

Abstract
The purpose of this study was to determine the role of the retinol dehydrogenase 12 (RDH12) gene in patients affected with Leber congenital amaurosis (LCA), autosomal recessive retinitis pigmentosa (arRP) and autosomal dominant/recessive cone-rod dystrophies (CORD). Changes in the promoter region, coding regions and exon/intron junctions of the RDH12 gene were evaluated using direct DNA sequencing of patients affected with LCA (n = 36 cases), RP (n = 62) and CORD (n = 21). The allele frequency of changes observed was assessed in a multiethnic control population (n = 159 individuals). Detailed biochemical and structural modeling analysis of the observed mutations were performed to assess their biological role in the inactivation of Rdh12. A comprehensive clinical assessment of retinal structure and function in LCA patients carrying mutations in the RDH12 gene was completed. Of the six changes identified, three were novel including a homozygous C201R change in a patient affected with LCA, a heterozygous A177V change in patients affected with CORD and a heterozygous G46G change in a patient affected with LCA. A novel compound heterozygote T49M/A269fsX270 mutation was also found in a patient with LCA, and both homozygous and heterozygous R161Q changes were seen in 26 patients affected with LCA, CORD or RP. These R161Q, G46G and the A177V sequence changes were shown to be polymorphic. We found that Rdh12 mutant proteins associated with LCA were inactive or displayed only residual activity when expressed in COS-7 and Sf9 cells, whereas those mutants that were considered polymorphisms were fully active. Thus, impairment of retinal structure and function for patients carrying these mutations correlated with the biochemical properties of the mutants.
Keywords: Visual cycle, Photoreceptors, Retina, RDH12, Retinol, LCA, CORD, Retinal diseases, Rhodopsin
1. Introduction
Leber congenital amaurosis (LCA)1 is an autosomal recessive, early onset severe retinal dystrophy and a leading cause of inherited childhood blindness. It accounts for 5% of all inherited retinal dystrophies (Cremers, van den Hurk, & den Hollander, 2002; Hanein et al., 2004; Perrault et al., 1999; Weleber, 2002). Mutations in eleven genes have been identified which together account for ~55% of LCA cases. These genes encode proteins involved in phototransduction, transcription, metabolic pathways, and other processes affecting retinal function. Mutations in genes which have been shown to cause LCA or related early-onset severe retinal dystrophies include CRX (Cone-Rod Homeobox) (Sohocki et al., 1998; Swaroop et al., 1999), CRB1 (Crumbs Homolog 1) (Cremers et al., 2002), AIPL1 (Aryl-hydrocarbon-interacting protein like 1) (Sohocki et al., 2000; Sohocki, Perrault et al., 2000), GUCY2D (Guanylate Cyclase E) (Perrault et al., 1996), TULP1 (Tubby-like protein 1) (Hanein et al., 2004), RPGRIP1 (Retinitis Pigmentosa GTPase Regulator Interaction Protein 1) (Cremers et al., 2002), IMPDH1 (inosine monophosphate dehydrogenase type I) (Bowne et al., 2006), RDH12 (retinol dehydrogenase 12) (Janecke et al., 2004; Perrault et al., 2004; Thompson et al., 2005), RPE65 (Retinal Pigment epithelium protein with molecular mass 65 kDa) (Gu et al., 1997; Morimura et al., 1998), LRAT (lecithin: retinol acyltransferase) (Thompson et al., 2001), RD3 (retinal degeneration 3) (Friedman et al., 2006), and CEP290 (a centrosomal protein) (den Hollander et al., 2006).
Janecke et al., identified five mutations in the RDH12 gene in patients affected by LCA (Janecke et al., 2004). The retinal structure of patients with RDH12 mutations was appreciably distorted, precluding identification of normal laminae (Jacobson et al., 2007). Subsequent studies have identified additional novel mutations in the RDH12 gene associated with early onset cone-rod dystrophy (CORD) and LCA (Perrault et al., 2004; Thompson et al., 2005) (Fig. 1). The RDH12 gene (chromosome 14q24) is approximately 12 kb in length (Haeseleer et al., 2002), consisting of seven exons (Fig. 1). The highest expression level of RDH12 is observed in the retina (Haeseleer et al., 2002). Rdh12 is a member of the short-chain dehydrogenase/reductase (SDR) family (Jornvall et al., 1995) and belongs to a sub-family of dual-specificity retinol dehydrogenases that metabolize both all-trans and cis-retinols/retinals (Haeseleer et al., 2002). Rdh12 is also involved in the metabolism of other non-retinoid alcohols/aldehydes (Belyaeva et al., 2005). In photoreceptor cells Rdh12 localizes to the inner segment of rod and cone photoreceptors (Haeseleer et al., 2002; Kurth et al., 2007; Maeda et al., 2006). Deletion of this gene in mice increased susceptibility to light-induced photoreceptor apoptosis (Maeda et al., 2006). Hence, we have speculated that light damage contributes to the severe visual impairment of individuals with null mutations in the RDH12 gene.
Fig. 1.

Intron/exon gene structure of human RDH12 and all identified genetic variants. A, B, and C correspond to three PCR fragments that overlap each other to cover the entire RDH12 gene. For comparison, genetic variants identified previously by other groups and variants identified in this study are listed below the structure. New genetic variants found in this study are denoted with a gray background.
To further assess the role of Rdh12 in retinal dystrophies, genomic DNA from 119 patients (LCA, RP, and CORD) were collected and analyzed for mutations of the coding sequences, intron/exon junction regions and promoter region using direct sequencing. Six different variants were observed of which three variants were novel. Detailed biochemical analyses were carried out on five of these variants to assess their biological implication in the inactivation of Rdh12. Comprehensive clinical examination of patients carrying these mutations provides further insight into the underlying mechanisms of visual impairment.
2. Materials and methods
2.1. Patient data collection
Patient information and DNA samples were collected respecting the regulation of the Health Information Portability and Accountability Act (HIPAA), the policies of Institutional Review Board of Case Western Reserve University, Cleveland, USA and that of the Research Ethics Board of Hospital for Sick Children, Toronto, Canada. This project respected the tenets of the Declaration of Helsinki. Consent and/or assent forms were obtained from all participants. Comprehensive eye examination included full field electroretinography (ERG) recorded according to the ISCEV standard (Marmor, Holder, Seeliger, & Yamamoto, 2004) and Goldmann visual field (GVF) testing. A Fourier-domain high-speed high–resolution OCT system (axial resolution was 4.5 μm; acquisition speed was 10,000 A – scans/frame, 9 frames/second) constructed at the UC Davis Medical Center combined with a hand-held scanner (Bioptigen, Inc.) was used for retinal layer image acquisition (Zawadzki et al., 2005). A total of 119 patients were included and carried the clinical diagnoses of either LCA (36 cases), arRP onset (62 cases) or CORD (21 cases). The exclusion criteria were the following: (1) non-availability of clinical information; (2) follow up was not possible; and (3) the nature of the retinal disease appeared not to be genetically determined. Genomic DNA from patient blood samples were extracted and purified using standard protocols.
2.2. Mutation screening
According to the RDH12 gene sequence, primer pairs were designed to generate three overlapping PCR fragments (Fig. 1). PCR was performed according to manufacturer’s suggestion (TripleMaster system, Eppendorf) and products were analyzed on a 0.8% agarose gel to verify production of a single band of expected length. Sequencing was performed utilizing primers designed for each exon, intron–exon junction region and the promoter region. Primer sequences for PCR and sequencing are included in Tables 1 and 2. Sequencing was performed on ABI 3730xl and all data were analyzed with Finch TV (www.geospiza.com/.nchtv/) and BioEdit 7.0.5.3 (www.mbio.ncsu.edu/BioEdit/bioedit.html) software.
Table 1.
Sequences of primers used in the experiments
| Name | Sequence 5′–3′ | Fa | Rb | Location | Usage |
|---|---|---|---|---|---|
| 5hRDH12-1 | TCTCTGTGACTTATGCCTATATCTGTCC | ✓ | Promoter region | Amplify PCR fragment a (Fig. 1) | |
| 3hRDH12-2 | CAGCAAATGTGTACTAACTACCGACTCT | ✓ | Intron 3 | Amplify PCR fragment a (Fig. 1) | |
| 5hRDH12-4 | CCCAAGCTCACTTACTATACCTCCTTTA | ✓ | Intron 2 | Amplify PCR fragment b (Fig. 1) | |
| 3hRDH12-4 | CTATAGGTTGAGCATCCCTTATCAGAA | ✓ | Intron 6 | Amplify PCR fragment b (Fig. 1) | |
| 5hRDH12-5 | AATGAGCAACTAGAGTCTGGGAGTAAAG | ✓ | Intron 6 | Amplify PCR fragment c (Fig. 1) | |
| 3hRDH12-7 | GACACTACAGAGAGGTGAGATGTAGC | ✓ | After exon 7 | Amplify PCR fragment c (Fig. 1) | |
| f6-2 | TTCCATCTTTGCAGTAGAGGTGG | ✓ | Promoter region | Sequencing | |
| r2 | TCCTGGGTGCTGAGCAGCAGC | ✓ | Exon 1 | Sequencing | |
| f11-3 | CTAAAAGGAGATACTAAGTGACAT | ✓ | Intron 2 | Sequencing | |
| r1 | CACAGTAGTACTGTTCAGTCTTC | ✓ | Intron 2 | Sequencing | |
| f13-2 | CCCAGTGACAATGCTTATTGG | ✓ | Intron 3 | Sequencing | |
| f15-2 | GAGGAATCCACAAACTCAGACC | ✓ | Intron 4 | Sequencing | |
| f20 | TGAAATAGAAGGACCTCCGAACC | ✓ | Intron 5 | Sequencing | |
| f29-2 | TTAGACCTGGTACTAGATCTTG | ✓ | Intron 6 | Sequencing | |
| f30-2 | GCCAGCTGGTGCTGCGAATCC | ✓ | Exon 7 | Sequencing |
F, forward.
R, reverse.
Table 2.
Primers for control population screening
| Mutations screened | Primer sequence | Product size (bp) | MgCl2 (mM) | Tm (°C) | Cycles | Other |
|---|---|---|---|---|---|---|
| Exon 2 (G46G/T49M) | CCGATAAACAGGCGACTACCTTTGGAAACACAAAGCAATATGGACA | 400 | 1.0 | 66 | 35 | Q solution |
| Exon 5 C201R (binds to mutant T) | TGTTGCAAAAAGCTACTGTGAAGCAGGGGTTTTGCCGATC | 347 | 1.0 | 57 | 39 | |
| Exon 5 A177V | TGTTGCAAAAAGCTACTGTGAGTTAATGTGTTCTCGGTAGT | 223 | 1.0 | 54 | 51 | |
| Exon 5 R160Q | AATTGGTTCACACCCAGAAGATGACTTCCCAAGTTCCTGTG | 398 | 1.0 | 66 | 35 | |
| Exon 5 for sequencing | TGTTGCAAAAAGCTACTGTGAAATTGGTTCACACCCAGAAGA | 594 | 1.0 | 57 | 39 | |
| Exon 6 c.806delCCCTG | CTTCTCCCCCTTTGTCAAGATCATCAGGCACAAACTCAGC | 199 | 1.0 | 56 | 38 |
2.3. Mutagenesis
Human RDH12 cDNA (Reference sequence is NM_152443 in NCBI database but G482 allele) was provided by Dr. F. Haeseleer (Department of Ophthalmology, University of Washington). Constructs were generated by using the QuickChange site-directed mutagenesis kit (Stratagene, La Jolla, CA). The RDH12 cDNA was subcloned into pcDNA3.1 and mutations corresponding to those identified in this study were introduced and confirmed by DNA sequencing. To eliminate the possibility of generating mutations in the vector sequence during mutagenesis, cDNA inserts were cut out with restriction enzymes and ligated into pcDNA3.1 (Invitrogen, Carlsbad, CA) for COS-7 cell expression and pFastBac HT (Invitrogen, Carlsbad, CA) for Sf9 cells expression. To produce baculovirus, pFastBac HT vector with inserts were transformed into DH10Bac competent cells. Positive clones were selected and PCR analyzed. Purified Bacmid was used for transfecting Sf9 cells and supernatant was collected as virus stock. Viral titer was determined via plaque assay.
2.4. Rdh12 expression and activity assay
Wild-type and mutant constructs of Rdh12 were transiently expressed in COS-7 cells and Sf9 cells. Fugene 6 (Roche Applied Science, Indianapolis, IN) was used for transfection (3 μl of reagent/1 μg of DNA per well of a 6-well plate) according to manufacturer’s instructions. Cells were collected two days post transfection and stored at −20 °C in storage buffer (140 mM NaCl, 3 mM KCl, 10 mM Na2HPO4, 2 mM KH2PO4, and pH 7.4, containing 1% BSA and 20% glycerol). Baculovirus infection was performed according to manufacturer’s instructions (Invitrogen, Carlsbad, CA). In each 35 mm well, 2 × 106 cells were seeded and one hour later were infected with baculovirus at a multiplicity of infection (MOI) of ~0.1–0.5. Sf9 cells were collected three days post-infection by centrifugation and stored at −20 °C in storage buffer. Protein was boiled and centrifuged before SDS–PAGE analysis.
Rdh12 activity assays were performed as previously described (Jang et al., 2001; Jang, McBee, Alekseev, Haeseleer, & Palczewski, 2000; Jang, Van Hooser et al., 2001). In brief, the reaction mixture contained cell lysate in 50 mM MES, pH 5.5, containing 1.2 mM NADPH, 20 μM all-trans-retinal, 0.1% Triton X-100, 0.5% bovine serum albumin (BSA) and 10% glycerol. The reaction was quenched by the addition of 300 μl ice-cold methanol 30 min after incubation at 37 °C. Following the addition of methanol, 300 μl of hexane was added to extract retinoids. Retinoids were separated by normal phase HPLC (Ultrasphere-Si, 4.6 μ 250 mm, Beckman, Fullerton, CA). The hexane solution was analyzed by HPLC using 10% ethyl acetate in hexane and monitored at 325 nm to confirm the production of retinol. Km and Vmax were derived from a plot of the inverse of the initial velocity of all-trans-retinol formation at six substrate concentrations and the inverse of the all-trans-retinal concentration. Reaction conditions were same as above, but were performed for 8 min at 37 °C.
3. Results
3.1. Patient recruitment and mutational analysis
One hundred nineteen patients of various ages were enrolled in this study (details in section 2, Table 3). Six variants (three novel) were identified in these patients afflicted with either LCA, arRP or CORD by direct sequencing of the RDH12 exon, intron–exon junctions and promoter regions (Table 3). Variants identified were located within the coding region and not in splice junctions or the promoter regions (Fig. 1). The three novel variants identified were: (1) a heterozygous 138C → T mutation, which is a silent mutation in exon 2 leaving Gly unchanged at amino acid position 46; (2) a heterozygous 530C → T mutation in exon 5 that changes Ala177 to Val; (3) homozygote 601T → C mutation in exon 5 that changes residue Cys201 to Arg. We also identified a novel compound heterozygote T49M/A269fsX270 in a patient with LCA. Other variants seen included the 146C → T mutation (T49M, exon 2); 482G → A mutation (R161Q, exon 5); 806del-CCCTG mutation (A269fsX270, exon 6); as well as the R161Q variant assumed to be a polymorphism (Thompson et al., 2005).
Table 3.
Mutations in the coding region of RDH12
| Location | Nucleotide changea | Change in amino acid residue | Number of patients | LCAb | arRPc | CORDd |
|---|---|---|---|---|---|---|
| Heterozygote | ||||||
| Exon 2 | 138C → T (new) | G46Ge | 1 | 1 | ||
| Exon 5 | 530C → T (new) | A177V | 2 | 2 | ||
| Compound heterozygote | ||||||
| Exon 2 | 146C → T | T49M | 1 (Patient 1) | 1 | ||
| Exon 6 | 806delCCCTG | A269fsX270 (missing C-terminal 47aa) | ||||
| Hetero- and homo-zygote | ||||||
| Exon 5 | 482G → A | R161Q | 26 | 7 | 13 | 6 |
| Homozygote | ||||||
| Exon 5 | 601T → C (new) | C201R | 1 (Patient 2) | 1 | ||
| Total patient number | LCA | arRP | CORD | |||
| 119 | 36 | 62 | 21 | |||
Nucleotides are numbered from the first base of the start (ATG) codon. Reference sequence is NM_152443 in NCBI database (with a change from A at nucleotide 482 in the reference sequence to a G. G482 allelle).
LCA, Leber congenital amaurosis.
arRP, autosomal recessive retinitis pigmentosa.
CORD, cone-rod dystrophy.
This mutation does not appear to generate an amino acid substitution, but it may result in the generation of an alternate splice acceptor site, yielding an alternatively spliced and likely truncated protein.
3.2. Impact of mutations on the Rdh12 structure
The mutations and polymorphisms found in this study (Table 3) comprise several different “classes” of mutations (Fig. 2). These mutations result in altered function due to defects in the substrate- or co-factor-binding site, whereas others can directly interfere with the catalysis of the reaction through disruption of the active site. The G46G silent mutation which on first glance appears to be a silent mutation, can lead to the generation of an altered splice acceptor site (sequence analyzed by ESE finder software, http://rulai.cshl.edu/tools/ESE/), resulting in the production of a truncated or malformed protein. The T49M mutation would likely interfere with the binding of NADP, as this residue is located in the phosphate-binding loop and the larger, more hydrophobic side chain of Met could perturb co-factor binding (Figs. 2 and 3). The R161Q polymorphism is far from the catalytic, substrate- or co-factor-binding sites or residues involved in the formation of the structure of the Rossmann fold, and thus unlikely to be able to influence the catalysis of Rdh12. The A177V mutation could disrupt the ability of the enzyme to bind substrate (retinal) as this residue is located in the putative retinol-binding site. The reason for the altered activity in the C201R mutant is based on its proximity to and likely disruption of the catalytic site. The location of the residue 201 is inside the catalytic motif (YXXXK), immediately after the Tyr residue (Fig. 2). Furthermore, the oligomeric state of SDR enzymes varies from tetramer to dimer to monomer and this residue is located in a region that at least in several other SDRs can be involved in oligomer formation (Jornvall et al., 1995).
Fig. 2.

Homology of Rdh12 among different species. Rdh12 from human, bovine, mouse and Drosophila are aligned together here to show the sequence homology. The putative transmembrane domain is located at the N-terminus. Regions corresponding to the Rossmann fold, phosphate-binding site and catalytic site are also labeled. Polymorphisms/mutations identified in this study are also indicated. The chromatogram figures show compound heterozygote of 146C → T/806delCCCTG (patient 1) and homozygote of 601T → C (patient 2).
Fig. 3.
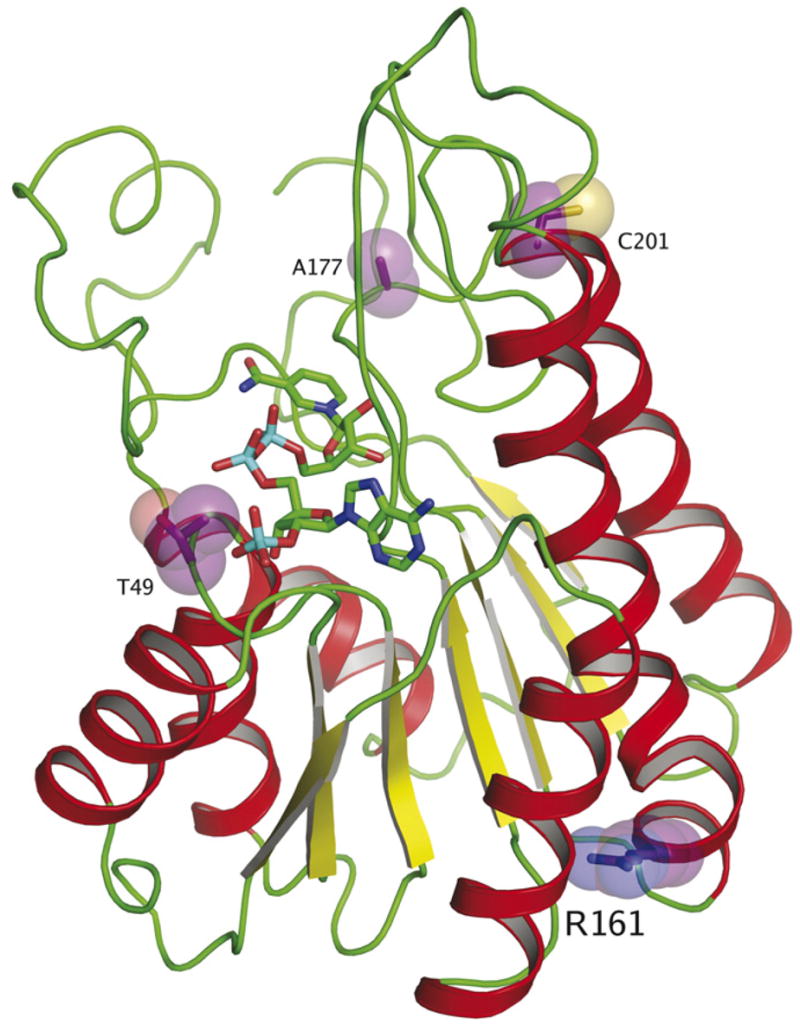
Homology model of Rdh12 showing sites of mutations/polymorphisms identified in this study. Residues 49, 161, 177, and 201 are shown. The T49M mutation can interfere with NADP binding, whereas change A177V may interfere with substrate binding. That the R161Q mutation does not appear to exhibit any changes in enzymatic activity is unsurprising, because the location is far from the active site and is likely not involved in catalysis. C201R, which lies close to the catalytic residues, can disrupt the formation of the active site, leading to a loss of enzymatic activity. The homology model was built using β-ketoacyl-[ACP]-reductase from Escherichia coli as a backbone model (PDB ID:1Q7B), using the Geno3D server (Combet et al., 2002) and O (Jones et al., 1991) for generation of an initial homology model and manual model building respectively. Figure was created with PYMOL (DeLano, W.L. The PyMOL Molecular Graphics System (2002) DeLano Scientific, San Carlos, CA, USA. http://www.pymol.org).
3.3. Impact of mutations on Rdh12 enzymatic activity
To test the enzymatic activity of the Rdh12 variants found in this study, site directed mutagenesis was used to create mutant constructs which were cloned into pcDNA3.1 and pFastBac HTa vectors, respectively. Total cell lysate was obtained two days after transient transfection in COS-7 cells or three days after virus infection in Sf9 cells. Immunoblotting with an anti-Rdh12 antibody (Maeda et al., 2006) confirmed protein expression of Rdh12 and its mutants. In COS-7 cells, C201R and A269fsX270 expression levels were not detectable, whereas other constructs showed polypeptides on SDS–PAGE of the predicted molecular weight of 38 kDa (data not shown). In Sf9 cells, expression levels of all mutant constructs were comparable to that of the wild-type with the exception of the C201R construct, which was expressed at ~30% of the wild type (Fig. 4A).
Fig. 4.
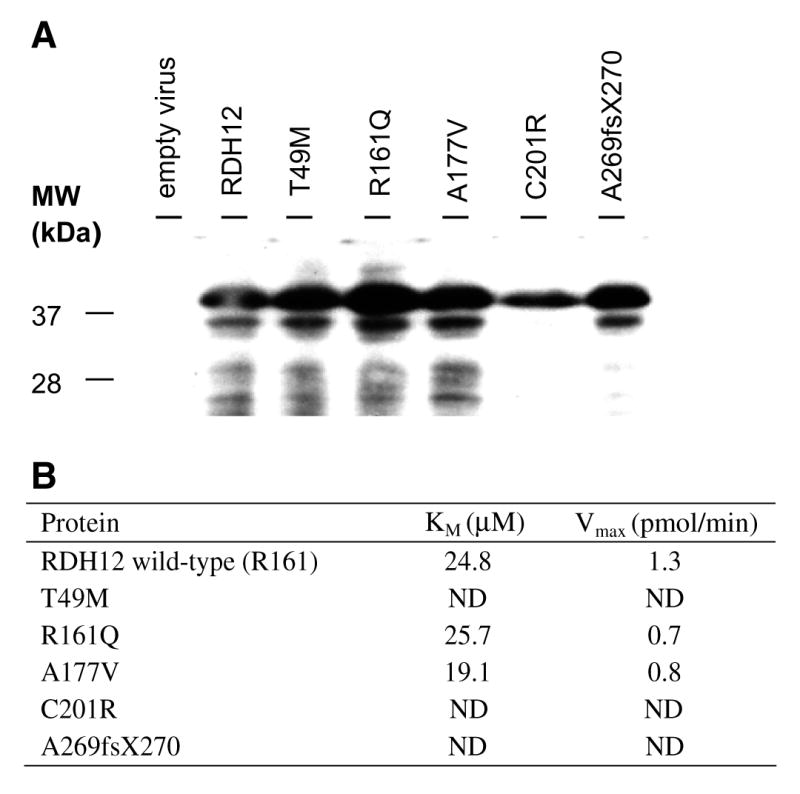
(A) Expression of Rdh12 and its mutants. Lanes were loaded with extracts from Sf9 cells infected with virus only, virus containing RDH12, T49M, R161Q, A177V, C201R, and A269fsX270 from left to right, respectively. Immunoblotting was carried out according to standard protocols using Immobilon-P to adsorb proteins (polyvinylidene difluoride; Millipore Corp.) (Ohguro et al., 1993). Mouse polyclonal antibodies against bacterially expressed full-length mouse Rdh12 were raised in BALB/c mice as described previously (Maeda et al., 2006). Secondary antibody is HRP conjugated anti-mouse IgG (Promega Corporation, Madison, WI) and signal was detected using SuperSignal West Pico Chemiluminescent Substrate (Pierce Biotechnology, Rockford, IL). (B) Calculated Km and Vmax values of Rdh12 constructs expressed in Sf9 cells. ND, not determined.
Testing of Rdh12 activity in COS-7 cells, showed a dramatic reduction in the ability to produce all-trans-retinol from all-trans-retinal (~95% less) for the T49M (17.8 pmol), C201R (0 pmol) and A269fsX270 (0 pmol) variants. The A177V (147.9 pmol) variant had similar production of all-trans-retinol compared to wild-type RDH12 (151.0 pmol) (Fig. 5). The R161Q ( 170.4 pmol) variant which we have putatively classified as a polymorphism also exhibited similar all-trans-RDH activity to wild-type RDH12 as described in previous studies (Janecke et al., 2004; Perrault et al., 2004). The amount of all-trans-retinol production was calculated by subtracting background of membranes isolated from cell transfected with the empty vector from raw data. Further characterization was carried out with proteins expressed in Sf9 cells. Profiles of the activity observed in Sf9 cells infected with virus carrying RDH12 and mutants was similar to that of COS-7 cells (Fig. 6). Comparable amounts of all-trans-retinol production was detected in the wild-type Rdh12 (27.2 pmol), R161Q mutant (19.2 pmol) and A177V mutant (17.9 pmol). Almost no production from all-trans-retinal to retinol was obtained from T49M (0.9 pmol), C201R (0 pmol) and A269fsX270 (0 pmol) mutants. Again, the production amount here was calculated by subtracting background (empty virus) from raw data. The Km and Vmax values were determined using proteins expressed in Sf9 cells. Km for the wild-type Rdh12, R161Q and A177V mutants were 24.8, 25.7, and 19.1 μM, respectively. Vmax for these proteins was 1.3, 0.7, and 0.8 pmol/min respectively (Fig. 4B).
Fig. 5.

HPLC analysis of the enzymatic activity of Rdh12 variants in COS-7 cells. Enzyme activity was detected by monitoring all-trans-retinol production via normal phase HPLC.
Fig. 6.

HPLC analysis of the enzymatic activity of Rdh12 variants in Sf9 cells. The reaction was carried out as described for the COS-7 cells.
3.4. Clinical manifestation of RDH12 sequence changes
The genotype–phenotype correlation was evaluated in two available patients with LCA (Table 4). The compound heterozygote mutation (T49M/A269fsX270) was identified in patient 1 who was from non-consanguineous parents of Italian ancestry with no known family history of eye diseases. We followed this patient from 6 to 21 years of age. Abnormal night vision and restricted side vision were first noticed at 2 years of age according to the parents. Visual acuity (VA) was reduced to 1.0 and 1.3 logarithm of the minimum angle of resolution (logMAR) with a small refractive error of +1.25 D (right eye) and +1.0 (left eye) at 6 years of age. At this age, his fundus had a diffuse retinopathy with pigment clumping and bone spiculae pigmentation, cystoid macular edema and attenuated blood vessels with mild optic atrophy in both eyes. Vision was stable at a second visit 9 years later. Goldmann visual field (GVF) testing revealed constricted fields to about 5° in diameter in both eyes with a thin peripheral almost circumferential island in his right eye. ERG was not recordable above noise for all tested stimuli at 15 years of age. VA improved slightly to 0.7logMAR using both eyes, under oral treatment with carbonic anhydrase inhibitor for the management of his macular edema, even though he lost the remaining peripheral field. Horizontal and vertical nystagmus was first documented at 19 years of age. VA was stable at his last visit at 21 years of age (Table 4). Fundus exam revealed atrophic macular changes, optic atrophy and bone spiculae pigmentation in both eyes at 21 years of age (Fig. 7).
Table 4.
Clinical phenotype of patients with RDH12 mutations
| Patient No. | Sex | Mutation | Age at test (years) | Nyctalopia | Photophobia | Nystagmus | Visual acuity (logMAR)a | Goldmann visual field (III4e)b | ERGc |
|---|---|---|---|---|---|---|---|---|---|
| 1 | M | Exon 2: 146C → T; T49M exon 6: 806delCCCTG; A269fsX270; compound heterozygote | 6 | + | − | 1.0/1.3 | NTd | NRe | |
| 15 | + | − | 0.7/1.0 | 5°/5° | |||||
| 17 | + | − | 0.7/0.7 | 5°/3° (plus peripheral island) | |||||
| 19 | + | − | + | 0.7/0.7 | |||||
| 21 | + | − | + | 0.7/0.7 | |||||
| 2 | M | Exon 5: 601T → C; C201R; homozygote | 29 | + | − | − | 2.3/2.3 | 10°/3° | NT |
| 32 | + | − | − | 2.3/2.3 |
LogMAR, logarithm of the minimum angle of resolution. Showed as right eye/left eye.
Stimulus sizes of III4e (10,000 apostilbs, 0 decibels, 4 mm2).
ERG, electroretinography.
NT, not tested.
NR, non-recordable above noise.
Fig. 7.

Fundus photography. A (right eye) and B (left eye) are fundus photography of patient 1 (21 years), with compound mutations (T49M/A269fsX270) and a LCA phenotype shows diffuse retinopathy with pigment clumping and bone spicule pigmentation, cystoid macular edema, attenuated blood vessels with mild optic atrophy in both eyes. C is fundus photography and corresponding FD-OCT images of patient 2 (32 years) who is homozygous for the C201R mutation and has a LCA phenotype. Lines are symbolizing area of FD-OCT scan acquisition. Both eyes show a macular pseudo-coloboma with dense pigmentation, optic atrophy, arteriole and venous attenuation and peripheral granular pigmentation. Foveal and extrafoveal FD-OCT images a–c demonstrate re-organization of retinal lamination and loss of photoreceptors and epiretinal membrane in the left eye.
The novel homozygous 601 T → C mutation (C201R) in exon 5 was seen in a single patient (patient 2) with LCA. This change was not seen in 318 control chromosomes. Patient 2 of Indian ancestry from a consanguineous family was first diagnosed with LCA at 5 years of age. He reported that he always had nyctalopia and restricted visual fields but was able to read printed text at a younger age. His VA and color perception had significantly deteriorated from 25 years of age. Cataract surgery was performed on both eyes at 30 years of age. At this age, he was only able to notice hand motion in both eyes, which is equivalent to 2.3logMAR (Schulze-Bonsel, Feltgen, Burau, Hansen, & Bach, 2006) (Table 4). Dilated fundus examination revealed a macular pseudo-coloboma with dense pigmentation, optic atrophy, arteriole and venous attenuation and peripheral granular pigmentation in the both eyes. Horizontal FD-OCT scans through the fovea revealed a thinned retina with re-organization of the retinal lamination. Structures of photoreceptors are not identifiable. Epiretinal membrane with moderate traction is visible in the scan through the left foveal region (Fig. 7).
4. Discussion
4.1. Rdh12 inactivation and polymorphisms
Among the five single missense mutations in the RDH12 gene identified in this study, the T49M and C201R mutations were catalytically inactive supporting their biological role in visual impairment. The T49M mutation was previously reported as a modifier (Thompson et al., 2005) with increased activity when combined with R161Q polymorphism. Four combinations of T49/R161, M49/R161, T49/Q161 and M49/Q161 were tested in COS-7 cells by Thompson et al. (2005), M49 showed 1.5 ~ 2 fold increase in reductase activity compared to T49 in either R161 or Q161 background. To the contrary, our finding showed that the T49M change on the R161 background did not display increased Rdh12 activity as published previously, but severely decreased RDH activity in both COS-7 and Sf9 cells (~95%). Our results are consistent with the clinical phenotype and the structural model of the enzyme (Figs. 2 and 3), which predicts that the larger Met residues will interfere with the binding of the 2′-phosphate group of NADP. The differences in activity of T49M mutant observed between our study and previous work could be due in part to differences in methodology for determining enzymatic activity.
As expected, the frameshift mutation A269fsX270 resulted in the formation of a truncated nonfunctional protein, because of the loss of the residues needed to form a seventh β-strand next to canonical sixth β-strand in the Rossmann fold is defective in enzymatic activity (Buehner, Ford, Moras, Olsen, & Rossmann, 1974) (Fig. 3). The C201R variant is also a loss of function mutation. The Cys residue is located within the catalytic motif (YXXXK), just after the catalytically important Tyr. The change from a neutral nonpolar residue to a basic polar residue within the catalytic motif can disrupt the proper alignment of residues involved in catalysis.
For the R161Q mutant, the amino acid change did not interfere with the Rdh12 activity because the location of the residue in the structure is far from the phosphate-binding site, catalytic site and substrate binding site (Figs. 2 and 3). Allele frequency from control population (12.5%) compared to allele frequency in patients (12.2%) further support the polymorphic nature of this variant. The A177V variant did also not alter enzyme activity, which did not support our prediction that it would alter activity considering that the location of the residue is in the putative retinal-binding site. The amino acid residues Ala and Val are very similar and the change only caused an alkyl extension of the side chain increasing hydrophobicity within the binding of hydrophobic second substrate. Furthermore, the residue may not be in direct contact with the retinol, which would also explain the apparent lack of effect on enzymatic activity.
4.2. Clinical correlations of RDH12 mutations to enzymatic activity
Two novel mutations in RDH12 were identified in patients affected with severe LCA (homozygote C201R (patient 2) and compound heterozygote T49M/A269fsX270 (patient 1)). These two mutations completely abrogate Rdh12 enzymatic activity based on our in vitro experiments which correlates with the severe phenotype. Both patients are legally blind from early onset photoreceptor degeneration. The dramatic loss of all-trans-RDH function of the C201R change at the catalytic motif correlates with the severe retinal degeneration that characterizes LCA. The T49M mutation was identified previously (Janecke et al., 2004) in a patient diagnosed with juvenile RP at 5 years old. This patient also carries 146C → T resulting in R62X, which must result in a loss of function mutation. Activity analysis performed by Thompson et al. (2005) suggested that the T49M led to a two-fold increase in Rdh12 activity, which we were not able to confirm in our study. Thus, the single enzyme activity of the T49M mutation clearly showed much less all-trans-RDH activity in this study. Similarly, our patient with the 146C → T mutation simultaneously carries 806delCCCTG variant resulting in A269fsX270 which has no all-trans-RDH activity in biochemical analysis. Both of the patients showed severe loss of retinal function with an advanced retinal dystrophy.
4.3. Role and clinical considerations related to LCA patients with mutations in the RDH12 gene: Animal model study and human physiology
Animal models lacking functional enzymes of the retinoid cycle exhibit varying forms of retinal degeneration and the differences observed in Rdh12, Rpe65, and Lrat null mice are apparent. All three proteins belong to the same group of retinoid cycle molecules involved in the regeneration of 11-cis-retinal; however, pathogenesis of retinal degeneration is different for all of the cases. In contrast to Rpe65−/− mice and Lrat−/− mice, which appear to develop retinal degeneration due to lack of 11-cis-retinal production and their retinal structure remains well organized at younger age, Rdh12−/− mice display unaffected 11-cis-retinal synthesis (Batten et al., 2005; Imanishi, Batten, Piston, Baehr, & Palczewski, 2004; Redmond et al., 1998). Gene therapy and pharmacological bypass with artificial retinoid chromophores were successful approaches in the rescue of photoreceptors in Rpe65−/− mice and Lrat−/− mice (Acland et al., 2005, 2001; Aleman et al., 2004; Batten et al., 2005; Jacobson et al., 2005; Van Hooser et al., 2000; Van Hooser et al., 2002) (reviewed in ref. (Travis, Golczak, Moise, & Palczewski, 2007)). Rdh12 null mice exhibit normal retinal structure at six weeks of age; however, they are highly susceptible to light induced retinal cell apoptosis in both cone and rod photoreceptors (Maeda et al., 2006), thereby light damage after birth can greatly contribute to severe retinal degeneration also in humans. The retinal structure observed for patient 2 with the RDH12 loss function mutation showed poor organization, especially in the foveal region which is exposed to the highest intensities of light. Avoiding exposure to intense light, suppression of the apoptotic pathway and slowing the retinoid cycle can all be candidate approaches for the prevention of retinal degeneration in patients with RDH12 mutations.
5. Conclusions
Mutations in RDH12 play a role in a broad range of phenotypes that include patients affected with LCA, RP, and CORD. Of the six changes identified, three mutations were novel. Our screening determined that the R160Q, A177V mutations are polymorphic variants. Genotype–phenotype correlation used biochemical studies and the assessment of retinal structure and function. Biochemical activity of Rdh12 mutant proteins associated with the LCA phenotype was severely impaired, whereas those mutants that are likely polymorphisms were fully active.
Acknowledgments
We thank Dr. F. Haeseleer (Deptartment of Ophthalmology, University of Washington) for the help provided to this study, Yesmino Elia for coordinating patient scheduling and Alex Levin for contributing to patient recruitment. We are grateful to John S. Werner and Robert J. Zawadzki (Deptartment of Ophthalmology and Vision Science, University of California Davis), who allowed the FDOCT imaging by creating and loaning the instrument and providing training and discussions. This research was supported in part by Grant EY009339 from the National Eye Institute, National Institutes of Health (K.P.), the Sick Kids Research Institute’s Restracomp Fund (C.G.) and the Foundation Fighting Blindness-Canada (E.H.).
Footnotes
Abbreviations used: Ar, autosomal recessive; CORD, cone-rod dystrophy; GVF, Goldmann visual field; LCA, leber congenital amaurosis; MES, 2-(Nmorpholino) ethane-sulfonic acid; FD-OCT, Fourier-domain optical coherence tomography; RDH, retinol dehydrogenase; RP, retinitis pigmentosa; VA, visual acuity.
Publisher's Disclaimer: This article was originally published in a journal published by Elsevier, and the attached copy is provided by Elsevier for the author's benefit and for the benefit of the author's institution, for non-commercial research and educational use including without limitation use in instruction at your institution, sending it to specific colleagues that you know, and providing a copy to your institution's administrator. All other uses, reproduction and distribution, including without limitation commercial reprints, selling or licensing copies or access, or posting on open internet sites, your personal or institution's website or repository, are prohibited. For exceptions, permission may be sought for such use through Elsevier's permissions site at: http://www.elsevier.com/locate/permissionusematerial
References
- Acland GM, Aguirre GD, Bennett J, Aleman TS, Cideciyan AV, Bennicelli J, et al. Long-term restoration of rod and cone vision by single dose rAAV-mediated gene transfer to the retina in a canine model of childhood blindness. Molecular Therapeutics. 2005;12(6):1072–1082. doi: 10.1016/j.ymthe.2005.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Acland GM, Aguirre GD, Ray J, Zhang Q, Aleman TS, Cideciyan AV, et al. Gene therapy restores vision in a canine model of childhood blindness. Nature Genetics. 2001;28(1):92–95. doi: 10.1038/ng0501-92. [DOI] [PubMed] [Google Scholar]
- Aleman TS, Jacobson SG, Chico JD, Scott ML, Cheung AY, Windsor EA, et al. Impairment of the transient pupillary light reflex in Rpe65(−/−) mice and humans with leber congenital amaurosis. Investigative Ophthalmology and Visual Science. 2004;45(4):1259–1271. doi: 10.1167/iovs.03-1230. [DOI] [PubMed] [Google Scholar]
- Batten ML, Imanishi Y, Tu DC, Doan T, Zhu L, Pang J, et al. Pharmacological and rAAV gene therapy rescue of visual functions in a blind mouse model of Leber congenital amaurosis. PLoS Medicine. 2005;2(11):e333. doi: 10.1371/journal.pmed.0020333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belyaeva OV, Korkina OV, Stetsenko AV, Kim T, Nelson PS, Kedishvili NY. Biochemical properties of purified human retinol dehydrogenase 12 (RDH12): Catalytic efficiency toward retinoids and C9 aldehydes and effects of cellular retinol-binding protein type I (CRBPI) and cellular retinaldehyde-binding protein (CRALBP) on the oxidation and reduction of retinoids. Biochemistry. 2005;44(18):7035–7047. doi: 10.1021/bi050226k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowne SJ, Sullivan LS, Mortimer SE, Hedstrom L, Zhu J, Spellicy CJ, et al. Spectrum and frequency of mutations in IMPDH1 associated with autosomal dominant retinitis pigmentosa and leber congenital amaurosis. Investigative Ophthalmology and Visual Science. 2006;47(1):34–42. doi: 10.1167/iovs.05-0868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buehner M, Ford GC, Moras D, Olsen KW, Rossmann MG. Structure determination of crystalline lobster D-glyceraldehyde-3-phosphate dehydrogenase. Journal of Molecular Biology. 1974;82(4):563–585. doi: 10.1016/0022-2836(74)90249-6. [DOI] [PubMed] [Google Scholar]
- Combet C, Jambon M, Deleage G, Geourjon C. Geno3D: Automatic comparative molecular modelling of protein. Bioinformatics. 2002;18(1):213–214. doi: 10.1093/bioinformatics/18.1.213. [DOI] [PubMed] [Google Scholar]
- Cremers FP, van den Hurk JA, den Hollander AI. Molecular genetics of Leber congenital amaurosis. Human Molecular Genetics. 2002;11(10):1169–1176. doi: 10.1093/hmg/11.10.1169. [DOI] [PubMed] [Google Scholar]
- den Hollander AI, Koenekoop RK, Yzer S, Lopez I, Arends ML, Voesenek KE, et al. Mutations in the CEP290 (NPHP6) gene are a frequent cause of Leber congenital amaurosis. American Journal of Human Genetics. 2006;79(3):556–561. doi: 10.1086/507318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman JS, Chang B, Kannabiran C, Chakarova C, Singh HP, Jalali S, et al. Premature truncation of a novel protein, RD3, exhibiting subnuclear localization is associated with retinal degeneration. American Journal of Human Genetics. 2006;79(6):1059–1070. doi: 10.1086/510021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu SM, Thompson DA, Srikumari CR, Lorenz B, Finckh U, Nicoletti A, et al. Mutations in RPE65 cause autosomal recessive childhood-onset severe retinal dystrophy. Nature Genetics. 1997;17(2):194–197. doi: 10.1038/ng1097-194. [DOI] [PubMed] [Google Scholar]
- Haeseleer F, Jang GF, Imanishi Y, Driessen CA, Matsumura M, Nelson PS, et al. Dual-substrate specificity short chain retinol dehydrogenases from the vertebrate retina. Journal of Biological Chemistry. 2002;277(47):45537–45546. doi: 10.1074/jbc.M208882200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanein S, Perrault I, Gerber S, Tanguy G, Barbet F, Ducroq D, et al. Leber congenital amaurosis: Comprehensive survey of the genetic heterogeneity, refinement of the clinical definition, and genotype–phenotype correlations as a strategy for molecular diagnosis. Human Mutation. 2004;23(4):306–317. doi: 10.1002/humu.20010. [DOI] [PubMed] [Google Scholar]
- Imanishi Y, Batten ML, Piston DW, Baehr W, Palczewski K. Noninvasive two-photon imaging reveals retinyl ester storage structures in the eye. Journal of Cell Biology. 2004;164(3):373–383. doi: 10.1083/jcb.200311079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson SG, Aleman TS, Cideciyan AV, Sumaroka A, Schwartz SB, Windsor EA, et al. Identifying photoreceptors in blind eyes caused by RPE65 mutations: Prerequisite for human gene therapy success. Proceedings of National Academy of Sciences of United States of America. 2005;102(17):6177–6182. doi: 10.1073/pnas.0500646102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson SG, Cideciyan AV, Aleman TS, Sumaroka A, Schwartz SB, Windsor EA, et al. RDH12 and RPE65, visual cycle genes causing Leber congenital amaurosis, differ in disease expression. Investigative Ophthalmology and Visual Science. 2007;48(1):332–338. doi: 10.1167/iovs.06-0599. [DOI] [PubMed] [Google Scholar]
- Janecke AR, Thompson DA, Utermann G, Becker C, Hubner CA, Schmid E, et al. Mutations in RDH12 encoding a photoreceptor cell retinol dehydrogenase cause childhood-onset severe retinal dystrophy. Natural Genetics. 2004;36(8):850–854. doi: 10.1038/ng1394. [DOI] [PubMed] [Google Scholar]
- Jang GF, Kuksa V, Filipek S, Bartl F, Ritter E, Gelb MH, et al. Mechanism of rhodopsin activation as examined with ring-constrained retinal analogs and the crystal structure of the ground state protein. Journal of Biological Chemistry. 2001;276(28):26148–26153. doi: 10.1074/jbc.M102212200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang GF, McBee JK, Alekseev AM, Haeseleer F, Palczewski K. Stereoisomeric specificity of the retinoid cycle in the vertebrate retina. Journal of Biological Chemistry. 2000;275(36):28128–28138. doi: 10.1074/jbc.M004488200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang GF, Van Hooser JP, Kuksa V, McBee JK, He YG, Janssen JJ, et al. Characterization of a dehydrogenase activity responsible for oxidation of 11-cis-retinol in the retinal pigment epithelium of mice with a disrupted RDH5 gene. A model for the human hereditary disease fundus albipunctatus. Journal of Biological Chemistry. 2001;276(35):32456–32465. doi: 10.1074/jbc.M104949200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones TA, Zou JY, Cowan SW, Kjeldgaard M. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallographica, Section A. 1991;47(Pt 2):110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- Jornvall H, Persson B, Krook M, Atrian S, Gonzalez-Duarte R, Jeffery J, et al. Short-chain dehydrogenases/reductases (SDR) Biochemistry. 1995;34(18):6003–6013. doi: 10.1021/bi00018a001. [DOI] [PubMed] [Google Scholar]
- Kurth I, Thompson DA, Ruther K, Feathers KL, Chrispell JD, Schroth J, et al. Targeted disruption of the murine retinal dehydrogenase gene Rdh12 does not limit visual cycle function. Molecular and Cellular Biology. 2007;27(4):1370–1379. doi: 10.1128/MCB.01486-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda A, Maeda T, Imanishi Y, Sun W, Jastrzebska B, Hatala DA, et al. Retinol dehydrogenase (RDH12) protects photoreceptors from light-induced degeneration in mice. Journal of Biological Chemistry. 2006;281(49):37697–37704. doi: 10.1074/jbc.M608375200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marmor MF, Holder GE, Seeliger MW, Yamamoto S. Standard for clinical electroretinography (2004 update) Documenta Ophthalmologica. 2004;108(2):107–114. doi: 10.1023/b:doop.0000036793.44912.45. [DOI] [PubMed] [Google Scholar]
- Morimura H, Fishman GA, Grover SA, Fulton AB, Berson EL, Dryja TP. Mutations in the RPE65 gene in patients with autosomal recessive retinitis pigmentosa or leber congenital amaurosis. Proceedings of National Academy of Sciences of United States of America. 1998;95(6):3088–3093. doi: 10.1073/pnas.95.6.3088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohguro H, Chiba S, Igarashi Y, Matsumoto H, Akino T, Palczewski K. Beta-arrestin and arrestin are recognized by autoantibodies in sera from multiple sclerosis patients. Proceedings of National Academy of Sciences of United States of America. 1993;90(8):3241–3245. doi: 10.1073/pnas.90.8.3241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrault I, Hanein S, Gerber S, Barbet F, Ducroq D, Dollfus H, et al. Retinal dehydrogenase 12 (RDH12) mutations in leber congenital amaurosis. American Journal of Human Genetics. 2004;75(4):639–646. doi: 10.1086/424889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrault I, Rozet JM, Calvas P, Gerber S, Camuzat A, Dollfus H, et al. Retinal-specific guanylate cyclase gene mutations in Leber’s congenital amaurosis. Nature Genetics. 1996;14(4):461–464. doi: 10.1038/ng1296-461. [DOI] [PubMed] [Google Scholar]
- Perrault I, Rozet JM, Gerber S, Ghazi I, Leowski C, Ducroq D, et al. Leber congenital amaurosis. Molecular Genetics and Metabolism. 1999;68(2):200–208. doi: 10.1006/mgme.1999.2906. [DOI] [PubMed] [Google Scholar]
- Redmond TM, Yu S, Lee E, Bok D, Hamasaki D, Chen N, et al. Rpe65 is necessary for production of 11-cis-vitamin A in the retinal visual cycle. Natural Genetics. 1998;20(4):344–351. doi: 10.1038/3813. [DOI] [PubMed] [Google Scholar]
- Schulze-Bonsel K, Feltgen N, Burau H, Hansen L, Bach M. Visual acuities “hand motion” and “counting fingers” can be quantified with the freiburg visual acuity test. Investigative Ophthalmology and Visual Science. 2006;47(3):1236–1240. doi: 10.1167/iovs.05-0981. [DOI] [PubMed] [Google Scholar]
- Sohocki MM, Bowne SJ, Sullivan LS, Blackshaw S, Cepko CL, Payne AM, et al. Mutations in a new photoreceptor–pineal gene on 17p cause Leber congenital amaurosis. Nature Genetics. 2000;24(1):79–83. doi: 10.1038/71732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sohocki MM, Perrault I, Leroy BP, Payne AM, Dharmaraj S, Bhattacharya SS, et al. Prevalence of AIPL1 mutations in inherited retinal degenerative disease. Molecular Genetics and Metabolism. 2000;70(2):142–150. doi: 10.1006/mgme.2000.3001. [DOI] [PubMed] [Google Scholar]
- Sohocki MM, Sullivan LS, Mintz-Hittner HA, Birch D, Heckenlively JR, Freund CL, et al. A range of clinical phenotypes associated with mutations in CRX, a photoreceptor transcription-factor gene. American Journal of Human Genetics. 1998;63(5):1307–1315. doi: 10.1086/302101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swaroop A, Wang QL, Wu W, Cook J, Coats C, Xu S, et al. Leber congenital amaurosis caused by a homozygous mutation (R90W) in the homeodomain of the retinal transcription factor CRX: Direct evidence for the involvement of CRX in the development of photoreceptor function. Human Molecular Genetics. 1999;8(2):299–305. doi: 10.1093/hmg/8.2.299. [DOI] [PubMed] [Google Scholar]
- Thompson DA, Janecke AR, Lange J, Feathers KL, Hubner CA, McHenry CL, et al. Retinal degeneration associated with RDH12 mutations results from decreased 11-cis retinal synthesis due to disruption of the visual cycle. Human Molecular Genetics. 2005;14(24):3865–3875. doi: 10.1093/hmg/ddi411. [DOI] [PubMed] [Google Scholar]
- Thompson DA, Li Y, McHenry CL, Carlson TJ, Ding X, Sieving PA, et al. Mutations in the gene encoding lecithin retinol acyltransferase are associated with early-onset severe retinal dystrophy. Nature Genetics. 2001;28(2):123–124. doi: 10.1038/88828. [DOI] [PubMed] [Google Scholar]
- Travis GH, Golczak M, Moise AR, Palczewski K. Diseases caused by defects in the visual cycle: Retinoids as potential therapeutic agents. Annual Review of Pharmacology and Toxicology. 2007;47:469–512. doi: 10.1146/annurev.pharmtox.47.120505.105225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Hooser JP, Aleman TS, He YG, Cideciyan AV, Kuksa V, Pittler SJ, et al. Rapid restoration of visual pigment and function with oral retinoid in a mouse model of childhood blindness. Proceedings of National Academy of Sciences of United States of America. 2000;97(15):8623–8628. doi: 10.1073/pnas.150236297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Hooser JP, Liang Y, Maeda T, Kuksa V, Jang GF, He YG, et al. Recovery of visual functions in a mouse model of Leber congenital amaurosis. Journal of Biological Chemistry. 2002;277(21):19173–19182. doi: 10.1074/jbc.M112384200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weleber RG. Infantile and childhood retinal blindness: A molecular perspective (The Franceschetti Lecture) Ophthalmic Genetics. 2002;23(2):71–97. doi: 10.1076/opge.23.2.71.2214. [DOI] [PubMed] [Google Scholar]
- Zawadzki RJ, Jones SM, Olivier SS, Zhao M, Bower BA, Izatt JA, et al. Adaptive-optics optical coherence tomography for high-resolution and high-speed 3D retinal in vivo imaging. Optics Express. 2005;13(21):8532–8546. doi: 10.1364/opex.13.008532. [DOI] [PMC free article] [PubMed] [Google Scholar]