

Abstract
Influenza infections induce a rapid, but transient, dendritic cell (DC) migration from the lungs to the lymph nodes (LNs) that is followed by substantial recruitment of DCs into the lungs without subsequent migration to the LNs. Given that peripheral DCs are primarily thought to be involved in the initiation of adaptive immunity after migration into lymphoid tissues, what role these newly lung-recruited DCs play in influenza virus immunity is unclear. In this study, we demonstrate that loss of non-LN migratory pulmonary DC subsets increases mortality, sustains higher viral titers, and impairs pulmonary CD8 T cell responses. Reconstitution of the lungs with pulmonary plasmacytoid DCs, CD8α+ DCs, or interstitial DCs restores CD8 T cell responses in a cell contact–, major histocompatability complex I–, and influenza peptide–dependent manner. Thus, after their initial activation in the LN, protective influenza-specific CD8 T cell responses require additional antigen-dependent interactions, specifically with DCs in the lungs.
DCs present in the airways and alveoli of the lungs play an integral role in the initiation of primary immune responses during pulmonary infections (1–3). During influenza virus infections, these respiratory DCs (rDCs) acquire viral antigens, mature, and migrate from the lungs to the LN, where they activate naive influenza-specific CD8 T cells (4–6). After the interaction of naive T cells with such antigen-bearing DCs, the CD8 T cells undergo activation and division in the LNs and migrate into the lungs to kill virally infected host cells, thereby eliminating the infection (7–10).
Recently, we have shown that pathogen-induced rDC migration from the lungs into the regional LNs occurs in a transient manner (6), as influenza virus infection triggers a rapid augmentation of rDC migration during the first 24–48 h after infection (a.i.). However, after this initial boost in rDC migration, trafficking returns to baseline levels and the remaining DCs in the lungs become refractory to additional migratory stimuli (6). Blockade of rDC migration or elimination of the initial trafficking DCs inhibits the subsequent activation of naive CD8 T cell responses (5, 6). Together, these results suggest that antigen trafficking from the lungs to the LN is dependent on rDCs, and that such movement of antigen into the LN occurs for only a very short time a.i.
The requirements for proper initiation of effector CD8 T cell responses by DCs in the LN have been the subject of many recent investigations. Together, these studies suggest that the developing CD8 T cell effector response is rapidly programmed (in some cases with as little as 2 h of antigen–DC contact), and that subsequent T cell expansion and effector ability can then follow an antigen-independent developmental program (11–13). However, although brief stimulation is sufficient for antigen-independent expansion of naive CD8 T cells in vitro, these cells undergo an abortive clonal expansion (13–15) or demonstrate expansion without effector commitment when subsequently transferred in vivo. This suggested that the continuation or maintenance of CD8 T cell programming was influenced by additional signals, and subsequent studies have demonstrated roles for IL-2, IL-12, IFN-α, and/or IFN-γ in regulating the expansion, differentiation, and contraction of effector CD8 T cell responses (14–19). To date, the majority of studies examining the requirements for CD8 T cell activation have focused on lymphoid organs and suggest that the T cell programming that occurs therein is sufficient to generate protective CD8 T cell immune responses. Therefore, in the context of influenza virus–specific immunity, the aforementioned studies suggest that by 48 h a.i., influenza-specific CD8 T cells should be capable of mounting a “programmed” protective effector immune response.
In contrast to the limited migration of rDCs from the lungs to the LN after influenza infection, DCs are continually recruited into the lungs throughout the course of many pulmonary challenges (2, 20–22). Because, as discussed in the previous paragraphs, these newly recruited lung DCs do not subsequently migrate to the LN, the role of these recruited pulmonary DCs remains unclear because they do not fit within the classical paradigm of peripheral DCs, which acquire antigens and then migrate into lymphoid tissues to initiate adaptive immune responses.
In this study, we demonstrate that selective depletion of DCs and macrophage populations from the airways and alveoli of the lungs after the conclusion of DC migration from the lungs to the LN results in increased mortality and viral titers, as well as impaired pulmonary CD8 T cell responses. Analysis of the APC populations present in the lungs during the course of infection revealed significantly decreased numbers of CD8α+ DCs and plasmacytoid DCs (pDCs) in the lungs of airway and alveolar DC (aDC)–depleted mice to day 6 a.i. Reconstitution of DC-depleted lungs with pulmonary CD8α+ DCs, pDCs, or interstitial DCs (iDCs) was able to restore CD8 T cell responses both in vitro and in vivo in a direct DC–T cell contact, MHC I–, and influenza viral antigen–specific manner. Together, our results suggest that full protective influenza-specific CD8 T cell responses are not conferred by the programming available in the regional LN and instead require additional antigen-specific interactions with DCs in the lungs. To our knowledge, this is the first study demonstrating that a secondary peripheral interaction of CD8 T cells with antigen-bearing DCs is necessary for effective antiviral immunity.
RESULTS
Influenza-induced DC recruitment into the lungs
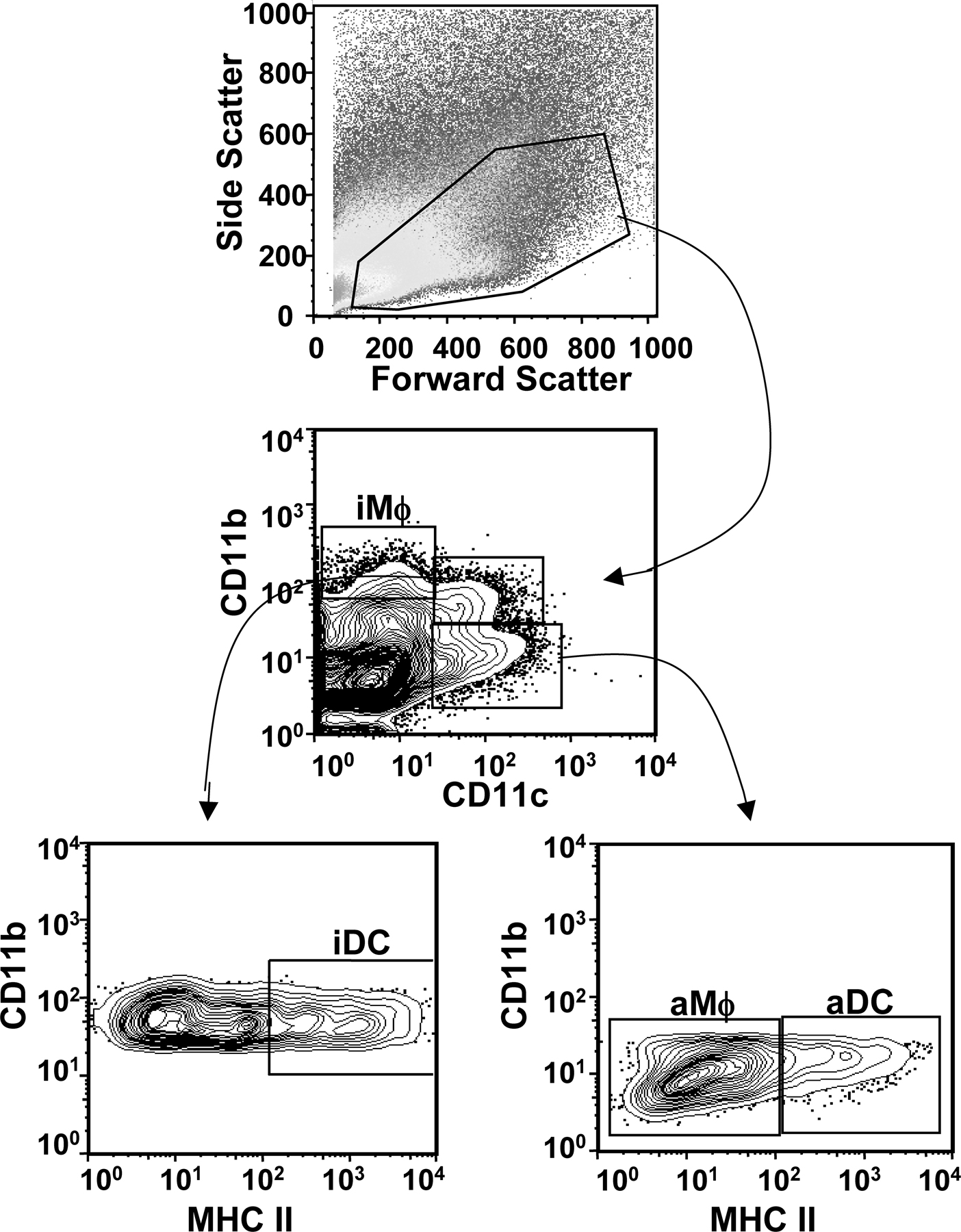
We and others (2, 6, 21–23) have previously observed a steady increase in the number of CD11c+, MHC II+ DCs in the lungs during respiratory challenges, including influenza virus infection. As is shown in Fig. 1 A, the number of DCs in the lungs increases 3–4-fold from day 0 to 6 a.i. Our previous studies have demonstrated that a lethal dose of influenza virus infection induces rapid, but transient, DC migration from the lungs to the LN (6). Consistent with our previous work, rDC migration into the LN after a sublethal influenza infection peaked at 18 h a.i. and returned to baseline by 48 h a.i. (Fig. 1 B). Traditionally, rDCs are thought to contribute to antiinfluenza immunity by trafficking from the lungs to the LN to initiate adaptive CD8 T cell responses. Therefore, given that this role is fulfilled by 48 h a.i. and the newly recruited lung DCs do not traffic to the LN (6), it was unclear what role the newly recruited lung DCs may play in influenza-specific immunity.
Figure 1.

Pulmonary DC dynamics during influenza virus infection. (A) Mice were infected with a sublethal dose of influenza and killed at the indicated days a.i. Total numbers of CD11c+MHC II+ DCs in the lungs were determined by flow cytometry. n = 5–9 mice/time point. Data are representative of two to three independent experiments. (B) Mice were administered i.n. CFSE and infected 6 h later. Mice were killed at the indicated times a.i., and the frequency of CFSE+ rDCs among total CD11c+MHC II+ LN resident (LN DC) in the lung draining LN was determined by flow cytometry. (C) Mice were infected with influenza, and then administered clodronate-liposomes (black bars, aDC depleted) or PBS-liposomes (gray bars) i.n. at 48 h a.i. Untreated mice served as an influenza-infected control (white bars). 6 h after treatment, lungs were examined by flow cytometry for the frequency (left) and total numbers (right) of aDCs (CD11c+CD11b−MHC II+Autofluorescenceneg) and iDCs (CD11c+CD11b+MHC II+). The data are the mean values ± the SEM (n = 4 mice/group) and are representative of two separate experiments. Clodronate-liposome depletion of aMφ is shown in Fig. S1. Fig. S1 is available at http://www.jem.org/cgi/content/full/jem.20080314/DC1.
Consequently, we sought to selectively deplete lung DCs and macrophages at a late time point (i.e., >2 d a.i.) after influenza virus infection to determine if a loss of these cells would alter the adaptive immune response and disease outcome. Bosio and Dow have recently used intratracheal administration of clodronate liposomes, a methodology frequently used for depletion of macrophages, for depletion of DCs from the lung airways and airspaces (24). Once in the airspaces, the clodronate-liposomes are taken up by phagocytic cells such as macrophages and DCs and degraded, releasing the clodronate to accumulate within the cell at high concentrations and induce apoptosis. Because the drug is encased in liposomes, it is unable to cross vascular barriers, and, if not phagocytosed, the liposomes are rapidly degraded and the clodronate is safely secreted by the renal system (25–27). Using a modification of the aforementioned technique, influenza-infected mice were treated intranasally (i.n.) with clodronate-liposomes at 48 h a.i. (after conclusion of normal DC trafficking from the lungs to the draining LN; Fig. 1 B). With this methodology, we were able to deplete the aDC populations (MHC II+AutofluorescencenegCD11c+ CD11b−) by ∼50% (Fig. 1 C) and alveolar macrophage (aMφ) populations by ∼60% (Fig. S1, available at http://www.jem.org/cgi/content/full/jem.20080314/DC1). The total number of iDCs (CD11c+MHC II+CD11b+) in the lungs were not significantly affected, a result that confirms that the liposomes do not deeply penetrate the lungs. Further, as seen in Fig. 1 C, we did not observe aDC depletion in control PBS-liposome–treated mice.
aDC depletion at 48 h a.i. results in increased mortality and viral titer
To determine how selective loss of aDCs (and aMφ) at 48 h a.i. might alter the course of influenza infection, mice were infected with influenza virus, treated with clodronate-liposomes at 48 h a.i., and then examined daily for morbidity (i.e., weight loss) and mortality (Fig. 2 A). Although the aDC-depleted mice did not show significantly increased morbidity compared with influenza-infected controls (not depicted), aDC depletion greatly increased mortality, with 100% of the mice succumbing by day 6–8 a.i. compared with only 10% of controls (Fig. 2 A). We next examined pulmonary viral titers in the lungs of aDC-depleted and control mice. The viral titers were significantly higher on day 4 and higher, although not statistically significant, on day 6 a.i. versus control mice (Fig. 2 B). Together, these results suggest that, in contrast to control mice, aDC-depleted mice appear to lack the necessary immune response to contain the viral infection.
Figure 2.

aDC depletion at 48 h a.i. results in increased mortality and pulmonary viral titers. (A) Mice were infected with influenza and aDC depleted (black triangles, aDC depleted) as in Fig 1, and then monitored daily for survival and weight loss as a sign of morbidity (not depicted) versus influenza-infected nontreated control mice (open circles). n = 10 mice/group. Data are representative of two independent experiments. (B) The lungs of influenza-infected, aDC-depleted (shaded bars), or control (white bars) mice were harvested on days 4 and 6 a.i., and pulmonary viral load was determined by infection of eggs. The data are the mean values ± the SEM (n = 4–7 mice/group) and are representative of two separate experiments.
aDC depletion at 48 h a.i. results in impaired CD8 T cell responses
Given the increased viral titers and disease severity observed in aDC-depleted mice, coupled with the known importance of influenza-specific CD8 T cells in mediating viral clearance and recovery during primary influenza infections (10), we next examined the magnitude of the influenza-specific CD8 T cell response in the lungs of aDC-depleted versus control mice. Whereas control mice exhibited a robust CD8 T cell response, aDC-depleted mice had significantly decreased numbers of influenza-specific CD8 T cells as measured by tetramer (Fig. 3 B, left) or intracellular cytokine staining (ICS) for IFN-γ (Fig. 3 B, right) on day 6 a.i. This effect was specific to aDC depletion and not liposome treatment, as no significant differences were observed between non–liposome-treated controls and empty PBS-liposome–treated mice. Next, to determine if such decreases in CD8 T cell numbers would have a consequence in elimination of viral-infected targets, in vivo cytotoxicity assays were performed. Although robust responses were observed in control mice, with ∼40% lysis of specific targets, we observed a nearly 50% decrease in the ability of aDC-depleted mice to kill virally infected targets (Fig. 3 C).
Figure 3.

aDC depletion at 48 h a.i. impairs pulmonary influenza-specific CD8 T cell responses. Mice were infected with influenza and aDC depleted as in Fig 1. The gating strategy for tetramer+ or IFN-γ+ cells among CD8a+ cells is shown in A. On day 6 a.i., lungs were harvested and analyzed by flow cytometry for total number (B) of influenza-specific tetramer+ or IFN-γ+ CD8 T cells. Control influenza infected (white bars) and naive (dark gray bars) mice were included as controls. The data are the mean values ± the SEM (n = 8–11 mice/group) and are representative of two to four separate experiments. In A, representative plots for individual mice are shown. (C) On day 6 a.i., in vivo cytotoxicity assays were performed on aDC-depleted (shaded bars) and control (white bars) mice. Samples were examined by flow cytometry for the percentage of specific lysis. The data are the mean values ± the SEM (n = 6–7 mice/group) and are representative of two separate experiments.
Because the liposome clodronate was administered at 48 h a.i., our expectation was that the decrease in antigen-specific CD8 T cells may not be caused by an altered response in the LN. To confirm this supposition and to further pinpoint the location of the defect in the CD8 T cell compartment of aDC-depleted mice, we used the Clone-4 (CL-4) T cell receptor transgenic CD8 T cell adoptive transfer system (28–31). CL-4 CD8 T cells recognize the HA533 epitope of A/PR/8/34, as well as the cross-reactive HA529 epitope of the A/JAPAN/305/57 virus and have been previously used to examine early CD8 T cell responses in the LN and lungs (7). Therefore, purified naive CL-4, Thy1.2+ CD8 T cells labeled with CFSE were adoptively transferred into Thy1.1+ mismatched mice and, 24 h later, the mice were infected with influenza virus. Half of these mice were depleted of aDCs at 48 h a.i., and the CL-4 T cell responses in the lungs and LN of all mice examined on days 3–5 a.i. As expected, because aDC depletion was not performed until after the conclusion of normal DC trafficking from the lungs to the LN (6), the early division profiles, cell numbers (Fig. 4, A and C), and activation marker expression (i.e., CD69 and CD25; not depicted) of CL-4 cells in the aDC-depleted LN appear similar to controls. Furthermore, i.n. liposome-clodronate administration does not alter the number of CD8α+ DCs, CD4+ DCs, CD8α−CD4− DCs, or the number of pDCs in the lung draining LNs on days 3–5 a.i. or change the ability of these DCs to stimulate naive CD8 T cells during in vitro co-cultures (unpublished data). These results suggest that the defect in the CD8 T cell compartment in aDC-depleted mice does not arise because of altered LN DC effector functions or inappropriate activation or expansion of the T cells in the LN. However, similar to the polyclonal T cell results discussed (Fig. 3 B), the number of CL-4 cells in the lungs of aDC-depleted mice was significantly decreased (Fig. 4 B). Further examination of the CFSE division profiles on day 5 a.i. (Fig. 4 D) revealed an accumulation of “intermediately divided” CD8 T cells in the lungs of aDC-depleted mice, a population that is not present in controls. These results suggest that although early activation of the T cells within the LN appears normal, influenza-specific CD8 T cells may require further interactions with DCs in the lungs to complete their normal program of expansion in the lungs.
Figure 4.

Influenza-specific CD8 T cell responses in the LNs and lungs of aDC-depleted mice. Influenza-specific Thy1.2+ CL-4 CD8 T cells were labeled and adoptively transferred into Thy1.1+ host mice. 24 h later, the host mice were infected with influenza, and aDC depleted as in Fig 1. On days 3–5 a.i., the LN (A and C) and lungs (B and D) of aDC-depleted (shaded bars and gray fill) and control (open bars or black line) mice were examined for total numbers (A and B) and CFSE division profiles (C and D) of CL-4 CD8 T cells. Representative histograms are shown in C and D. The data are the mean values ± the SEM (n = 4–6 mice/group) and are representative of two to three separate experiments. Mice where reduced numbers of CL-4 cells were adoptively transferred before infection showed a similar result (Fig. S2). Fig. S2 is available at http://www.jem.org/cgi/content/full/jem.20080314/DC1.
aDC depletion at 48 h a.i. results in impaired DC recruitment to the lungs
As discussed, pulmonary infections with pathogens like influenza virus result in increased numbers of DCs in the lungs (6, 21, 22). However, few detailed analyses have been performed to determine what DC subsets are recruited into the lungs during infection. Given the dramatic changes and the fact that many DC subsets have known roles in antiviral and CD8 T cell immune responses, we next determined what effect aDC depletion has on the numbers and types of DCs recruited into the lungs compared with control mice. Mice were infected with influenza virus, aDCs depleted at 48 h a.i., and the DC subsets present in the lungs were examined on days 4 and 6 a.i. In control mice, we observed significant increases in the numbers of lung-resident APC populations, including aDC, iDC, aMφ, and interstitial macrophage (iMφ; Fig. 5 B). We also examined the lungs for recruitment of DC populations that are not resident in great numbers within naive mice. Although we did not observe significant recruitment of CD4+ DC, suggesting that this subset does not play an important role in the lungs during influenza infection, we did observe recruitment of pDCs and CD8α+ DCs into the lungs throughout the course of infection (Fig. 5, A and B). Whereas pDCs have been previously documented in the lungs, to our knowledge, this is the first description of CD8α+ DCs being present in the lungs (Fig. 5 C). It was interesting that these subsets were recruited to the lungs during influenza infection, as both are known to play important antiviral roles; pDCs through production of type I IFNs and their ability to stimulate potent CD8 T cell responses, particularly in the context of influenza infection (32–34), and CD8α+ DCs through their ability to cross-present viral antigens (35, 36) and their role as potent stimulators of CD8 T cell responses.
Figure 5.

DC recruitment into the lungs of aDC-depleted mice is decreased. Mice that were infected with influenza virus and aDC depleted as in Fig. 1 were killed on days 4 and 6 a.i., and their lungs were analyzed by flow cytometry for the frequency (A) and total numbers (B and C) of the indicated DC subsets. Representative plots for individual mice of CD11c+MHC II+ cells are shown in A. The data are the mean values ± the SEM (n = 5–9 mice/group) and are representative of 2–3 independent experiments. For DC gating strategies please see Figs. S3–S5. Figs. S3–S5 are available at http://www.jem.org/cgi/content/full/jem.20080314/DC1.
When the DCs present in the lungs of aDC-depleted and control mice were compared, we observed significantly reduced numbers of total CD11c+MHC II+ DCs in the lungs of aDC-depleted mice compared with control mice on all days examined, with a global decrease in most DCs and macrophage subtypes (Fig. 5). However, we noted particularly pronounced decreases in the numbers of pDCs, CD8α+ DCs, aDCs, and aMφ in the lungs of aDC-depleted mice (Fig. 5, B and C; 75, 73, 81, and 65%, respectively, day 6 a.i.).
Reconstitution of aDC-depleted mice with CD8α+ DCs, pDCs, and iDCs is able to restore CD8 T cell responses in the lungs
Because aDCs, pDCs, and CD8α+ DCs play important antiviral roles in other experimental models, we hypothesized that these DC subsets were likewise integral to the immune response in the lungs during influenza infections. Therefore, to determine which cells were necessary or sufficient for productive pulmonary CD8 T cell immunity, mice were infected and aDC depleted as before, and then reconstituted i.n. on day 3 a.i. (i.e., 24 h after depletion) with pulmonary DC subsets purified from the lungs of day 6 influenza-infected donors. When the CD8 T cell response was measured on day 5 a.i. in mice reconstituted with aDCs or aMφ, no restoration of antigen-specific tetramer+ or IFN-γ+ CD8 T cells (Fig. 6 A) was observed. However, when mice were reconstituted with physiological numbers of purified pDCs, CD8α+ DCs, or iDCs, we did observe restoration of numbers of antigen-specific CD8 T cells to levels similar to nontreated controls. Interestingly, all three subsets appeared to mediate an equal effect on CD8 T cell responses, as there were no significant differences observed between mice reconstituted with pDCs, CD8α+ DCs, or iDCs.
Figure 6.

pDCs, CD8α+ DCs, and iDCs restore influenza-specific CD8 T cell responses in aDC-depleted mice. (A) Mice were infected and aDC depleted as described in Fig 1. 24 h after depletion (i.e., 3 d a.i.), aDC-depleted mice were reconstituted i.n. with 2.5 × 104 of the indicated APC subset. On day 5 a.i., the number of influenza-specific CD8 T cells in the lungs was determined by tetramer- (left) or IFN-γ ICS–based (right) flow cytometry. The data are the mean values ± the SEM (n = 6–8 mice/group) and are representative of 2–3 separate experiments. (B) Naive Thy1.2+ CL-4 CD8 T cells were adoptively transferred into naive Thy1.1+ hosts mice that were subsequently infected with influenza virus. Some groups of these mice were then aDC depleted at 48 h a.i. On day 4 a.i., CL-4 CD8 T cells were purified from control (black filled) or aDC-depleted (black line) lungs, CFSE labeled, and placed in co-culture with the indicated purified pulmonary DC subsets for 48 h. Proliferation of the T cells (i.e., CD8α+Vβ8+CD90.2+ gated cells) was then measured by CFSE dilution. CFSE-labeled CD8 T cells that were fixed immediately after CFSE staining were used as undivided controls (gray filled). Data are representative of four to five separate experiments. (C) CL-4 CD8 T cells and pulmonary DC subsets were purified as described in B. On day 4 a.i., CL-4 CD8 T cells from day 4 infected aDC-depleted mice were CFSE labeled and cultured for 48 h with the indicated DC subsets that were either live (open), fixed (light gray filled), or on opposite sides of a transwell (gray filled) from the T cells. Proliferation of the T cells (i.e., CD8α+Vβ8+CD90.2+ gated cells) was then measured by CFSE dilution. CFSE-labeled CD8 T cells that were fixed immediately after CFSE staining were used as undivided controls (gray filled). The data are the mean values ± the SEM and are representative of 4–5 separate experiments.
Because the clodronate-liposome treatment specifically depletes aDCs and aMφ from the lungs, it was unexpected that reconstitution of aDC-depleted lungs with these populations had no effect on the T cell response. It was possible that the lack of restorative ability was caused by impaired trafficking of these cell subsets in vivo after adoptive transfer rather than their inherent T cell stimulatory capacity. Therefore, we next performed in vitro co-culture experiments using purified DC subtypes cultured with purified CFSE-labeled, CL-4 CD8 T cells from the lungs of aDC-depleted and nontreated control mice. These experiments confirmed that, like the in vivo results, aDCs or aMφ were unable to promote normal expansion of “aDC-depleted” CD8 T cells from aDC-depleted hosts (Fig. 6 B). However, similar to our in vivo results, pDCs, CD8α+ DCs, and iDCs were able to induce proliferation of both control and aDC-depleted CD8 T cells (Fig. 6 B).
There are several potential mechanisms through which the pulmonary pDCs, CD8α+ DCs, and iDCs could restore CD8 T cell responses in aDC-depleted mice. Pulmonary DCs could be providing soluble cytokine signals to the T cells; signals such as costimulatory molecules, additional antigen, etc., via direct cell–cell contact with the T cells; or some combination thereof. Therefore, to delineate the mechanism by which these DC populations were able to restore pulmonary CD8 T cell responses, we again performed in vitro co-culture experiments with purified DC subsets and activated CD8 T cells purified from the lungs of aDC-depleted mice. First, we co-cultured T cells with fixed DCs. Although the fixed DCs would still provide cell–cell contact signals to the T cells, including costimulatory interactions and MHC-peptide complexes present on their surface, they would be unable to secrete cytokines or up-regulate additional surface molecules after interaction with the T cells. In these fixed DC co-culture experiments, we observed only a slight decrease in the ability of DCs to restore CD8 T cell responses relative to live DCs (Fig. 6 C), suggesting that the interactions between the DCs and CD8 T cells do not require cytokines or additional T-dependent up-regulation of molecules on the DC cell surface. Further, when DCs and T cells were separated by a transwell during the co-culture, a situation where cytokine could pass freely from the DCs to the CD8 T cells but no cell–cell contact would occur, we observed a drastic decrease in the ability of the DCs to restore full CD8 T cell responses as measured by CFSE dilution (Fig. 6 C). Together, these results suggest that DC rescue of aDC-depleted CD8 T cells requires direct contact with DCs.
DC rescue of CD8 T cell responses in aDC-depleted mice occurs in a MHC class I and viral antigen–specific manner
Because cell–cell contact was required for full DC-stimulated CD8 T cell activity in our in vitro system, we hypothesized that interactions between these cells may require MHC I. Therefore, to investigate the requirement for MHC I in restoring CD8 T cell responses to aDC-depleted mice in vivo, we used pulmonary DCs purified from day 6 influenza-infected β2M−/− mice, which express only very low levels of MHC I on their cell surface. In contrast to mice reconstituted with wild-type pDCs, CD8α+ DCs, or iDCs, those mice given β2M−/− DC subsets were unable to restore normal control levels of influenza-specific tetramer+ or IFN-γ+ CD8 T cells (Fig. 7 A). This result suggests that the interactions between aDC-depleted CD8 T cells and the reconstituted DC subsets occur in a MHC I–dependent manner and that effective CD8 T cell immunity in vivo requires DC–T cell contact in the lungs, in addition to the initial contact that occurs in the lung draining LN.
Figure 7.

In vivo reconstitution of aDC-depleted CD8 T cell responses by pulmonary DC subsets requires MHC I and influenza antigen. (A) Host mice were infected and aDC depleted as in Fig 1. 24 h after depletion, mice were reconstituted with the indicated pulmonary donor DC subsets purified from day 6 infected donor wild-type or β2M−/− (KO) DCs. On day 5 a.i., the number of antigen-specific CD8 T cells (tetramer [left] or ICS for IFN-γ [right]) was enumerated in host lungs by flow cytometry. The data are the mean values ± the SEM (n = 6–8 mice/group) and are representative of two separate experiments. (B) Using the reconstitution system described in A, aDC-depleted mice were reconstituted with the indicated pulmonary DC populations purified from day 6 B/Lee-infected donors. Separate groups of aDC-depleted mice received DCs that were purified from B/Lee-infected hosts that had subsequently been pulsed in vitro with 1 μM of the influenza peptides HA204, HA529, and NP147 (+ Peptide). On day 5 a.i., the number of antigen-specific CD8 T cells (tetramer [left] or ICS for IFN-γ [right]) was enumerated in host lungs by flow cytometry. The data are the mean values ± the SEM (n = 8–11 mice/group) and are representative of three separate experiments.
Because MHC I molecules function in presenting self- and foreign antigens to TCR on CD8 T cells, we next determined if DC rescue of influenza-specific T cell responses was occurring in an influenza viral antigen–independent or –dependent manner. If the interactions were to occur in an influenza viral antigen–specific manner, this would suggest that only influenza-specific CD8 T cells would expand after DC reconstitution. Alternatively, this interaction could occur in a self-antigen–MHC I–dependent manner, in which case all activated effector CD8 T cells, not just influenza-specific T cells, would increase in response to DC reconstitution. The protein sequences of the HA and NP proteins of influenza type B strains vary drastically from those of type A influenza, thereby leading to distinct CD8 T cell epitopes. Therefore, our expectation was that adoptive transfer of DCs carrying peptides from influenza type B would be unable to restore T cell responses in influenza A–infected, aDC-depleted mice if the T cell–DC interactions were viral antigen–specific. Our results show that transfer of pDCs and CD8α+ DCs from influenza B mice did not result in a significant increase in influenza A–specific CD8 T cell responses as measured by tetramer or ICS for IFN-γ (Fig. 7 B). These results suggest that for the pDC and CD8α+ DC subsets, DCs induced rescue of the influenza-specific response appears to require viral antigens. iDC-mediated T cell rescue appears to be more complex, as the T cell response appears to be rescued in a MHC I–dependent, but influenza A virus peptide–independent, manner that leads to restoration of effector functions but not to a tetramer-binding phenotype.
Given the inability of pDC and CD8α+ DC subsets from type B–infected mice to stimulate CD8 T cell responses, we next determined whether DCs from influenza B–infected mice were capable of driving a CD8 T cell response when provided with the correct influenza type A peptides. If the lack of a T cell response to the transferred DC subsets from type B–infected mice was caused by an influenza antigen–specific interaction and not simply by an inherent DC difference after type A versus B influenza infection, one would expect increased antigen-specific CD8 T cell responses when aDC-depleted mice are reconstituted with influenza type B DCs that have been pulsed in vitro with the correct influenza A CD8 epitopes before transfer. As shown in Fig. 7 B, reconstitution of aDC-depleted mice with influenza A peptide-pulsed influenza B donor DCs allows significant increases in numbers of pulmonary antigen-specific IFN-γ+ and tetramer+ CD8 T cells. Together, the results suggest that the inability of the DC subsets purified from influenza B–infected mice to reconstitute CD8 T cell responses in aDC-depleted mice is caused by a lack of the correct influenza A peptides. Further, these results indicate that CD8 T cells require MHC I–dependent, influenza viral antigen–specific interactions with DCs in the lungs to confer an effective antiviral response.
DISCUSSION
The results presented herein suggest that generation of an effective influenza-specific CD8 T cell response requires activated CD8 T cells to interact with pulmonary pDCs, CD8α+ DCs, or iDCs in a MHC class I–, viral epitope–dependent manner once they enter the lungs. This secondary interaction is in addition to the initial DC–T cell interactions that occur in the LN during activation of naive CD8 T cells. To our knowledge, this is the first study detailing a critical role for peripheral DC–CD8 T cell interactions after initial programming of primary effector T cells in the LN. This suggests that the influenza-specific CD8 T cell response may be regulated by a “two-hit” model of development and that the magnitude, and possibly phenotype, of the peripheral CD8 T cells generated may be related to both the initial programming that occurs in the LN, but also by secondary contacts with DC subsets at the site of the infection. Of note, we have observed differential magnitudes of recruitment into the lungs of the various DC subsets described in this study during i.n. infections with various substrains of influenza A viruses, as well as during high and low dose infections with the same strain (unpublished data). These results suggest that the characteristics and subsets of the pulmonary DCs present in the lungs during respiratory challenges could potentially have a profound effect on the ensuing immune response and its outcome.
A two-hit model would allow the immune system to titrate, in an antigen-specific way, the magnitude and duration of the T cell response generated. This would be critically important in a vital organ such as the lungs, where T cell responses sufficient to control an infection are required, but, if overly robust, are known to induce harmful immunopathology (37). Therein, a two-hit mechanism would allow the immune response to balance the benefit and cost of pulmonary CD8 T cell immunity.
Whether interactions of DCs with T cells in nonlymphoid tissues is a common or more global mechanism through which the immune system titrates the magnitude of the immune response in peripheral tissues is unclear at this time. However, a recent study has described a similar peripheral interaction of DCs and memory CD8 T cells in the dorsal root ganglia (38). After reactivation of latent herpes simplex virus, a direct DC–memory T cell interaction in this nonlymphoid tissue allows the peripheral expansion of the T cells to reestablish control of the virus. Further, Smit et al. have recently shown that during respiratory syncytial virus (RSV) infections, increased numbers of pulmonary pDCs in the lungs enhance the magnitude to the RSV-specific CD8 T cell response (39). Although this study did not directly show interaction of the CD8 T cells and pDCs in the lungs, their results would be consistent with those observed in this study where lung-resident pDCs increase the magnitude of the virus-specific pulmonary CD8 T cell response.
Interestingly, influenza virus infections lead to the recruitment of CD8α+ DCs and pDCs into the lungs. Both of these subsets have well-established roles in viral infection (33–36). The results described herein show an as of yet unknown direct role for these DCs in amplification of influenza-specific CD8 T cell immune responses. Although many reports have described pDC recruitment into the lungs after respiratory challenge (40, 41), to our knowledge, this is the first study describing recruitment of CD8α+ DCs into the lungs. CD8α+ DCs are potent cross-presenters of viral antigens and are normally associated with lymphoid tissues. The mechanism controlling the recruitment of these cells and their locations within the infected lungs remains unknown. Given the affinity of CD8α+ DCs for lymphoid tissue, it is possible that these cells are being recruited into the recently described inducible bronchus-associated lymphoid tissue (42), where naive T cell–DC interactions have been described (42). Lung-recruited pDCs, on the other hand, are often associated with the alveolar interstitium (43), suggesting that the pDCs and CD8α+ DCs could be mediating their effects in distinct locations in the lungs.
The source of the recruited pDCs and CD8α+ DCs currently remains unclear. Whereas aDCs and iDCs are thought to differentiate from blood-derived monocytes (44–47) or precursors that reside in the lungs (48), pDCs and CD8α+ DCs are found differentiated within the blood and/or other tissues before infection and can develop from a single precursor (49, 50). In a preliminary experiment, where liposome-clodronate was administered i.v. (38, 46, 51, 52) instead of i.n. at 48 h a.i., we observed a significant decrease in pulmonary aDCs, iDCs, pDCs, and CD8α+ DCs, but no change in aMφ numbers (unpublished data). Because i.v. liposome-clodronate administration alters cell populations in the blood, bone marrow, spleen, and liver, but not in the lungs, this result suggests that the pDCs and CD8α+ DCs, or their precursors, most likely are recruited from one of these tissues.
The mechanism by which pDCs, CD8α+ DCs, and iDCs drive pulmonary influenza–specific T cell expansion is MHC I dependent and appears to be largely dependent on viral antigens. This increase in the magnitude of the T cell response could potentially occur through three processes: (a) increasing antigen-specific T cell migration into the lungs; (b) inducing subsequent T cell proliferation; and/or (c) protecting the cells from apoptosis. Because influenza-specific T cell numbers are similar in the blood and spleens of control and aDC-depleted mice (not depicted) and transfer of influenza B DCs only leads to a limited, at best, increase in antigen-specific CD8 T cell numbers in the lungs (Fig. 7 B), altered T cell migration to the lungs does not appear to be the dominant pathway involved in DC rescue of T cell responses. However, our results do show that an intermediate CFSE division phenotype exists in some aDC-depleted T cells found in the lungs on day 5 a.i. (Fig. 4 C), and in vitro co-culture of the aDC-depleted T cells with the DCs induces proliferation. These results suggest that a second round of DC-induced proliferation of the T cells may occur after the T cells enter the lungs (53). Finally, recent studies have suggested that peripheral expression of costimulatory ligands such as OX40L and 4-1BBL may protect CD8 T cells from apoptosis (54). To date, we have found no difference in expression of the classical costimulation molecules (i.e., CD40, CD80, and CD86; unpublished data) on those DCs, which increase the magnitude of the T cell response. However, the potential individual contributions of the various DCs expressed costimulatory molecules in the rescue of the T cell response awaits more detailed analysis.
The fact that the aDC population, depleted during the initial liposome-clodronate treatment, did not restore pulmonary T cell responses to control levels was unexpected, given that these DCs are one of the primary populations that traffic from the lungs to the LNs during an influenza virus infection and participate in the initial activation of naive CD8 T cells in the regional LNs (1, 5, 6). This, and the fact that aDCs can stimulate control T cells to proliferate during in vitro cultures (Fig. 6 B), suggests that the aDCs should have the necessary factors needed to stimulate the aDC-depleted T cells. Interestingly, Belz et al. recently showed a similar memory T cell ignorance of aDCs in the LNs. In this study, although the aDCs and CD8α+ DCs in the LNs could activate naive CD8 T cells, only the CD8α+ DC subset could drive secondary expansion of the memory T cells (4). Further, although we have demonstrated that MHC I and influenza viral antigens are necessary to confer a full CD8 T cell response, it is likely that these factors alone are not sufficient, given that virally infected MHC I+ epithelial cells are not limiting during the course of an influenza infection, yet these cells are unable to provide the necessary signals to influenza-specific CD8 T cells in the lungs. Moreover, adoptive transfer of pDCs purified from the spleens of influenza virus–infected animals, unlike transfer of the aforementioned pulmonary pDCs, does not drive rescue of influenza-specific CD8 T cell responses (Fig. S6, available at http://www.jem.org/cgi/content/full/jem.20080314/DC1). Together, these studies suggest pulmonary pDC-, CD8α+ DC–, and iDC-mediated rescue of T cells may require additional molecules, such as CD70 (4), in addition to viral antigen and MHC class I.
The results presented herein largely suggest that the reduced influenza-specific CD8 T cells numbers in the lungs are responsible for the increased disease severity. Consistent with this idea, reconstitution of the lungs with pDCs, CD8α+ DCs, or iDCs leads to an increased T cell response, and therein reduced pulmonary virus titers (unpublished data). However, because the i.n. liposome-clodronate treatment eliminates pulmonary APC populations (i.e., aMφ and aDC) and alters DC recruitment (i.e., pDCs and CD8α+ DCs), we cannot completely rule out that loss of these cells alters some form of APC direct control of the virus infection (55) or at least decreases clearance of virus-related debris, thereby increasing mortality. When we have reconstituted the depleted lungs with aMφ or aDCs (Fig. 6 A), we have not seen an abatement of influenza-associated morbidity or mortality (not depicted), suggesting that these cells, independent of full CD8 T cell responses, cannot alleviate the increased disease severity. It is of interest, however, that recent studies have identified an early role for aMφ in immunity to RSV infections (56) and a highly pathogenic 1918 HA/NA–containing influenza virus (55). In these studies, differences in the production of cytokines and chemokines (TNF-α, IL-6, CCL3, IFN-α, and CCL5), as well as NK cell activation, after RSV infection and control of virus titers/morbidity during 1918 HA/NA–containing influenza virus infections of aMφ-depleted animals were limited to day 1 a.i. (i.e., RSV) or required the depletion of aMφ before infection (i.e., 1918 HA/NA–containing influenza virus). In this study, the aMφ were not depleted until 48 h a.i. (i.e., after this first 24-h a.i. window), therefore it remains to be seen if changes in chemokine and cytokine production or NK activation have occurred. Regardless, the aMφ, unlike pDCs, CD8α+ DCs, and iDCs, appear to have no direct role in amplifying the influenza-specific CD8 T cell response, and therein allow the elimination of the virus from the lungs.
Herein, we have shown that influenza virus infections induce the recruitment or expansion of CD8α+ pDCs and iDCs in the lungs. In the absence of these DC subsets, the influenza-specific CD8 T cell response is dramatically inhibited. This inhibition of T cell immunity is not caused by altered activation of naive T cells in the LN, as CD25, CD69, and T cell division (Fig. 4), as well as DC subset cell numbers and ability of day 3 and 5 a.i. purified LN DCs to activate naive CD8 T cells, appears identical in the aDC-depleted and control mice. Furthermore, after activation in the LN, the influenza-specific T cells are found in the blood and spleen, but not the lungs, at normal levels. Reconstitution with pDCs, CD8α+ DCs, and iDCs increases the numbers of influenza-specific CD8 T cells present in a DC–T cell contact–, MHC I–, and viral epitope–dependent manner. Collectively, our results suggest that pulmonary influenza–specific CD8 T cells require at least two antigen-specific interactions with DCs, one as naive cells in the LN, and a second as activated cells after arriving in the lungs.
MATERIALS AND METHODS
Mice
BALB/c mice were purchased from the National Cancer Institute. Clone 4 (CL-4) TCR transgenic mice (H-2d; Thy-1.2) specific to the HA533-541 epitope of H1 and H2 Influenza A viruses were a gift from L. Sherman (Scripps Research Institute, La Jolla, CA). BALB/c Thy1.1 congenic mice were a gift from R. Enelow (Dartmouth College, Hanover, NH) and J. Harty (University of Iowa, Iowa City, IA). All mice were maintained in the animal care facility at the University of Iowa. Experiments were conducted according to federal and institutional guidelines and were approved by the University of Iowa Animal Care and Use Committee.
Virus infection
Mice were anesthetized by halothane or isoflurane inhalation and infected i.n. with either a 0.1-LD50 (3.67 × 104 EIU) or a 0.01-LD50 (3.67 × 103 EIU) dose of mouse-adapted A/JAPAN/305/57 (H2N2) or 7.50 × 104 EIU of mouse-adapted B/Lee/40 in 50 μl of Iscove's media. Viruses were grown as previously described (9).
Lung virus titer
Pulmonary viral titers were performed as previously described (9).
Clodronate-liposome treatment
Pulmonary DCs and macrophage depletion was performed by treatment with liposomes containing dichloromethylene bisphosphonate (clodronate). Clodronate was a gift from Roche. It was encapsulated in liposomes, as previously described (27). Phosphatidylcholine (LIPOID E PC) was obtained from Lipoid GmbH. Cholesterol was purchased from Sigma-Aldrich. At 48 h a.i., mice were anesthetized by halothane or isoflurane inhalation and administered 75 μl of clodronate-liposomes or PBS-liposomes i.n.
Peptides
Influenza virus peptides HA204-212 (LYQNVGTYV), HA529-537 (IYATVAGSL), and NP147-155 (TYQRTRALV) were synthesized by BioSynthesis Inc.
MHC I tetramers
Tetramers HA204 (H-2K(d)/LYQNVGTYV), HA529 (H-2K(d)/IYATVAGSL), and NP147 (H2K(d)/TYQRTRALV) were obtained from the National Institute of Allergy and Infectious Disease MHC Tetramer Core Facility.
Preparation of cells
Lungs, LNs, and spleens were pressed through wire mesh to obtain a single-cell suspension, which were enumerated by trypan blue exclusion. For DC preparations, lungs were digested for 25 min at 25°C in media containing 1 mg/ml Collagenase (Sigma-Aldrich) and 0.02 mg/ml DNase (Sigma-Aldrich) before single-cell preparation.
Flow cytometry
The following monoclonal antibodies were used for these studies: rat anti–mouse CD3ε (145-2C11), rat anti–mouse CD4 (CT-CD4), rat anti–mouse CD8α (53–6.7), rat anti–mouse IFN-γ (XMG1.2), rat anti–mouse IA/IE (M5/114.15.2), mouse anti–mouse Vβ8 (F23.1), rat anti–mouse CD11b (M1/70), hamster anti–mouse CD11c (HL3; all Becton Dickinson); mouse anti–mouse CD90.2 (5a-8), rat anti–mouse CD45R (RA3-6B2), rat anti–mouse NK cells (DX5), rat anti–mouse Gr-1 (RB6-8C5; all from Caltag). For surface staining, isolated cells (106) cells were stained with antibody or tetramer, and then fixed using BD FACS Lysing Solution (BD Biosciences). For ICS, fixed cells were permeabilized and labeled with antibodies in FACS buffer containing 0.5% Saponin (Acros Organics). All flow cytometry data were acquired on a BD FACSCalibur (BD Biosciences) in CellQuest (BD Biosciences) and analyzed using FlowJo software (Tree Star, Inc.).
In vivo cytotoxicity assays
In vivo cytotoxicity assays were performed on day 6 aDC-depleted and control mice, as previously described (9).
Purification and adoptive transfer of cell populations
Naive CD8α+ CL-4 T cells.
Single-cell suspension of splenic cells from CL-4 mice were labeled with CD8α Microbeads (MBs; Miltenyi Biotech) and purified according to the manufacturer's instructions (Miltenyi Biotec). Purified Thy1.2+ CL-4 cells were labeled with 2.5 μM CFSE (Invitrogen) and adoptively transferred (107) i.v. into BALB/c Thy1.1 congenic mice. 24 h later, the host mice were infected with influenza virus.
Activated pulmonary CD8α+ CL-4 T cells.
Activated pulmonary donor Thy1.2+CD8+ CL-4 T cells obtained from the lungs of day 4 infected Thy1.1+ host mice were enriched to 80–85% purity by sequential positive selection based upon their expression of CD8α+, Vβ8+, and Thy1.2+ using MACS MBs according to the manufacturer's instructions. For multiple positive selection steps, MBs were removed from cells using MACS Multisort Release Reagent according to the manufacturer's instructions. Purified CL-4 T cells were then labeled with 2.5 μM CFSE (Invitrogen), washed, and co-cultured with purified DCs.
Dendritic cells.
Purification of pulmonary DC populations by FACS or MACS. All DC populations were purified from the lungs of wild-type or β2M−/−A/JAPAN/305/57-infected or B/Lee/40-infected mice on day 6 a.i.
pDC populations were sequentially enriched based upon their expression of CD8α+ (positive selection, anti-CD8 MBs), CD3ε− (negative selection, anti-PE MBs) and CD45R+ (positive selection, anti-CD45R MBs). CD8α+ DC populations were sequentially enriched based upon their expression of CD8α+ (positive selection, anti-CD8 MBs), CD3ε− (negative selection, anti-PE MBs), and CD45R− (negative selection, anti-CD45R MBs). For multiple positive selection steps, MBs were removed from cells using MACS Multisort Release Reagent from the MACS anti-PE Multisort kit according to the manufacturer's instructions.
aDCs were FACS sorted based on their expression of CD11c+MHC II+CD11b−Autofluorescenceneg or MACS sorted sequentially based upon their expression of CD11c+ (positive selection, anti-CD11c MBs), MHC II+ (positive selection, anti-PE MBs), and CD11b− (negative selection, anti-biotin MBs). iDC populations were FACS sorted based on their expression of CD11c+MHCII+CD11b+.
Autofluorescenceneg were MACS sorted sequentially based upon their expression of CD11c+ (positive selection, anti-CD11c MBs), MHC II+ (positive selection, anti-PE MBs), and CD11b+ (positive selection, anti-biotin MBs). aMφ populations were FACS sorted based on their expression of CD11c+MHC II−CD11b−Autofluorescence+ or MACS sorted sequentially based on their expression of CD11c+ (positive selection, anti-CD11c MBs), MHC II− (negative selection, anti-PE MBs), and CD11b− (negative selection, anti-biotin MBs). For multiple positive selection steps, MBs were removed from cells using MACS Multisort Release Reagent from the MACS anti-PE Multisort kit according to the manufacturer's instructions.
All pDC and CD8α+ DC populations were enriched to 80–85% purity by MACS (Miltenyi Biotec). aDC, iDC, and aMφ populations were enriched to at least 80–85% purity either by FACS sorting or by MACS sorting. Purified DCs (2.5 × 104) were adoptively transferred i.n. to aDC-depleted mice on day 3 a.i. (i.e., 24 h after depletion).
In vitro cell co-cultures
Purified DC populations were co-cultured for 48 h at a 1:5 ratio with activated pulmonary CFSE-labeled CL-4 CD8 T cells purified from day 4 infected control or aDC-depleted mice.
Statistical analysis
Statistical significance of the difference between two sets of data were assessed using an unpaired one-tailed Student's t test or a paired Student's t test for control and experimental data groups that could be paired. Differences were considered to be statistically significant at P < 0.05.
Online supplemental material
Fig. S1 shows the depletion of aMφ by i.n. administration of clodronate-liposomes at 48 h a.i. Fig. S2 shows a similar inhibition in the pulmonary influenza-specific CD8 T cell response of aDC-depleted mice after adoptive transfer of lower numbers (104) of CL-4 influenza-specific CD8 T cells. Fig. S3 shows the gating strategy used to identify pulmonary aDCs, iDCs, aMφ, and iMφ. Fig. S4 shows the gating strategy used to identify pulmonary total pDCs, CD8+ pDCs, and CD8α+ DCs. Fig. S5 shows the kinetics of total pDC and CD8+ pDC recruitment into the lungs after a sublethal influenza virus infection. Fig. S6 shows the influenza-specific CD8 T cell response in aDC-depleted mice after i.n. reconstitution with splenic or pulmonary pDCs or CD8α+ DC. The online version of this article is available at http://www.jem.org/cgi/content/full/jem.20080314/DC1.
Supplementary Material
Acknowledgments
We thank J. Harty, T Griffith, S. Varga, and T. Waldschmidt for critical reading of this manuscript.
This work was supported by grants from the National Institutes of Health (AI071085 and AI076989 to K.L. Legge).
The authors declare no competing financial interests.
Abbreviations used: aDC, airway and alveolar DC; a.i., after infection; aMφ, alveolar macrophage; ICS, intracellular cytokine staining; iDC, interstitial DC; iMφ, interstitial macrophage; i.n., intranasally; MB, Microbead; pDC, plasmacytoid DC; rDC, respiratory DC; RSV, respiratory syncytial virus.
References
- 1.Vermaelen, K.Y., I. Carro-Muino, B.N. Lambrecht, and R.A. Pauwels. 2001. Specific migratory dendritic cells rapidly transport antigen from the airways to the thoracic lymph nodes. J. Exp. Med. 193:51–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Xia, W., C.E. Pinto, and R.L. Kradin. 1995. The antigen-presenting activities of Ia+ dendritic cells shift dynamically from lung to lymph node after an airway challenge with soluble antigen. J. Exp. Med. 181:1275–1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Holt, P.G., P.A. Stumbles, and A.S. McWilliam. 1999. Functional studies on dendritic cells in the respiratory tract and related mucosal tissues. J. Leukoc. Biol. 66:272–275. [DOI] [PubMed] [Google Scholar]
- 4.Belz, G.T., S. Bedoui, F. Kupresanin, F.R. Carbone, and W.R. Heath. 2007. Minimal activation of memory CD8(+) T cell by tissue-derived dendritic cells favors the stimulation of naive CD8(+) T cells. Nat. Immunol. 8:1060–1066. [DOI] [PubMed] [Google Scholar]
- 5.Belz, G.T., C.M. Smith, L. Kleinert, P. Reading, A. Brooks, K. Shortman, F.R. Carbone, and W.R. Heath. 2004. Distinct migrating and nonmigrating dendritic cell populations are involved in MHC class I-restricted antigen presentation after lung infection with virus. Proc. Natl. Acad. Sci. USA. 101:8670–8675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Legge, K.L., and T.J. Braciale. 2003. Accelerated migration of respiratory dendritic cells to the regional lymph nodes is limited to the early phase of pulmonary infection. Immunity. 18:265–277. [DOI] [PubMed] [Google Scholar]
- 7.Lawrence, C.W., and T.J. Braciale. 2004. Activation, differentiation, and migration of naive virus-specific CD8+ T cells during pulmonary influenza virus infection. J. Immunol. 173:1209–1218. [DOI] [PubMed] [Google Scholar]
- 8.Lawrence, C.W., R.M. Ream, and T.J. Braciale. 2005. Frequency, specificity, and sites of expansion of CD8+ T cells during primary pulmonary influenza virus infection. J. Immunol. 174:5332–5340. [DOI] [PubMed] [Google Scholar]
- 9.Legge, K.L., and T.J. Braciale. 2005. Lymph node dendritic cells control CD8+ T cell responses through regulated FasL expression. Immunity. 23:649–659. [DOI] [PubMed] [Google Scholar]
- 10.Topham, D.J., R.A. Tripp, and P.C. Doherty. 1997. CD8+ T cells clear influenza virus by perforin or Fas-dependent processes. J. Immunol. 159:5197–5200. [PubMed] [Google Scholar]
- 11.Wong, P., and E.G. Pamer. 2001. Cutting edge: antigen-independent CD8 T cell proliferation. J. Immunol. 166:5864–5868. [DOI] [PubMed] [Google Scholar]
- 12.Kaech, S.M., and R. Ahmed. 2001. Memory CD8+ T cell differentiation: initial antigen encounter triggers a developmental program in naive cells. Nat. Immunol. 2:415–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Van Stipdonk, M.J., E.E. Lemmens, and S.P. Schoenberger. 2001. Naive CTLs require a single brief period of antigen stimulation for clonal expansion and differentiation. Nat. Immunol. 2:423–429. [DOI] [PubMed] [Google Scholar]
- 14.van Stipdonk, M.J., G. Hardenberg, M.S. Bijker, E.E. Lemmens, N.M. Droin, D.R. Green, and S.P. Schoenberger. 2003. Dynamic programming of CD8+ T lymphocyte responses. Nat. Immunol. 4:361–365. [DOI] [PubMed] [Google Scholar]
- 15.Wong, P., and E.G. Pamer. 2004. Disparate in vitro and in vivo requirements for IL-2 during antigen-independent CD8 T cell expansion. J. Immunol. 172:2171–2176. [DOI] [PubMed] [Google Scholar]
- 16.Mescher, M.F., J.M. Curtsinger, P. Agarwal, K.A. Casey, M. Gerner, C.D. Hammerbeck, F. Popescu, and Z. Xiao. 2006. Signals required for programming effector and memory development by CD8+ T cells. Immunol. Rev. 211:81–92. [DOI] [PubMed] [Google Scholar]
- 17.Williams, M.A., and M.J. Bevan. 2007. Effector and memory CTL differentiation. Annu. Rev. Immunol. 25:171–192. [DOI] [PubMed] [Google Scholar]
- 18.Liu, L., R.C. Fuhlbrigge, K. Karibian, T. Tian, and T.S. Kupper. 2006. Dynamic programming of CD8+ T cell trafficking after live viral immunization. Immunity. 25:511–520. [DOI] [PubMed] [Google Scholar]
- 19.Badovinac, V.P., and J.T. Harty. 2006. Programming, demarcating, and manipulating CD8+ T-cell memory. Immunol. Rev. 211:67–80. [DOI] [PubMed] [Google Scholar]
- 20.Holt, P.G., D.J. Nelson, and A.S. McWilliam. 1995. Population dynamics and functions of respiratory tract dendritic cells in the rat. Adv. Exp. Med. Biol. 378:177–181. [DOI] [PubMed] [Google Scholar]
- 21.McWilliam, A.S., S. Napoli, A.M. Marsh, F.L. Pemper, D.J. Nelson, C.L. Pimm, P.A. Stumbles, T.N. Wells, and P.G. Holt. 1996. Dendritic cells are recruited into the airway epithelium during the inflammatory response to a broad spectrum of stimuli. J. Exp. Med. 184:2429–2432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McWilliam, A.S., D. Nelson, J.A. Thomas, and P.G. Holt. 1994. Rapid dendritic cell recruitment is a hallmark of the acute inflammatory response at mucosal surfaces. J. Exp. Med. 179:1331–1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.De Heer, H.J., H. Hammad, T. Soullie, D. Hijdra, N. Vos, M.A. Willart, H.C. Hoogsteden, and B.N. Lambrecht. 2004. Essential role of lung plasmacytoid dendritic cells in preventing asthmatic reactions to harmless inhaled antigen. J. Exp. Med. 200:89–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bosio, C.M., and S.W. Dow. 2005. Francisella tularensis induces aberrant activation of pulmonary dendritic cells. J. Immunol. 175:6792–6801. [DOI] [PubMed] [Google Scholar]
- 25.van Rooijen, N. 1992. Liposome-mediated elimination of macrophages. Res. Immunol. 143:215–219. [DOI] [PubMed] [Google Scholar]
- 26.Van Rooijen, N. 1989. The liposome-mediated macrophage ‘suicide’ technique. J. Immunol. Methods. 124:1–6. [DOI] [PubMed] [Google Scholar]
- 27.Van Rooijen, N., and A. Sanders. 1994. Liposome mediated depletion of macrophages: mechanism of action, preparation of liposomes and applications. J. Immunol. Methods. 174:83–93. [DOI] [PubMed] [Google Scholar]
- 28.Kreuwel, H.T., S. Aung, C. Silao, and L.A. Sherman. 2002. Memory CD8(+) T cells undergo peripheral tolerance. Immunity. 17:73–81. [DOI] [PubMed] [Google Scholar]
- 29.Morgan, D.J., H.T. Kreuwel, S. Fleck, H.I. Levitsky, D.M. Pardoll, and L.A. Sherman. 1998. Activation of low avidity CTL specific for a self epitope results in tumor rejection but not autoimmunity. J. Immunol. 160:643–651. [PubMed] [Google Scholar]
- 30.Morgan, D.J., H.T. Kreuwel, and L.A. Sherman. 1999. Antigen concentration and precursor frequency determine the rate of CD8+ T cell tolerance to peripherally expressed antigens. J. Immunol. 163:723–727. [PubMed] [Google Scholar]
- 31.Hernandez, J., S. Aung, K. Marquardt, and L.A. Sherman. 2002. Uncoupling of proliferative potential and gain of effector function by CD8+ T cells responding to self-antigens. J. Exp. Med. 196:323–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schlecht, G., S. Garcia, N. Escriou, A.A. Freitas, C. Leclerc, and G. Dadaglio. 2004. Murine plasmacytoid dendritic cells induce effector/memory CD8+ T-cell responses in vivo after viral stimulation. Blood. 104:1808–1815. [DOI] [PubMed] [Google Scholar]
- 33.Fonteneau, J.F., M. Gilliet, M. Larsson, I. Dasilva, C. Munz, Y.J. Liu, and N. Bhardwaj. 2003. Activation of influenza virus-specific CD4+ and CD8+ T cells: a new role for plasmacytoid dendritic cells in adaptive immunity. Blood. 101:3520–3526. [DOI] [PubMed] [Google Scholar]
- 34.Cella, M., F. Facchetti, A. Lanzavecchia, and M. Colonna. 2000. Plasmacytoid dendritic cells activated by influenza virus and CD40L drive a potent TH1 polarization. Nat. Immunol. 1:305–310. [DOI] [PubMed] [Google Scholar]
- 35.Belz, G.T., C.M. Smith, D. Eichner, K. Shortman, G. Karupiah, F.R. Carbone, and W.R. Heath. 2004. Cutting edge: conventional CD8 alpha+ dendritic cells are generally involved in priming CTL immunity to viruses. J. Immunol. 172:1996–2000. [DOI] [PubMed] [Google Scholar]
- 36.Smith, C.M., G.T. Belz, N.S. Wilson, J.A. Villadangos, K. Shortman, F.R. Carbone, and W.R. Heath. 2003. Cutting edge: conventional CD8 alpha+ dendritic cells are preferentially involved in CTL priming after footpad infection with herpes simplex virus-1. J. Immunol. 170:4437–4440. [DOI] [PubMed] [Google Scholar]
- 37.Bruder, D., A. Srikiatkhachorn, and R.I. Enelow. 2006. Cellular immunity and lung injury in respiratory virus infection. Viral Immunol. 19:147–155. [DOI] [PubMed] [Google Scholar]
- 38.Wakim, L.M., J. Waithman, N. van Rooijen, W.R. Heath, and F.R. Carbone. 2008. Dendritic cell-induced memory T cell activation in nonlymphoid tissues. Science. 319:198–202. [DOI] [PubMed] [Google Scholar]
- 39.Smit, J.J., D.M. Lindell, L. Boon, M. Kool, B.N. Lambrecht, and N.W. Lukacs. 2008. The balance between plasmacytoid DC versus conventional DC determines pulmonary immunity to virus infections. PLoS ONE. 3:e1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Grayson, M.H., M.S. Ramos, M.M. Rohlfing, R. Kitchens, H.D. Wang, A. Gould, E. Agapov, and M.J. Holtzman. 2007. Controls for lung dendritic cell maturation and migration during respiratory viral infection. J. Immunol. 179:1438–1448. [DOI] [PubMed] [Google Scholar]
- 41.Hammad, H., and B. Lambrecht. 2006. Recent progress in the biology of airway dendritic cells and implications for understanding the regulation of asthmatic inflammation. J. Allergy Clin. Immunol. 118:331–336. [DOI] [PubMed] [Google Scholar]
- 42.Moyron-Quiroz, J.E., J. Rangel-Moreno, K. Kusser, L. Hartson, F. Sprague, S. Goodrich, D.L. Woodland, F.E. Lund, and T.D. Randall. 2004. Role of inducible bronchus associated lymphoid tissue (iBALT) in respiratory immunity. Nat. Med. 10:927–934. [DOI] [PubMed] [Google Scholar]
- 43.de Heer, H.J., H. Hammad, M. Kool, and B.N. Lambrecht. 2005. Dendritic cell subsets and immune regulation in the lung. Semin. Immunol. 17:295–303. [DOI] [PubMed] [Google Scholar]
- 44.Landsman, L., C. Varol, and S. Jung. 2007. Distinct differentiation potential of blood monocyte subsets in the lung. J. Immunol. 178:2000–2007. [DOI] [PubMed] [Google Scholar]
- 45.Varol, C., L. Landsman, D.K. Fogg, L. Greenshtein, B. Gildor, R. Margalit, V. Kalchenko, F. Geissmann, and S. Jung. 2007. Monocytes give rise to mucosal, but not splenic, conventional dendritic cells. J. Exp. Med. 204:171–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jakubzick, C., F. Tacke, F. Ginhoux, A.J. Wagers, N. van Rooijen, M. Mack, M. Merad, and G.J. Randolph. 2008. Blood monocyte subsets differentially give rise to CD103+ and CD103- pulmonary dendritic cell populations. J. Immunol. 180:3019–3027. [DOI] [PubMed] [Google Scholar]
- 47.Schneeberger, E.E., Q. Vu, B.W. LeBlanc, and C.M. Doerschuk. 2000. The accumulation of dendritic cells in the lung is impaired in CD18−/− but not in ICAM-1−/− mutant mice. J. Immunol. 164:2472–2478. [DOI] [PubMed] [Google Scholar]
- 48.Wang, H., N. Peters, V. Laza-Stanca, N. Nawroly, S.L. Johnston, and J. Schwarze. 2006. Local CD11c+ MHC class II- precursors generate lung dendritic cells during respiratory viral infection, but are depleted in the process. J. Immunol. 177:2536–2542. [DOI] [PubMed] [Google Scholar]
- 49.Naik, S.H., P. Sathe, H.Y. Park, D. Metcalf, A.I. Proietto, A. Dakic, S. Carotta, M. O'Keeffe, M. Bahlo, A. Papenfuss, et al. 2007. Development of plasmacytoid and conventional dendritic cell subtypes from single precursor cells derived in vitro and in vivo. Nat. Immunol. 8:1217–1226. [DOI] [PubMed] [Google Scholar]
- 50.Shortman, K., and S.H. Naik. 2007. Steady-state and inflammatory dendritic-cell development. Nat. Rev. Immunol. 7:19–30. [DOI] [PubMed] [Google Scholar]
- 51.Le Borgne, M., N. Etchart, A. Goubier, S.A. Lira, J.C. Sirard, N. van Rooijen, C. Caux, S. Ait-Yahia, A. Vicari, D. Kaiserlian, and B. Dubois. 2006. Dendritic cells rapidly recruited into epithelial tissues via CCR6/CCL20 are responsible for CD8+ T cell crosspriming in vivo. Immunity. 24:191–201. [DOI] [PubMed] [Google Scholar]
- 52.Tacke, F., F. Ginhoux, C. Jakubzick, N. van Rooijen, M. Merad, and G.J. Randolph. 2006. Immature monocytes acquire antigens from other cells in the bone marrow and present them to T cells after maturing in the periphery. J. Exp. Med. 203:583–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.McGill, J.M., and K.L. Legge. 2008. Continued proliferation of influenza-specific T cell in the lungs during the early stages of influenza virus infections. In Options for the Control of Influenza. In press.
- 54.Marrack, P., and J. Kappler. 2004. Control of T cell viability. Annu. Rev. Immunol. 22:765–787. [DOI] [PubMed] [Google Scholar]
- 55.Tumpey, T.M., A. Garcia-Sastre, J.K. Taubenberger, P. Palese, D.E. Swayne, and C.F. Basler. 2004. Pathogenicity and immunogenicity of influenza viruses with genes from the 1918 pandemic virus. Proc. Natl. Acad. Sci. USA. 101:3166–3171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pribul, P.K., J. Harker, B. Wang, H. Wang, J.S. Tregoning, J. Schwarze, and P.J. Openshaw. 2008. Alveolar macrophages are a major determinant of early responses to viral lung infection but do not influence subsequent disease development. J. Virol. 82:4441–4448. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}