

Abstract
Plasmid pLNBIV was used to overexpress the biosynthetic pathway of nucleoside-diphosphate (NDP)-activatedl-digitoxose in the mithramycin producer Streptomyces argillaceus. This led to a “flooding” of the biosynthetic pathway of the antitumor drug mithramycin (MTM) with NDP-activated deoxysugars, which do not normally occur in the pathway, and consequently to the production of the four new mithramycin derivatives 1-4 with altered saccharide patterns. Their structures reflect that NDP sugars produced by pLNBIV, namely, l-digitoxose and its biosynthetic intermediates, influenced the glycosyl transfer to positions B, D, and E, while positions A and C remained unaffected. All four new structures have unique, previously not found sugar decoration patterns, which arise from either overcoming the substrate specificity or inhibition of certain glycosyltransferases (GTs) of the MTM pathway with the foreign NDP sugars expressed by pLNBIV. An apoptosis TUNEL (=terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling) assay revealed that compounds 1 (demycarosyl-3D-β-d-digitoxosyl-MTM) and 3 (deoliosyl-3C-β-d-mycarosyl-MTM) show improved activity (64.8 ± 2% and 50.3 ± 2.5% induction of apoptosis, respectively) against the estrogen receptor (ER)-positive human breast cancer cell line MCF-7 compared with the parent drug MTM (37.8 ± 2.5% induction of apoptosis). In addition, compounds 1 and 4 (3A-deolivosyl-MTM) show significant effects on the ER-negative human breast cancer cell line MDA-231 (63.6 ± 2% and 12.6 ± 2.5% induction of apoptosis, respectively), which is not inhibited by the parent drug MTM itself (2.6 ± 1.5% induction of apoptosis), but for which chemotherapeutic agents are urgently needed.
Mithramycin (MTM), a clinically used aureolic acid-type anti-cancer drug and calcium-lowering agent produced by Streptomyces argillaceus as well as various other streptomycetes, consists of a polyketide-derived tricyclic core with a highly functionalized pentyl side chain at the 3-position and five deoxysugars linked as trisaccharide and disaccharide chains in the 2- and 6-positions, respectively. Its aglycon including the 3-side chain is biosynthesized from 10 acyl-CoA units by a type II polyketide synthase, and all five deoxyhexose moieties are successively attached to the tetracyclic intermediate premithramycinone, resulting in the fully glycosylated intermediate premithramycin B via premithramycin A3,1-7 before an oxidative cleavage reaction, catalyzed by Baeyer-Villigerase MtmOIV,3,5,7 followed by the reduction of a side chain keto group by MtmW, finishes the biosynthesis (Scheme 1). The glycosylation steps remain partly obscure, although the sequence could be established unambiguously: initially, the trisaccharide chain is formed through successive glycosylation steps, before the disaccharide chain is attached, also in a stepwise sequential order. It was shown that the glycosyltransferases (GTs) MtmGIII and MtmGIV are involved in the trisaccharide chain formation, while MtmGI and MtmGII catalyze the attachment of the disaccharide chain (Scheme 1).2,6,8,9
Scheme 1.

Key Steps of the Mithramycin Biosynthetic Pathway, Showing the Sequential Order of the Five Glycosyltransfer Steps along with the Catalyzing Enzymes
Since genes for only four GTs were found in the mtm gene cluster, which are responsible for five glycosylation steps, it was suggested from indirect evidence8 that MtmGIV catalyzes the attachment of both sugars C (proven) and E (likely), while MtmGIII catalyzes the attachment of oliose D. MtmGI and MtmGII are responsible for the attachment of olivoses A and B, respectively. It is well-known that the glycosylation pattern of DNA-binding anticancer drugs has an essential impact on the biological activity.10-18 Therefore, we wanted to generate new mithramycin analogues with altered saccharide patterns. We were particularly interested in the construction of alternative molecules with variations in the trisaccharide chain (maintaining three sugars there) and changing or shortening the disaccharide moiety.
Keeping the lower sugar side chain as a trisaccharide is important because several studies showed that all three sugars in this trisaccharide chain are necessary for optimal DNA interaction.16-18 In contrast, no SAR (structure-activity relationship) conclusions regarding the upper disaccharide chain of mithramycin have ever been drawn because MTM derivatives with alterations in the upper chain were not available. To achieve an alteration of MTM’s saccharide pattern, we heterologously overexpressed deoxysugar biosynthesis pathways into the mithramycin producer S. argillaceus using our recently assembled deoxysugar plasmids. Here we report the results of using plasmid pLNBIV, which encodes the biosynthesis of nucleosyldiphosphate (NDP)-l-digitoxose, but also its NDP-d- and l-4-ketosugar intermediates.15,19
Results and Discussion
Four new mithramycin derivatives were found to be accumulated in S. argillaceus (pLNBIV), partially with quite unexpected structures. These were demycarosyl-3D-β-d-digitoxosyl-MTM (1), deoliosyl-3C-α-l-digitoxosyl-MTM (2), deoliosyl-3C-β-d-mycarosyl-MTM (3), and 3A-deolivosyl-MTM (4).
These four products were selected on the basis of their HPLC-UV and HPLC-MS data, which indicated a mithramycin (not premithramycin) chromophore (UV data) and the presence of at least four sugar residues (mass data). These selections were made from previous SAR studies showing that all tetracyclic premithramycin derivatives do not bind to DNA and therefore were biologically inactive, in contrast to tricyclic mithramycin derivatives, in which the alkyl side chains at the 3- and 7-positions were established.4,20,21 In addition, a complete trisaccharide chain appears to be essential for DNA interactions.16-18 Production of selected compounds were 4.47 mg/L 1, 1.81 mg/L 2, 1.03 mg/L 3, and 3.41 mg/L 4. These four products were isolated and structurally characterized by 1D- and 2D-NMR spectroscopy as well as mass spectrometry. The type and connectivity of all deoxysugar units were determined using various 1D- and 2D-NMR spectra, including 1D-TOCSY and 1H,1H DQF-COSY (to unambiguously assign the protons of each deoxysugar moiety), 1H,1H NOESY (to determine sugar linkages and to distinguish sugar chair conformations), and CIGAR-HMBC22 (to determine connections between sugar moieties).
For compound 1, a molecular formula of C51H74O24 was deduced from the HRFAB mass spectrum (m/z [M + Na]+: calcd, 1093.4468; found, 1093.4447), which indicated a molecular mass of 14 amu smaller than that of the parent compound MTM. The 1H and 13C NMR spectra showed that the aglycon moiety, the disaccharide side chain (sugars A and B), and sugar units C and D of the trisaccharide chain of this molecule were identical with the corresponding moieties of MTM,23 while the difference was found in sugar E, which apparently was a non-C-methylated dideoxy sugar. This was also evident from the MS fragmentation pattern (see Experimental Section). Analysis of the 1H NMR data showed that the β-d-mycarose unit of MTM was replaced by a β-d-digitoxose in 1, most evidently indicated by the equatorially positioned H-3E signal at δH 4.45 (ddd, J = 3.0, 3.0, and 3.0 Hz), which showed e,a couplings with H-2Eax (2.00, ddd, J = 12, 9.6 and 3.0 Hz) and H-4E (3.62, dd, J = 9.2 and 3.0 Hz) and an e,e coupling with H-2Eeq (2.45 ddd, J = 12, 3.0 and 2.0 Hz). The 13C NMR data also showed that the 3E-CH3 was missing and significantly altered carbon signals for the 2E, 3E, and 4E positions. The connectivity and conformation of the β-d-digitoxose moiety in 1 was deduced from the 1H,13C CIGAR-HMBC and 1H,1H NOESY interactions (see Table 1), respectively, showing a coupling between its anomeric proton (H-1E, at δH 5.62) and C-3D (δC 77.2) and through-space couplings between the three axial protons H-1E (δH 5.62), H-3E (δH 4.45), and H-5E (δH 4.69), respectively, which confirm the 4C1 conformation of sugar E. Overall, the structure of 1 was deduced as demycarosyl-3D-β-d-digitoxosyl-MTM.
Table 1.
1H (400 MHz) and 13C (75.4 MHz) NMR Spectroscopic Data (pyridine-d5) of Demycarosyl-3D-β-d-digitoxosyl-MTM (1) and Deoliosyl-3C-α-l-digitoxosyl-MTM (2)
| 1 |
2 |
|||||||
|---|---|---|---|---|---|---|---|---|
| pos. | δC, mult. | δH (J in Hz) | HMBC | NOESY | δC | δH (J in Hz) | HMBC | NOESY |
| 1 | 204.3, qC | 204.2 | ||||||
| 2 | 78.7, CH | 4.96 d (11.6) | C-1, C-1C, C-3, C-4, C-1′ | H-1C, H-3, H-4ax, H-4eq | 77.9 | 4.93 d (11.6) | C-1, C-1C, C-3, C-4, C-1′ | H-1C, H-3, H-4ax, H-4eq |
| 3 | 42.8, CH | 3.48 m | C-2, C-4, C-1′ | H-2, H-4ax, H-4eq, H-1′ | 42.8 | 3.41 m | C-2, C-4, C-1′ | H-2, H-4ax, H-4eq, H-1′ |
| 4ax | 28.5, CH2 | 3.11 dd (15.9 and 3.6) | C-2, C-3, C-1′ | H-3, H-2 | 28.2 | 3.08 dd (15.9 and 3.6) | C-2, C-3, C-1′ | H-3, H-2 |
| 4eq | 3.27 dd (15.9 and 13.0) | H-3, H-2 | 3.26 dd (15.9 and 13.0) | |||||
| 4a | 137.2, qC | 137.2 | ||||||
| 5 | 102.2, CH | 7.05 s | C-6, C-8a, C-7, C-10 | H-10, H-1A, H-5A | 102.1 | 7.04 s | C-6, C-8a, C-7, C-10 | H-10, H-1A, H-5A |
| 6 | 160.4, qC | 160.4 | ||||||
| 7 | 111.7, qC | 111.6 | ||||||
| 7-CH3 | 9.4, CH3 | 2.47 s | C-6, C-7, C-8 | 9.4 | 2.47 s | C-6, C-7, C-8 | ||
| 8 | 156.9, qC | 157.0 | ||||||
| 8a | 109.4, qC | 109.4 | ||||||
| 9 | 165.7, qC | 165.7 | ||||||
| 9a | 108.9, qC | 108.9 | ||||||
| 10 | 117.4, CH | 6.63 s | C-5, C-8a, C-9a, C-10a | H-5, H-1A, H-4ax, H-4eq | 117.4 | 6.61 s | C-5, C-8a, C-9a, C-10a | H-5, H-1A, H-4ax, H-4eq |
| 10a | 139.5, qC | 139.4 | ||||||
| 1′ | 83.1, CH | 5.49 d (1.2) | C-2, C-3, C-4, 1′-OCH3, C-2′ | H-2, H-3, 1′-OCH3 | 83.1 | 5.46 d (1.2) | C-2, C-3, C-4, 1′-OCH3, C-2′ | H-2, H-3, 1′-OCH3 |
| 1′-OCH3 | 59.1, CH3 | 3.69 s | C-1′, C-2′, C-3 | 59.4 | 3.69 s | C-1′, C-2′, C-3 | ||
| 2′ | 213.5, qC | 213.4 | ||||||
| 3′ | 81.4, CH | 4.70 d (2.8) | C-2′, C-4′, C-5′ | H-4′, H-5′ | 81.3 | 4.71 d (2.8) | C-2′, C-4′, C-5′ | H-4′, H-5′ |
| 4′ | 69.6, CH | 4.84 dq (6.2 and 2.8) | C-2′, C-3′, C-5′ | H-5′, H-3′ | 69.6 | 4.84 dq (6.2 and 2.8) | C-2′, C-3′, C-5′ | H-5′, H-3′ |
| 5′ | 19.7, CH3 | 1.60 d (6.2) | C-3′, C-4′ | H-4′, H-3′ | 21.1 | 1.58 d (6.2) | C-3′, C-4′ | H-4′, H-3′ |
| 1A | 97.9, CH | 5.68 dd (9.6 and 2.0) | C-6 | H-3A, H-5A, H-2Aax, H-2Aeq, H-5, H-10 | 97.9 | 5.67 dd (9.6 and 2.0) | C-6 | H-5, H-10, H-3A, H-5A, H-2Aax, H-2Aeq |
| 2Aax | 38.0, CH2 | 2.25 ddd (12, 12 and 9.6) | C-1A, C-3A, C-4A | H-1A, H-4A, H-2Aeq | 37.7 | 2.24 ddd (12, 12 and 9.6) | C-1A, C-3A | H-1A, H-4A |
| 2Aeq | 2.72 ddd (12, 5.0 and 2.0) | H-1A, H-3A, H-2Aax | 2.73 ddd (12, 5.0 and 2.0) | H-1A | ||||
| 3A | 80.1, CH | 4.29 ddd (12, 9.0 and 5.0) | C-1B, C-4A | H-1A, H-1B, H-5A, H-2Aax, H-2Aeq | 80.1 | 4.29 ddd (12, 9.0 and 5.0) | C-1B, C-4A | H-1A, H-1B, H-5A, H-2Aeq |
| 4A | 76.1, CH | 3.63 t (9.0 Hz) | C-3A, C-5A, C-6A | H-2Aax, H3-6A | 76.1 | 3.63 t (9.0) | C-3A, C-5A, C-6A | H-2Aax, H3-6A |
| 5A | 73.9, CH | 4.00 dq (9.0 and 6.0) | C-1A, C-3A, C-4A, C-6A | H-1A, H-3A, H3-6A | 73.9 | 4.00 dq (9.0 and 6.0) | C-4A, C-6A | H-1A, H-3A, H3-6A |
| 6A | 19.4, CH3 | 1.71 d (6.0) | C-4A, C-5A | H-4A, H-5A | 19.6 | 1.71 d (6.0) | C-4A, C-5A | H-4A, H-5A |
| 1B | 99.4, CH | 5.05 dd (9.6 and 2.0) | C-3A | H-2Bax, H-2Beq, H-3A, H-3B, H-5B | 99.3 | 5.05 dd (9.6 and 2.0) | C-3A | H-3A, H-3B, H-5B, H-2Bax, H-2Beq |
| 2Bax | 41.6, CH2 | 2.15 ddd (12, 12 and 9.6) | C-1B, C-3B, C-4B | H-1B, H-4B | 41.6 | 2.17 ddd (12, 12 and 9.6) | C-1B, C-3B | H-1B, H-4B |
| 2Beq | 2.65 ddd (12, 5.0 and 2.0) | H-1B | 2.65 ddd (12, 5.0 and 2.0) | H-1B | ||||
| 3B | 72.4, CH | 4.16 ddd (12, 9.0 and 5.0) | C-4B | H-2Beq, H-1B, H-5B | 72.4 | 4.15 ddd (12, 9.0 and 5.0) | C-1B, C-4B | H-1B, H-5B |
| 4B | 78.7, CH | 3.58 t (9.0) | C-3B, C-5B, C-6B | H-2Bax, H3-6B | 78.7 | 3.58 t (9.0) | C-3B, C-5B, C-6B | H-2Bax, H-3B, H-5B, H3-6B |
| 5B | 74.6, CH | 3.75 dq (9.0 and 6.0) | C-1B, C-3B, C-4B, C-6B | H-1B, H-3B, H3-6B | 74.0 | 3.75 dq (9.0 and 6.2) | C-4B, C-6B | H-1B, H-3B, H-4B, H3-6B |
| 6B | 19.1, CH3 | 1.64 d (6.0) | C-4B, C-5B | H-5B | 19.3 | 1.64 d (6.2) | C-4B, C-5B | H-4B, H-5B |
| 1C | 101.9, CH | 5.39 dd (9.6 and 2.0) | C-2 | H-2, H-3C, H-5C, H-2Ceq | 101.9 | 5.37 dd (9.6 and 2.0) | C-2 | H-2, H-3C, H-5C, H-2Ceq, H-2Cax |
| 2Cax | 38.7, CH2 | 2.02 ddd (12, 12 and 9.6) | C-1C, C-3C, C-4C | H-4C | 37.7 | 1.93 ddd (12, 12 and 9.6) | C-3C | H-4C |
| 2Ceq | 2.99 ddd (12, 5.0 and 2.0) | H-1C | 2.98 ddd (12, 5.0 and 2.0) | H-1C, H-1D, H-3C, H-2Cax | ||||
| 3C | 81.7, CH | 4.06 ddd (12, 9.0 and 5.0) | C-1D, C-4C | H-1C, H-1D, H-5C, H-2Ceq | 77.9 | 4.08 ddd (12, 9.0 and 5.0) | C-1D, C-4C | H-1C, H-5C, H-4C, H-2Ceq |
| 4C | 76.3, CH | 3.46 t (9.0) | C-3C, C-5C, C-6C | H-2Cax, H3-6C | 76.5 | 3.48 t (9.0) | C-3C, C-6C | H-2Cax, H-5C, H-6C |
| 5C | 73.5, CH | 3.68 dq (9.0 and 6.0) | C-4C, C-6C | H-3C, H-1C, H3-6C | 73.4 | 3.68 dq (9.0 and 6.0) | C-4C, C-6C | H-1C, H-3C, H-4C, H3-6C |
| 6C | 19.7, CH3 | 1.59 d (6.0) | C-4C, C-5C | H-5C, H-4C | 19.4 | 1.57 d (6.0) | C-4C, C-5C | H-4C, H-5C |
| 1D | 100.4, CH | 4.78 dd (9.6 and 2.0) | C-3C | H-3D, H-5D, H-2Deq, H-3C | 94.9 | 5.22 dd (4.0 and 2.3) | C-3C | H-3D, H-2Ceq, H-2Deq, H-2Dax |
| 2Dax | 33.3, CH2 | 2.42 ddd (12, 12 and 9.6) | C-1D, C-3D, C-4D | H-1D | 34.6 | 2.02 ddd (14.4, 4.0 and 3.0) | C-1D, C-3D | H-1D, H-3D, H-2Deq, |
| 2Deq | 2.18 ddd (12, 5.0 and 2.0) | 2.35 ddd (14.4, 3.0 and 2.3) | H-1D, H-3D, H-2Dax | |||||
| 3D | 77.2, CH | 4.22 ddd (12, 5.0 and 2.5) | C-1E, C-4D | H-1D, H-2Deq, H-5D, H-1E | 75.2 | 4.39 ddd (3.0 3.0 and 3.0) | C-1D, C-4D, C-5D, C-1E | H-1D, H-2Dax, H-2Deq, H-1E |
| 4D | 70.2, CH | 4.10 br d (2.5) | C-3D, C-5D | H3-6D | 72.8 | 3.67 ddd (9.0 and 3.0) | C-5D, C-6D | H-5D, H3-6D |
| 5D | 74.6, CH | 3.71 dq (6.0 and 2.5) | C-4D, C-6D | H-1D, H-3D, H3-6D | 67.4 | 4.69 dq (9.0 and 6.2) | C-4D, C-6D | H-1D, H-4D, H3-6D |
| 6D | 19.4, CH3 | 1.54 d (6.0) | C-4D, C-5D | H-5D | 18.9 | 1.54 d (6.2) | C-4D, C-5D | H-4D, H-5D |
| 1E | 97.8, CH | 5.62 dd (9.6 and 2.0) | C-3D | H-5E, H-2Eax, H-2Eeq, H-3D | 98.7 | 5.44 dd (9.6 and 2.0) | C-3D | H-2Eax, H-2Eeq, H-3D, H-5E |
| 2Eax | 40.3, CH2 | 2.00 ddd (12, 9.6 and 3.0) | C-1E, C-3E, C-4E | H-1E, H-2Eax | 45.5 | 1.86 dd (13.0, and 9.0) | C-1E, C-3E | H-1E, H-4E, |
| 2Eeq | 2.45 ddd (12, 3.0 and 2.0) | 2.41 dd (13.0 and 2.0) | H-1E | |||||
| 3E | 69.1, CH | 4.45 ddd (3.0, 3.0, and 3.0) | C-4E | H-4E | 71.4 | |||
| 3E-CH3 | 28.2 | 1.44 s | C-3E, C-4E, C-2E | H-4E | ||||
| 4E | 72.4, CH | 3.62 dd (9.2 and 3.0) | C-3E, C-5E | H-2Eax, H-3E, H3-6E | 77.8 | 3.31 d (9.2) | 3E-CH3, C-5E, C-6E | H-2Eax, H3-3E, H-5E, H3-6E |
| 5E | 71.2, CH | 4.42 dq (9.2 and 6.0) | C-1E, C-4E, C-6E | H3-6E, H-1E | 72.2 | 4.22 dq (9.2 and 6.0) | H-1E, H3-6E | |
| 6E | 17.9, CH3 | 1.61 d (6.0) | C-4E, C-5E | H-5E | 19.1 | 1.60 d (6.0) | C-4E, C-5E | H-4E, H-5E |
The molecular formula of compound 2 was deduced as C52H76O24 from the +ve ESIMS, showing the molecular ion peak of m/z 1107 [M + Na]+ and from the HRFABMS (calcd for [M + Na]+ m/z 1107.4624, found 1107.4603). Comparison of the 1H and 13C NMR NMR data of this compound with those of MTM revealed identical sugar patterns, except for sugar D, for which the data suggested an β-l-digitoxosyl moiety instead of the β-d-oliosyl residue found in MTM. In the 1H NMR spectrum the anomeric proton H-1D (δH 5.22) showed two small coupling constants (dd, J = 4.0 and 2.3 Hz), typical for an α-glycosidically linked 2-deoxyhexose, usually an l-sugar. Furthermore, the observed couplings of the three methine protons in 3-, 4-, and 5-position of this sugar moiety, H-3D (δH 4.39, ddd, J = 3.0, 3.0 and 3.0 Hz), H-4D (δH 3.67, dd, J = 9.0 and 3.0 Hz), and H-5D (δH 4.69, dq, J = 9.0 and 6.2 Hz), showed H-3 in equatorial, but H-4 and H-5 in axial positions, which overall indicated a digitoxose stereochemistry. This all suggested that sugar D must be α-l-digitoxose. The position of this α-l-digitoxose as sugar D of the trisaccharide was confirmed through 1H,13C CIGAR-HMBC, in which cross-peaks were observed between H-3C (δ 4.06) and C-1D (δ 100.4), between H-3D (δ 4.22) and C-1E (δ 97.8), as well as between the anomeric protons H-1D (δ 4.78) and C-3C (δ 81.7), and between H-1E (δ 5.62) and C-3D (δ 77.2). All NMR assignments were verified with 1H,13C HSQC, CIGAR-HMBC,22 1H,1H DQF-COSY, and NOESY spectra. Overall, structure 2 could be deduced as deoliosyl-3C-α-l-digitoxosyl-MTM (Table 1).
Compounds 3 and 4 were analyzed through HRFABMS, and molecular formulas of C46H66O21 (m/z [M + Na+]: calcd, 977.3994; found, 977.3948) and C46H66O21 (m/z [M + H+]: calcd, 955.4175; found, 955.4208), respectively, were deduced. These data suggest that both compounds lack one 2,6-dideoxysugar unit when compared with MTM. The comparison of the NMR data of compound 3 with MTM showed that this compound contains two disaccharide chains and that MTM’s d-oliose moiety (sugar D in MTM) of the trisaccharide chain was missing. The d-mycarose moiety (normally sugar E) was directly attached to the d-olivose in the lower saccharide chain of 3, as revealed by the HMBC long-range couplings (see Table 2). Thus compound 3 was identified as deoliosyl-3C-β-d-mycarosyl-MTM. In contrast, the NMR data of compound 4 revealed that this analogue lacks the second sugar normally found in MTM’s disaccharide chain (sugar B), while the rest of the molecule was identical with MTM. Thus, compound 4 was identified as 3A-deolivosyl-MTM. Both structures 3 and 4 were confirmed by extensive NMR work including 1H,13C HSQC, CIGAR-HMBC, 1H,1H DQF-COSY, and NOESY experiments (see Table 2).
Table 2.
1H (400 MHz) and 13C (75.4 MHz) NMR Spectroscopic Data (pyridine-d5) of Demycarosyl-3C-β-d-mycarosyl-MTM (3) and 3A-Deolivosyl-MTM (4)
| 3 |
4 |
|||||||
|---|---|---|---|---|---|---|---|---|
| pos. | δC, mult. | δH (J in Hz) | HMBC | NOESY | δC | δH (J in Hz) | HMBC | NOESY |
| 1 | 204.2, qC | 204.4 | ||||||
| 2 | 77.9, CH | 4.96 d (11.6) | C-1, C-1C, C-3, C-4, C-1′ | H-1C, H-3, H-4ax, H-4eq | 77.9 | 4.96 d (11.6) | C-1, C-1C, C-3, C-4, C-1′ | H-1C, H-3, H-4ax, H-4eq |
| 3 | 42.6, CH | 3.48 m | C-2, C-4, C-1′ | H-2, H-4ax, H-4eq, H-1′ | 42.6 | 3.48 m | C-2, C-4, C-1′ | H-2, H-4ax, H-4eq, H-1′ |
| 4ax | 28.2, CH2 | 3.10 dd (15.9 and 3.6) | C-2, C-3, C-1′ | H-3, H-2 | 28.1 | 3.12 dd (15.9 and 3.6) | C-2, C-3, C-1′ | H-3, H-2 |
| 4eq | 3.27 dd (15.9 and 13.0) | H-3, H-2 | 3.26 dd (15.9 and 13.0) | H-3, H-2 | ||||
| 4a | 137.2, qC | 137.2 | ||||||
| 5 | 102.2, CH | 7.04 s | C-6, C-8a, C-7, C-10 | H-10, H-1A, H-5A | 102.2 | 7.04 s | C-6, C-7, C-8a, C-10 | H-10, H-1A, H-5A |
| 6 | 160.5, qC | 160.6 | ||||||
| 7 | 111.2, qC | 111.7 | ||||||
| 7-CH3 | 8.9, CH3 | 2.47 s | C-6, C-7, C-8 | 8.9 | 2.48 s | C-6, C-7, C-8 | ||
| 8 | 157.0, qC | 157.0 | ||||||
| 8a | 109.3, qC | 109.4 | ||||||
| 9 | 166.0, qC | 165.9 | ||||||
| 9a | 108.2, qC | 108.9 | ||||||
| 10 | 117.4, CH | 6.62 s | C-5, C-8a, C-9a, C-10a | H-5, H-1A, H-4ax, H-4eq | 117.4 | 6.62 s | C-5, C-8a, C-9a, C-10a | H-5, H-1A, H-4ax, H-4eq |
| 10a | 139.4, qC | 139.5 | ||||||
| 1′ | 83.0, CH | 5.49 d (1.2) | C-2, C-3, C-4, 1′-OCH3, C-2′ | H-2, H-3, 1′-OCH3 | 83.0 | 5.49 d (1.2) | C-2, C-3, C-4, 1′-OCH3, C-2′ | H-2, H-3, 1′-OCH3 |
| 1′-OCH3 | 59.1, CH3 | 3.71 s | C-1′, C-2′, C-3 | 59.1 | 3.69 s | C-1′, C-2′, C-3 | ||
| 2′ | 213.2, qC | 213.5 | ||||||
| 3′ | 81.0, CH | 4.71 d (2.8) | C-2′, C-4′, C-5′ | H-4′, H-5′ | 81.1 | 4.70 d (2.8) | C-2′, C-4′, C-5′ | H-4′, H-5′ |
| 4′ | 69.3, CH | 4.84 dq (6.2 and 2.8) | C-2′, C-3′, C-5′ | H-5′, H-3′ | 69.4 | 4.84 dq (6.0 and 2.8) | C-2′, C-3′, C-5′ | H-5′, H-3′ |
| 5′ | 20.7, CH3 | 1.60 d (6.2 Hz) | C-3′, C-4′ | H-4′, H-3′ | 20.7 | 1.59 d (6.0) | C-3′, C-4′ | H-4′, H-3′ |
| 1A | 97.9, CH | 5.67 d (9.6 and 2.0) | C-6 | H-5, H-5A, H-3A, H-2Aeq, H-10 | 98.0 | 5.63 dd (9.6 and 2.0) | C-6 | H-5, H-3A, H-5A, H-2Aeq |
| 2Aax | 37.6, CH2 | 2.25 ddd (12, 12 and 9.6) | C-1A, C-3A | H-4A, H-2Aeq | 36.7 | 2.00 ddd (12.0, 12.0 and 9.6) | C-1A, C-3A, C-4A | H-4A, H-2Aeq |
| 2Aeq | 2.72 ddd (12, 5.0 and 2.0) | H-1A, H-3A, H-2Aax | 2.73 ddd (12.0, 5.0 and 2.0) | H-1A, H-2Aax | ||||
| 3A | 80.0, CH | 4.28 ddd (12, 9.0 and 5.0) | C-1B, C-4A | H-1A, H-1B, H-5A | 74.2 | 4.35 ddd (12, 9.2 and 5.0) | C-2A, 4A | H-1A, H-5A |
| 4A | 75.9, CH | 3.63 t (9.0) | C-3A, C-5A, C-6A | H-2Aax, H3-6A | 75.4 | 3.63 t (9.2) | C-3A, C-5A, C-6A | H-2Aax, H3-6A |
| 5A | 73.8, CH | 3.99 dq (9.0 and 6.0) | C-4A, C-6A | H-1A, H-3A, H3-6A | 73.8 | 4.01 dq (9.2 and 6.0 Hz) | C-1A, C-3A, C-4A, C-6A | H-1A, H-3A, H3-6A |
| 6A | 19.0, CH3 | 1.71 d (6.0) | C-5A, C-4A | H-4A, H-5A | 19.4 | 1.70 d (6.0) | C-4A, C-5A | H-4A, H-5A |
| 1B | 99.4, CH | 5.05 dd (9.6 and 2.0) | C-3A | H-3B, H-3A, H-2Bax, H-2Beq, H-5B | ||||
| 2Bax | 41.2, CH2 | 2.17 ddd (12, 12 and 9.6) | C-1B, C-3B | H-1B, H-4B | ||||
| 2Beq | 2.65 ddd (12, 5.0 and 2.0) | H-1B | ||||||
| 3B | 72.1, CH | 4.14 ddd (12, 9.0 and 5.0) | C-4B | H-1B, H-5B | ||||
| 4B | 78.5, CH | 3.58 t (9.0) | C-5B, C-6B | H-2Bax | ||||
| 5B | 73.7, CH | 3.75 dq (9.0 and 6.0) | C-4B, C-6B | H-1B, H-3B, H3-6B | ||||
| 6B | 18.9, CH3 | 1.64 d (6.0) | C-4B, C-5B | H-5B | ||||
| 1C | 101.6, CH | 5.40 dd (9.6 and 2.0) | C-2 | H-2, H-3C, H-5C, H-2Ceq | 101.7 | 5.40 dd (9.6 and 2.0) | C-2 | H-2, H-5C, H-3C, H-2Ceq |
| 2Cax | 38.5, CH2 | 2.02 ddd (12, 12 and 9.6) | C-3C | H-4C | 38.4 | 2.02 ddd (12, 12 and 9.6) | C-1C, C-3C, C-4C | H-4C |
| 2Ceq | H-1C2.99 ddd (12, 5.0 and 2.0) | 2.99 ddd (12, 5.0 and 2.0) | ||||||
| 3C | 81.1, CH | 4.03 ddd (12, 9.0 and 5.0) | C-1D, C-4C | H-1C, H-1D, H-5C, H-2Ceq | 81.6 | 4.06 ddd (12, 9.0 and 5.0) | C-1D, C-4C | H-1C, H-5C, H-1D |
| 4C | 76.3, CH | 3.48 t (9.0) | C-3C, C-5C | H-2Cax, H3-6C | 76.2 | 3.47 t (9.0) | C-3C, C-5C, C-6C | H-2Cax, H3-6C |
| 5C | 73.6, CH | 3.69 dq (9.0 and 6.0) | C-4C, C-6C | H-3C, H-1C, H3-6C | 73.3 | 3.67 dq (9.0 and 6.0) | C-1C, C-4C, C-6C | H-1C, H-3C, H3-6C |
| 6C | 18.7, CH3 | 1.58 d (6.0) | C-4C, C-5C | H-4C, H-5C | 18.9 | 1.60 d (6.0) | C-4C, C-5C | H-5C |
| 1D | 98.9, CH | 5.46 dd (9.6 and 2.0) | C-3C | H-3C, H-5D | 100.3 | 4.79 dd (9.6 and 2.0) | C-3C | H-3C, H-3D, H-5D |
| 2Dax | 45.2, CH2 | 1.86 dd (13.0 and 9.6) | C-1D, C-3D | H-2Deq | 33.1 | 2.42 ddd (12, 12 and 9.6) | C-1D, C-3D, C-4D | |
| 2Deq | 2.33 dd (13.0 and 2.0) | H-2Dax | 2.18 ddd (12, 5.0 and 2.0) | |||||
| 3D | 71.2, CH | 77.0 | 4.21 ddd (12, 5.0 and 2.5) | C-1E, C-4D | H-1D, H-5D, H-1E | |||
| 3D-CH3 | 27.9, CH3 | 1.50 s | C-2D, C-4D, C-3D | H-4D | ||||
| 4D | 77.4, CH | 3.35 d (9.2) | C-3D, C-5D | 69.9 | 4.10 br d (2.5) | C-3D, C-5D | H-5D, H3-6D | |
| 5D | 72.1, CH | 4.29 dq (9.2 and 6.2) | C-4D, C-6D | H-1D | 72.1 | 3.75 dq (6.2 and 2.5) | C-1D, C-4D, C-6D | H-1D, H-3D, H-4D |
| 6D | 19.0, CH3 | 1.61 d (6.2 Hz) | C-4D, C-5D | H-5D, 3D-CH3 | 17.5 | 1.54 d (6.2) | C-4D, C-5D | H-4D, H-5D |
| 1E | 98.4 | 5.55 dd (9.6 and 2.0) | C-3D | H-2Eax, H-2Eeq, H-5E, H-3D | ||||
| 2Eax | 45.5 | 1.89 dd (13.0 and 9.6) | C-1E, C-3E, C-4E | H-1E, H-4E | ||||
| 2Eeq | 2.32 dd (13.0 and 2.0) | H-1E | ||||||
| 3E | 71.2 | - | - | - | ||||
| 3E-CH3 | 28.2 | 1.50 s | C-2E, C-4E, C-3E | H-4E | ||||
| 4E | 77.7 | 3.38 d (9.2) | C-3E, C-5E | H3-3E, H3-6E, H-2Eax | ||||
| 5E | 71.9 | 4.29 dq (9.2 and 6.0) | C-1E, C-4E, C-6E | H-1E, H3-6E | ||||
| 6E | 19.0 | 1.61 d (6.0) | C-4E, C-5E | H-4E, H-5E | ||||
The structural analysis showed that three of the new MTM derivatives (compounds 1, 2, and 4) had desired modifications in their sugar decoration pattern, namely, either an altered trisaccharide chain or only one sugar instead of the disaccharide chain in the 6-position, while the important sugar E16-18 is missing in structure 3. Compound 1 contains unexpectedly a d-digitoxosyl residue as sugar E instead of the d-mycarose normally found in this position in MTM. This sugar could arise from the intermediate NDP-4-keto-d-olivose, which can be inverted in its 3-position through tautomerism to NDP-4-keto-d-digitoxose,19 before being transferred as 4-ketosugar into the E-position by MtmGIV, as suggested earlier.6,8 The reduction to the equatorial 4-OH group would be achieved afterward through ketoreductase MtmTIII.8 Because of the l-sugar incorporation, structure 2 is clearly the most unusual new MTM analogue. It contains the pLNBIV-encoded sugar l-digitoxose in position D. On first view, this appears to reveal a remarkable sugar donor substrate tolerance of MtmGIII: the GT that normally places a d-oliose into this position. However, it is likely that this l-sugar is transferred in a 4C1-conformation resembling the d-oliose normally occupying position D and switches later to the 1C4-conformation found in 2 (Scheme 3). Structure 3 could be either explained again by sugar donor substrate tolerance of MtmGIII, now transferring d-mycarose instead of d-oliose (may be due to a lack of sufficient amounts of its natural sugar donor substrate NDP-d-oliose) or by acceptor substrate tolerance of MtmGIV, which can also establish the d-mycarose in the D- instead of in the E-position. Again, the provision of NDP-d-oliose must be somewhat limited in S. argillaceus (pLNBIV) to explain this result, most likely because MtmGIII is partly inhibited through an unnatural NDP sugar provided by pLNBIV, e.g., NDP-l-digitoxose, which might be only a poor substrate for this enzyme, or by one of its intermediates. To explain structure 4, a partial inhibition of MtmGII, the GT normally responsible for the attachment of sugar B, is most likely. This is again likely caused by a wrong NDP-sugar donor accumulated by pLNBIV, either NDP-l-digitoxose or, more likely, one of its 4-keto-intermediates (see Scheme 3). Like in MTM, all intersugar linkages of the new compounds were 1,3. The assignment of d- or l- configurations of sugar moieties in this study was based on the fact that so far all glycosyltransferases (GTs) characterized from Streptomycetes that were associated with secondary metabolism, including all the MTM-GTs, belong to the GT-1 family of inverting enzymes.24-26 Such GTs establish a β-glycosidic bond in an SN2-like fashion using an NDP-activated sugar in which the NDP group was α-linked, or vice versa. With the known biosynthetic pathways of deoxyhexoses, such GTs establish β-glycosidic links for all d-sugars and α-glycosidic links for all l-sugars (Scheme 3).
Scheme 3.

Biosynthetic Pathway to NDP-l-digitoxose Encoded by Plasmid pLNBIV, Illustrating Also How Products and Intermediates May Have Influenced the Production of Structures 1-4
An initial assessment of the antitumor activity of the new MTM analogues 1-4 using the SRB (sulforhodamine B) cytotoxicity assay at 100 μM concentrations27 against human and murine breast and lung cancer cell lines showed that all four new MTM analogues were active (data not shown). To further focus on the observed antibreast cancer activity of MTM and its new derivatives, we performed an apoptotic assay, namely, the TUNEL assay (=terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling).28-30 In this, estrogen receptor-positive (ER+) MCF-7 and the estrogen receptor-negative (ER-) MDA-231 breast cancer cells were treated with different concentrations of mithramycin and its derivatives (15, 20, 25, 30 μM) for 24 h. Mithramycin induced 37.77% apoptosis (37.77% TUNEL-positive cells) in ER+ MCF-7 cells at a concentration of 15 μM, while the mithramycin derivatives (compounds 1, 2, 3, 4) induced 64.8%, 3.9%, 50.29%, and 26.3% apoptosis, respectively. In the ER- MDA-231 cells, mithramycin induced 2.6% apoptosis and the derivatives (1-4) induced 63.6%, 6.46%, 7.29%, and 12.6% respectively, at a concentration of 20 μM (Figure 1).
Figure 1.
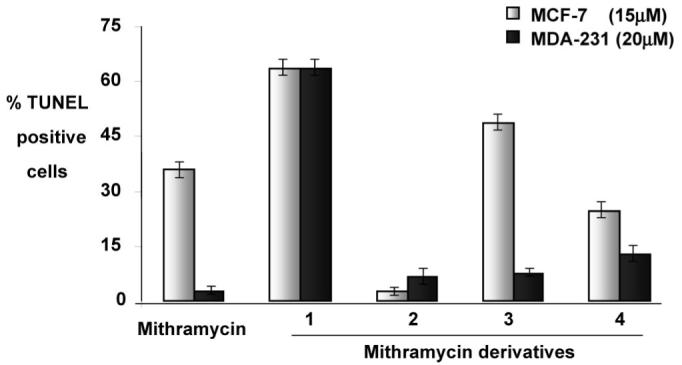
Mithramycin and its derivatives induced apoptosis in breast cancer cells. Breast cancer cells (MCF-7, 15 μM; MDA-231, 20 μM) were treated with mithramycin and derivatives 1-4. Apoptotic cells were scored after 24 h by the TUNEL (=terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling) assay and confocal microscopy. The baseline apoptosis in the untreated group was normalized with data on the treated group. The data shown are representative of the combined means ± standard error from three independent experiments.
These results suggest that derivatives 1 and 3 exhibit more potent anticancer activity than mithramycin on the ER-positive breast cancer cell line MCF-7, and all new derivatives, particularly compound 1, show a better effect on the ER-negative breast cancer cell line MDA-231. More than 60% of the early stage breast tumors are ER-positive and respond well to endocrine therapy. In contrast, ER-negative breast cancer is highly invasive and has a poor prognostic outlook. The standard therapeutic protocol for ER-negative breast cancer is limited; therefore better drugs are very much in need.31-33 Our results suggest that compound 1, the derivative most similar to mithramycin (only the 3E-methyl group is missing), significantly induced apoptosis in both the ER+ MCF-7 and the ER- MDA 231 cells when compared to mithramycin. Thus, this compound may serve as an effective therapeutic aid in the management of breast cancer. Also compound 3 significantly induced apoptosis in the ER+ MCF-7 cells compared to MTM despite its significantly shortened lower saccharide chain, while compound 4 (the derivative in which mithramycin’s sugar B is missing) displayed a less significant but moderate apoptotic induction in both of these cancer cell lines. Only compound 2 failed to induce apoptosis in MCF-7 cells and has only a very weak effect on MDA-231 cells, suggesting that the incorporation of an l-sugar into the trisaccharide chain with the consequential shortening and distortion of that chain has a detrimental effect on the biological activity. The results from the study underscore the possible potentiality of compound 1 in the treatment and management of breast cancer. Further in vitro and in vivo investigations are required to completely determine the antitumor efficacy of these compounds.
In summary, combinatorial biosynthesis is a powerful tool to generate new drugs. Here, a gene recombination experiment was performed with a cassette encoding pathways of complete NDP-activated deoxysugars, namely, NDP-activated l-digitoxose and its biosynthetic intermediates. As a consequence of the “flooding” of the MTM pathway with a plasmid encoding these “foreign” NDP sugars, the production of four new MTM derivatives with unprecedented sugar patterns was observed. The formation of these new compounds could be explained either by relaxed substrate specificity of GTs of the MTM pathway toward their NDP sugar donor substrates, resulting in the incorporation of one of the plasmid-encoded sugars, or by inhibiting certain GTs (here MtmGIII, MtmGII) of the MTM pathway with these “foreign” NDP sugars. The results also reflect indirectly some substrate tolerance of oxygenase MtmOIV (Scheme 1),15,19 which is necessary to generate biologically active mithramycin-type drugs from inactive premithramycins. The studies also show again20,21,34 that it is possible to improve the biological activity of a parent natural product (here mithramycin) through combinatorial biosynthesis. Subtle modifications in the deoxysugar pattern appear to be very profitable. While previous activity enhancements were achieved through modifications of the 3-side chain of mithramycin,20,34 the mithramycin derivatives described in this work are the first examples where an increase of biological activity was achieved through modification of the saccharide moieties. The high concentrations used in the initial cytotoxiciy assays resulted in a 100% cell kill, thereby preventing an appreciation of either their absolute or relative potencies.
Experimental Section
General Experimental Procedures
Optical rotations were recorded on a Perkin-Elmer 241 polarimeter. UV spectra were recorded on a Shimadzu UV-2501 PC spectrometer. IR spectra were obtained from a pure sample of KBr discs using a Nicolet Magna IR-560 spectrometer. All NMR data were recorded in pyridine-d5, using a 400 MHz Varian Inova instrument, except the 13C broadband spectra, which were recorded at 75.4 MHz on a 300 MHz Varian Mercury spectrometer. δ values were adjusted to the solvent peaks (δ 8.74 and 150.35 ppm for 1H and 13C NMR, respectively). The electrospray ionization mass spectra (ESIMS), showing MS fragmentations of pseudomolecular ions in both +ve and -ve mode, were acquired using a Thermo Finnigan LCQ mass spectrometer, with sample introduction by direct infusion. The high-resolution positive-mode fast atom bombardment mass spectrometry (FAB) spectra were acquired using a model VG70SQ double-focusing magnetic sector MS instrument. HPLC-MS analyses were carried out using a Waters ZQ2000 HPLC-MS system, with acetonitrile (CH3CN) and 0.1% trifluoroacetic acid (TFA) in H2O as solvents and a reversed-phase column (Symmetry C18, 2.1 × 150 mm, Waters). A linear gradient from 10% to 100% CH3CN in 30 min, at a flow rate of 1 mL/min, was used. Detection and spectroscopic characterization of peaks were performed with a photodiode array detector and MassLynx 4.0 software (Waters). MS analyses were carried out using electrospray ionization in the positive mode, with a capillary voltage of 3 kV and cone voltages of 20 and 100 V. For chromatography, a μBondapak C18 radial compression cartridge (PrepPak Cartridge, 25 × 100 mm, Waters) and semipreparative columns (Symmetry C18, 7.8 × 300 mm, Waters) were used.
Bioassays
The SRB (sulforhodamine B) cytotoxicity assays were performed for MTM and its analogues using a standard protocol developed by the NCI.27 The TUNEL apoptosis assay was carried out as described.28-30
Plasmid pLNBIV
E. coli/Streptomyces shuttle plasmid pLNBIV was constructed as described earlier.15 It contains genes involved in the biosynthesis of the l-oleandrose moiety of oleandomycin from Streptomyces antibioticus ATCC11891 in which the 4-ketoreductase (KR)-encoding gene oleU was replaced by eryBIV, a KR-encoding gene from the erythromycin pathway.15 All these genes are under control of the strong ermEp* promoter, which allows overexpression in Streptomyces. The plasmid encodes the biosynthesis of NDP-l-digitoxose and its biosynthetic intermediates.19
Expression, Purification, and Characterization of MTM Analogues
Plasmid pLNBIV was introduced into the MTM producer Streptomyces argillaceus ATCC 12956 by protoplast transformation according to standard procedures for Streptomyces.35 Transformants were selected with thiostrepton (50 μg/mL final concentration). A thiostrepton-resistant colony was selected for further characterization (strain S. argillaceus (pLNBIV)).
For HPLC analysis of mithramycin-related compounds, S. argillaceus (pLNBIV) was grown on R5A solid medium,36 for 5 days at 28 °C. For HPLC analyses (Waters ZQ 2000 system), a reversed-phase column (Symmetry C18, 2.1 × 150 mm) was used. EtOAc extracts were dried in vacuo and resolved in the HPLC solvents (CH3CN and 0.1% TFA in H2O). Samples were eluted with 10% CH3CN during the first 4 min, followed by a linear gradient from 10 to 88% acetonitrile over 26 min, at a flow rate of 0.25 mL/min. Detection and spectroscopic characterization of peaks were performed with a photodiode array detector, and MS analyses were done by electrospray ionization in the positive mode.
For purification of compounds, strain S. argillaceus (pLNBIV) was grown in R5A medium in a two-step culture method, as previously described.36 In the production step, eight 2 L Erlenmeyer flasks, each containing 400 mL of medium, were incubated for 5 days. The cultures were centrifuged (12 000 rpm, 30 min) and filtered through a solid-phase extraction cartridge (Sep Vac 35 cm3, Waters). The fractions obtained were analyzed by HPLC, and those containing mithramycin-related compounds (eluting between 18 and 22 min) were pooled and dried in vacuo. This extract was redissolved and chromatographed using a μBondapak C18 radial compression cartridge (PrepPak Cartridge, 25 × 100 mm). An isocratic elution with a mixture of CH3CN and 0.1% TFA in H2O (42:58), at 10 mL/min, was used. Compounds 1 and 4 were repurified with a semipreparative column (Symmetry C18, 7.8 × 300 mm) using isocratic elution with CH3CN and 0.1% TFA in H2O (37:63), at 3 mL/min. Compounds 2 and 3 were also repurified with the same column and solvents, but changing the solvent mixture to 45:55. In all cases, after every purification step, the collected compounds were diluted 4-fold with H2O and then desalted and concentrated by solid-phase extraction, being finally lyophilized. Yields obtained were 14.3 mg of compound 1, 5.8 mg of 2, 3.3 mg of 3, and 10.9 mg of compound 4.
The new MTM derivatives were characterized by using NMR spectroscopy, mass spectrometry, and UV and IR spectroscopy. Different mass spectrometric techniques were used to determine the molecular weight and formulas of the metabolites, and MS fragmentations of pseudomolecular ions gave comprehensive sugar splitting patterns (see MS data below), which helped to define the saccharide chains and types of sugars. All NMR assignments were based on 1H and 13C spectra using 1H, 13C-HSQC, CIGAR-HMBC,22 1H,1H-DQ-COSY, and NOESY spectra, allowing an unambiguous assignment of all NMR signals. Compound 1 was additionally investigated with 1D-TOCSY spectra to identify and understand the sugar spin patterns separately.
Demycarosyl-3D-β-d-digitoxosyl MTM (1)
yellow, amorphous solid; [α]25D -13 (c 0.027, MeOH); UV (MeOH) λmax (∊) 231 (15 600), 283 (56 300), 315 (6900), 330 (sh), 430 (10 900) nm; IR (KBr) νmax 3420 (OH), 2920 (CH), 2848 (CH), 1716 (C=O), 1631 (C=O), 1514 (C=C), 1382, 1130, 1064 cm-1; 1H and 13C NMR (pyridine-d5) see Table 1; negative ESIMS m/z (relative intensity) 1069 ([M - H]-, 100), 1105/1107 ([M + Cl]-, 22), 939 ([M - H - {sugar A}], 7); positive ESIMS m/z 1109 ([M + K]+, 11), 1093 ([M + Na]+, 100), 833 ([M - {sugar A and B} + Na]+, 7), 811 ([M + H - {sugar A and B}], 16), 681 ([M + H - {trisaccharide chain}], 14), 421 ([M + H - {tri- and disaccharide}], 18); HRFABMS m/z 1093.4447 ([M + Na]+, calcd for C51H74O24Na, 1093.4468, 100).
Deoliosyl-3C-α-l-digitoxosyl-MTM (2)
yellow, amorphous solid; [α]25D -21 (c 0.014, MeOH); UV (MeOH) λmax (∊) 229 (17 100), 283 (54 200), 316 (7300), 330 (sh), 430 (11 400) nm; IR (KBr) νmax 3409 (OH), 2924 (CH), 2850 (CH), 1716 (C=O), 1634 (C=O), 1514 (C=C), 1374, 1126, 1064 cm-1; 1H and 13C NMR (pyridine-d5), see Table 1; negative ESIMS m/z 1083 ([M - H]-, 100), 1119/1121 ([M + Cl]-, 26); positive ESIMS m/z 1123 ([M + K]+, 13), 1107 ([M + Na]+, 100), 847 ([M - {disaccharide chain} + Na]+, 5), 825 ([M + H - {disaccharide chain], 27), 681 ([M + H - {sugars A, B, and E}], 11), 551 ([M + H - {sugars A, B, D, and E}], 34), and 421 ([M + H - {tri- and disaccharide}], 22); HRFABMS m/z 1107.4603 ([M + Na]+, calcd for C52H76O24Na, 1107.4624, 100).
Deoliosyl-3C-β-d-mycarosyl-MTM (3)
yellow, amorphous solid; [α]25D -24 (c 0.016, MeOH); UV (MeOH) λmax (∊) 230 (14 500), 283 (49 300), 316 (6300), 330 (sh), 429 (10 300) nm; IR (KBr) νmax 3425 (OH), 2924 (CH), 2850 (CH), 1716 (C=O), 1631 (C=O), 1514 (C=C), 1374, 1122, 1064 cm-1; 1H and 13C NMR (pyridine-d5), see Table 2; negative ESIMS m/z 953 ([M - H]-, 100); positive ESIMS m/z 993 ([M + K]+, 5), 977 ([M + Na]+, 100), 695 ([M + H - {sugars A and B}], 8), 681 ([M - {sugars B and D}], 10), and 421 ([M + H - {bis- disaccharide}], 20); HRFABMS m/z 977.3948 ([M + Na]+, calcd for C46H66O21Na, 977.3994, 100).
3A-Deolivosyl-MTM (4)
yellow, amorphous solid; [α]25D -28 (c 0.014, MeOH); UV (MeOH) λmax (∊) 229 (16 600), 283 (53 300), 316 (6900), 330 (sh), 430 (11 100) nm; IR (KBr) νmax 3421 (OH), 2924 (CH), 2850 (CH), 1716 (C=O), 1634 (C=O), 1514 (C=C), 1374, 1122, 1060 cm-1; 1H and 13C NMR (pyridine-d5), see Table 2; negative ESIMS m/z 989/991 ([M + Cl]-, 9), 953 ([M - H]-, 100), 823 ([M - sugar A], 5), 809 ([M + H - {sugar E}], 8); positive ESIMS m/z 993 ([M + K]+, 10), 977 ([M + Na]+, 100), 833 ([M - {sugar E} + Na]+, 13), 825 ([M + H - {sugar A}], 11), 681 ([M + H - {sugars A and E}], 25), 551 ([M + H - {sugars A, D, and E}], 50), and 421 ([M + H - tri- and monosaccharide], 33); HRFABMS m/z 955.4208 ([M + H]+, calcd for C46H67O21, 955.4175, 100%).
Supplementary Material
Scheme 2.
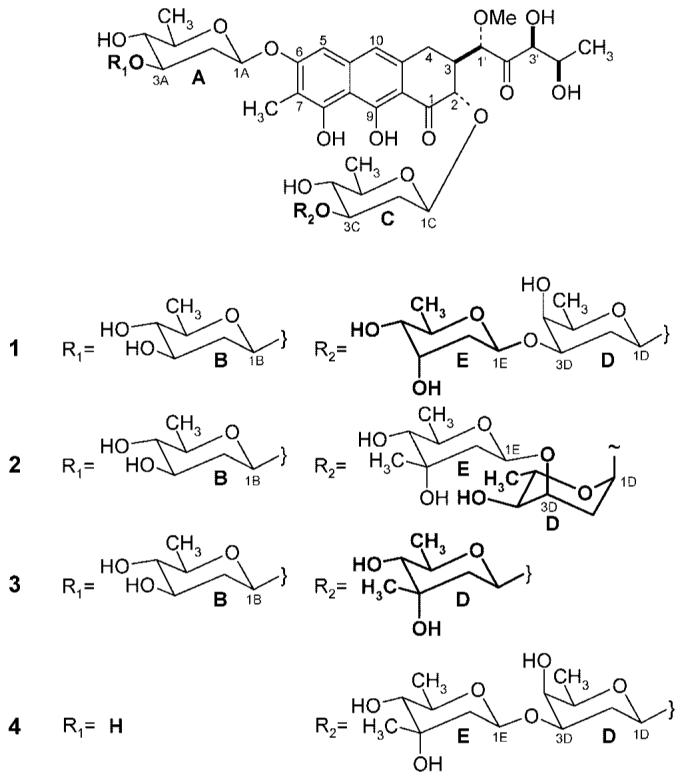
Chemical Structures of the New Mithramycin Analogues 1-4 (the moieties different from mithramycin are highlighted in bold)
Acknowledgment
This work was supported by grants of the NIH (CA 91901) to J.R., the Spanish Ministry of Education and Science (BIO2005-04115) to C.M., and Red Temática de Investigación Cooperativa de Centros de Cáncer (Ministry of Health, Spain; ISCIII-RETIC RD06/0020) to J.A.S. We thank the NMR and MS Core Facilities of the University of Kentucky for the use of their instruments and the Nebraska Center for Mass Spectrometry for HRFAB mass spectra. M.P. was the recipient of a predoctoral fellowship of the Spanish Ministry of Science and Technology.
References and Notes
- (1).Rohr J, Méndez C, Salas JA. Bioorg. Chem. 1999;27:41–54. [Google Scholar]
- (2).Nur-e-Alam M, Méndez C, Salas JA, Rohr J. ChemBioChem. 2005;6:632–636. doi: 10.1002/cbic.200400309. [DOI] [PubMed] [Google Scholar]
- (3).Gibson M, Nur-e-Alam M, Oliveira MA, Rohr J. J. Am. Chem. Soc. 2005;127:17594–17595. doi: 10.1021/ja055750t. [DOI] [PubMed] [Google Scholar]
- (4).Rohr J, Weissbach U, Beninga C, Künzel E, Siems K, Bindseil KU, Lombó F, Prado L, Braña AF, Méndez C, Salas J. [Google Scholar]
- (5).Prado L, Fernández E, Weissbach U, Blanco G, Quirós LM, Braña AF, Méndez C, Rohr J, Salas JA. Chem. Biol. 1999;6:19–30. doi: 10.1016/s1074-5521(99)80017-9. [DOI] [PubMed] [Google Scholar]
- (6).Fernández E, Weissbach U, Sánchez Reillo C, Braña AF, Méndez C, Rohr J, Salas JA. J. Bacteriol. 1998;180:4929–4937. doi: 10.1128/jb.180.18.4929-4937.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Rodriguez D, Quirós LM, Braña AF, Salas JA. J. Bacteriol. 2003;185:3962–3965. doi: 10.1128/JB.185.13.3962-3965.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Remsing LL, Garcia-Bernardo J, Gonzalez A, Künzel E, Rix U, Braña AF, Bearden DW, Méndez C, Salas JA, Rohr J. J. Am. Chem. Soc. 2002;124:1606–1614. doi: 10.1021/ja0105156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Blanco G, Fernández E, Fernández MJ, Braña AF, Weissbach U, Künzel E, Rohr J, Méndez C, Salas JA. Mol. Gen. Genet. 2000;262:991–1000. doi: 10.1007/pl00008667. [DOI] [PubMed] [Google Scholar]
- (10).Méndez C, Salas JA. Trends Biotechnol. 2001;19:449–456. doi: 10.1016/s0167-7799(01)01765-6. [DOI] [PubMed] [Google Scholar]
- (12).Rupprath C, Schumacher T, Elling L. Curr. Med. Chem. 2005;12:1637–1675. doi: 10.2174/0929867054367167. [DOI] [PubMed] [Google Scholar]
- (12).Walsh C, Freel Meyers CL, Losey HC. J. Med. Chem. 2003;46:3425–3436. doi: 10.1021/jm030257i. [DOI] [PubMed] [Google Scholar]
- (13).Langenhan JM, Griffith BR, Thorson JS. J. Nat. Prod. 2005;68:1696–1711. doi: 10.1021/np0502084. [DOI] [PubMed] [Google Scholar]
- (14).Salas JA, Méndez CJ. Mol. Microbiol. Biotechnol. 2005;9:77–85. doi: 10.1159/000088838. [DOI] [PubMed] [Google Scholar]
- (15).Rodriguez L, Aguirrezabalaga I, Allende M, Braña AF, Méndez C, Salas JA. Chem. Biol. 2002;9:721–729. doi: 10.1016/s1074-5521(02)00154-0. [DOI] [PubMed] [Google Scholar]
- (16).Sastry M, Fiala R, Patel DJ. J. Mol. Biol. 1995;251:674–689. doi: 10.1006/jmbi.1995.0464. [DOI] [PubMed] [Google Scholar]
- (17).Sastry M, Patel DJ. Biochemistry. 1993;32:6588–6604. doi: 10.1021/bi00077a012. [DOI] [PubMed] [Google Scholar]
- (18).Chakrabarti S, Bhattacharyya B, Dasgupta D. J. Phys. Chem. B. 2002;106:6947–6953. [Google Scholar]
- (19).Fischer C, Rodríguez L, Patallo EP, Lipata F, Braña AF, Méndez C, Salas JA, Rohr J. J. Nat. Prod. 2002;65:1685–1689. doi: 10.1021/np020112z. [DOI] [PubMed] [Google Scholar]
- (20).Remsing LL, Bahadori HR, Carbone GM, McGuffie EM, Catapano CV, Rohr J. Biochemistry. 2003;42:8313–8324. doi: 10.1021/bi034091z. [DOI] [PubMed] [Google Scholar]
- (21).Albertini V, Jain A, Vignati S, Napoli S, Rinaldi A, Kwee I, Nur-e-Alam M, Bergant J, Bertoni F, Carbone GM, Rohr J, Catapano CV. Nucleic Acids Res. 2006;34:1721–1734. doi: 10.1093/nar/gkl063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Reynolds WF, Enriquez RG. J. Nat. Prod. 2002;65:221–244. doi: 10.1021/np010444o. [DOI] [PubMed] [Google Scholar]
- (23).Wohlert SE, Künzel E, Machinek R, Mendez C, Salas JA, Rohr J. J. Nat. Prod. 1999;62:119–121. doi: 10.1021/np980355k. [DOI] [PubMed] [Google Scholar]
- (24).Rix U, Fischer C, Remsing LL, Rohr J. Nat. Prod. Rep. 2002;19:542–580. doi: 10.1039/b103920m. [DOI] [PubMed] [Google Scholar]
- (25).Campbell JA, Davies GJ, Bulone V, Henrissat B. Biochem. J. 1998;329:719. doi: 10.1042/bj3290719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Campbell JA, Davies GJ, Bulone V, Henrissat B. Biochem. J. 1997;326:929–939. doi: 10.1042/bj3260929u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Rubinstein LV, Shoemaker RH, Paull KD, Simon RM, Tosini S, Skehan P, Scudiero DA, Monks A, Boyd MR. J. Natl. Cancer Inst. 1990;82:1113–1118. doi: 10.1093/jnci/82.13.1113. [DOI] [PubMed] [Google Scholar]
- (28).Srinivasan S, Ranga RS, Burikhanov R, Han SS, Chendil D. Cancer Res. 2007;67:246–253. doi: 10.1158/0008-5472.CAN-06-2430. [DOI] [PubMed] [Google Scholar]
- (29).Ranga S, Sowmyalakshmi S, Burikhanov R, Akbarsha MA, Chendil D. Mol. Cell. Biochem. 2005;280:125–133. doi: 10.1007/s11010-005-8518-3. [DOI] [PubMed] [Google Scholar]
- (30).Sowmyalakshmi S, Nur-e-Alam M, Akbarsha MA, Thirugnanam S, Rohr J, Chendil D. Planta. 2005;220:910–918. doi: 10.1007/s00425-004-1405-4. [DOI] [PubMed] [Google Scholar]
- (31).Sotiriou C, Neo SY, McShane LM, Korn EL, Long PM, Jazaeri A, Martiat P, Fox SB, Harris AL, Liu ET. Proc. Natl. Acad. Sci. USA. 2003;100:10393–10398. doi: 10.1073/pnas.1732912100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).World Cancer Report of the World Health Organization . In: Steward B, Kleihus P, editors. IARC Press; Lyon: 2003. pp. 188–233. [Google Scholar]
- (33).Berry DA, Cirrincione C, Henderson IC, Citron ML, Budman DR, Goldstein LJ, Martino S, Perez EA, Muss HB, Norton L, Hudis C, Winer EP. J. Am. Med. Assoc. 2006;295:1658–1667. doi: 10.1001/jama.295.14.1658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Remsing LL, González AM, Nur-e-Alam M, Fernández-Lozano MJ, Braña AF, Rix U, Oliveira MA, Méndez C, Salas JA, Rohr J. J. Am. Chem. Soc. 2003;125:5745–5753. doi: 10.1021/ja034162h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Kieser T, Bibb MJ, Buttner MJ, Chater KF, Hopwood DA. Practical Streptomyces Genetics. The John Innes Foundation; Norwich, UK: 2000. [Google Scholar]
- (36).Fernández E, Weissbach U, Reillo CS, Braña AF, Méndez C, Rohr J, Salas JA. J. Bacteriol. 1998;180:4929–4937. doi: 10.1128/jb.180.18.4929-4937.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.