprised of CTA1 (residues N1-R192), the amino-terminal ADP ribosyltransferase/NAD+ glycohydrolase enzymatic domain (3;4) and CTA2 (residues S193-E240), the carboxyl-terminal helical domain. The enzymatic domain consists of three regions: CTA11 (residues N1-N142), a globular region of mixed β-sheet and -helices; region CTA12 (residues R143-G161), that forms an extended bridge; and CTA13 (residues Y162-R192), a largely hydrophobic αregion of non-regular structure (11).
CTA1 and CTA2 are covalently linked by a protease-labile loop and a disulfide bond between C187 and C199 (10). Dissociation of CTA1 from CTA2 requires both proteolytic cleavage between R192 and S193 and reduction of the C187–C199 disulfide bond (12). The CTA2 domain forms a helical structure along the interface with CTA1, with the C-terminal end protruding through the central pore of the B5 subunits. This anchors the enzymatic CTA1 domain to the B5 subunits through non-covalent interactions.
During the process of internalization into the host cell and activation, CTA1 dissociates from the B5 and CTA2 subunits and by retrotranslocation is then transported as the active fragment from the endoplasmic reticulum (ER) to the cytosol (8;13–19). Entry of the holotoxin into the host cell is initiated through the interaction of the B5 with the oligosaccharide domain of a membrane glycolipid receptor GM1, which triggers endocytosis and trafficking of the holotoxin to the ER (8;20). Dissociation of the B5 subunits exposes a C-terminal KDEL motif within CTA1A2 which is recognized by the receptor ERD2 and promotes trafficking from the Golgi apparatus to the ER (8;10). Recent studies indicate that mutations to the KDEL sequence abolish transport of CTA1A2 into the ER (21). Reduction of the disulfide bond and dissociation of CTA1 from CTA2 in the ER is catalyzed by protein disulfide isomerase (PDI), and transport of CTA1 to the cytosol occurs via the protein-conducting channel Sec61 (22) and other components of the ER-associated degradation (ERAD) pathway (18;19). Additionally, studies have suggested that, while the free enzyme is recognized as a misfolded protein by the ERAD pathway (17;23), a paucity of lysines in CTA1 minimizes its ubiquitination and subsequent proteasomal degradation (24).
CT is a member of a growing family of intracellular-acting bacterial toxins and exo-enzymes whose activity is regulated by protein-protein interactions with host cell factors (7;9;25). The ADP-ribosyltransferase activity of CTA1 in the cytosol is enhanced by protein-protein interactions with the GTP-bound form of eukaryotic adenosine diphosphate (ADP)-ribosylation factor 6 (ARF6) to form a CTA1:ARF6-GTP complex (23;26). The crystal structure of the CTA1:ARF6-GTP complex shows that the ARF6-GTP binding site overlaps with the site vacated by CTA2 prior to retrotranslocation (27). Differences in the crystal structure data for the holotoxin and the CTA1:ARF6-GTP complex (27) have led to the model that ARF6-GTP binding elicits dramatic changes in CTA1 loop regions, exposing the toxin’s active site. This suggests ARF6-GTP functions as an allosteric activator of CTA1’s active site (27). However, understanding how free CTA1 functions as an intermediate species between the holotoxin and the CTA1:ARF6-GTP complex will explain the activation mechanism of CT.
The mechanism underlying ARF6-GTP stimulation of CTA1 enzymatic activity remains poorly understood in part because of the lack of structural information for “free” CTA1 (e.g. not associated within CTA2/B5 or with ARF6-GTP). Structural studies have been challenging because CTA1 is prone to aggregation and resists crystallization (27; 28). To circumvent these challenges, we used nuclear magnetic resonance (NMR) spectroscopic and biophysical methods to probe structural features of the CTA1 enzymatic domain that underlie its activation mechanism.
Results
Recombinant CTA1 binds both ARF6-GTP and CTA2
In order to evaluate structural differences in CTA1 in the absence and presence of ARF6-GTP and CTA2, we over-expressed recombinant forms of CTA1 (Supplementary Figure 1) as recombinant proteins in Escherichia (E.) coli. We expressed 15N-labeled recombinant CTA1 (N1-G186), which was truncated at G186 to remove the free thiol group of C187; CTA1 partitioned primarily into inclusion bodies. After solubilization in 6 M guanidine (Gdm)-HCl, the protein was dialyzed against native buffer and then eluted as a monomeric peak from preparative Gel Filtration FPLC chromatography (Figure 1a). During optimization of our refolding protocol, we found that a considerably larger fraction of CTA1 eluted from the gel filtration column as a higher molecular-mass species in urea-solubilized samples (Supplementary Figure 2) than in Gdm-HCl-solubilized samples. Moreover, we consistently detected a dimeric shoulder peak in the urea-solubilized samples. Hence, guanidine-mediated solubilization was used in further studies. Notably, the use of chemical denaturants (6 M urea) and reducing agents are routinely used in the purification of active CTA1 from the native holotoxin produced by Vibrio cholerae (29).
Figure 1. Preparative Gel Filtration Chromatographs and 1H/15N HSQC Spectra.
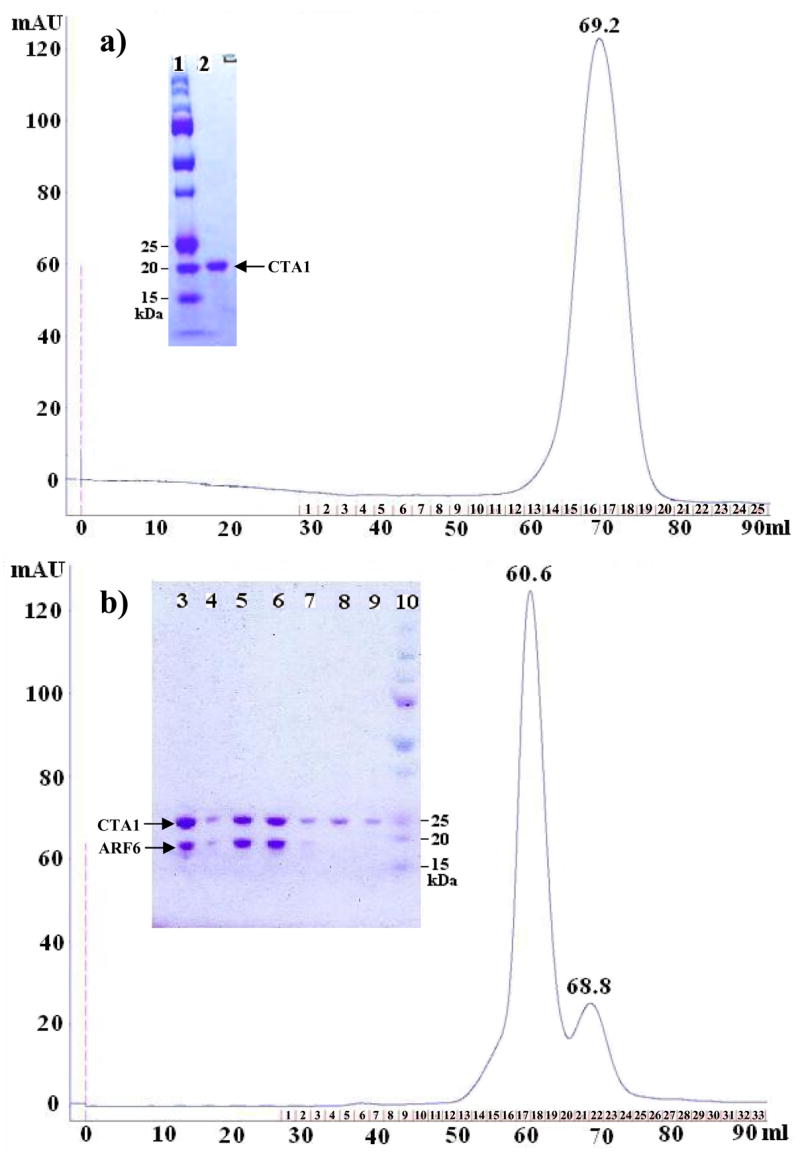
a) Refolded free CTA1 (1–186); b) CTA1:ARF6-GTP complex (13C/15N-labeled CTA1 and unlabeled ARF6), with inserts of SDS-PAGE of the Size Exclusion Chromatography fractions for each sample preparation. Lanes 1 and 10: Molecular Mass (MM) markers (Bio-Rad). Lane 2: pooled fractions of the peak eluted at 69.2 mL from the column. Lane 3: Ni-NTA purified CTA1:ARF6-GTP. Lanes 4–7: fractions 16–19; lanes 8–9: fractions 21–22. Lanes 5–6: peak at 60.6 mL, and lane 8: shoulder CTA1 peak at 68.8mL (fraction 21). The expected MM of free CTA1 (1–186) is 21.8 kDa; NhisCTA1 and ARF6 of the active complex are 23.8 kDa and 20.1 kDa, respectively, as calibrated using molecular mass standards.
We prepared a soluble form of CTA1 comprising residues N1-R192 with an N-terminal 6xHis affinity tag sequence by co-expression with ARF6 in E. coli, followed by Ni-NTA affinity purification of the CTA1:ARF6-GTP complex (27). Extended dialysis of the complex against a native EDTA buffer (to chelate Mg2+ from ARF6 upon dissociation of CTA1 and ARF6-GTP) resulted in soluble free enzyme that was then affinity purified. This approach yielded low amounts (6 μM) of free NhisCTA1 (E110:112D), but allowed us for the first time to directly compare a soluble form of CTA1 not subjected to chemical denaturants against a CTA1 sample which had been refolded from inclusion bodies. Comparative 1D NMR spectra of both the soluble expressed and inclusion body refolded CTA1 show similar profiles within their amide regions (Supplementary Figure 3). This shows that the fold adopted by CTA1 was consistent between both purification protocols, indicating that refolding from inclusion bodies generates a native-like protein analogous to the conventional method used to obtain CTA1 by dialysis of the holotoxin against 6 M urea and reducing agents (23;29).
To investigate if refolded recombinant CTA1 binds ARF6-GTP, we used an additional full-length gene construct with an N-terminal 6xHis tag (NhisCTA1, N1-R192). To form a selectively labeled CTA1:ARF6-GTP in vitro-generated active complex, we lysed unlabeled ARF6 cell paste in the presence of refolded 15N/13C-labeled NhisCTA1. Following affinity purification, gel filtration FPLC analysis of the mixture (Figure 1b) revealed a dominant species with a shorter retention time than observed for either free CTA1 or ARF6. SDS-PAGE analysis indicated this major peak was composed of an equimolar mixture of CTA1 and ARF6 (Figure 1b). Thus, our protocol purified an active CTA1:ARF6-GTP complex without the presence of the inactivating (E110:112D) mutations which are required for co-expression of both CTA1 and ARF6 in E. coli (27). An alternate protocol of separately purifying each component prior to mixing did not form a complex (data not shown), as the purified ARF6 was most likely in its inactive GDP-bound form (30).
To further validate the functional integrity of refolded recombinant CTA1, we measured the capacity of CTA1 to recognize and bind CTA2, its physiological partner in the holotoxin, by performing surface plasmon resonance studies of CTA1 (N1-G186) with a peptide corresponding to residues D201–S229 of CTA2. Binding was identified by the increase of response units (RU) in the presence of the ligand (Fig. 2a); the kinetic parameters are shown in Table 1. These data indicates tight (nM Kd) binding between the two domains, even in the absence of a disulfide bond.
Figure 2. Binding of CTA1 to CTA2 and Circular Dichroism Spectra of CTA Constructs.

a) Surface Plasmon Resonance (SPR) traces of CTA1 (200 nM) in HBS buffer pH 7.4, binding to surface amine tethered CTA2 at 21°C; fitted data is shown in red. b) Far-UV CD spectra of CTA1A2 (blue circles) and CTA1 (red diamonds) at 288 K.
Table 1.
SPR Binding Parameters of CTA1 binding CTA2 peptides
| kon (M−1 s−1) | koff (s−1) | Rmax (RU) | KD (nM) |
|---|---|---|---|
| 260,000 ± 12,000 | 0.014 ± 0.0001 | 220 ± 1 | 53 ± 4 |
Circular dichroism (CD) spectroscopy was used to estimate the secondary structure content of both the recombinant CTA1 (N1-G186) and CTA1A2 (N1-L240). The CTA1 spectrum revealed a minima (singular) at 205 nm and a weak shoulder minima at 219 nm when compared to recombinant CTA1A2 at 206 nm and 221 nm, respectively (Figure 2b). Based on the molar ellipticity at 222 nm, we calculated α-helical structure (31) to be approximately 13 % for CTA1 and 18 % for CTA1A2. Thus, when measured under the same conditions, the circular dichroism spectrum of CTA1 indicated a decreased helicity when compared to CTA1A2. This decrease in helical structure is consistent with the truncation of CTA2, which forms an extended α-helix in the holotoxin crystal structure (10). Our CD spectra of CTA1A2 closely resembles the published CD spectra for the A domain of the shiga AB5 toxin (32) and with the recently published CD spectra for CTA1 and CTA1A2 (33).
Free CTA1 is partially unfolded and refolds upon ARF6-GTP binding
The 2D NMR 1H/15N HSQC spectrum of recombinant CTA1 (N1-G186) (Figure 3a) revealed that CTA1 is partially unfolded and displays the characteristics of a molten globule in solution [for review, see (34)]. Notably, only 20% of the total amide protons resonated either up-field or down-field when compared to the random coil chemical shift region between 7.5 and 8.5 ppm (34–36). Moreover, there is considerable variation in peak intensity within the 1H/15N HSQC spectrum, likely due to intermediate exchange broadening.
Figure 3. NMR Spectral Comparison of Free CTA1 with Both the Active and Inactive CTA1:ARF6-GTP Complexes.
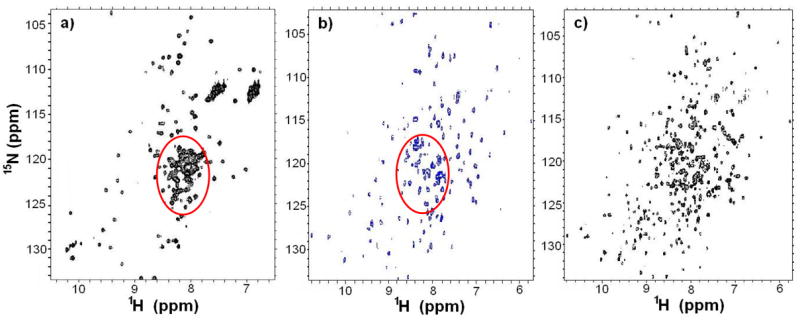
2D 1H/15N HSQC spectra of a) the full length CTA1 (N1-G186) {32 scans at 298K of a 0.2 mM 1H/15N-labeled sample}; b) the selectively 1H/15N/13C CTA1 active CTA1:ARF6-GTP complex; and c) the 15N uniformly labeled inactive CTA1:ARF6-GTP (E110:112D) complex. All spectra were acquired on a Bruker 800 MHz NMR spectrometer. The red circles in panels a) and b) indicate the increased chemical shift dispersion of the backbone resonances upon association of CTA1 with ARF6-GTP.
To evaluate whether ARF6-GTP binding is sufficient to induce the transition to a fully folded complex, we collected NMR spectra of the selectively-labeled CTA1 in the active CTA1:ARF6-GTP complex to compare against both the inactive CTA1:ARF6-GTP (E110:112D) complex (27) and free CTA1 (Figure 3). The 1H/15N HSQC spectra of wild type and mutant (E110:112D) forms of the CTA1:ARF6-GTP complex were similar, indicating that these active site residue mutations do not significantly affect the structural changes induced in CTA1 by the formation of a complex with ARF6-GTP. The 1H/15N HSQC spectra of the complex samples demonstrate better overall line shape and increased dispersion relative to the spectrum of the free CTA1 sample. Many of the resonances present in the central random-coil region of the free enzyme are dispersed in the NMR spectrum for the active complex sample where CTA1 is selectively labeled. Thus, the binding of ARF6-GTP ligand to CTA1 results in an ordering (folding) of partially unfolded regions within the free enzymatic domain, indicating an allosteric coupling between these interaction partners (37).
Unfolding within CTA1 is largely localized to the C-terminal CTA12 and CTA13 regions
Crystal structures of CT holotoxin (10) and CTA1:ARF6-GTP complex (E110:112D) (27) revealed that the CTA1 carboxyl-terminus CTA13 region (residues Y162-R192) extends away from the globular CTA11 domain. To evaluate whether the unfolded regions detected within full-length CTA1 are localized to the carboxyl-terminus, we generated and analyzed a mutant form of CTA1 without the carboxyl-terminal region, called CTA1-T2 (N1-F167). For CTA1-T2, the sequence was truncated between F167↓P168P169 as P168P169 make extensive contacts with the CTA2 linker in the holotoxin crystal structure (10) and are likely to destabilize the free enzymatic domain. Spectra of the refolded CTA1-T2 sample show similar chemical shift (δ) values and dispersion to the CTA1 sample (Supplementary Figure 4). Most of the changes in the 1H/15N HSQC spectrum corresponded to a decrease in the number of resonances within the random coil region, while leaving unaffected the dispersed resonances, suggesting that the structure of the catalytic core domain was unaffected by the truncation of the C-terminus. The 1H/15N HSQC spectra of CTA1-T4 (N1-G144) shows an additional decrease in the number of resonances in the central cluster, with only small shifts in the dispersed resonances evident (Supplementary Figure 4).
As an alternate method of exploring the partial unfolding of the C-terminus and the refolding of the CTA1 domain upon ARF6-GTP binding, we performed 4,4′-bis(1-anilinonaphthalene 8-sulfonate) (bis-ANS) fluorescence experiments. The bis-ANS fluorescence reflects the extent of hydrophobic exposure (38) of CTA1 when compared to both the CTA1:ARF6-GTP complex and Gdm-HCl denatured samples (Figure 4). These data indicate bis-ANS fluorescence of the full length CTA1 sample is reduced in the truncated CTA1-T2 sample, and further reduced in the CTA1:ARF6-GTP sample or in the presence of chemical denaturants. These results indicate a loss of hydrophobic bis-ANS binding sites upon ARF6-GTP binding to the enzymatic domain, when fully unfolded or when truncated at the C-terminus. Therefore, ARF6-GTP binding is sufficient to induce profound changes in CTA1: from a partially disordered state in the free enzyme to an ordered state within the CTA1:ARF6-GTP complex.
Figure 4. Bis-ANS Fluorescence Emission Spectra.
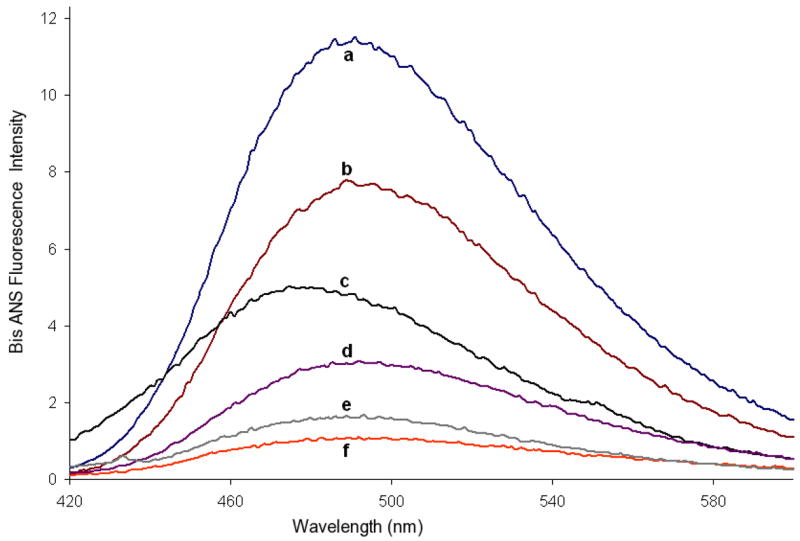
Fluorescence emission spectra of 10μM bis-ANS with a) CTA1 (N1-G186), b) CTA1-T2 (N1-F167), c) CTA1:ARF6-GTP, d) CTA1 + 2 M Gdm-HCl, e) CTA1 + 6 M Gdm-HCl, and f) bis-ANS blank. All samples were in buffer of 10μM bis-ANS, 20 mM Tris, pH 7.2 and 50 mM NaCl. Concentration of each protein sample was 10μM.
NMR sequence specific assignment of CTA1
Triple resonance NMR spectra on a 0.2 mM 2H/13C/15N uniformly labeled CTA1-T2 sample were acquired, which enabled sequential assignment of 81 % of the backbone 1H and 15N resonances (129 out of 159 non-proline amide resonances) (Supplementary Figure 5). Sample concentration was limited to 200μM, as aggregation was evident in more concentrated samples and NMR spectra were marked by enhanced line-broadening. The dispersed resonances in the 1H/15N HSQC spectra that originate from a structured core of the protein show significantly reduced peak volumes when compared to those from the random coil region (Supplementary Table 1). Structural mapping of assigned crosspeaks that show increased peak volumes indicates that they predominantly localize to residues at or near the C-terminal CTA12 and CTA13 regions (Supplementary Figure 6). Most residues that lie within the CTA11 region show significantly reduced peak volumes, which suggest that residues within a residual structured core show decreased internal motions when compared to those at or near the unstructured C-terminus.
Activation loop helix is present in both the free CTA1 and CTA1:ARF6-GTP complex
Previous crystallographic data (27) suggested that ARF6-GTP may function as an allosteric activator for the CTA1 active site by inducing a loop to helix transition for residues R25-F31 within the activation loop (residues R25-N40). This activation loop helix formation breaks an interaction between R11 and H55, leading to an opening of the active site and increased flexibility of the active site loop (28). To test this hypothesis, we evaluated secondary structure within CTA1-T2 by using 13Cα and 13Cβ chemical shift deviations from their statistical random coil values (Δδcoil) (39;40). These data indicate that the overall secondary structure of free CTA1-T2 correlates to the CTA1 domain within CTA1:ARF6-GTP crystal structure (27). Residues R25-Y30 from the activation loop show strong helical propensity, analogous to this region with the CTA1:ARF6-GTP complex, while they are non-regular in the holotoxin (10) (Figure 5). Thus, CTA1 binding to ARF6-GTP is not essential for inducing a helical transition within the activation loop, but instead may occur once the free enzyme is dissociated from CTA2.
Figure 5. Secondary Structure Mapping of CTA1-T2.

a) Weighted CSI (59) versus residue plot, illustrating the secondary structure in the free CTA1-T2 enzyme, with the activation loop helix (R25-Y30) highlighted in yellow; secondary structural elements of CTA1 that were determined from CSI as displayed on a ribbon diagram of b) the inactive holotoxin (10) (PDB# 1XTC) and c) the ARF6-GTP complex (27) (PDB# 2A5F). For each structure, the activation loop helix (highlighted in yellow) and the active site loop (R46-D57, highlighted in magenta) are labeled. All other α-helices are shown in red, β-strands are cyan, and the truncated C-terminal region is green.
The CSI data for CTA1-T2 (N1-F167) indicates approximately 40 residues (24 %) are in β-structure and 28 residues (17 %) are in helix. These data are consistent with the CTA1 (N1-G186) circular dichroism spectra, from which we calculate 24 CTA1 residues (13 %) to be helical. One notable difference between the NMR and crystallographic data is within residues E137-H140, which are predicted to be α-helical from the CSI plot (Figure 5), while this region is part of a β-strand within the CT crystal structures. This suggests there is a local structural rearrangement of these residues within the free enzyme.
Formation of the CTA1:ARF6-GTP complex does not promote NAD+ binding to CTA1
To further evaluate the role of ARF6-GTP allostery in CTA1 activation, we used NMR spectroscopy to determine whether ARF6-GTP binding increases CTA1 affinity for its substrate, NAD+. In these experiments, we monitored changes in the chemical shifts of amide protons in the 1H/15N HSQC spectrum of free CTA1-T2 or the CTA1:ARF6-GTP complex as a function of NAD+ concentration (Figure 6). The binding affinity of NAD+ to CTA1-T2 (Kd = 2.0 ± 0.3 mM) was similar to that reported earlier for CTA1 (Kd = 4.0 ± 0.4 mM (41)), while the CTA1:ARF6-GTP complex bound NAD+ with only a modest increase in affinity (Kd = 1.2 ± 0.2 mM) (Figure 6). These data show that ARF6-GTP does not function strictly as an allosteric activator of the active site to greatly increase the affinity of CTA1 for its substrate, NAD+.
Figure 6. NAD+ Substrate Titrations with Free CTA1 and Active CTA1:ARF6-GTP Complex.
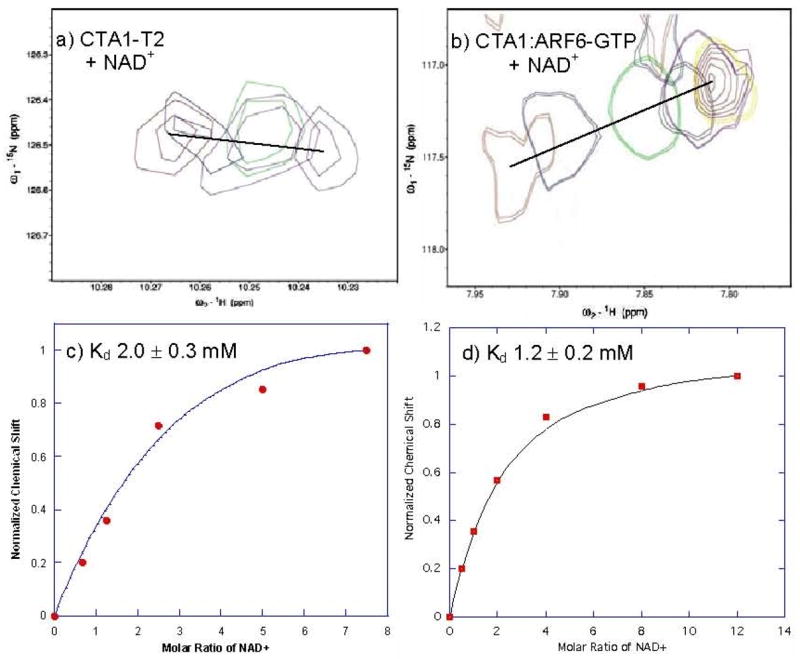
a) Overlay of peaks from residue D9 of CTA1-T2, with increasing concentrations of NAD+. b) Overlay of a selected resonance of CTA1:ARF6-GTP, with increasing concentrations of NAD+. c) Plot of normalized chemical shift as a function of the molar ratio [NAD+]:[CTA1-T2]. d) Plot of the normalized chemical shift as a function of the molar ratio of [NAD+]:[CTA1:ARF6-GTP]. Lines drawn through the points in c) and d) are best fit curves to the standard binding equation described in methods and materials. The Kd value was estimated as 2.0 ± 0.3 mM for CTA1-T2, and a Kd value of 1.2 ± 0.2 mM for CTA1:ARF6-GTP.
Discussion
This work provides a conceptual framework for understanding the structural changes that accompany the activation of cholera toxin during intoxication of host cells. Our NMR data indicate that the free CTA1 enzymatic domain is partially unfolded, in contrast to when it is complexed with either CTA2 in the holotoxin or with ARF6-GTP following retrotranslocation. Further strengthening this argument was that the refolded recombinant CTA1 is catalytically active (23) and, due to its thermal instability, CTA1 is a substrate for export to the cytosol via the ERAD pathway (33).
Teter et al. showed that C-terminal CTA13 truncation decreases catalytic activity by preventing association of CTA1 with ARF6-GTP, but does not prevent retrotranslocation from the ER to the cytosol (23). Our NMR analysis of the free enzyme indicates CTA1 remains partially unfolded, even when both the CTA13 (CTA1-T2) and the CTA12 (CTA1-T4) are truncated (Supplementary Figure 4). These data provide a rationale for why a CTA13 truncated enzymatic domain may still act as an ERAD pathway substrate, even though binding to ARF6-GTP is disrupted.
Our results also highlight the allosteric role of ARF6-GTP binding to CTA1. We show helix formation in the activation loop of the free enzyme, which suggests an opening of the active site occurs upon dissociation of CTA2, and prior to association of ARF6-GTP. A preformed active site in free enzyme also explains why NAD+ substrate binding is not greatly enhanced by the formation of a complex with ARF6-GTP (Figure 6). Additionally, binding of NAD+ did not enhance the peak dispersion within the 1H/15N HSQC spectra (data not shown), indicating that substrate binding does not induce refolding of the free enzyme. Hence, CT is unlike that of the enzymatic molten globule chorismate mutase, where substrate binding does induce conformational ordering (42). The finding that the binding affinity of NAD+ for CTA1 is nearly identical in the presence or absence of ARF6-GTP further indicates an alternative role for its host factor in enhancing the activation of CTA1. We propose that the primary role of ARF6-GTP binding is in site-to-site allostery (37) that couples the refolding of the C-terminus of CTA1 with the formation of a high-affinity CTA1:ARF6-GTP complex.
We show CTA1 displays partial unfolding, primarily at both the C-terminal CTA12 and CTA13 regions, while the active site adopts a conformation that is catalytically competent prior to ARF6-GTP binding. This allows the toxin’s free CTA1 domain to activate its catalytic site while “playing dead” so that it can recruit proteins from the host cell’s ERAD pathway that are required for retrotranslocation from the ER to the cytosol. Thus, CTA1 undergoes an ordered (holotoxin) to disordered (free CTA1) to ordered (CTA1:ARF6-GTP complex) structural transition (Figure 7) to accommodate the dual requirements of retrotranslocation and activation. This model proposes that the partially unfolded free enzyme recruits proteins from the ERAD pathway to promote its own retrotranslocation from the ER to the cytosol. Subsequently, the model predicts that CTA1 forms a globular complex with ARF6-GTP, which escapes degradation of the chaperonin/proteasomal machinery of the host cell to ADP-ribosylate its target Gsα.
Figure 7. Structural Rearrangements of CTA1.
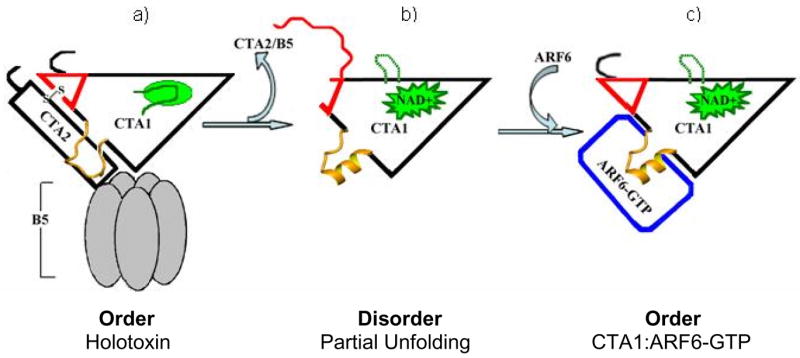
a) Order: holotoxin; with a folded CTA1 C-terminal region (red triangle), and an inactive catalytic site (yellow activation loop and green active site loop); b) Disorder: free CTA1, where the C-terminal region is disordered and the catalytic site is active; and c) Order: the CTA1:ARF6-GTP complex, with a folded CTA1 C-terminal region and an active catalytic site.
This model of order–disorder–order transitions illustrates how the formation of a complex with an intracellular host factor may represent a general mechanism for the activation of AB5 toxins. These host factors for the AB5 toxin family include: ARF6, which binds CT (15;43–45); ARF3, which binds heat labile enterotoxin (46;47); and calmodulin, which binds pertussis toxin (48;49), all of which are essential in enhancing the activity of the toxin’s enzymatic domain. Notably, disorder-order structural transitions have been increasingly demonstrated to accompany high affinity interactions between endogenous cellular protein-protein binding partners (50–52). Moreover, other intracellular acting toxins (such as the diphtheria and botulinum toxins) also adopt enzymatically active partially unfolded structures to accommodate their dual requirements of activation and membrane translocation in a host cell (7;53). Our results underscore the elegant molecular interplay that regulates the action of intracellular bacterial toxins within a host cell.
Materials and Methods
CTA subunit gene constructs
A synthetic gene construct CTA1, which encoded residues N1-G186, was inserted into the Nde I site in pET-20b(+) (Novagen, Inc.). The QuikChange® II Site-Directed Mutagenesis Kit (Stratagene) was used to generate genes encoding mutant forms of CTA1 with carboxyl-terminal truncations. CTA1-T2 and CTA1-T4 were generated by the introduction of stop codons after the F167 and G144 codons, respectively. Additionally, a synthetic CTA1A2 gene (encoding residues N1-L240) was ligated into pET-20b(+), replacing the Nde I/Hind III fragment, thus encoding a recombinant protein with a carboxyl-terminal 6xHis affinity tag sequence.
CTA1 protein expression and purification
CTA1 and CTA1-T2 were over-expressed in E. coli BL21(DE3) transformed with plasmids harboring the genes encoding these proteins. For unlabeled samples, E. coli BL21(DE3) was cultured in Luria Broth supplemented with 50 μg/mL ampicillin. 15N-labeled and 15N/13C-labeled protein samples were generated by culturing E. coli BL21(DE3) in M9 minimal media supplemented with 50 μg/mL ampicillin and containing 15NH4Cl, 15(NH4)2SO4 and 13C-Glucose (Sigma-Aldrich) as the sole nitrogen and carbon sources, respectively.
2H/15N/13C-labeled CTA1-T2 was generated by first culturing transformed E. coli BL21(DE3) in 10 mL of M9 minimal media supplemented with 50 μg/mL ampicillin and containing 15NH4Cl, 15(NH4)2SO4 and 13C-Glucose. After incubation for 4 h at 37 °C, the cells were harvested by centrifugation at 4,000 rpm in a F21B-8x50Y rotor (FIBERLite, Piramoon Technologies, Inc.); the available pellet was resuspended in M9 in 2H2O and added to 1 L of M9 in 2H2O with 50 μg/mL ampicillin. Cells were cultured to mid log phase (OD600nm approximately 0.6), expression was induced with 0.1 mM of IPTG, and expression continued overnight at 37 °C.
Cells were harvested by centrifugation at 4,000 rpm, resuspended in homogenization buffer (50 mM Tris, pH 8.0, 2 mM EDTA, 2 mM DTT, 200 μg/mL lysozyme (Sigma-Aldrich) and 10 % B-PER (Thermo Fisher Scientific, Pierce Product). The resuspended cells were sonicated on ice and centrifuged at 13,000 rpm; the pellets were washed several times with homogenization buffer with a final wash in ultrapure water. The inclusion bodies were solubilized by suspending the pellets in 6 M Gdm-HCl, 50 mM Tris, pH 8.0, and allowed to incubate overnight at 4 °C with mixing. The solubilized proteins were renatured by dialysis against refolding buffer (20 mM Tris, pH 7.2, 100 mM NaCl, and 5 % glycerol) at 4 °C. The refolded protein samples were centrifuged as previously described, filtered (0.45 μm) and purified at 4 °C using the ÄKTA™ FPLC System with a HiLoad™ 16/60 Superdex™ 75 prep grade Gel Filtration Column (GE Healthcare).
CTA1A2 affinity purification and refolding
CTA1A2 was expressed as described for CTA1, and the harvested bacterial pellet was suspended in homogenization buffer. The cells were lysed by sonication; additional washes resulted in a clean inclusion body pellet. The pellet was solubilized in 6 M Gdm-HCl, 20 mM Tris pH 7.5, 200 mM NaCl, and 1 mM EDTA. Renaturation and purification of the solubilized proteins was achieved by on-column refolding using the HisTrap™ HP affinity column (GE Healthcare) according to the manufacturer’s instructions. The native refolding buffer was 20 mM Tris, pH 7.5, 200 mM NaCl, and 1 mM EDTA; elution from the column was achieved using 300 mM imidazole in 20 mM Tris, pH 7.5, 200 mM NaCl. Further purification of the protein was completed using gel filtration chromatography essentially as described for CTA1.
NhisCTA1 and ARF6 constructs and in vitro CTA1:ARF6-GTP complex purification
NhisCTA1 (N1-R192), ARF6 and the NhisCTA1/ARF6 co-expression constructs were a kind gift of Dr. Randall K. Holmes (University of Colorado Health Sciences Center). The NhisCTA1 construct was mutated (C187S) to enhance the stability of the recombinant protein for NMR studies. An 1H/15N-labeled sample of inactive CTA1:ARF6-GTP (E110:112D) complex was co-expressed in BL21(DE3) cells in M9 minimal media as previously described and purified by the published protocol (27). In vitro purification of the active CTA1:ARF6-GTP complex was performed using 15N/13C-isotopically labeled NhisCTA1 protein and unlabeled ARF6 protein, both expressed in BL21(DE3) cells. Supplementation of the cells with 0.1 mM IPTG was used in the over-expression of ARF6 protein, which expressed in the soluble fraction. The NhisCTA1 over-expressed as inclusion bodies with a yield of ~80% of the total cellular protein, which were purified and denatured as earlier described for CTA1. Following denaturation, the refolding and purification of NhisCTA1 was performed on the HisTrap column, using the ÄKTA™ FPLC System for gradient buffer exchange into 20 mM Tris, pH 7.4, 500 mM NaCl, 5 % glycerol, 20 mM imidazole. Following refolding, NhisCTA1 was eluted using 600 mM Imidazole, 20 mM Tris, pH 7.4, 500 mM NaCl, and further dialyzed against 20 mM Tris, pH 8.2, 1 mM MgCl2, and 200 mM NaCl to remove the imidazole. The refolded free NhisCTA1 sample was used to resuspend the frozen ARF6 cell pellet harvested from 500 mL LB. The resuspended cells were sonicated on ice for 10 minutes every 60 minutes and stirred continuously for 4 h at 4 °C. The sample was centrifuged to separate soluble proteins from the cellular debris, and the cleared lysate was harvested and filtered as previously described here. Subsequent purification of the in vitro-generated CTA1:ARF6-GTP complex was performed essentially as described (27). Purified fractions containing CTA1:ARF6-GTP complex were pooled, concentrated and buffer exchanged into 20 mM, dTris-HCl, pH 7.4, 200 mM NaCl, 1 mM EDTA, 0.0 5 % NaN3 and 6 % 2H2O for NMR analysis.
NMR experiments
All NMR spectra of CTA1, CTA1-T2 and the CTA1:ARF6-GTP complex were recorded at 15°C on a Bruker Avance 800 MHz NMR spectrometer equipped with a cryogenic Z-gradient triple-resonance TXI probe. NMR spectra acquired include 2D 15N/1H TROSY, 3D 15N/1H TROSY-HNCA, 3D 15N/1H TROSY-HN(CO)CA, 3D 15N/1H TROSY-HNCO, 3D 15N/1H TROSY-HNCACB and 3D 15N/1H TROSY-HN(CO)CACB (54;55). Spectra were typically 1024 complex points in the direct 1H dimension and 35–70 and 32 complex points in the 13C and 15N dimensions, respectively. NMRPIPE (56) and SPARKY (57) were used to process and analyze the data for sequence specific assignment of CTA1-T2.
NAD+ titrations by NMR
Titration experiments were performed aliquotting NAD+ into the 15N-labeled CTA1-T2 and CTA1:ARF6-GTP complex (CTA1 15N/13C-labeled). NAD+ was dissolved in the same buffer used for the protein samples. The protein sample concentrations used for titration in these experiments were 0.2 mM of 15N-labeled CTA1-T2 and 0.1 mM of 15N/13C-labeled active CTA1:ARF6-GTP complex. For each step of the titration, a 1D 1H spectrum and a 2D 1H/15N HSQC or 2D 1H/15N TROSY was acquired at 15 °C for CTA1-T2 and at 37 °C for CTA1:ARF6-GTP on a Bruker Avance 800 MHz NMR spectrometer. Changes in average amide chemical shifts (Δδavg) were calculated using Equation 1:
| (Equation 1) |
Where ΔδN and ΔδH are the amide nitrogen and amide proton chemical shift changes from the free-state to the NAD+ bound states (58). NMR chemical shift changes were fit using KaleidaGraph 3.52 (Synergy Software) to a bimolecular binding model through non-linear regression analysis.
CD experiments
The CD spectra of both CTA1 and CTA1A2 were acquired using an Olis RSM1000 instrument, from 200 to 260 nm (CTA1A2) and from 197.5 to 260 nm (CTA1), using an integration time of 5 seconds. Each sample, 40μM CTA1A2 and 20μM CTA1, was in 20 mM Tris pH 7.5, 200 mM NaCl, and 1 mM EDTA and spectra were acquired 288 K. Each spectrum was an average of 3 scans, smoothed over 5 data points. The % of α-helical content was estimated by the following Equation 2 based on the molar ellipticity [θ] at 222 nm (31):
| (Equation 2) |
CTA1 binding CTA2 SPR experiments
All SPR experiments were performed on a BIAcore 2000 instrument (GE Healthcare). 1μM synthetic CTA2 peptide sample, D201-S229 (DEKTQSLGVKFLDEYQ SKVKRQIFSGYQS) in 5 mM sodium acetate pH 5.0 was applied to a CM5 chip using the NHS/EDC amine coupling kit according to the manufacturer’s protocol. A second cell was used as a blank to allow for baseline correction. A 200 nM CTA1 sample in HBS buffer (10 mM HEPES, pH 7.4, 150 mM NaCl, 3 mM EDTA, 0.005% Surfactant P20) was injected onto the chip at a flow rate of 10μL/min. Data was calculated from six runs, with the chip re-equilibrated between each run by washing with two 100μL injections of 10 mM glycine pH 2.0. The maximal binding response (Rmax) was calculated using the following Equation 3:
| (Equation 3) |
where MMA=21.1 kDa, MML=4.31 kDa, Sm=1 and RU=45. Baseline corrected sensorgrams for surface plasmon resonance measurements were processed by BIAevaluation software (GE Healthcare) and analyzed using a 1:1 Langmuir model by Scrubber (v 2.0; Center for Biomolecular Interaction Analysis, University of Utah).
Bis-ANS binding experiments
Bis-ANS fluorescence experiments were performed using a Varian Cary Eclipse spectrofluorimeter with an excitation wavelength of 395 nm (slit width 2.5 nm) and an emission wavelength of 495 nm (slit width 5 nm), scanned from 420 to 600 nm. Solution buffer was 10μM bis-ANS, 20 mM Tris pH 7.2, and 50 mM NaCl; protein concentration was 10μM; and sample size was 1 mL. Experiments samples included were CTA1 (N1-G186), CTA1-T2 (N1-F167), CTA1:ARF6-GTP, CTA1 + 2 M Gdm-HCl, and CTA1 + 6 M Gdm-HCl, and sample buffer.
NMR assignment of CTA1
The chemical shift assignments for CTA1-T2 were deposited in the BMRB under accession #15162.
Supplementary Material
Acknowledgments
We thank Dr. Youlin Xia and the UH Keck/IMD NMR Center for help with the NMR experimentation, Wei-hong Jin, who assisted in the assembly of the synthetic gene encoding CTA1, Dr. Ana I. Medrano for help with CTA1:ARF6-GTP and Roberto Carreño for help with the SPR experiments. We also thank Dr. Randall K. Holmes (University of Colorado Health Sciences Center) for the CT and ARF6 clones and the reviewer for suggesting the bis-ANS experiments. This work is supported by Welch Foundation Grants and a NASA Grant to GBL and SRB. RSA was supported by an NIH training fellowship from the Keck Center Pharmacoinformatics Training Program of the Gulf Coast Consortia.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- 1.Sack DA, Sack RB, Nair GB, Siddique AK. Cholera. Lancet. 2004;363:223–233. doi: 10.1016/s0140-6736(03)15328-7. [DOI] [PubMed] [Google Scholar]
- 2.Stewart-Tull DE. Vaba, Haiza, Kholera, Foklune or Cholera: in any language still the disease of seven pandemics. J Appl Microbiol. 2001;91:580–591. doi: 10.1046/j.1365-2672.2001.01493.x. [DOI] [PubMed] [Google Scholar]
- 3.Gill DM, Meren R. ADP-ribosylation of membrane proteins catalyzed by cholera toxin: basis of the activation of adenylate cyclase. Proc Natl Acad Sci U S A. 1978;75:3050–3054. doi: 10.1073/pnas.75.7.3050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sharp GW, Hynie S. Stimulation of intestinal adenyl cyclase by cholera toxin. Nature. 1971;229:266–269. doi: 10.1038/229266a0. [DOI] [PubMed] [Google Scholar]
- 5.Schafer DE, Lust WD, Sircar B, Goldberg ND. Elevated concentration of adenosine 3′:5′-cyclic monophosphate in intestinal mucosa after treatment with cholera toxin. Proc Natl Acad Sci U S A. 1970;67:851–856. doi: 10.1073/pnas.67.2.851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Randazzo PA, Terui T, Sturch S, Kahn RA. The amino terminus of ADP-ribosylation factor (ARF) 1 is essential for interaction with Gs and ARF GTPase-activating protein. J Biol Chem. 1994;269:29490–29494. [PubMed] [Google Scholar]
- 7.Lemichez E, Boquet P. To be helped or not helped, that is the question. J Cell Biol. 2003;160:991–992. doi: 10.1083/jcb.200303032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lencer WI, Tsai B. The intracellular voyage of cholera toxin: going retro. Trends Biochem Sci. 2003;28:639–645. doi: 10.1016/j.tibs.2003.10.002. [DOI] [PubMed] [Google Scholar]
- 9.Moss J, Vaughan M. Activation of toxin ADP-ribosyltransferases by eukaryotic ADP-ribosylation factors. Mol Cell Biochem. 1999;193:153–157. [PubMed] [Google Scholar]
- 10.Zhang RG, Scott DL, Westbrook ML, Nance S, Spangler BD, Shipley GG, Westbrook EM. The three-dimensional crystal structure of cholera toxin. J Mol Biol. 1995;251:563–573. doi: 10.1006/jmbi.1995.0456. [DOI] [PubMed] [Google Scholar]
- 11.Jobling MG, Holmes RK. Identification of motifs in cholera toxin A1 polypeptide that are required for its interaction with human ADP-ribosylation factor 6 in a bacterial two-hybrid system. Proc Natl Acad Sci U S A. 2000;97:14662–14667. doi: 10.1073/pnas.011442598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mekalanos JJ, Collier RJ, Romig WR. Enzymic activity of cholera toxin. II Relationships to proteolytic processing, disulfide bond reduction, and subunit composition. J Biol Chem. 1979;254:5855–5861. [PubMed] [Google Scholar]
- 13.Stein PE, Boodhoo A, Armstrong GD, Cockle SA, Klein MH, Read RJ. The crystal structure of pertussis toxin. Structure. 1994;2:45–57. doi: 10.1016/s0969-2126(00)00007-1. [DOI] [PubMed] [Google Scholar]
- 14.Fraser ME, Fujinaga M, Cherney MM, Melton-Celsa AR, Twiddy EM, O’Brien AD, James MN. Structure of shiga toxin type 2 (Stx2) from Escherichia coli O157:H7. J Biol Chem. 2004;279:27511–27517. doi: 10.1074/jbc.M401939200. [DOI] [PubMed] [Google Scholar]
- 15.De Haan L, Hirst TR. Cholera toxin: a paradigm for multi-functional engagement of cellular mechanisms (Review) Mol Membr Biol. 2004;21:77–92. doi: 10.1080/09687680410001663267. [DOI] [PubMed] [Google Scholar]
- 16.Sandvig K, van Deurs B. Transport of protein toxins into cells: pathways used by ricin, cholera toxin and Shiga toxin. FEBS Lett. 2002;529:49–53. doi: 10.1016/s0014-5793(02)03182-4. [DOI] [PubMed] [Google Scholar]
- 17.Hazes B, Read RJ. Accumulating evidence suggests that several AB-toxins subvert the endoplasmic reticulum-associated protein degradation pathway to enter target cells. Biochemistry. 1997;36:11051–11054. doi: 10.1021/bi971383p. [DOI] [PubMed] [Google Scholar]
- 18.Teter K, Allyn RL, Jobling MG, Holmes RK. Transfer of the cholera toxin A1 polypeptide from the endoplasmic reticulum to the cytosol is a rapid process facilitated by the endoplasmic reticulum-associated degradation pathway. Infect Immun. 2002;70:6166–6171. doi: 10.1128/IAI.70.11.6166-6171.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tsai B, Ye Y, Rapoport TA. Retro-translocation of proteins from the endoplasmic reticulum into the cytosol. Nat Rev Mol Cell Biol. 2002;3:246–255. doi: 10.1038/nrm780. [DOI] [PubMed] [Google Scholar]
- 20.Lencer WI. Retrograde transport of cholera toxin into the ER of host cells. Int J Med Microbiol. 2004;293:491–494. doi: 10.1078/1438-4221-00293. [DOI] [PubMed] [Google Scholar]
- 21.Hagiwara Y, Kawamura YI, Kataoka K, Rahima B, Jackson RJ, Komase K, Dohi T, Boyaka PN, Takeda Y, Kiyono H, McGhee JR, Fujihashi K. A second generation of double mutant cholera toxin adjuvants: enhanced immunity without intracellular trafficking. J Immunol. 2006;177:3045–3054. doi: 10.4049/jimmunol.177.5.3045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schmitz A, Herrgen H, Winkeler A, Herzog V. Cholera toxin is exported from microsomes by the Sec61p complex. J Cell Biol. 2000;148:1203–1212. doi: 10.1083/jcb.148.6.1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Teter K, Jobling MG, Sentz D, Holmes RK. The cholera toxin A1(3) subdomain is essential for interaction with ADP-ribosylation factor 6 and full toxic activity but is not required for translocation from the endoplasmic reticulum to the cytosol. Infect Immun. 2006;74:2259–2267. doi: 10.1128/IAI.74.4.2259-2267.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rodighiero C, Tsai B, Rapoport TA, Lencer WI. Role of ubiquitination in retro-translocation of cholera toxin and escape of cytosolic degradation. EMBO Rep. 2002;3:1222–1227. doi: 10.1093/embo-reports/kvf239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kahn RA, Gilman AG. ADP-ribosylation of Gs promotes the dissociation of its alpha and beta subunits. J Biol Chem. 1984;259:6235–6240. [PubMed] [Google Scholar]
- 26.Osborne JC, Jr, Stanley SJ, Moss J. Kinetic mechanisms of two NAD:arginine ADP-ribosyltransferases: the soluble, salt-stimulated transferase from turkey erythrocytes and choleragen, a toxin from Vibrio cholerae. Biochemistry. 1985;24:5235–5240. doi: 10.1021/bi00340a042. [DOI] [PubMed] [Google Scholar]
- 27.O’Neal CJ, Jobling MG, Holmes RK, Hol WG. Structural basis for the activation of cholera toxin by human ARF6-GTP. Science. 2005;309:1093–1096. doi: 10.1126/science.1113398. [DOI] [PubMed] [Google Scholar]
- 28.O’Neal CJ, Amaya EI, Jobling MG, Holmes RK, Hol WG. Crystal structures of an intrinsically active cholera toxin mutant yield insight into the toxin activation mechanism. Biochemistry. 2004;43:3772–3782. doi: 10.1021/bi0360152. [DOI] [PubMed] [Google Scholar]
- 29.Mekalanos JJ, Collier RJ, Romig WR. Purification of cholera toxin and its subunits: new methods of preparation and the use of hypertoxinogenic mutants. Infect Immun. 1978;20:552–558. doi: 10.1128/iai.20.2.552-558.1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kahn RA, Gilman AG. The protein cofactor necessary for ADP-ribosylation of Gs by cholera toxin is itself a GTP binding protein. J Biol Chem. 1986;261:7906–7911. [PubMed] [Google Scholar]
- 31.Woody RW. Circular dichroism. Methods Enzymol. 1995;246:34–71. doi: 10.1016/0076-6879(95)46006-3. [DOI] [PubMed] [Google Scholar]
- 32.LaPointe P, Wei X, Gariepy J. A role for the protease-sensitive loop region of Shiga-like toxin 1 in the retrotranslocation of its A1 domain from the endoplasmic reticulum lumen. J Biol Chem. 2005;280:23310–23318. doi: 10.1074/jbc.M414193200. [DOI] [PubMed] [Google Scholar]
- 33.Pande AH, Scaglione P, Taylor M, Nemec KN, Tuthill S, Moe D, Holmes RK, Tatulian SA, Teter K. Conformational instability of the cholera toxin A1 polypeptide. J Mol Biol. 2007;374:1114–1128. doi: 10.1016/j.jmb.2007.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dyson HJ, Wright PE. Equilibrium NMR studies of unfolded and partially folded proteins. Nat Struct Biol. 1998;5(Suppl):499–503. doi: 10.1038/739. [DOI] [PubMed] [Google Scholar]
- 35.Wishart DS, Bigam CG, Yao J, Abildgaard F, Dyson HJ, Oldfield E, Markley JL, Sykes BD. 1H, 13C and 15N chemical shift referencing in biomolecular NMR. J Biomol NMR. 1995;6:135–140. doi: 10.1007/BF00211777. [DOI] [PubMed] [Google Scholar]
- 36.Redfield C. Using nuclear magnetic resonance spectroscopy to study molten globule states of proteins. Methods. 2004;34:121–132. doi: 10.1016/j.ymeth.2004.03.009. [DOI] [PubMed] [Google Scholar]
- 37.Hilser VJ, Thompson EB. Intrinsic disorder as a mechanism to optimize allosteric coupling in proteins. Proc Natl Acad Sci U S A. 2007;104:8311–8315. doi: 10.1073/pnas.0700329104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Smoot AL, Panda M, Brazil BT, Buckle AM, Fersht AR, Horowitz PM. The binding of bis-ANS to the isolated GroEL apical domain fragment induces the formation of a folding intermediate with increased hydrophobic surface not observed in tetradecameric GroEL. Biochemistry. 2001;40:4484–4492. doi: 10.1021/bi001822b. [DOI] [PubMed] [Google Scholar]
- 39.Spera S, Bax A. Empirical Correlation Between Protein Backbone Conformation and C-Alpha and C-Beta C-13 Nuclear-Magnetic-Resonance Chemical-Shifts. Journal of the American Chemical Society. 1991;113:5490–5492. [Google Scholar]
- 40.Wishart DS, Sykes BD. The 13C chemical-shift index: a simple method for the identification of protein secondary structure using 13C chemical-shift data. J Biomol NMR. 1994;4:171–180. doi: 10.1007/BF00175245. [DOI] [PubMed] [Google Scholar]
- 41.Galloway TS, van Heyningen S. Binding of NAD+ by cholera toxin. Biochem J. 1987;244:225–230. doi: 10.1042/bj2440225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vamvaca K, Vogeli B, Kast P, Pervushin K, Hilvert D. An enzymatic molten globule: efficient coupling of folding and catalysis. Proc Natl Acad Sci U S A. 2004;101:12860–12864. doi: 10.1073/pnas.0404109101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Morinaga N, Kaihou Y, Vitale N, Moss J, Noda M. Involvement of ADP-ribosylation factor 1 in cholera toxin-induced morphological changes of Chinese hamster ovary cells. J Biol Chem. 2001;276:22838–22843. doi: 10.1074/jbc.M101184200. [DOI] [PubMed] [Google Scholar]
- 44.Moss J, Vaughan M. Structure and function of ARF proteins: activators of cholera toxin and critical components of intracellular vesicular transport processes. J Biol Chem. 1995;270:12327–12330. doi: 10.1074/jbc.270.21.12327. [DOI] [PubMed] [Google Scholar]
- 45.Spangler BD. Structure and function of cholera toxin and the related Escherichia coli heat-labile enterotoxin. Microbiol Rev. 1992;56:622–647. doi: 10.1128/mr.56.4.622-647.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhu X, Kim E, Boman AL, Hodel A, Cieplak W, Kahn RA. ARF binds the C-terminal region of the Escherichia coli heat-labile toxin (LTA1) and competes for the binding of LTA2. Biochemistry. 2001;40:4560–4568. doi: 10.1021/bi002628s. [DOI] [PubMed] [Google Scholar]
- 47.Zhu X, Kahn RA. The Escherichia coli heat labile toxin binds to Golgi membranes and alters Golgi and cell morphologies using ADP-ribosylation factor-dependent processes. J Biol Chem. 2001;276:25014–25021. doi: 10.1074/jbc.M100923200. [DOI] [PubMed] [Google Scholar]
- 48.Guo Q, Shen Y, Lee YS, Gibbs CS, Mrksich M, Tang WJ. Structural basis for the interaction of Bordetella pertussis adenylyl cyclase toxin with calmodulin. EMBO J. 2005;24:3190–3201. doi: 10.1038/sj.emboj.7600800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ladant D, Ullmann A. Bordatella pertussis adenylate cyclase: a toxin with multiple talents. Trends Microbiol. 1999;7:172–176. doi: 10.1016/s0966-842x(99)01468-7. [DOI] [PubMed] [Google Scholar]
- 50.Love JJ, Li X, Chung J, Dyson HJ, Wright PE. The LEF-1 high-mobility group domain undergoes a disorder-to-order transition upon formation of a complex with cognate DNA. Biochemistry. 2004;43:8725–8734. doi: 10.1021/bi049591m. [DOI] [PubMed] [Google Scholar]
- 51.Dyson HJ, Wright PE. Coupling of folding and binding for unstructured proteins. Curr Opin Struct Biol. 2002;12:54–60. doi: 10.1016/s0959-440x(02)00289-0. [DOI] [PubMed] [Google Scholar]
- 52.Dyson HJ, Wright PE. Intrinsically unstructured proteins and their functions. Nat Rev Mol Cell Biol. 2005;6:197–208. doi: 10.1038/nrm1589. [DOI] [PubMed] [Google Scholar]
- 53.Kukreja R, Singh B. Biologically active novel conformational state of botulinum, the most poisonous poison. J Biol Chem. 2005;280:39346–39352. doi: 10.1074/jbc.M508463200. [DOI] [PubMed] [Google Scholar]
- 54.Salzmann M, Pervushin K, Wider G, Senn H, Wuthrich K. TROSY in triple-resonance experiments: new perspectives for sequential NMR assignment of large proteins. Proc Natl Acad Sci U S A. 1998;95:13585–13590. doi: 10.1073/pnas.95.23.13585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sattler M, Schleucher J, Griesinger C. Heteronuclear multidimensional NMR experiments for the structure determination of proteins in solution employing pulsed field gradients. Prog NMR Spect. 1999;34:93–158. [Google Scholar]
- 56.Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. Nmrpipe - A Multidimensional Spectral Processing System Based on Unix Pipes. Journal of Biomolecular Nmr. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 57.Goddard T, Kneller DG. 3. SPARKY 3 University of California; San Francisco: 2006. pp. 10–13. [Google Scholar]
- 58.Hall DA, Vander Kooi CW, Stasik CN, Stevens SY, Zuiderweg ER, Matthews RG. Mapping the interactions between flavodoxin and its physiological partners flavodoxin reductase and cobalamin-dependent methionine synthase. Proc Natl Acad Sci U S A. 2001;98:9521–9526. doi: 10.1073/pnas.171168898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Weighted CSI Program. http://nmr.uhnres.utoronto.ca/ikura/csi/
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.