

Abstract
Absorption of a photon by an opsin pigment causes isomerization of the chromophore from 11-cis-retinaldehyde to all-trans-retinaldehyde. Regeneration of visual chromophore following light exposure is dependent on an enzyme pathway called the retinoid or visual cycle. Our understanding of this pathway has been greatly facilitated by the identification of disease-causing mutations in the genes coding for visual cycle enzymes. Defects in nearly every step of this pathway are responsible for human-inherited retinal dystrophies. These retinal dystrophies can be divided into two etiologic groups. One involves the impaired synthesis of visual chromophore. The second involves accumulation of cytotoxic products derived from all-trans-retinaldehyde. Gene therapy has been successfully used in animal models of these diseases to rescue the function of enzymes involved in chromophore regeneration, restoring vision. Dystrophies resulting from impaired chromophore synthesis can also be treated by supplementation with a chromophore analog. Dystrophies resulting from the accumulation of toxic pigments can be treated pharmacologically by inhibiting the visual cycle, or limiting the supply of vitamin A to the eyes. Recent progress in both areas provides hope that multiple inherited retinal diseases will soon be treated by pharmaceutical intervention.
Keywords: photoreceptors, vertebrate/metabolism, retinol (vitamin A), retinoid (visual) cycle, retina, Leber congenital amaurosis/therapy, macular degeneration/drug therapy
BACKGROUND AND SCOPE
The vertebrate retina contains two types of photosensitive cells, rods and cones. Both cell types are polarized and contain a nonmotile cilium connected to an outer segment (OS), which comprises a stack of membranous discs. The light-receptive proteins in these cells are the opsins, which are members of the G protein-coupled receptor (GPCR) family. The chromophore for most opsins is 11-cis-retinaldehyde (11-cis-RAL), which is covalently coupled to a lysine residue via a Schiff-base linkage (11-cis-retinylidene) (1). Absorption of a photon by a rhodopsin or cone-opsin pigment causes isomerization of 11-cis-RAL to all-trans-retinaldehyde (all-trans-RAL). This isomerization induces a sequence of short-lived changes in opsin conformation until reaching the activated state, metarhodopsin II (MII) (2). MII stimulates transducin, the α-subunit of a heterotrimeric G protein, as the first event in visual transduction (3). Stimulation of the transduction cascade results in closure of cGMP-gated cation channels in the plasma membrane of the OS and hyperpolarization of the photoreceptor cell. After a brief period of activation, MII is inactivated via phosphorylation and binding to arrestin, and subsequently decays to yield apo-opsin and free all-trans-RAL. To maintain continuous vision, vertebrates have evolved an enzymatic pathway that regenerates 11-cis-RAL (structure shown in Figure 1a) from all-trans-RAL to reform visual pigment. This pathway, called the visual cycle or retinoid cycle, takes place mainly in the retinal pigment epithelium (RPE), a single layer of cells adjacent to the photoreceptors (Figure 2). Important differences exist between rods and cones with respect to photoreception, phototransduction, and chromophore regeneration. These differences permit vision over a range of background light intensities spanning nine logs.
Figure 1.

Structures of visual retinoids (a), N-retinylidene-N-retinyl ethanolamine (A2E) (b), and inhibitors of A2E biogenesis (c).
Figure 2.

Visual cycle in retinal pigment epithelium (RPE) cells. Absorption of a photon (hv) by a rhodopsin pigment molecule induces isomerization of 11-cis-RAL to all-trans-RAL resulting in activated metarhodopsin II (MII). Decay of MII yields apo-opsin and free all-trans-RAL. Translocation of all-trans-RAL from the disc membrane to the cytoplasmic space is facilitated by an ATP-binding cassette transporter called ABCR or ABCA4. Mutations in the human ABCA4 gene cause recessive Stargardt macular degeneration. The all-trans-RAL is reduced to all-trans-retinol (all-trans-ROL) by two or more all-trans-ROL dehydrogenases (all-trans-RDHs). Mutations in the gene for one (RDH12) cause Leber congenital amaurosis (LCA) (85). The all-trans-ROL is released by photoreceptors and taken up by RPE cells where it is esterified to a fatty acid by lecithin:retinol acyl transferase (LRAT). Mutations in the LRAT gene cause a severe form of recessive retinitis pigmentosa (RP) (86). The isomerase, RPE-specific 65 kDa protein (Rpe65) uses an all-trans-retinyl ester as substrate to form 11-cis-retinol (11-cis-ROL) plus a free fatty acid. Mutations in the RPE65 gene also cause LCA (87, 88). The Rpe65 isomerase activity may be regulated by the non-photoreceptor opsin, RGR (131). Mutations in the RPE-retinal G protein-coupled receptor (RGR) gene also cause recessive RP (93). 11-cis-ROL binds to the cellular retinaldehyde-binding protein (CRALBP). Mutations in the gene for CRALBP (RLBP1) cause still another form of recessive RP (137). 11-cis-ROL may be esterified by LRAT to yield 11-cis-retinyl esters, a stable storage-form of pre-isomerized retinoid. 11-cis-retinyl esters are hydrolyzed to 11-cis-ROL for synthesis of chromophore by 11-cis-retinyl ester hydrolase (11-cis-REH), which has not yet been cloned. 11-cis-ROL is oxidized to 11-cis-RAL by several 11-cis-ROL dehydrogenases (11-cis-RDHs), including RDH5 and RDH11. Mutations in the gene coding for RDH5 cause delayed dark adaptation and a mild retinal dystrophy called fundus albipunctatus in humans (141). 11-cis-RAL is also bound to CRALBP before being released by the RPE and taken up by the photoreceptor where it recombines with opsin to form rhodopsin.
Ensuring an adequate supply of chromophore is critical for maintaining vision and preserving the health of photoreceptors. The genes for many enzymes and retinoid-binding proteins of the visual cycle have been implicated in recessively inherited blinding diseases such as Leber congenital amaurosis (LCA), Stargardt macular degeneration, congenital cone-rod dystrophy, and retinitis pigmentosa (RP). The severity and high penetrance of these diseases underscores the essential nature of the retinoid cycle for maintaining a functional visual system. The phenotypes of these diseases have been successfully replicated in experimental animals with engineered or spontaneous mutations in retinoid cycle genes. These animal models have provided insight into the etiology of the different diseases and served as a starting point in the development of therapies.
THE RETINOID (VISUAL) CYCLE
Transport of all-trans-RAL from OS Discs
Following inactivation of MII, all-trans-RAL dissociates from the opsin protein into the bilayer of OS disc membranes. In vitro, all-trans-RAL partitions rapidly between phospholipid vesicles in aqueous media (4, 5); hence, disc membranes are not a barrier to its diffusion. Retinal also readily condenses with phosphatidylethanolamine (PE) to form the imine, N-retinylidene-PE (N-ret-PE). Although this reaction is reversible, clearance of all-trans-RAL from the interior of discs is delayed by the formation of N-ret-PE on the inner leaflet of disc membranes. A retina-specific ATP-binding cassette transporter called ABCR (also known as ABCA4) has been described in the discs of rod and cone OS (6–8). Mice with a knockout mutation in the abcr gene show delayed clearance of all-trans-RAL and elevated N-ret-PE in the retina following exposure to light (9, 10). In vitro biochemical studies suggest that ABCR is an outwardly directed flippase for N-ret-PE (11, 12), consistent with the phenotype in abcr−/−mice (9, 10). Thus, ABCR appears to facilitate the transfer of all-trans-RAL from disc membranes to the cytoplasmic space for subsequent reduction to all-trans-retinol (all-trans-ROL).
Reduction of all-trans-RAL to all-trans-ROL
The first catalytic step in the visual cycle involves enzymatic reduction of all-trans-RAL to all-trans-ROL (vitamin A) by retinol dehydrogenases (RDHs) (Figure 2). This reaction is carried out in rods and cones by members of the short-chain dehydrogenase/reductase (SDR) family. Several SDRs that catalyze reduction of all-trans-RAL in vitro using NADPH as a cofactor have been identified in photoreceptors (13–15). Mice with a knockout mutation in the gene for photoreceptor RDH (prRDH, RDH8) exhibited a several-fold slower clearance of all-trans-RAL from retinas following a bright flash (16). However, the kinetics of rhodopsin regeneration in these mice were indistinguishable from wild-type (WT) mice. These observations suggest that mouse photoreceptors contain at least one additional, functionally significant all-trans-ROL dehydrogenase besides RDH8.
Transfer of all-trans-ROL from Photoreceptor to RPE Cell
Newly formed all-trans-ROL is rapidly released by photoreceptor cells to the extracellular space, also called the interphotoreceptor matrix (IPM). Interphotoreceptor retinoid-binding protein (IRBP) is a 140-kDa glycoprotein secreted by photoreceptors and present at micromolar concentrations in the IPM (17–19). IRBP contains binding sites for both 11-cis- and all-trans-retinoids (20). The endogenous retinoid ligands for IRBP include all-trans-ROL, 11-cis-RAL, and 11-cis-retinol (11-cis-ROL) (21, 22). In vitro, IRBP has been shown to accelerate the removal of all-trans-ROL from bleached photoreceptors (23). Retinoids bound to IRBP are protected from both oxidation and isomerization (24, 25). Within RPE cells, cellular retinol-binding protein-1 (CRBP1) binds all-trans-ROL with a 100-fold higher affinity than does IRBP (26, 27). CRBP1 thus promotes the uptake of all-trans-ROL into the RPE. RPE cells may also take up all-trans-ROL by endocytosis of holo-IRBP (28) and by phagocytosis of shed OS (29). Finally, all-trans-ROL is taken up from blood in the choroidal circulation through the basal membranes of RPE cells.
Esterification of All-trans-ROL in RPE Cells
The major retinyl ester synthase in RPE cells is lecithin:retinol acyl transferase (LRAT), which catalyzes the transfer of a fatty acyl group from phosphatidylcholine to all-trans-ROL (30, 31) (Figure 2). LRAT preferentially uses all-trans-ROL bound to CRBP1 as substrate (32). Consistently, mice with a knockout mutation in the crbp1 gene have increased all-trans-ROL and reduced all-trans-retinyl esters in the RPE (33). A second retinyl ester synthase activity called acyl-CoA:retinol acyltransferase (ARAT) has been identified in RPE (30, 34) and cone-dominant chicken and ground-squirrel retinas (35, 36). In contrast to LRAT, ARAT uses an activated fatty acid such as palmitoyl coenzyme A (palm CoA) as an acyl donor to all-trans-ROL. ARAT has not been purified or cloned. However, acyl-CoA:diacylglycerol acyltransferase type-1 (DGAT1), which catalyzes palm CoA-dependent synthesis of triglycerides from diacylglycerol, was recently shown also to catalyze palm CoA-dependent synthesis of all-trans-retinyl esters from all-trans-ROL (37–39), and thus may contribute to ARAT activity in some tissues. Mice with a knockout mutation in the lrat gene contain only trace retinyl esters in ocular tissues (40). The virtual absence of retinyl esters despite the presumed presence of ARAT in lrat−/− eyes may be due to the approximately tenfold-higher KM of ARAT for all-trans-ROL (34). Both retinyl ester synthases are strongly inhibited by apo-CRBP1 (36). Interestingly, lrat−/− mice have higher than normal levels of retinyl esters in adipose tissue (39), which is likely due to ARAT activity. All-trans-retinyl esters represent a stable storage form of vitamin A that is non-cytotoxic and resistant to oxidation and thermal isomerization. These properties are due to the very low water-solubility of retinyl esters. The all-trans-retinyl esters are stored within internal membranes, and as oil-droplet structures associated with adipose differentiation-related protein in RPE cells (41).
Synthesis of 11-cis-ROL
The isomerase in RPE cells catalyzes the energetically unfavorable conversion of a planar all-trans-retinoid to the sterically constrained 11-cis configuration (Figure 2). The isomerase uses all-trans-retinyl esters as substrate (42–44). The abundant protein, RPE-specific 65-kDa protein (Rpe65), was recently identified as the isomerase in RPE cells (45–47). Consistently, mice with a knockout mutation in the rpe65 gene contain high levels of all-trans-retinyl esters in ocular tissues and no detectable 11-cis-retinoids (48). Rpe65 bears sequence similarity to β-carotene oxygenases found in mammals, insects, and plants (49), and to apocarotene oxygenase (ACO) in cyanobacteria (50). X-ray diffraction analysis showed that ACO has a seven-bladed β-propeller structure, with an Fe2+-4-His sphere at its axis (50). The four His residues that define the Fe2+-binding site are conserved in all members of the carotenoid cleavage enzyme family including Rpe65 (50). Rpe65 was shown to bind Fe2+, which is required for its catalytic activity (51). Rpe65 was also reported to be reversibly palmitoylated at three Cys residues, which may modulate its association with membranes (52).
The isomerization reaction is inhibited by its product, 11-cis-ROL (53, 54). This inhibition is relieved by the specific binding of 11-cis-ROL to cellular retinal-binding protein (CRALBP) in RPE cells (55, 56). CRALBP also binds 11-cis-RAL (56). A newly synthesized molecule of 11-cis-ROL has two potential fates. As discussed below, it can be oxidized to 11-cis-RAL for use as visual chromophore. Alternatively, it can be esterified by LRAT to form an 11-cis-retinyl ester (57). 11-cis-retinyl esters represent a storage form of pre-isomerized chromophore precursor. Hydrolysis of 11-cis-retinyl esters is catalyzed by 11-cis-retinyl ester hydrolase (11-cis-REH) in the plasma membrane of RPE cells (58, 59). 11-cis-REH is activated by apo-CRALBP (60), suggesting a regulatory mechanism to match the production of 11-cis-ROL with demand.
Oxidation of 11-cis-ROL to 11-cis-RAL
The final catalytic step in the visual cycle is oxidation of 11-cis-ROL to 11-cis-RAL (Figure 2). The first enzyme shown to catalyze this reaction in RPE cells is 11-cis-ROL dehydrogenase type 5 (RDH5) (61). Mice with a knockout mutation in the rdh5 gene show accumulation of 11-cis-ROL and 11-cis-retinyl esters in the RPE, and delayed recovery of rod sensitivity following exposure to bright light (62). Synthesis of 11-cis-RAL in rdh5−/− mice indicates the presence of at least one additional 11-cis-RDH activity in RPE. RDH11 is another SDR in RPE cells that oxidizes 11-cis-ROL to 11-cis-RAL (15). RDH11 may act in concert with RDH5 to generate visual chromophore. Double-knockout mice with disruptions in both the rdh5 and rdh11 genes still synthesize 11-cis-RAL, albeit more slowly than rdh5−/− or rdh11−/−single-knockout mice (63). This observation indicates the presence of at least one additional 11-cis-RDH activity in the RPE. The mechanism of 11-cis-RAL release from RPE cells into the IPM is not well understood. 11-cis-RAL is bound to CRALBP in RPE cells (56). CRALBP binds to ezrin-radixin-moesin (ERM)-binding phosphoprotein 50 (EBP50), a PDZ-domain protein associated with the cytoskeleton adjacent to the plasma membrane of RPE and Muller cells (64, 65). This interaction may target 11-cis-RAL to the apical plasma membrane for release into the IPM. IRBP binds 11-cis-RAL at higher affinity than it binds all-trans-ROL (66). Both in vitro and in cell culture, IRBP has been shown to remove 11-cis-RAL efficiently from RPE membranes (67, 68). Given the seeming importance of IRBP in the bidirectional flow of retinoids between photoreceptors and RPE, it is surprising that mice with a knockout mutation in the irbp gene exhibit only slight slowing of rhodopsin regeneration after a bright flash (69, 70). This retinoid transport may be due to the presence of other retinoid-binding proteins in the IPM such as retinol-binding protein (RBP) (19).
Regeneration of Visual Pigments
Although qualitatively similar, rods and cones differ greatly in sensitivity, dynamic range, and speed of the photoresponse. For example, rods detect single photons (71) and show response saturation at 500 photoisomerizations per second (72). Cones, on the other hand, are 100-fold less sensitive than rods (73) but can still respond to light flashes at levels of background illumination that produce 106 photoisomerizations per second (74). Also, the response of cones is several-fold faster than rods (75). These differences reflect intrinsic properties of the rod and cone visual pigments. Rhodopsin is exceedingly quiet, with a spontaneous activation rate of one thermal isomerization per 2000 years (76). Cone opsins are much noisier than rhodopsin for two reasons. First, the spontaneous isomerization rate of red-cone opsin is 10,000-fold higher than that of rhodopsin (77). Second, 11-cis-RAL spontaneously dissociates from cone-opsin. A dark-adapted red cone contains approximately 10% apo-cone-opsin due to spontaneous dissociation of chromophore (78). In contrast, 11-cis-RAL combines irreversibly with opsin in rods. Chromophore-free (apo-) opsin weakly activates transducin (79). These effects contribute to partial activation of the transduction cascade in cones independent of light. This “dark noise” is responsible for the reduced sensitivity and faster photoresponse observed in cones.
ALTERNATE PATHWAY FOR REGENERATING VISUAL CHROMOPHORE IN CONES
Rods require 11-cis-RAL from the RPE to regenerate rhodopsin pigment. Cones, however, are not dependent on the RPE (80, 81) and can use both 11-cis-RAL and 11-cis-ROL to regenerate cone-opsin pigments (82). An NADP+/NADPH-dependent 11-cis-RDH activity has been identified in cones, but not rods, which may explain the capacity of cones to utilize 11-cis-ROL as a chromophore precursor (35). Cone-dominant ground-squirrel and chicken retinas contain a retinoid isomerase activity distinct from Rpe65 that converts all-trans-ROL directly to 11-cis-ROL without formation of an all-trans-retinyl ester intermediate (35, 36). This isomerization reaction appears to be driven by secondary esterification of the 11-cis-ROL product (35, 36). The ester synthase in cone-dominant retinas uses palm CoA as an acyl donor, and therefore is a type of ARAT (35, 36). Cultured Muller cells take up all-trans-ROL and release 11-cis-ROL (83), suggesting that the retinol isomerase and coupled palm CoA-dependent ester synthase are located in Muller cells. These activities, plus the 11-cis-RDH in cones, appear to define catalytic steps of an alternate visual cycle in cone-dominant retinas (Figure 3). This proposed pathway may permit cones to escape competition from rods for visual chromophore under conditions of bright light. Confirmation of this pathway awaits molecular cloning of the enzymes for these three catalytic activities.
Figure 3.

Hypothesized alternate visual cycle for regeneration of cone pigments. Absorption of a photon (hv) induces 11-cis to all-trans isomerization of the retinaldehyde chromophore in a cone opsin pigment. After dissociation from apo-cone-opsin, the resulting all-trans-RAL is reduced to all-trans-ROL by the NADP+/NADPH-dependent all-trans-ROL dehydrogenase (all-trans-RDH) and released from the photoreceptor outer segment (OS), where it is bound to interstitial retinol binding protein (IRBP) (not shown). The all-trans-ROL released by rods and cones is taken up by Muller cells where it is isomerized to 11- cis-ROL by an isomerase, and esterified to form an 11-cis-retinyl ester by acyl-CoA:retinol acyltransferase (ARAT). These enzyme activities are kinetically coupled and named “isomerosynthase” (35, 36). An 11-cis-retinyl ester is hydrolyzed by 11-cis-retinyl ester hydrolase (REH) to yield 11-cis-ROL, which binds to cellular retinaldehyde-binding protein (CRALBP). 11-cis-REH is regulated by the ratio of apo- to holo-CRALBP in the retinal pigment epithelium (RPE) cells (60). A similar regulatory mechanism is suggested here in Muller cells. 11- cis-ROL released by the Muller cell is bound to IRBP (not shown) and taken up by the cone outer-segment, which contains an NADP+/NADPH-dependent 11-cis-ROL dehydrogenase (11-cis-RDH) activity (35). This activity oxidizes 11-cis-ROL to 11-cis-RAL, which combines with apo-cone-opsin to form a new cone visual pigment. The presence of reciprocal NADP+/NADPH-dependent all-trans-ROL and 11-cis-RDHs in cones affords a self-renewing supply of dinucleotide cofactor at all rates of photoisomerization.
EYE DISEASES ASSOCIATED WITH DEFECTIVE METABOLISM OF VISUAL RETINOIDS
Vision in vertebrates is dependent on a sustained supply of 11-cis-RAL chromophore. Accordingly, mutations in the genes for retinoid cycle proteins frequently cause impaired vision (Table 1). Most of these diseases are recessively inherited due to loss of the catalytic or retinoid-binding function. In contrast, diseases caused by mutations affecting structural or signaling proteins typically exhibit dominant inheritance (84). The phenotypic severity of retinoid cycle mutations is strongly influenced by the degree of functional redundancy at each step in the pathway. For example, the RPE contains multiple enzymes that catalyze oxidation of 11-cis-ROL to 11-cis-RAL (63), but only one enzyme (Rpe65) that catalyzes isomerization of all-trans-retinyl esters to 11-cis-ROL. Accordingly, mutations in RDH5 cause only a mild clinical phenotype, whereas mutations in RPE65 cause the severe blinding disease, LCA. Mutations in the RDH12 and LRAT genes also cause LCA (85–88). This phenomenon of phenotypic convergence is called nonallelic heterogeneity. In contrast, mutations in the same gene may produce a variety of clinical phenotypes. For example, different mutations in ABCR can cause Stargardt macular degeneration (6), cone-rod dystrophy (89), or recessive RP (90, 91). Also, heterozygous mutations in ABCR increase the risk of developing age-related macular degeneration (92). This phenomenon of phenotypic divergence is called allelic heterogeneity.
Table 1.
Human eye diseases associated with defective metabolism of visual retinoids
| Traditional name | Gene name and locus | Biochemical properties | Gene defects associated with eye diseases; inheritance | Phenotype | Ref. |
|---|---|---|---|---|---|
| ABCR, rim membrane protein (RMP), Stargardt disease protein (STGD1), ABC10, CORD3, RP19 | Retinal-specific ATP-binding cassette transporter (ABCR) member 4; ABCA4; 1p22.1-p21 | Multi-spanning membrane-bound protein found along the rim of disc in rods and cones, possess preferable activity toward N-retinylidene-phosphatidylethanolamine (N-ret-PE) removing all-trans-RAL from intradiscal space | 1. Stargardt disease (STGD), also known as fundus flavimaculatus (FFM); recessive | Juvenile- and late-age-onset macular dystrophy, alterations of the peripheral retina, and subretinal deposition of lipofuscin-like material | (6) |
| 2. Risk factor in age-related macular degeneration (controversial) | Age-related macular degeneration (ARMD) is a degenerative condition of the macula (the central retina). Age-related macular degeneration occurs in two forms: wet (occurs when abnormal blood vessels behind the retina start to grow under the macula) and dry (when cones of the macula degenerate) | (217) | |||
| 3. Autosomal recessive cone-rod dystrophy | A progressive retinal degenerative disease that causes deterioration of the cones and rods in the retina, eventually leading to blindness | (218) | |||
| 4. Autosomal recessive retinitis pigmentosa (RP) | RP refers to a group of inherited disorders that slowly lead to blindness owing to abnormalities of primarily the rods followed by cone death | (90, 219) | |||
|
| |||||
| RDH12, LCA3 gene | Retinol dehydrogenase (RDH) 12 (all-trans and 9-cis); RDH12; 14q24.1 | RDH12 is a microsomal short-chain alcohol dehydrogenase, expressed preferentially in the retina, that catalyzes the redox reaction with high preference toward NADP (NADPH), but displays dual-specificity toward both all-trans-and cis-ROLs. | Autosomal recessive childhood-onset severe retinal dystrophy | Severe, progressive rod-cone dystrophy with severe macular atrophy but no or mild hyperopia (farsightedness) | (116) |
|
| |||||
| LRAT; lecithin:retinol acyltransferase | Lecithin: retinol acyltransferase; LRAT; 4q32.1 | Single helix-anchorage membrane-bound ubiquitously expressed phosphatidylcholine–retinol O-acyltransferase with highest levels in the liver and RPE | Autosomal recessive early-onset severe retinal dystrophy (symptoms similar to or identical with Leber’s Congenital Amaurosis, congenital retinal blindness, hereditary epithelial aplasia, and retinal aplasia) | Congenital or early severe and progressive rod-cone dystrophy with progressive retinal atrophy, although in some cases vision can remain stable through young adult life, searching or pendular nystagmus—an involuntary, rhythmical, repeated movement of the eyes, photophobia, sluggish pupillary responses; the electroretinographic (ERG) response is characteristically “nondetectable” or severely subnormal; characteristic poking, rubbing, and/or pressing of the eyes (oculo-digital signs). Fundus may have a normal appearance in infancy and progressively pigmented later in life | (86) |
| Rpe65, Retinal pigment epithelium (RPE)-specific protein 65 kDa, LCA2, RP20; membrane-bound Rpe65 | RPE-specific protein 65 kDa; Rpe65; 1p31 | Membrane associated, RPE specific 65 kDa proteins, Rpe65 possesses Fe2+-dependent isomerization activity of all-trans-retinyl esters to 11-cis-ROL. Based on the sequence, Rpe65 belongs to carotenoid oxygenases. Rpe65 binds retinoids with a high affinity | Leber congenital amaurosis (LCA), recessive | Progressive visual loss, infant born blind or with highly attenuated vision, lose vision within a few months after birth, early-onset severe rod-cone dystrophy, in infants blindness in dark settings and light perception at bright light, attenuated retinal vessels and a variable amount of retinal pigmentation in older patients and a reduced or nondetectable electroretinogram at all ages | (88) |
| Autosomal recessive RP | Photoreceptor degeneration with good central vision within the first decade of life, attenuated retinal vessels and a variable amount of retinal pigmentation in older patients, and a reduced or nondetectable electroretinogram at all ages | (220) | |||
|
| |||||
| RGR, Retinal G protein– coupled receptor; RPE retinal G protein– coupled receptor | Retinal G protein–coupled receptor; RGR; 10q23 | RGR belongs to a superfamily of G protein–coupled receptors (heptahelical transmembrane receptor) is an invertebrate rhodopsin homologue found exclusively in the RPE and Muller cells. Possible proposed roles are as a retinal photoisomerase or in GPCR signaling in yet unidentified pathway | Autosomal dominant and recessive RP | Severely constricted visual fields, attenuated retinal vessels, diffuse depigmentation of the RPE, and intraretinal pigment deposits in the periphery. The depigmented patches involved the central macula associated with severely decreased acuity | (93) |
|
| |||||
| CRALBP, Cellular retinal-binding protein | Retinaldehyde binding protein 1, RLBP1; 15q26 | Abundant, soluble protein of the (RPE) and retinal Muller cells, where it binds 11-cis-ROL and 11-cis-RAL | Autosomal recessive retinitis punctata albescens, autosomal recessive RP Bothnia type | Bothnia dystrophy is a retinal dystrophy with striking similarity to the rod-cone dystrophies and has a high prevalence in northern Sweden. Retinitis punctata albescens is clinically similar to RP and results in progressive visual field loss, night-blindness, and retinal vascular attenuation | (136, 137) |
|
| |||||
| RDH5; 11-cis-ROL dehydrogenase. | Retinol dehydrogenase (RDH) 5 (11-cis and 9-cis); RDH5; 12q13-q14 | RDH5 is a microsomal short-chain alcohol dehydrogenase, ubiquitously expressed with a high expression in the RPE. The enzyme catalyzes the redox reaction with the highest preference toward NAD (NADH), and 11-cis- and 9-cis-ROL | Autosomal recessive fundus albipunctatus | This form of fleck retinal disease is characterized by discrete uniform white dots over the entire fundus with greatest density in the midperiphery and no macular involvement. Night blindness (delay dark adaptation) occurs with or without cone dystrophy. Rod and cone resensitization was extremely delayed following full bleaches; the rate of cone recovery was slower than rods | (141) |
A powerful approach to understanding the etiologies of retinoid cycle diseases has been the creation and characterization of animal models (Table 2). These animal models are also useful for developing genetic and pharmacologic treatments. However, phenotypic differences exist between animals and humans carrying mutations in the same gene. For example, loss-of-function mutations in the human RPE retinal G protein–coupled receptor (RGR) gene cause RP leading to photoreceptor degeneration and blindness (93), whereas rgr−/− knockout mice show no signs of photoreceptor degeneration (94, 95). A similar situation exists with recessive Stargardt macular degeneration, which causes visual loss in children (96), and abcr−/− knockout mice, where photoreceptors degenerate only very slowly (97). These apparent differences in phenotypic severity may reflect the much longer lifespan of humans. At the optical center of the human retina is a structure called the macula, which contains a very high density of photoreceptors. Rods and cones within the macula are unusually vulnerable to cell death in a group of blinding diseases called the macular degenerations. Photoreceptors of the peripheral retina are affected late in the course of several macular degenerations. The retinas of most non-primate animals, including mice, do not contain a macula. The entire retina of these animals histologically resembles the peripheral retina of humans. The absence of a central macula is another explanation for the milder phenotypes in animals with mutations in retinoid cycle genes. Despite these differences, animal models with engineered mutations in visual cycle genes are valuable for studying the function of the encoded protein and for developing new treatments.
Table 2.
Animal models of human eye diseases associated with defective metabolism of visual retinoids
| Gene Name | Animal Model | Phenotype | Ref. |
|---|---|---|---|
| Retinal-specific ATP-binding cassette transporter member 4 (ABCA4) | abcr−/− mice | The two lines of abcr−/− mice exhibit accumulation of high levels of lipofuscin within the RPE cells; increased levels of retinal in the outer segment (OS) and delayed dark adaptation | (9, 210) |
|
| |||
| Lecithin: retinal acyltransferase (LRAT) | lrat−/− mice | The lrat−/− mice have no detectable levels of retinyl esters within the RPE and show severely attenuated rod and cone visual responses. Other tissues such as liver, kidney, and lung have trace levels of retinyl esters, but there are increased levels of retinyl esters in adipose tissue. A second line of lrat−/− mice that have the Neo cassette removed following recombination show a similar phenotype | (39, 40, 221) |
|
| |||
| RPE-specific protein 65 kDa; Rpe65 | rpe65−/− mice | The rpe65−/− mice show severely attenuated rod and cone responses, progressive retinal degeneration owing to block in regeneration of 11-cis-RAL, consistent with the role of Rpe65 in the isomerization of all-trans-ROL. High levels of all-trans-retinyl esters accumulate in lipid vacuoles in the RPE of rpe65−/− mice | (88) |
| rd12 mice | The rd12 mice have a naturally occurring nonsense mutation in exon 3 of murine Rpe65. There are no detectable levels of 11-cis-RAL or rhodopsin and lipid droplets accumulate in the RPE. At 5 months of age, small white dots appear on the retinas of rd12 mice | (123) | |
| Swedish Briard/Briard Beagle dogs with naturally occurring mutation | A homozygous 4bp deletion in exon 5 of canine Rpe65 leads to a frameshift mutation and truncation of the resulting polypeptide. These dogs exhibit early onset, progressive retinal dystrophy and accumulation of lipid vacuoles in their RPE | (105, 106) | |
|
| |||
| Retinal G protein–coupled receptor; RG | rgr−/− mice | The rgr−/− mice have reduced levels of 11-cis-RAL and rhodopsin, and slightly reduced electroretinographic (ERG) responses. The rates of rhodopsin regeneration are similar to WT mice | (94, 95) |
|
| |||
| Retinal-binding protein 1, RLBP1 | rlbp1−/− mice | rlbp1−/− mice have delayed rates of rhodopsin regeneration and recovery of chromophore following a bleach and accumulation of retinyl esters | (140) |
|
| |||
| 11-cis-ROL dehydrogenase (RDH); RDH5 | rdh5−/− mice | The rdh5−/− mice exhibit accumulation of cis-retinyl esters in their RPE. In contrast to the human disease caused by mutations in RDH5, fundus albipunctatus, the rdh5−/− mice do not exhibit delayed dark adaptation or the formation of the characteristic white dots | (144) |
RP Caused by Mutations in the Gene for Rhodopsin
Mutations in the gene for rhodopsin are the most common cause of RP and lead to both autosomal dominant RP (ADRP) and autosomal recessive RP (ARRP). Mutant rhodopsin can cause disease through several different pathologic mechanisms, such as cellular toxicity owing to improperly folded or constitutively activated protein in ADRP, or impaired activation or reduced opsin level leading to ARRP. Mice with a knockout mutation in the rod opsin gene fail to form rod OS and have no rod electroretinographic (ERG) response, but show a cone response early in life (98). Cone photoreceptors disappear at three months of age. Heterozygous opsin+/− mice have 40% less rhodopsin and consequently a 40% reduction in 11-cis-RAL (99).
Supplementation with natural or synthetic retinoids may slow photoreceptor degeneration owing to opsin mutations (103). A naturally occurring mutation in dogs causing a Thr4Arg rhodopsin substitution leads to autosomal dominantly inherited retinal degeneration (104). The degeneration resembles that of the Thr4Lys rhodopsin substitution that affects some ADRP patients. Retinal degeneration in dogs with Thr4Arg-substituted rhodopsin is due to its constitutive activation. This mechanism was demonstrated by crossing dogs carrying Thr4Arg-substituted rhodopsin with Briard dogs with a null mutation in the gene for Rpe65 (105, 106). These double heterozygous-mutant dogs exhibited greatly accelerated degeneration (107). High levels of apo-opsin owing to loss of the Rpe65-isomerase, combined with the presence of Thr4Arg-substituted rhodopsin, caused severe overstimulation of the transduction cascade and rapid photoreceptor death. Retinoid supplementation may be a viable approach to slow photoreceptor degeneration in human diseases associated with critical apo-opsin accumulation, as discussed below.
Stargardt Macular Degeneration
Autosomal recessive Stargardt disease is the most common inherited maculopathy of the young, with an estimated prevalence of 1:10,000 (108). The first symptoms of Stargardt disease are blurred vision not correctable with glasses, and delayed dark adaptation. Later, Stargardt disease causes progressive loss of central vision with central blind spots and a diminished ability to perceive colors. Ultimately, the disease Funduscopic examination of the retina in Stargardt patients shows a macular lesion surrounded by irregular yellow-white flecks or spots, which give rise to the alternate name for this disease, fundus flavimaculatus. Histologically, Stargardt disease is characterized by the accumulation of fluorescent lipofuscin pigments in cells of the RPE, degeneration of the RPE, and death of photoreceptors (96, 110). Stargardt disease patients commonly show a “dark choroid” during fluorescein angiography due to this light-absorbing lipofuscin material in the RPE (111). Stargardt disease patients also show fundus autofluorescence by scanning laser ophthalmoscopy, again owing to these lipofuscin pigments (112). Stargardt disease is caused by mutations in the ABCR (ABCA4) gene (6). Approximately 400 alleles of ABCR have been identified in patients with inherited macular and retinal degenerations. ABCR is also highly polymorphic in the normal population. This heterogeneity presents a challenge to define disease-causing alleles. Biochemical analysis of postmortem samples from Stargardt disease patients showed elevated PE and N-ret-PE in the retina, and elevated lipofuscin fluorophores such as A2PE-H2 and A2E in the RPE (10).
The abcr−/− knockout mouse is an animal model for Stargardt disease and ABCR-mediated retinal degenerations (9). Several features of the Stargardt disease phenotype are present in these mice. For example, PE and N-ret-PE are elevated in abcr−/− retinas (9, 10). Clearance of all-trans-RAL from OS is delayed in abcr−/− mice following light exposure. Accumulated lipofuscin pigments are visible by electron microscopy in the RPE of abcr−/− mice. The lipofuscin fluorophores, A2E and A2PE-H2, are also dramatically elevated in RPE (9, 10). Physiologically, abcr−/− mice showed delayed recovery of rod sensitivity following light exposure (9), again similar to patients with Stargardt disease. Loss of ABCR activity causes increased N-ret-PE and all-trans-RAL in OS discs. This initial reaction favors secondary condensation of N-ret-PE with all-trans-RAL to yield A2PE-H2. A2PE-H2 in OS is probably not toxic to photoreceptors; however, the distal ~10% of photoreceptor OS are phagocytosed daily by the RPE as part of the normal disc-renewal process (29). A2PE-H2 is converted to A2E and A2E-epoxides upon exposure to short-wavelength light in the acidic and oxidizing conditions of RPE phagolysosomes (10, 113, 114).
LCA Due to Mutations in the Gene for all-trans-RDH
All-trans-RAL is reduced to less-toxic all-trans-ROL by members of the short-chain alcohol dehydrogenases, including RDH8 and RDH12. Recently, mutations in RDH12 were shown to cause LCA (115, 116). No mutation in RDH8 has been associated with a retinal dystrophy in humans. Mice with a knockout mutation in the rdh8 gene show normal kinetics of rhodopsin regeneration and delayed recovery of sensitivity following exposure to bright light (16). An identical pattern is seen in abcr−/− mice (9). ABCR and all-trans-RDH act sequentially in the visual cycle to remove all-trans-RAL following a photobleach (Figure 2). Delayed dark adaptation in rdh8−/− and abcr−/− mice is probably due to noncovalent association of all-trans-RAL with opsin to form a “noisy” photoproduct that activates transducin (117).
Mutations in the LRAT Gene Cause Early LCA
Loss-of-function mutations in the LRAT gene have been reported in three individuals with LCA (86). These patients are otherwise healthy, suggesting that LRAT is not required for the uptake or storage of all-trans-ROL in extraocular tissues when the intake of dietary vitamin A is normal. The lrat−/− knockout mouse is an animal model for LRAT-mediated retinal dystrophy. These animals contain only trace retinoids in ocular tissues (40). Accordingly, vision is extremely impaired from birth. OS are shortened in lrat−/− mice, and photoreceptors degenerate very slowly.
Mutations in RPE65 Cause LCA and Severe Recessive RP
Mutations in the gene for Rpe65 account for a significant percentage (11.4%) of early-onset retinal degenerations in humans (118). Numerous loss-of-function alleles in RPE65 have been described. Rod and cone function is absent or severely compromised at birth, as evidenced by extinguished or barely detectable responses by ERG.
The rpe65−/− mouse was the first animal model of a human disease caused by mutations in a retinoid cycle gene (48). These mice show massive accumulation of all-trans-retinyl esters and absent 11-cis-retinoids in ocular tissue, consistent with the role of Rpe65 as a retinoid isomerase (45–47). A lack of 11-cis-RAL chromophore leads to the virtual absence of rhodopsin and a nearly undetectable ERG response. Young rpe65−/− mice have a relatively normal retinal anatomy (48). The OSs of these mice contain a large amount of apo-opsin, which helps stabilize the structure of OS discs, in contrast to opsin−/− mice with severe OS disorganization (98). Age-dependent photoreceptor degeneration is seen in rpe65−/− mice, with the loss of cones preceding the loss of rods (119). This degeneration may be due to constitutive activation of the visual transduction cascade with a simultaneous reduction in intracellular Ca2+. In contrast to rhodopsin, which is “silent,” chromophore-free apo-opsin weakly activates transducin (120). This weak activation becomes significant in an OS populated largely or exclusively by apo-opsin (121). With prolonged dark-rearing, rpe65−/−mice gradually acquire light-sensitivity as assessed by ERG. This transient recovery of vision, which only survives one round of photoisomerization, is due to the formation of iso-rhodopsin (122). Iso-rhodopsin contains 9-cis-RAL chromophore instead of 11-cis-RAL. The origin of this 9-cis-RAL is unknown. It may represent a precursor of 9-cis-retinoic acid, a putative transcriptional regulatory molecule, which is trapped upon combination with apo-opsin.
Other animal models of RPE65-mediated retinal degenerations include the Swedish Briard dog (105, 106) and the rd12 mouse (123). Both spontaneous mutations result in premature truncation of the Rpe65 protein and cause severely reduced visual function. Interestingly, rd12 mice have small white dots throughout their retinas, similar to humans with fundus albipunctatus (123). The Briard dog and rd12 mouse phenotypes have been successfully rescued by gene therapy involving the viral-mediated transfer of a WT RPE65 gene to RPE cells (124–126).
Dominant and Recessive RP Due to Mutations in the RGR Gene
RGR is a non-photoreceptor opsin in RPE and Muller cells (127, 128). The function of RGR is unknown. In the dark, RGR preferentially binds all-trans-RAL, which forms a Schiff base with a lysine residue in the seventh transmembrane helix, similar to rhodopsin (129). Light exposure induces photoconversion of this bound all-trans-RAL to 11-cis-RAL (129). This photoisomerization has led to the suggestion that RGR functions as a “reverse” photoisomerase to regenerate visual chromophore in light, similar to retinochrome in the squid (130). Evidence against this proposed function for RGR is the observation of similar ERGs and 11-cis-RAL levels in rpe65−/−single and rpe65−/−, rgr−/− double knockout mice (95). More recently, RGR was suggested to activate the isomerase in a light-independent fashion (131), although no direct effect of RGR on isomerase activity was shown. Frame-shift mutations in the human RGR gene are associated with ADRP (93). Homozygous and compound-heterozygous mutations in RGR additionally have been associated with ARRP in a consanguineous Spanish family (132). Dominant and recessive inheritance of a retinal disease phenotype with mutations in the same gene have also been described for opsin (133, 134) and ABCR (6, 92).
The rgr−/− knockout mouse represents an animal model of RGR-mediated ARRP. The phenotype in rgr−/− mice is mild (94). These mice show reduced 11-cis-RAL and rhodopsin levels in the retina following light exposure, and increased all-trans-and 13-cis-retinyl esters in the RPE (94, 95, 135). Slightly decreased ERG responses are also seen, commensurate with the decreased levels of rhodopsin. Further study is required to elucidate the function of RGR and to explain the phenotype in humans with RGR-mediated RP.
Retinitis Punctata Albescens Caused by Mutations in the RLBP1 Gene Coding for CRALBP
Mutations in RLBP1 are responsible for a form of recessive RP called retinitis punctata albescens, characterized by subretinal punctate white-yellow deposits found during funduscopic exams (136, 137). Clinically, retinitis punctata albescens caused by mutations in RLBP1 is associated with night blindness and progressively impaired visual acuity during childhood (138). Although the rod response is undetectable by routine ERG, it reverts to normal following prolonged dark adaptation (139). Reductions in cone function are seen in older patients owing to photoreceptor degeneration.
Mice with a knockout mutation in the rlbp1 gene synthesize 11-cis-RAL chromophore very slowly and show massively delayed recovery of rod sensitivity following light exposure (140), consistent with the clinical phenotype in humans. As discussed above, binding of 11-cis-ROL by CRALBP is required to avoid product-inhibition of the isomerase (53, 54). In the absence of CRALBP, synthesis of 11-cis-ROL, and hence 11-cis-RAL chromophore, is greatly slowed. Accumulation of all-trans-retinyl esters is also seen in rlbp1−/− mice due to reduced consumption of substrate by the isomerase (140). Contrary to humans with RLBP1 mutations, photoreceptor degeneration is not seen in the rlbp1−/− mice.
Fundus Albipunctatus Caused by Mutations in RDH5
Fundus albipunctatus is a mild and rare form of congenital stationary night-blindness with a characteristic appearance on funduscopic exam of white dots scattered throughout the retina. Visual acuity is normal in patients with fundus albipunctatus, and the condition is only mildly progressive. This phenotype suggests a slowing of chromophore synthesis with no photoreceptor degeneration. Fundus albipunctatus is a recessive disease caused by mutations in the RDH5 gene for an 11-cis-RDH in RPE cells (141, 142).
Mice with a knockout mutation in the rdh5 gene also have a mild phenotype. Dark adaptation is normal in rdh5−/− mice and white spots are not visible on the retinas (62, 143). Thus, RDH5 represents a minor contributor to total 11-cis-RDH activity in humans and mice. The most prominent feature of the rdh5−/− phenotype is the accumulation of 13-cis-retinyl esters in the RPE (62). This accumulation may be due to low activity of the remaining 11-cis-RDHs toward 13-cis-ROL, followed by esterification of this aberrant substrate into 13-cis-retinyl esters by LRAT. In humans, the white dots may represent local accumulations of 11- and 13-cis-retinyl esters (144).
THERAPEUTIC STRATEGIES FOR DISEASES CAUSED BY DEFECTS IN THE VISUAL CYCLE
Three strategies to treat diseases caused by mutations in retinoid cycle genes have been explored. The first and conceptually simplest strategy is to replace the defective gene by viral gene therapy. RPE cells take up and express recombinant viruses at high efficiency, which represents an important advantage of this approach. The second strategy involves pharmacologic replacement of missing chromophore. This strategy may be applicable to diseases with a primary deficit in chromophore biosynthesis. Examples include LCA owing to mutations in the LRAT and RPE65 genes. The third strategy is to slow the synthesis of chromophore by inhibiting steps in the visual cycle or limiting availability of all-trans-ROL precursor. This strategy is applicable to diseases associated with accumulation of toxic lipofuscin fluorophores such as A2E. As discussed below, A2E accumulation is seen in multiple forms of retinal and macular degenerations.
Gene Therapy Approach
The major goal of gene therapy is to incorporate a functional gene into a target cell, restoring production of the affected protein. Considerable progress has been made in developing efficient viral delivery systems and overcoming problems with undesirable immune responses (for reviews see References 145, 146). Successful gene therapy is dependent on efficient transduction of the target cell and sustained expression of the recombinant virus at a sufficient level. Adeno-associated virus (AAV), a nonpathogenic parvovirus, has been the most successful vector owing to its ability to transduce a variety of non-dividing cell types. AAV vectors contain no viral coding regions and therefore have low cytotoxicity. AAV has been employed for gene delivery to muscle, brain, liver, lung, RPE, and neural retina. Disadvantages of AAV vectors include a small cargo size (<5 kb) and the immune reaction elicited by the viral capsid, which limits the number of possible recurrent treatments (147, 148). The immune-privileged status of the eye helps with this problem.
An initial study of viral gene-transfer into the eye was performed on transgenic mice and rats expressing Pro23His-substituted rhodopsin. Viral-mediated expression of ribozymes that targeted the transgene mRNA reduced expression of the mutant rhodopsin and slowed the rate of photoreceptor degeneration (149, 150). Following this initial success, gene therapy was employed for treating RPE65-mediated LCA in the Briard dog (105). These dogs suffer from early and severe visual impairment, similar to humans with LCA. AAV vectors carrying WT human or canine RPE65 were delivered subretinally to rpe65-mutant Briard dogs (124, 126, 151, 152). ERG measurements showed dramatic improvements in the light sensitivity of rods and cones. Immunohistochemical analysis showed expression of Rpe65 in RPE cells surrounding the site of subretinal injection. Retinoid analysis confirmed that rescue of Rpe65 expression leads to production of significant 11-cis-RAL in the treated eyes. Importantly, expression of Rpe65 and restoration of vision in the treated dogs was stable over the four-year study period. Finally, the rescued photoreceptors in these dogs were protected from the degeneration normally associated with LCA. According to the latest studies, more than 50 Briard dogs have been treated by gene therapy, with 95% showing restored vision (153). Similar results were seen with gene therapy of rpe65−/− mice, although here the restoration of vision only lasted a few months (154). The AAV-mediated gene therapy of LCA caused by RPE65 mutations is ready for clinical studies, having completed both proof-of-concept and biosafety studies in dogs, mice, and monkeys. The first LCA patients for the Phase I trials are expected to be enrolled during 2006 (153).
More recently, lrat−/− knockout mice were treated with recombinant AAV carrying a WT mouse lrat cDNA (155). These animals showed increased light sensitivity by ERG. Expression of exogenous LRAT was detected in the RPE cell layer near the injection site. Restoration of the retinoid cycle was detected by the appearance of 11-cis-RAL and a significant amount of regenerated rhodopsin (50% of WT) in the treated eye. Additionally, rescue of pupillary response in treated mice demonstrated intact neural signaling to the brain. These mice were also recipients of combined therapy involving gene therapy and chromophore supplementation by oral administration. Mice receiving both AAV-lrat gene therapy and 9-cis-retinyl acetate supplementation showed greater functional improvement than mice that received gene therapy alone. Such a combined-treatment approach might be applicable to humans.
Chromophore Supplementation Approach
The first experiments aimed at bypassing the biochemical defect caused by the absence of Rpe65 were performed by oral gavage of rpe65−/− mice with 9-cis-RAL (156, 157). 9-cis-RAL combines with opsin to form light-sensitive iso-rhodopsin (158). 9-cis-RAL was selected for the initial studies because of its lower cost and higher stability compared with 11-cis-RAL. Moreover, iso-rhodopsin has an absorbance maximum of 494 nm versus 502 nm for rhodopsin. This permits identification of the reconstituted iso-rhodopsin experimentally (122). Dietary supplementation of rpe65−/− mice with 9-cis-RAL restored light sensitivity to levels found in WT animals, as assayed by single-cell recordings and ERG. The amplitude of the saturating response increased with the number of doses. The improvement of rod functions has been observed up to six months after treatment (Figure 4a). Simultaneously, administration of 9-cis-RAL helped to preserve the morphology of the retina by improving interface contact between RPE and rod OS. Treatment with 9-cis-RAL decreased the content of apo-opsin in rpe65−/− mice, reducing constitutive activation of the phototransduction cascade, which led to slower photoreceptor degeneration. Similar recovery of visual function was observed following intraperitoneal injection of 11-cis-RAL into rpe65−/−mice (159) (see side bars Discovery of Vitamin A and Function of Vitamin A for additional information).
Figure 4.
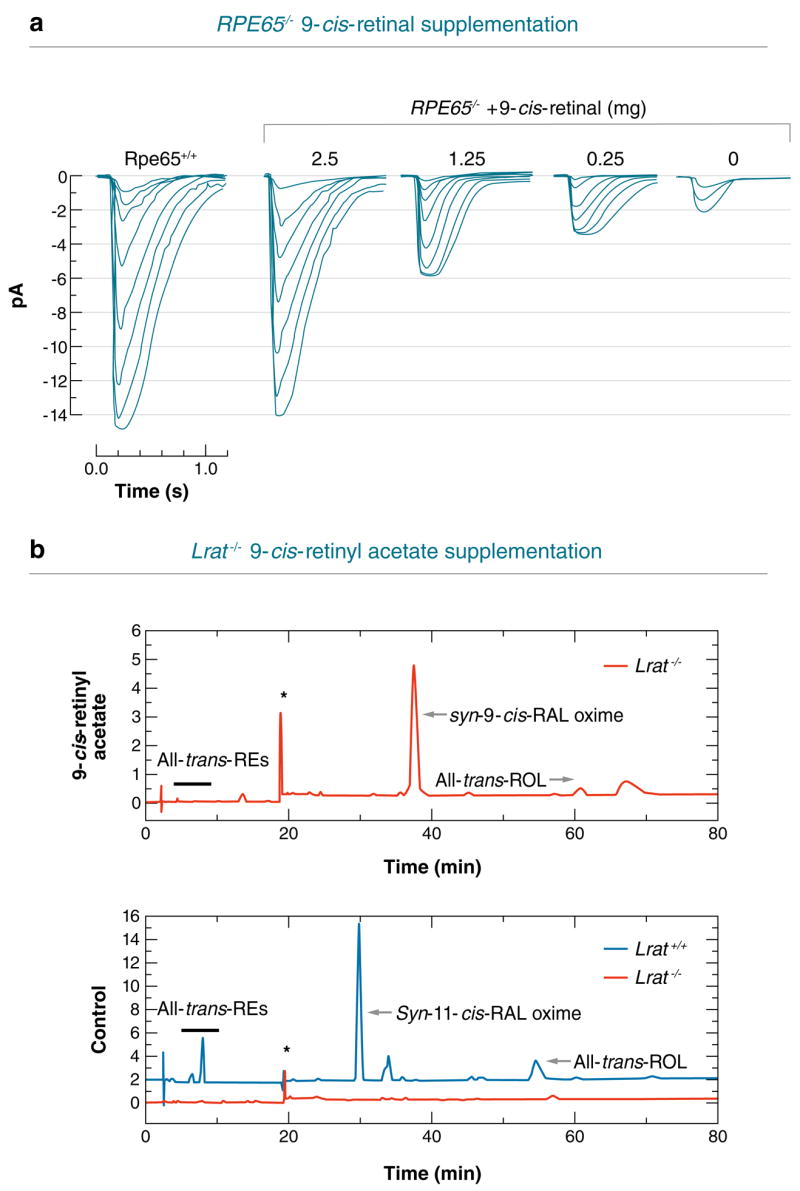
Effects of chromophore supplementation and gene therapy on vision in rpe65−/− and lrat−/−knockout mice. (a) Single-cell recording stimulus response families of rpe−/− mice supplemented with 9-cis-RAL compared with rpe+/+ mice. (b) High performance liquid chromatography (HPLC) analysis of retinoids present in the eyes of lrat−/− mice treated with 9-cis-retinyl acetate. The presented results were reproduced from References 155 and 157.
The therapeutic success of 9-cis- and 11-cis-RAL supplementation in rpe65−/−mice suggests that this approach may be useful to treat LCA caused by mutations affecting other visual cycle proteins such as LRAT. 9-cis-retinyl esters, 9-cis-RAL, and 9-cis-ROL were all effective in the rescue of visual pigments in lrat−/− mice by oral gavage (155) (Figure 4b). The 9-cis-retinyl esters, 9-cis-retinyl succinate and 9-cis-retinyl acetate are better suited for oral administration owing to their stability and low reactivity compared with 9-cis-RAL. Although light sensitivity was eightfold lower in treated lrat−/− versus WT mice, the response increased with multiple doses. This observation suggests that in the absence of LRAT, the 9-cis-chromophore may be recycled following phagocytosis of iso-rhodopsin containing OS by the RPE cells. The visual pigment was restored in lrat−/− mice 4–5 h after gavage and it was retained at the level of 50% of the initial amount for 120 days after a single dose. The formation of 9-cis-retinyl palmitate in the liver of 9-cis-retinyl acetate-gavaged lrat−/−mice points to the existence of alternate pathways of formation of retinyl esters such as ARAT or a retinyl trans-esterase (34, 37, 39).
DISCOVERY OF VITAMIN A
The effect of vitamin A on cures of dietary deficiency diseases has been known since ancient times. Since that time, the recommended treatment of the first symptoms of vitamin A deficiency, known as night-blindness, involved “continuous eating of… liver of goats.” The scientific approach that ultimately led to the discovery of vitamin A was given by E.V. McCollum. In 1913, performing nutrition experiments, he found that young rats on a diet of pure milk sugar, minerals, and olive oil failed to grow, while addition of butter fat or egg yolk extract restored their health. He proved the existence of the fat-soluble factor (then called fat-soluble factor A, as opposed to other dietary factors water-soluble B) by adding either butter extract to the olive oil, which became sufficient to support animal growth. In attempts to isolate and identify the factor, T.B. Osborne and L.B. Mendel, as well as H. Steenbock, confirmed the biological activity of the yellowish butter, egg yolk, and carrot extracts. However, they noticed that colorless liver and kidney extracts exhibited similar properties of growth promotion. This observation led to the hypothesis that fat-soluble factor A was only associated with yellow pigment and was being converted into an active colorless form. Ten years later, in 1930, T. Moore showed conversion of the yellow pigment (carotene) into colorless (retinol) by feeding rats with crystalline carotene and examining of their livers. He concluded that carotene was the precursor of vitamin A. At the same time, P. Karrer et al. isolated and determined the chemical structure of carotene and retinol. In 1947, O. Isler et al. performed the first total synthesis of retinol.
High doses of retinoids have been shown to be toxic in numerous studies (for a review, see Reference 160). One mode of retinoid toxicity results from the oxidation of retinol or retinaldehyde to retinoic acid by retinal dehydrogenase types 1, 2, 3, and 4 (161). All-trans-retinoic acid and its 9-cis isomer are important regulators of gene expression via the RAR and RXR nuclear receptors (162). Stimulation of these receptors leads to undesirable effects, including teratogenicity. However, acute and prolonged treatment of mice with 9-cis-retinoids did not cause obvious adverse effects such as reduced litter size, abnormal development, or growth retardation. This lack of apparent toxicity can be explained by the low levels of all-trans-retinoic acid detected in lrat−/− mice after administration of 9-cis-retinyl esters. The increased all-trans-retinoic acid levels observed after treatment usually disappeared within a day. Resistance to the potential toxicity of all-trans-retinoic acid seems to be related to inefficient esterification (storage) of the retinoids, leading to their rapid removal by oxidation and secretion. Although more toxicological studies are needed, the above described observation raises the hope for a trial of chromophore supplementation in humans.
FUNCTIONS OF VITAMIN A
Early observation based on the physiological response to a depletion of β-carotene or retinol pointed to three important functions of vitamin A in the processes of vision, epithelial differentiation, and growth. L.S. Fridericia & E. Holm scientifically confirmed the visual functions of vitamin A in 1925 by showing slower rates of visual purple regeneration in vitamin A–deficient rats. However, it was G. Wald who found that visual purple of the retina, called rhodopsin, contains the chromophore “retinene” (later identified by R. A. Morton to be retinaldehyde). Wald was able to show that one 11-cis isomer of retinaldehyde is preferentially bound to opsin and serves as a light acceptor undergoing light isomerization to the all-trans isomer. He also characterized molecular components of the visual cycle that leads to regeneration of 11-cis-RAL. Wald’s exceptional impact on understanding of the molecular basis of vitamin A action was awarded the Nobel Prize in Physiology and Medicine in 1967. Parallel to vision, S. B. Wolbach & P. R. Howe first reported the influence of vitamin A on epithelial cell differentiation in 1925. Moreover, the pathology of a vitamin A deficiency is manifested by reduction of bone growth and male and female reproduction. In the 1950s and 1960s, identification of plasma and cellular retinoid binding proteins by D.S. Goodman, F. Chytil & D.E. Ong had a great impact on the understanding of vitamin A transport and metabolism. The real breakthrough came with the discovery of the retinoic acid receptor in cell nuclei by P. Chambon & R.M. Evans in 1987. Activation or inhibition of specific gene transcription by the retinoic acid receptors explains at the molecular level many metabolic functions of vitamin A with regards to embryonic development, differentiation, and growth.
Retinoid Inhibitors of A2E Formation
Accumulation of fluorescent lipofuscin pigments in cells of the RPE is an important pathological feature of Stargardt disease (96, 110). These pigments are responsible for the “dark choroid” seen during fluorescein angiography (111) and the fundus autofluorescence seen by scanning laser ophthalmoscopy (112). The maximum emission wavelength of lipofuscin fluorescence in Stargardt patients is 650 nm (163), which corresponds to the emission maximum of A2PE-H2, an abundant A2E precursor in abcr−/− mice (10, 97). The cytotoxicity of A2E in RPE cells is well established. A2E has been shown to sensitize RPE cells to blue-light damage (164–166), impair the degradation of phospholipids from phagocytosed OS (167), induce the release of pro-apoptotic proteins from the mitochondria (168, 169), and destabilize cellular membranes through its properties as a cationic detergent (170–172). Further, irradiation of A2E with blue (430 nm) light resulted in a series of oxirane products containing up to nine epoxide rings, formed by the addition of singlet oxygen to double bonds along the polyene chains (113). A2E oxiranes were shown to induce DNA fragmentation by forming adducts with purines and pyrimidines in cultured ARPE-19 cells (173, 174), representing still another mechanism of A2E cytotoxicity. As expected, lipofuscin accumulation, indicated by fundus autofluorescence, precedes macular degeneration and visual loss in Stargardt patients (175). Hence, the likely sequence for photoreceptor degeneration in Stargardt disease is (a) lipofuscin accumulates in cells of the RPE; (b) RPE cells begin to function abnormally and ultimately degenerate owing to A2E-mediated cytotoxicity; and (c) the photoreceptors die secondary to loss of the RPE support-role (176), resulting in blindness. In Stargardt disease, the greatest concentration of lipofuscin is seen in RPE cells overlying the perifoveal region of the macula (96). The macula is located at the optical center of the retina and contains the highest density of photoreceptors. The vulnerability of the macula in Stargardt and other macular degenerations is due to the increased ratio of photoreceptors to RPE cells (177) and high incident light in this region.
Lipofuscin accumulation in the RPE is not limited to Stargardt disease. Increased fundus autofluorescence by scanning laser ophthalmoscopy is commonly seen in patients with age-related macular degeneration (AMD) (178–180). The fluorescent material that accumulates in the aged RPE has spectral properties similar to A2PE-H2, observed biochemically in abcr−/− mice (10, 181). Strong fundus autofluorescence is also seen in patients with Best vitelliform macular dystrophy and a subset of patients with cone-rod dystrophy (182). Patients with dominant Stargardt disease, caused by mutations in the ELOVL4 gene, show a dark choroid by fluorescein angiography, again owing to lipofuscin in RPE cells (183, 184). In contrast, patients with LCA owing to mutations in the RPE65 gene show undetectable fundus autofluorescence (185), consistent with the role of Rpe65 as a retinoid isomerase (45–47) and the virtual absence of chromophore and lipofuscin in rpe65−/− mutants (48, 222). Lipofuscin accumulation in RPE cells has been observed in multiple animal models of inherited retinal and macular degeneration besides the abcr−/− mouse. For example, mice with a knockout mutation in the rdh8 gene, encoding an all-trans-RDH in photoreceptors, contain several-fold higher levels of A2E compared with WT eyes (16). Transgenic mice with a mutation in the elovl4 gene contain very high levels of A2E in the RPE (184). The Royal College of Surgeons (RCS) rat, with a mutation in the mertk gene for a receptor tyrosine kinase required for phagocytosis of shed OS (186), also accumulates A2E and its precursors in the RPE (187, 188). Humans with MERTK mutations have the inherited blinding disease, RP (189). Transgenic mice that express a mutant form of cathepsin D (mcd) in RPE cells manifest many features of AMD including photoreceptor degeneration, basal laminar and basal linear deposits, and autofluorescent lipofuscin pigments in the RPE (190, 191). Mice with knockout mutations in the genes for monocyte chemoattractant protein-1 (Ccl-2), or its cognate chemokine receptor-2 (Ccr-2), also show features of AMD, including photoreceptor degeneration, basal deposits, thickening of Bruch’s membrane, choroidal neovascularization, and A2E accumulation in RPE cells (192).
It is easy to understand why mutations in the abcr or rdh8 genes lead to elevated A2E. Both genetic defects cause delayed clearance of all-trans-RAL (9, 16), the primary reactant in A2E biogenesis (193). But how can we explain A2E accumulation in retinal degenerations caused by mutations in the genes for proteins with no role in retinoid processing? Examples include genes for the Ca2+-activated Cl− channel, bestrophin (194); the putative fatty-acid elongation factor, ELOVL4 (184); the receptor kinase, MERTK (186); the lysosomal aspartate proteinase, cathepsin D (190, 191); and the monocyte chemoattractant protein plus its receptor, Ccl-2 and Ccr-2 (192). The answer to this question may be found in an unusual interaction between photoreceptors and RPE cells. Each morning, with the onset of daylight, the distal 10% of rod and cone OS break off and are phagocytosed by the overlying RPE (29). This means that nearly 10% of the total ocular retinoid pool passes daily through the RPE phagolysosomal system. One mouse eye contains ~500 pmol of rhodopsin (114, 195), thus ~50 pmol retinal transits through RPE phagolysosomes per day. However, the total content of A2E in a five-month-old WT mouse is less than 2 pmol per eye (9). Thus, only a tiny fraction of retinaldehyde phagocytosed by the RPE normally accumulates as A2E. The RPE must therefore be very efficient at blocking net synthesis of this toxic fluorophore. The RPE becomes progressively less efficient at eliminating or preventing the formation of A2E after being compromised by a genetic, immunologic, or environmental insult. Given its cytotoxicity, A2E accumulation is probably a self-accelerating process.
Accumulation of A2E with subsequent poisoning of the RPE appears to be an etiologic factor in retinal and macular degenerations of multiple causes. Inhibiting A2E formation should therefore slow the progression of visual loss in diseases associated with lipofuscin autofluorescence. Several approaches have been tested or proposed to slow accumulation of A2E. All have an effect on visual-cycle retinoids and ultimately limit the formation of all-trans-RAL (Figure 5). The first approach was to prevent photoisomerization of 11-cis-RAL by raising abcr−/− mice in total darkness. After 18 weeks, levels of A2E in WT and abcr−/− mice were similar, while A2E levels were 10-fold higher in the eyes of abcr−/− mice raised under cyclic light (10). Also, when abcr−/− mice were reared under cyclic light for 12 weeks, then transferred to total darkness for an additional 16 weeks, A2E levels in these 28-week-old mice were identical to the levels in 12-week-old abcr−/− mice reared under cyclic light (10). These environmental manipulations established that A2E synthesis could be blocked by inhibiting formation of all-trans-RAL. Levels of all-trans-RAL are increased only about two-fold in abcr−/− versus WT retinas following light exposure despite massive accumulation of A2E (9, 10). Thus, only a modest decrease in all-trans-RAL levels is required to slow dramatically the rate of A2E accumulation.
Figure 5.

Simplified visual cycle showing sites of action for the inhibitors of N-retinylidene-N-retinyl ethanolamine (A2E) formation. All-trans-RAL released by photoactivated rhodopsin condenses with phosphatidylethanolamine (PE) in disc membranes to form N-retinylidene-PE (N-ret-PE). Secondary condensation of N-ret-PE with another all-trans-RAL, followed by electrocyclization, hydrolysis of the phosphate ester, and oxidation yields A2E. Pharmacologic interventions that reduce light-dependent formation of all-trans-RAL greatly reduce the probability of this secondary condensation and inhibit formation of A2E. N-(4-hydroxyphenyl)retinamide (HPR) acts to reduce levels of holo-retinol binding protein (RBP) in serum. Rpe65-mediated isomerization can be blocked by retinylamine (Ret-NH2), the farnesyl-containing analogues, TDH and TDT, and isotretinoin. Isotretinoin also inhibits 11-cis-ROL dehydrogenase (11-cis-RDH).
A more practical approach to limiting light-dependent formation of all-trans-RAL is to slow synthesis of 11-cis-RAL by inhibiting the visual cycle pharmacologically. The first attempt was with isotretinoin (13-cis-retinoic acid or Accutane®), a drug commonly used for the treatment of acne. An occasional side effect of Accutane® is reduced night vision (196). This effect is due to its inhibitory effect on 11-cis-RDH in RPE cells (197, 198) (Figure 5). Isotretinoin also binds to Rpe65 (199) and therefore might also inhibit the isomerase. Treatment of WT albino rats with isotretinoin prevented light-induced retinal damage, prompting the suggestion that this visual-cycle inhibitor may be useful for treating retinal or macular degenerations in humans (211). Treatment of abcr−/− mice with isotretinoin completely blocked new synthesis of A2PE-H2, A2E, and A2E-oxiranes (114, 200). Electron microscopic analysis showed reduced lipofuscin pigments in the treated animals (200). ERG of the treated animals showed delayed recovery of rod sensitivity following exposure to bright light, but otherwise normal visual function. These data validated the strategy of inhibiting the visual cycle pharmacologically as a mechanism to slow A2E accumulation in Stargardt and other lipofuscin-based diseases. Unfortunately, isotretinoin at doses sufficient to inhibit the visual cycle has unacceptable side effects. It is therefore unsuitable for long-term treatment of patients with macular degeneration.
N-(4-hydroxyphenyl)retinamide (HPR) (Figure 1c) is a retinoid analog that has been used for approximately 20 years as a chemotherapeutic agent to treat cancer. One action of HPR is to reduce serum levels of vitamin A by competing for binding sites on RBP (201). HPR binding to RBP prevents its interaction with transthyretin, causing loss of RBP to glomerular filtration (202). Unlike other tissues, the eye is highly dependent on RBP to deliver all-trans-ROL from serum. Mice with a knockout mutation in the rbp gene have a predominantly ocular phenotype (203). Treatment of abcr−/− mice with HPR at doses similar to those used in humans for treating cancer (2.5–10.0 mg kg−1 day−1) arrested the accumulation of A2E and its precursors (204) (Figure 5). Lipofuscin autofluorescence in the RPE, assessed by laser-scanning microscopy, was also dramatically reduced in HPR-treated mice (204). ERG of HPR-treated WT and abcr−/− mice showed slightly delayed recovery of rod sensitivity (delayed dark adaptation) following exposure to bright light, but otherwise normal visual function (204). Given the established safety of HPR in humans during clinical trials (205), these observations suggest that HPR may be useful for treatment of retinal and macular degenerations associated with lipofuscin accumulation, such as Stargardt disease. RBP-deficient mice have impaired retinal function early in life owing to low circulating levels of retinol (206). On a vitamin A–sufficient diet they acquire normal vision by 5 months of age. RBP is expressed by many tissues, including the eye and liver. Liver-derived RBP is responsible for the mobilization of retinol from hepatic stores. Ectopic, muscle-specific expression of human RBP on a rbp−/− background is sufficient to recover visual function (207). The role of RPE-expressed RBP in the visual cycle or retinol transport is not clear.
Positively charged retinoids, such as retinylamine (Ret-NH2), are inhibitors of the isomerization reaction in bovine RPE microsomes (208). Consistently, mice treated with all-trans-Ret-NH2 showed normal dark-adapted visual function by ERG, but delayed dark adaptation following light exposure. Ret-NH2 is reversibly N-acylated by LRAT to form inactive retinylamides (209), which prolongues the inhibitory effect. These observations suggest that Ret-NH2 may also be useful to inhibit A2E formation in maculopathies caused by lipofuscin accumulation. Mice treated with Ret-NH2 were resistant to light-induced retina damage.
Two farnesyl-containing isoprenoids (TDT and TDH) have been shown to bind Rpe65 in vitro (210). Administration of TDT and TDH to abcr−/− mice caused delayed dark adaptation by ERG and reduced A2E levels (210). Nothing is known about the toxicity or potential carcinogenicity of these agents. If safe, however, these data suggest that TDT and TDH may also reduce lipofuscin accumulation in humans.
What deficits in visual function might be expected in humans taking visual-cycle inhibitors? Sensitivity of the visual system in vertebrates is only limited by the efficiency of photon capture (quantum catch) in very dim light. Thus, a patient with a partially inhibited visual cycle would notice no changes in daylight vision. In dim light, the rate of photoisomerization, by definition, is very slow. Hence, a fully dark-adapted patient on visual-cycle inhibitors would start with a “full tank” of rhodopsin and would notice no change in visual performance as long as he remained in dim light. The effect of visual cycle inhibitors would be felt during transitions from bright to dim light. With pharmacologically slowed rhodopsin regeneration, a patient on treatment would take longer to dark-adapt. This would be felt, for example, upon entering a darkened theater or driving during daytime into a dimly lit tunnel. The severity of this effect would be influenced by the site of pharmacologic inhibition in the visual cycle. Isotretinoin, which effects the final catalytic step in the visual cycle (Figure 5), dramatically slowed dark adaptation in WT and abcr−/− mice (114, 200). The milder delay in dark-adaptation observed in WT and abcr−/− mice treated with HPR versus isotretinoin (200) may arise because HPR does not inhibit or antagonize any proteins in the visual cycle (Figure 5). Instead, HPR reduces the amount of all-trans-ROL entering the RPE, thereby lowering the steady-state levels of all intraocular retinoids. If these drugs are effective at slowing the progression of blindness in patients with retinal or macular degenerations associated with lipofuscin-accumulation, the functional penalty of delayed dark adaptation will probably be deemed worthwhile.
CONCLUSIONS AND PERSPECTIVES
Our understanding of the mechanisms involved in visual-pigment regeneration is progressing rapidly. This understanding has stimulated the development of new approaches to treat inherited retinal diseases caused by defects in visual cycle proteins. The availability of experimental animals with spontaneous or engineered mutations in the genes for these proteins has been invaluable to this progress. Three therapeutic strategies have been considered.
Gene therapy involves the introduction of a normal gene within a recombinant virus for expression in a population of cells affected by a disease-causing mutation. Fortunately, most steps of the visual cycle take place within the RPE, a cell type that is easy to target owing to its high phagocytotic potential. Gene therapy has been successful in the treatment of mouse and dog models for LCA caused by mutations in RPE65. Clinical trials in humans with RPE65-mediated LCA are set to begin soon. However, the total number of patients with this subtype of LCA is very small. Successful gene therapy with RPE65 will represent an exciting proof of concept, but at best will only restore vision to a tiny subset of patients that suffer from inherited blindness.
Chromophore replacement therapy involves administration of natural or artificial chromophore or chromophore precursor. This strategy is analogous to treating an inborn error of metabolism by providing a metabolite downstream of the blocked step. It is suitable for diseases caused by impaired chromophore biogenesis, such as RPE65-mediated LCA. A hypothetical drawback of this approach is the accumulation of toxic or teratogenic retinoic acid with prolonged retinoid supplementation. Mice that received prolonged supplementation with 9-cis-retinoids exhibited no signs of toxicity.
Therapeutic inhibition of the visual cycle involves the use of retinoid analogs to slow the biosynthesis of 11-cis-RAL chromophore. By partially depleting rhodopsin, the amount of all-trans-RAL released by light exposure is reduced. This approach has been shown to arrest the accumulation of toxic lipofuscin fluorophores in the RPE of abcr−/− and WT mice (114, 200, 204). The functional penalty for treating with visual cycle inhibitors is delayed dark adaptation, although this effect was mild in mice. A hypothetical drawback of slowing rhodopsin regeneration is that the presence of apoopsin could lead to constitutive activation of the visual transduction cascade and might cause photoreceptor degeneration (79). However, no photoreceptor degeneration was seen in mice following prolonged treatment with isotretinoin or HPR (114, 200, 204). This lack of degeneration is probably due to the modest reduction in rhodopsin required to inhibit A2E accumulation. On the other hand, visual cycle inhibitors prevented light-induced damage to the retina (211), suggesting a possible protective effect of these drugs.
The present accomplishments in this research field were made possible by the availability of mice with modified or disrupted genes for retinoid cycle proteins. In most cases, these mice are close models of the cognate human diseases. However, retinal anatomies differ between experimental animals and humans with respect to the presence of a macula and the distribution of rods and cones. Also, quantitative differences are seen in the levels of visual retinoids and the specific activities of retinoid-processing enzymes.
The discovery of a cone-specific pathway for chromophore regeneration operating in diurnal animals has important implications for human disease. With the advent of artificial lighting, the functioning of cones is far more important than rods for useful vision in humans. Rods, however, are important for night vision and are necessary for cone survival. Preserving cone function by maintaining rod survival at the expense of rod function could be a major theme in the future development of therapies for macular degeneration, Stargardt disease, and RP.
Several routes of drug delivery are possible in the treatment of ocular diseases with retinoid analogs. As discussed, retinoids can be delivered orally, with excellent delivery to the eye. Potentially, retinoid drugs could also be delivered by eye drops, intraocular injection into different compartments of the eye, or periorbital injections into the fat surrounding the eye. Finally, the drugs could be released from slow-release devices that encapsulate the drug. These approaches will lower potential toxicity compared with the drugs delivered systemically.
Until recently, the retina has not been accessible for high-resolution images. New applications of two-photon microscopy that exploit the intrinsic fluorescence of retinoids permit visualization of RPE-cell structures in live animals (41, 212). With further development, these techniques may provide new information about retinoid metabolism and the response to treatments in humans.
The health of the RPE is affected by intense long-term exposure to light, which can induce damage to both the retina and the RPE (213). The continuous exposure of the RPE and photoreceptors to light, the large consumption of oxygen by these cells, the daily shedding and phagocytosis of photoreceptor OS, and the high levels of docosahexaenoate (DHA)-containing lipids in the retina all contribute to the formation of free radicals and oxidative injury. Free radical–induced oxidation of DHA-containing lipids generates ω-(2-carboxyethyl)pyrrole (CEP) protein adducts that are more abundant in ocular tissues and plasma from AMD patients than normal human donors (214, 215). These observations suggest that antioxidative treatments should be effective against retinal diseases. Thus far, the only approved pharmacological treatment of the AMD is a combination of antioxidants (216). Sclera penetrative antioxidants would be very helpful in the maintenance of the RPE and protect against the oxidative stress.
In summary, the understanding of the visual processes on a molecular level, generation of animal models, breakthroughs in analytical methods, and the focus of many laboratories on diseases of the retina and RPE suggest that we will see remarkable progress toward the treatment of these diseases in the coming years.
UNRESOLVED ISSUES
The molecular mechanisms of isomerization of all-trans-retinoids to 11-cis-retinoids should be determined.
High-resolution structures of retinoid cycle enzymes should be obtained to understand their specificity and mechanisms of action.
New drugs of low toxicity and high efficacy should be developed and tested for the treatment of retinoid-cycle diseases.
A greater understanding of cellular flow of retinoids in photoreceptor and RPE cell is required.
A greater molecular understanding of the difference and similarities between rod and cone chromophore regeneration is needed.
SUMMARY
Vision is based on the light-induced isomerization of 11-cis- to all-trans-RAL, which leads to a conformational change in rhodopsin or cone opsin, and activation of the phototransduction cascade.
The retinoid cycle is essential for the conversion of all-trans- to 11-cis-RAL, regeneration of rod and cone visual pigment, and maintenance of continuous vision.
Regeneration of 11-cis-RAL requires the cooperation of RPE and photoreceptor cells and involves several enzymes and transport proteins.
Intrinsic differences exist in the ways that cone and rod photoreceptors acquire 11-cis-RAL and regenerate visual pigment.
Deleterious mutations that affect the enzymes involved in the retinoid cycle can lead to blinding diseases.
The etiology of diseases caused by an impaired retinoid cycle has been studied in knockout animal models and animals affected by naturally occurring mutations.
In some pathological conditions resulting from an impaired retinoid flux, the accumulation of all-trans-RAL leads to formation of toxic products, such as N-retinylidene-N-retinyl ethanolamine (A2E).
Animal models of disease caused by an impaired retinoid cycle have been successfully treated using either gene-therapy and/or by supplementation of chromophore through a pharmacological approach.
The accumulation of all-trans-RAL and production of A2E can be prevented using pharmacological therapies aimed at reducing the rate of the retinoid cycle and/or limiting uptake of serum retinol into the RPE.
Possible future therapeutic interventions could involve the regulation of the rate of the retinoid cycle to match the level of chromophore needed.
Acknowledgments
We gratefully acknowledge Roxana Radu for her valuable comments on the manuscript. This research was supported by NIH grants EY11713, EY015844, EY11373, and EY09339, and the Foundation Fighting Blindness. G.H.T. is the Charles Kenneth Feldman and Jules & Doris Stein Research to Prevent Blindness Professor.
- Rod cell
photoreceptor cell specialized to function in dim light and subserves monochromatic vision
- Cone cell
photoreceptor cell that functions in bright light and, based on the expression of the particular cone opsin gene (blue, green, or red), responds to different wavelengths of light
- Outer segment (OS)
a membranous structure of rod and cone photoreceptors comprising a stack of flattened discs, which are the sites of photon-capture and the reactions of visual transduction
- G protein-coupled receptor (GPCR)
family of integral membrane proteins characterized by seven membrane-spanning regions involved in signal transduction across cell membranes
- Chromophore
any chemical group that absorbs light. In vision of vertebrates 11-cis-RAL is utilized as a light-absorbing compound
- Schiff base
a carbon-nitrogen double bond where nitrogen is connected to aryl or alkyl groups but not to hydrogen
- Photoreceptor cell
cell specialized in sensing light and conversion of the rate of photon absorption into nervous signal
- Leber Congenital Amaurosis (LCA)
degenerative disease resulting in severe loss of vision owing to abnormal development of photoreceptor cells in the retina or premature degeneration of the retinal cells
- Stargardt macular degeneration
an inherited disease of children causing progressive loss of central vision and caused by mutations in the ABCA4 gene
- Photoisomerization
light-induced changes of conformation resulting in a switch from one isomer to another
- Macula
the central part of the retina responsible for perceiving fine visual details
- Electroretinography (ERG)
technique used to measure electrical response of rods, cones, and inner retinal neurons. It is employed for diagnosis of various retinal diseases as well as verification of photoreceptor cells functions
- Retinal dystrophy
eye disease leading to impairment of vision and loss of color vision, followed by night blindness and decrease of visual field leads to bilateral visual loss (109).
- Retinal pigment epithelium (RPE)
the pigment cell monolayer forming the outer blood-retinal barrier that nourishes the retinal cells and plays a central role in retinal physiology by supporting the function of the photoreceptors
Footnotes
The Annual Review of Pharmacology and Toxicology is online at http://pharmtox.annualreviews.org
DISCLOSURE STATEMENT
G.H.T. is a consultant for Sirion Therapeutics, Inc.
The University of Washington and Acucela Inc. and the University of Washington and QLT Inc. may commercialize some of the technology described in this work. K.P. is a consultant for both Acucela Inc. and QLT Inc.
LITERATURE CITED
- 1.Filipek S, Stenkamp RE, Teller DC, Palczewski K. G protein-coupled receptor rhodopsin: a prospectus. Annu Rev Physiol. 2003;65:851–79. doi: 10.1146/annurev.physiol.65.092101.142611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Okada T, Ernst OP, Palczewski K, Hofmann KP. Activation of rhodopsin: new insights from structural and biochemical studies. Trends Biochem Sci. 2001;26:318–24. doi: 10.1016/s0968-0004(01)01799-6. [DOI] [PubMed] [Google Scholar]
- 3.Arshavsky VY, Lamb TD, Pugh EN. G proteins and phototransduction. Annu Rev Physiol. 2002;64:153–87. doi: 10.1146/annurev.physiol.64.082701.102229. [DOI] [PubMed] [Google Scholar]
- 4.Rando RR, Bangerter FW. The rapid intermembraneous transfer of retinoids. Biochem Biophys Res Commun. 1982;104:430–36. doi: 10.1016/0006-291x(82)90655-6. [DOI] [PubMed] [Google Scholar]
- 5.Groenendijk GW, De Grip WJ, Daemen FJ. Rod outer segment membranes: good acceptors for retinoids? Vision Res. 1984;24:1623–27. doi: 10.1016/0042-6989(84)90320-1. [DOI] [PubMed] [Google Scholar]
- 6.Allikmets R, Singh N, Sun H, Shroyer NF, Hutchinson A, et al. A photoreceptor cell-specific ATP-binding transporter gene (ABCR) is mutated in recessive Stargardt macular dystrophy. Nat Genet. 1997;15:236–46. doi: 10.1038/ng0397-236. [DOI] [PubMed] [Google Scholar]
- 7.Illing M, Molday LL, Molday RS. The 220-kDa rim protein of retinal rod outer segments is a member of the ABC transporter superfamily. J Biol Chem. 1997;272:10303–10. doi: 10.1074/jbc.272.15.10303. [DOI] [PubMed] [Google Scholar]
- 8.Azarian SM, Travis GH. The photoreceptor rim protein is an ABC transporter encoded by the gene for recessive Stargardt’s disease (ABCR) FEBS Lett. 1997;409:247–52. doi: 10.1016/s0014-5793(97)00517-6. [DOI] [PubMed] [Google Scholar]
- 9.Weng J, Mata NL, Azarian SM, Tzekov RT, Birch DG, Travis GH. Insights into the function of Rim protein in photoreceptors and etiology of Stargardt’s disease from the phenotype in abcr knockout mice. Cell. 1999;98:13–23. doi: 10.1016/S0092-8674(00)80602-9. [DOI] [PubMed] [Google Scholar]
- 10.Mata NL, Weng J, Travis GH. Biosynthesis of a major lipofuscin fluorophore in mice and humans with ABCR-mediated retinal and macular degeneration. Proc Natl Acad Sci USA. 2000;97:7154–59. doi: 10.1073/pnas.130110497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sun H, Molday RS, Nathans J. Retinal stimulates ATP hydrolysis by purified and reconstituted ABCR, the photoreceptor-specific ATP-binding cassette transporter responsible for Stargardt disease. J Biol Chem. 1999;274:8269–81. doi: 10.1074/jbc.274.12.8269. [DOI] [PubMed] [Google Scholar]
- 12.Beharry S, Zhong M, Molday RS. N-retinylidene-phosphatidyl-ethanolamine is the preferred retinoid substrate for the photoreceptor-specific ABC transporter ABCA4 (ABCR) J Biol Chem. 2004;279:53972–79. doi: 10.1074/jbc.M405216200. [DOI] [PubMed] [Google Scholar]
- 13.Haeseleer F, Huang J, Lebioda L, Saari JC, Palczewski K. Molecular characterization of a novel short-chain dehydrogenase/reductase that reduces all-trans-retinal. J Biol Chem. 1998;273:21790–99. doi: 10.1074/jbc.273.34.21790. [DOI] [PubMed] [Google Scholar]
- 14.Rattner A, Smallwood PM, Nathans J. Identification and characterization of all-trans-retinol dehydrogenase from photoreceptor outer segments, the visual cycle enzyme that reduces all-trans-retinal to all-trans-retinol. J Biol Chem. 2000;275:11034–43. doi: 10.1074/jbc.275.15.11034. [DOI] [PubMed] [Google Scholar]
- 15.Haeseleer F, Jang GF, Imanishi Y, Driessen CA, Matsumura M, et al. Dual-substrate specificity short chain retinol dehydrogenases from the vertebrate retina. J Biol Chem. 2002;277:45537–46. doi: 10.1074/jbc.M208882200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Maeda A, Maeda T, Imanishi Y, Kuksa V, Alekseev A, et al. Role of photoreceptor-specific retinol dehydrogenase in the retinoid cycle in vivo. J Biol Chem. 2005;280:18822–32. doi: 10.1074/jbc.M501757200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Adler AJ, Evans CD. Some functional characteristics of purified bovine interphotoreceptor retinol-binding protein. Invest Ophthalmol Vis Sci. 1985;26:273–82. [PubMed] [Google Scholar]
- 18.Redmond TM, Wiggert B, Robey FA, Nguyen NY, Lewis MS, et al. Isolation and characterization of monkey interphotoreceptor retinoid-binding protein, a unique extracellular matrix component of the retina. Biochemistry. 1985;24:787–93. doi: 10.1021/bi00324a038. [DOI] [PubMed] [Google Scholar]
- 19.Adler AJ, Edwards RB. Human interphotoreceptor matrix contains serum albumin and retinol-binding protein. Exp Eye Res. 2000;70:227–34. doi: 10.1006/exer.1999.0780. [DOI] [PubMed] [Google Scholar]
- 20.Shaw NS, Noy N. Interphotoreceptor retinoid-binding protein contains three retinoid binding sites. Exp Eye Res. 2001;72:183–90. doi: 10.1006/exer.2000.0945. [DOI] [PubMed] [Google Scholar]
- 21.Lin ZS, Fong SL, Bridges CD. Retinoids bound to interstitial retinol-binding protein during light and dark-adaptation. Vision Res. 1989;29:1699–709. doi: 10.1016/0042-6989(89)90152-1. [DOI] [PubMed] [Google Scholar]
- 22.Adler AJ, Spencer SA. Effect of light on endogenous ligands carried by interphotoreceptor retinoid-binding protein. Exp Eye Res. 1991;53:337–46. doi: 10.1016/0014-4835(91)90239-b. [DOI] [PubMed] [Google Scholar]
- 23.Tsina E, Chen C, Koutalos Y, Ala-Laurila P, Tsacopoulos M, et al. Physiological and microfluorometric studies of reduction and clearance of retinal in bleached rod photoreceptors. J Gen Physiol. 2004;124:429–43. doi: 10.1085/jgp.200409078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Crouch RK, Hazard ES, Lind T, Wiggert B, Chader G, Corson DW. Interphotoreceptor retinoid-binding protein and alpha-tocopherol preserve the isomeric and oxidation state of retinol. Photochem Photobiol. 1992;56:251–55. doi: 10.1111/j.1751-1097.1992.tb02154.x. [DOI] [PubMed] [Google Scholar]
- 25.Pepperberg DR, Okajima TL, Wiggert B, Ripps H, Crouch RK, Chader GJ. Interphotoreceptor retinoid-binding protein (IRBP). Molecular biology and physiological role in the visual cycle of rhodopsin. Mol Neurobiol. 1993;7:61–85. doi: 10.1007/BF02780609. [DOI] [PubMed] [Google Scholar]
- 26.Bok D, Ong DE, Chytil F. Immunocytochemical localization of cellular retinol binding protein in the rat retina. Invest Ophthalmol Vis Sci. 1984;25:877–83. [PubMed] [Google Scholar]
- 27.Edwards RB, Adler AJ. Exchange of retinol between IRBP and CRBP. Exp Eye Res. 1994;59:161–70. doi: 10.1006/exer.1994.1094. [DOI] [PubMed] [Google Scholar]
- 28.Cunningham LL, Gonzalez-Fernandez F. Internalization of interphotoreceptor retinoid-binding protein by the Xenopus retinal pigment epithelium. J Comp Neurol. 2003;466:331–42. doi: 10.1002/cne.10861. [DOI] [PubMed] [Google Scholar]
- 29.Young RW, Bok D. Participation of the retinal pigment epithelium in the rod outer segment renewal process. J Cell Biol. 1969;42:392–403. doi: 10.1083/jcb.42.2.392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Saari JC, Bredberg DL. CoA- and non-CoA-dependent retinol esterification in retinal pigment epithelium. J Biol Chem. 1988;263:8084–90. [PubMed] [Google Scholar]
- 31.MacDonald PN, Ong DE. Evidence for a lecithin-retinol acyltransferase activity in the rat small intestine. J Biol Chem. 1988;263:12478–82. [PubMed] [Google Scholar]
- 32.Yost RW, Harrison EH, Ross AC. Esterification by rat liver microsomes of retinol bound to cellular retinol-binding protein. J Biol Chem. 1988;263:18693–701. [PubMed] [Google Scholar]
- 33.Saari JC, Nawrot M, Garwin GG, Kennedy MJ, Hurley JB, et al. Analysis of the visual cycle in cellular retinol-binding protein type I (CRBPI) knockout mice. Invest Ophthalmol Vis Sci. 2002;43:1730–35. [PubMed] [Google Scholar]
- 34.Kaschula CH, Jin MH, Desmond-Smith NS, Travis GH. Acyl CoA:retinol acyltransferase (ARAT) activity is present in bovine retinal pigment epithelium. Exp Eye Res. 2005;82(1):111–21. doi: 10.1016/j.exer.2005.05.010. [DOI] [PubMed] [Google Scholar]
- 35.Mata NL, Radu RA, Clemmons R, Travis GH. Isomerization and oxidation of vitamin A in cone-dominant retinas. A novel pathway for visual-pigment regeneration in daylight. Neuron. 2002;36:69–80. doi: 10.1016/s0896-6273(02)00912-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mata NL, Ruiz A, Radu RA, Bui TV, Travis GH. Chicken retinas contain a retinoid isomerase activity that catalyzes the direct conversion of all-trans-retinol to 11-cis-retinol. Biochemistry. 2005;44:11715–21. doi: 10.1021/bi050942m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yen CL, Monetti M, Burri BJ, Farese RVJ. The triacylglycerol synthesis enzyme DGAT1 also catalyzes the synthesis of diacylglycerols, waxes, and retinyl esters. J Lipid Res. 2005;46:1502–11. doi: 10.1194/jlr.M500036-JLR200. [DOI] [PubMed] [Google Scholar]
- 38.Orland MD, Anwar K, Cromley D, Chu CH, Chen L, et al. Acyl coenzyme A dependent retinol esterification by acyl coenzyme A: diacylglycerol acyltransferase 1. Biochim Biophys Acta. 2005;1737:76–82. doi: 10.1016/j.bbalip.2005.09.003. [DOI] [PubMed] [Google Scholar]
- 39.O’Byrne SM, Wongsiriroj N, Libien J, Vogel S, Goldberg IJ, et al. Retinoid absorption and storage is impaired in mice lacking lecithin:retinol acyltransferase (LRAT) J Biol Chem. 2005;280:35647–57. doi: 10.1074/jbc.M507924200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Batten ML, Imanishi Y, Maeda T, Tu DC, Moise AR, et al. Lecithin-retinol acyltransferase is essential for accumulation of all-trans-retinyl esters in the eye and in the liver. J Biol Chem. 2004;279:10422–32. doi: 10.1074/jbc.M312410200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Imanishi Y, Gerke V, Palczewski K. Retinosomes: new insights into intracellular managing of hydrophobic substances in lipid bodies. J Cell Biol. 2004;166:447–53. doi: 10.1083/jcb.200405110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gollapalli DR, Rando RR. All-trans-retinyl esters are the substrates for isomerization in the vertebrate visual cycle. Biochemistry. 2003;42:5809–18. doi: 10.1021/bi0341004. [DOI] [PubMed] [Google Scholar]
- 43.Moiseyev G, Crouch RK, Goletz P, Oatis JJ, Redmond TM, Ma JX. Retinyl esters are the substrate for isomerohydrolase. Biochemistry. 2003;42:2229–38. doi: 10.1021/bi026911y. [DOI] [PubMed] [Google Scholar]
- 44.Mata NL, Moghrabi WN, Lee JS, Bui TV, Radu RA, et al. Rpe65 is a retinyl ester binding protein that presents insoluble substrate to the isomerase in retinal pigment epithelial cells. J Biol Chem. 2004;279:635–43. doi: 10.1074/jbc.M310042200. [DOI] [PubMed] [Google Scholar]
- 45.Jin M, Li S, Moghrabi WN, Sun H, Travis GH. Rpe65 is the retinoid isomerase in bovine retinal pigment epithelium. Cell. 2005;122:449–59. doi: 10.1016/j.cell.2005.06.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Moiseyev G, Chen Y, Takahashi Y, Wu BX, Ma JX. Rpe65 is the isomerohydrolase in the retinoid visual cycle. Proc Natl Acad Sci USA. 2005;102:12413–18. doi: 10.1073/pnas.0503460102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Redmond TM, Poliakov E, Yu S, Tsai JY, Lu Z, Gentleman S. Mutation of key residues of Rpe65 abolishes its enzymatic role as isomerohydrolase in the visual cycle. Proc Natl Acad Sci USA. 2005;102:13658–63. doi: 10.1073/pnas.0504167102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Redmond TM, Yu S, Lee E, Bok D, Hamasaki D, et al. Rpe65 is necessary for production of 11-cis-vitamin A in the retinal visual cycle. Nat Genet. 1998;20:344–51. doi: 10.1038/3813. [DOI] [PubMed] [Google Scholar]
- 49.Redmond TM, Gentleman S, Duncan T, Yu S, Wiggert B, et al. Identification, expression, and substrate specificity of a mammalian beta-carotene 15,15′-dioxygenase. J Biol Chem. 2001;276:6560–65. doi: 10.1074/jbc.M009030200. [DOI] [PubMed] [Google Scholar]
- 50.Kloer DP, Ruch S, Al-Babili S, Beyer P, Schulz GE. The structure of a retinal-forming carotenoid oxygenase. Science. 2005;308:267–69. doi: 10.1126/science.1108965. [DOI] [PubMed] [Google Scholar]
- 51.Moiseyev G, Takahashi Y, Chen Y, Gentleman S, Redmond TM, et al. Rpe65 is an Iron(II)-dependent isomerohydrolase in the retinoid visual cycle. J Biol Chem. 2005;281:2835–40. doi: 10.1074/jbc.M508903200. [DOI] [PubMed] [Google Scholar]
- 52.Xue L, Gollapalli DR, Maiti P, Jahng WJ, Rando RR. A palmitoylation switch mechanism in the regulation of the visual cycle. Cell. 2004;117:761–71. doi: 10.1016/j.cell.2004.05.016. [DOI] [PubMed] [Google Scholar]
- 53.Winston A, Rando RR. Regulation of isomerohydrolase activity in the visual cycle. Biochemistry. 1998;37:2044–50. doi: 10.1021/bi971908d. [DOI] [PubMed] [Google Scholar]
- 54.McBee JK, van Hooser JP, Jang GF, Palczewski K. Isomerization of 11-cis-retinoids to all-trans-retinoids in vitro and in vivo. J Biol Chem. 2001;276:48483–93. doi: 10.1074/jbc.M105840200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bunt-Milam AH, Saari JC. Immunocytochemical localization of two retinoid-binding proteins in vertebrate retina. J Cell Biol. 1983;97:703–12. doi: 10.1083/jcb.97.3.703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Saari JC, Bredberg DL. Photochemistry and stereoselectivity of cellular retinaldehyde-binding protein from bovine retina. J Biol Chem. 1987;262:7618–22. [PubMed] [Google Scholar]
- 57.Saari JC, Bredberg DL, Farrell DF. Retinol esterification in bovine retinal pigment epithelium: reversibility of lecithin:retinol acyltransferase. Biochem J. 1993;291(Pt 3):697–700. doi: 10.1042/bj2910697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Blaner WS, Das SR, Gouras P, Flood MT. Hydrolysis of 11-cis- and all-trans-retinyl palmitate by homogenates of human retinal epithelial cells. J Biol Chem. 1987;262:53–58. [PubMed] [Google Scholar]
- 59.Mata NL, Tsin AT, Chambers JP. Hydrolysis of 11-cis- and all-trans-retinyl palmitate by retinal pigment epithelium microsomes. J Biol Chem. 1992;267:9794–99. [PubMed] [Google Scholar]
- 60.Stecher H, Gelb MH, Saari JC, Palczewski K. Preferential release of 11-cis-retinol from retinal pigment epithelial cells in the presence of cellular retinaldehyde-binding protein. J Biol Chem. 1999;274:8577–85. doi: 10.1074/jbc.274.13.8577. [DOI] [PubMed] [Google Scholar]
- 61.Driessen CA, Janssen BP, Winkens HJ, van Vugt AH, de Leeuw TL, Janssen JJ. Cloning and expression of a cDNA encoding bovine retinal pigment epithelial 11-cis retinol dehydrogenase. Invest Ophthalmol Vis Sci. 1995;36:1988–96. [PubMed] [Google Scholar]
- 62.Driessen C, Winkens HJ, Hoffmann K, Kuhlmann LD, Janssen BPM, et al. Disruption of the 11-cis-retinol dehydrogenase gene leads to accumulation of cis-retinols and cis-retinyl esters. Mol Cellular Biol. 2000;20:4275–87. doi: 10.1128/mcb.20.12.4275-4287.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kim TS, Maeda A, Maeda T, Heinlein C, Kedishvili N, et al. Delayed dark adaptation in 11-cis-retinol dehydrogenase-deficient mice: a role of RDH11 in visual processes in vivo. J Biol Chem. 2005;280:8694–704. doi: 10.1074/jbc.M413172200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Nawrot M, Liu T, Garwin GG, Crabb JW, Saari JC. Scaffold proteins and visual pigment regeneration. Photochem Photobiol. 2006 doi: 10.1562/2006-01-25-RA-784. In press. [DOI] [PubMed] [Google Scholar]
- 65.Nawrot M, West K, Huang J, Possin DE, Bretscher A, et al. Cellular retinaldehyde-binding protein interacts with ERM-binding phosphoprotein 50 in retinal pigment epithelium. Invest Ophthalmol Vis Sci. 2004;45:393–401. doi: 10.1167/iovs.03-0989. [DOI] [PubMed] [Google Scholar]
- 66.Chen Y, Noy N. Retinoid specificity of interphotoreceptor retinoid-binding protein. Biochemistry. 1994;33:10658–65. doi: 10.1021/bi00201a013. [DOI] [PubMed] [Google Scholar]
- 67.Carlson A, Bok D. Promotion of the release of 11-cis-retinal from cultured retinal pigment epithelium by interphotoreceptor retinoid-binding protein. Biochemistry. 1992;31:9056–62. doi: 10.1021/bi00152a049. [DOI] [PubMed] [Google Scholar]
- 68.Edwards RB, Adler AJ. IRBP enhances removal of 11-cis-retinaldehyde from isolated RPE membranes. Exp Eye Res. 2000;70:235–45. doi: 10.1006/exer.1999.0781. [DOI] [PubMed] [Google Scholar]
- 69.Palczewski K, van Hooser JP, Garwin GG, Chen J, Liou GI, Saari JC. Kinetics of visual pigment regeneration in excised mouse eyes and in mice with a targeted disruption of the gene encoding interphotoreceptor retinoid-binding protein or arrestin. Biochemistry. 1999;38:12012–19. doi: 10.1021/bi990504d. [DOI] [PubMed] [Google Scholar]
- 70.Ripps H, Peachey NS, Xu X, Nozell SE, Smith SB, Liou GI. The rhodopsin cycle is preserved in IRBP “knockout” mice despite abnormalities in retinal structure and function. Vis Neurosci. 2000;17:97–105. doi: 10.1017/s095252380017110x. [DOI] [PubMed] [Google Scholar]
- 71.Baylor DA, Lamb TD, Yau KW. Responses of retinal rods to single photons. J Physiol. 1979;288:613–34. [PMC free article] [PubMed] [Google Scholar]
- 72.Baylor DA, Nunn BJ, Schnapf JL. The photocurrent, noise and spectral sensitivity of rods of the monkey Macaca fascicularis. J Physiol. 1984;357:575–607. doi: 10.1113/jphysiol.1984.sp015518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Pugh EN, Lamb TD. Phototransduction in vertebrate rods and cones: molecular mechanisms of amplification, recovery and light adaptation. In: Stavenga DG, DeGrip WJ, Pugh EN Jr, editors. Handbook of Biological Physics. Amsterdam: Elsevier Sci. B.V; 2000. pp. 184–255. [Google Scholar]
- 74.Schnapf JL, Nunn BJ, Meister M, Baylor DA. Visual transduction in cones of the monkey Macaca fascicularis. J Physiol. 1990;427:681–713. doi: 10.1113/jphysiol.1990.sp018193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Baylor DA. Photoreceptor signals and vision. Proctor lecture. Invest Ophthalmol Vis Sci. 1987;28:34–49. [PubMed] [Google Scholar]
- 76.Baylor DA, Matthews G, Yau KW. Two components of electrical dark noise in toad retinal rod outer segments. J Physiol. 1980;309:591–621. doi: 10.1113/jphysiol.1980.sp013529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kefalov V, Fu Y, Marsh-Armstrong N, Yau KW. Role of visual pigment properties in rod and cone phototransduction. Nature. 2003;425:526–31. doi: 10.1038/nature01992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kefalov VJ, Estevez ME, Kono M, Goletz PW, Crouch RK, et al. Breaking the covalent bond—a pigment property that contributes to desensitization in cones. Neuron. 2005;46:879–90. doi: 10.1016/j.neuron.2005.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Fain GL, Matthews HR, Cornwall MC, Koutalos Y. Adaptation in vertebrate photoreceptors. Physiol Rev. 2001;81:117–51. doi: 10.1152/physrev.2001.81.1.117. [DOI] [PubMed] [Google Scholar]
- 80.Goldstein EB, Wolf BM. Regeneration of the green-rod pigment in the isolated frog retina. Vis Res. 1973;13:527–34. doi: 10.1016/0042-6989(73)90022-9. [DOI] [PubMed] [Google Scholar]
- 81.Hood DC, Hock PA. Recovery of cone receptor activity in the frog’s isolated retina. Vis Res. 1973;13:1943–51. doi: 10.1016/0042-6989(73)90065-5. [DOI] [PubMed] [Google Scholar]
- 82.Jones GJ, Crouch RK, Wiggert B, Cornwall MC, Chader GJ. Retinoid requirements for recovery of sensitivity after visual-pigment bleaching in isolated photoreceptors. Proc Nat Acad Sci USA. 1989;86:9606–10. doi: 10.1073/pnas.86.23.9606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Das SR, Bhardwaj N, Kjeldbye H, Gouras P. Muller cells of chicken retina synthesize 11-cis-retinol. Biochem J. 1992;285:907–13. doi: 10.1042/bj2850907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Dryja TP. Molecular genetics of Oguchi disease, fundus albipunctatus, and other forms of stationary night blindness: LVII Edward Jackson Memorial Lecture. Am J Ophthalmol. 2000;130:547–63. doi: 10.1016/s0002-9394(00)00737-6. [DOI] [PubMed] [Google Scholar]
- 85.Thompson DA, Janecke AR, Lange J, Feathers KL, Hubner CA, et al. Retinal degeneration associated with RDH12 mutations results from decreased 11-cis retinal synthesis due to disruption of the visual cycle. Hum Mol Genet. 2005;14:3865–75. doi: 10.1093/hmg/ddi411. [DOI] [PubMed] [Google Scholar]
- 86.Thompson DA, Li Y, McHenry CL, Carlson TJ, Ding X, et al. Mutations in the gene encoding lecithin retinol acyltransferase are associated with early-onset severe retinal dystrophy. Nat Genet. 2001;28:123–24. doi: 10.1038/88828. [DOI] [PubMed] [Google Scholar]
- 87.Marlhens F, Bareil C, Griffoin JM, Zrenner E, Amalric P, et al. Mutations in Rpe65 cause Lebers congenital amaurosis. Nat Genet. 1997;17:139–41. doi: 10.1038/ng1097-139. [DOI] [PubMed] [Google Scholar]
- 88.Gu SM, Thompson DA, Srikumari CR, Lorenz B, Finckh U, et al. Mutations in RPE65 cause autosomal recessive childhood-onset severe retinal dystrophy. Nat Genet. 1997;17:194–97. doi: 10.1038/ng1097-194. [DOI] [PubMed] [Google Scholar]
- 89.Cremers FP, van de Pol DJ, van Driel M, den Hollander AI, van Haren FJ, et al. Autosomal recessive retinitis pigmentosa and cone-rod dystrophy caused by splice site mutations in the Stargardt’s disease gene ABCR. Hum Mol Genet. 1998;7:355–62. doi: 10.1093/hmg/7.3.355. [DOI] [PubMed] [Google Scholar]
- 90.Martinez-Mir A, Paloma E, Allikmets R, Ayuso C, del Rio T, et al. Retinitis pigmentosa caused by a homozygous mutation in the Stargardt disease gene ABCR. Nat Genet. 1998;18:11–12. doi: 10.1038/ng0198-11. [DOI] [PubMed] [Google Scholar]
- 91.Zhang Q, Zulfiqar F, Xiao X, Riazuddin SA, Ayyagari R, et al. Severe autosomal recessive retinitis pigmentosa maps to chromosome 1p13.3-p21.2 between D1S2896 and D1S457 but outside ABCA4. Hum Genet. 2005;118:356–65. doi: 10.1007/s00439-005-0054-4. [DOI] [PubMed] [Google Scholar]
- 92.Allikmets R. Further evidence for an association of ABCR alleles with age-related macular degeneration. The International ABCR Screening Consortium. Am J Hum Genet. 2000;67:487–91. doi: 10.1086/303018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Morimura H, Saindelle-Ribeaudeau F, Berson EL, Dryja TP. Mutations in RGR, encoding a light-sensitive opsin homologue, in patients with retinitis pigmentosa. Nat Genet. 1999;23:393–94. doi: 10.1038/70496. [DOI] [PubMed] [Google Scholar]
- 94.Chen P, Hao W, Rife L, Wang XP, Shen D, et al. A photic visual cycle of rhodopsin regeneration is dependent on Rgr. Nat Genet. 2001;28:256–60. doi: 10.1038/90089. [DOI] [PubMed] [Google Scholar]
- 95.Maeda T, van Hooser JP, Driessen CA, Filipek S, Janssen JJ, Palczewski K. Evaluation of the role of the retinal G protein-coupled receptor (RGR) in the vertebrate retina in vivo. J Neurochem. 2003;85:944–56. doi: 10.1046/j.1471-4159.2003.01741.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Birnbach CD, Jarvelainen M, Possin DE, Milam AH. Histopathology and immunocytochemistry of the neurosensory retina in fundus flavimaculatus. Ophthalmology. 1994;101:1211–19. doi: 10.1016/s0161-6420(13)31725-4. [DOI] [PubMed] [Google Scholar]
- 97.Mata NL, Tzekov RT, Liu XR, Weng J, Birch DG, Travis GH. Delayed dark-adaptation and lipofuscin accumulation in abcr ± mice: implications for involvement of ABCR in age-related macular degeneration. Invest Ophthal Vis Sci. 2001;42:1685–90. [PubMed] [Google Scholar]
- 98.Humphries MM, Rancourt D, Farrar GJ, Kenna P, Hazel M, et al. Retinopathy induced in mice by targeted disruption of the rhodopsin gene. Nat Genet. 1997;15:216–19. doi: 10.1038/ng0297-216. [DOI] [PubMed] [Google Scholar]
- 99.Liang Y, Fotiadis D, Maeda T, Maeda A, Modzelewska A, et al. Rhodopsin signaling and organization in heterozygote rhodopsin knockout mice. J Biol Chem. 2004;279:48189–96. doi: 10.1074/jbc.M408362200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Harper WS, Gaillard ER. Studies of all-trans-retinal as a photooxidizing agent. Photochem Photobiol. 2001;73:71–76. doi: 10.1562/0031-8655(2001)073<0071:soatra>2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 101.Rozanowska M, Wessels J, Boulton M, Burke JM, Rodgers MA, et al. Blue light-induced singlet oxygen generation by retinal lipofuscin in nonpolar media. Free Radic Biol Med. 1998;24:1107–12. doi: 10.1016/s0891-5849(97)00395-x. [DOI] [PubMed] [Google Scholar]
- 102.Dillon J, Gaillard ER, Bilski P, Chignell CF, Reszka KJ. The photochemistry of the retinoids as studied by steady-state and pulsed methods. Photochem Photobiol. 1996;63:680–85. doi: 10.1111/j.1751-1097.1996.tb05673.x. [DOI] [PubMed] [Google Scholar]
- 103.Li T, Sandberg MA, Pawlyk BS, Rosner B, Hayes KC, et al. Effect of vitamin A supplementation on rhodopsin mutants threonine-17 → methionine and proline-347 → serine in transgenic mice and in cell cultures. Proc Natl Acad Sci USA. 1998;95:11933–38. doi: 10.1073/pnas.95.20.11933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Kijas JW, Cideciyan AV, Aleman TS, Pianta MJ, Pearce-Kelling SE, et al. Naturally occurring rhodopsin mutation in the dog causes retinal dysfunction and degeneration mimicking human dominant retinitis pigmentosa. Proc Natl Acad Sci USA. 2002;99:6328–33. doi: 10.1073/pnas.082714499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Aguirre GD, Baldwin V, Pearce-Kelling S, Narfstrom K, Ray K, Acland GM. Congenital stationary night blindness in the dog: common mutation in the RPE65 gene indicates founder effect. Mol Vis. 1998;4:23. [PubMed] [Google Scholar]
- 106.Veske A, Nilsson SE, Narfstrom K, Gal A. Retinal dystrophy of Swedish briard/briard-beagle dogs is due to a 4-bp deletion in RPE65. Genomics. 1999;57:57–61. doi: 10.1006/geno.1999.5754. [DOI] [PubMed] [Google Scholar]
- 107.Zhu L, Jang GF, Jastrzebska B, Filipek S, Pearce-Kelling SE, et al. A naturally occurring mutation of the opsin gene (T4R) in dogs affects glycosylation and stability of the G protein-coupled receptor. J Biol Chem. 2004;279:53828–39. doi: 10.1074/jbc.M408472200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Blacharski PA. Fundus flavimaculutus. New York: Raven Press; 1988. pp. 135–59. [Google Scholar]
- 109.Fishman GA, Farbman JS, Alexander KR. Delayed rod dark adaptation in patients with Stargardt’s disease. Ophthalmology. 1991;98:957–62. doi: 10.1016/s0161-6420(91)32196-1. [DOI] [PubMed] [Google Scholar]
- 110.De Laey JJ, Verougstraete C. Hyperlipofuscinosis and subretinal fibrosis in Stargardt’s disease. Retina. 1995;15:399–406. doi: 10.1097/00006982-199515050-00005. [DOI] [PubMed] [Google Scholar]
- 111.Schwoerer J, Secretan M, Zografos L, Piguet B. Indocyanine green angiography in Fundus flavimaculatus. Ophthalmologica. 2000;214:240–45. doi: 10.1159/000027498. [DOI] [PubMed] [Google Scholar]
- 112.Lois N, Halfyard AS, Bird AC, Holder GE, Fitzke FW. Fundus autofluorescence in Stargardt macular dystrophy-fundus flavimaculatus. Am J Ophthalmol. 2004;138:55–63. doi: 10.1016/j.ajo.2004.02.056. [DOI] [PubMed] [Google Scholar]
- 113.Ben-Shabat S, Itagaki Y, Jockusch S, Sparrow JR, Turro NJ, Nakanishi K. Formation of a Nonaoxirane from A2E, a lipofuscin fluorophore related to macular degeneration, and evidence of singlet oxygen involvement. Angew Chem Int Ed Engl. 2002;41:814–17. doi: 10.1002/1521-3773(20020301)41:5<814::aid-anie814>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- 114.Radu RA, Mata NL, Bagla A, Travis GH. Light exposure stimulates formation of A2E oxiranes in a mouse model of Stargardt’s macular degeneration. Proc Natl Acad Sci USA. 2004;101:5928–33. doi: 10.1073/pnas.0308302101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Perrault I, Hanein S, Gerber S, Barbet F, Ducroq D, et al. Retinal dehydrogenase 12 (RDH12) mutations in leber congenital amaurosis. Am J Hum Genet. 2004;75:639–46. doi: 10.1086/424889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Janecke AR, Thompson DA, Utermann G, Becker C, Hubner CA, et al. Mutations in RDH12 encoding a photoreceptor cell retinol dehydrogenase cause childhood-onset severe retinal dystrophy. Nat Genet. 2004;36:850–54. doi: 10.1038/ng1394. [DOI] [PubMed] [Google Scholar]
- 117.Jager S, Palczewski K, Hofmann KP. Opsin/all-trans-retinal complex activates transducin by different mechanisms than photolyzed rhodopsin. Biochemistry. 1996;35:2901–8. doi: 10.1021/bi9524068. [DOI] [PubMed] [Google Scholar]
- 118.Thompson DA, Gyurus P, Fleischer LL, Bingham EL, McHenry CL, et al. Genetics and phenotypes of RPE65 mutations in inherited retinal degeneration. Invest Ophthalmol Vis Sci. 2000;41:4293–99. [PubMed] [Google Scholar]
- 119.Znoiko SL, Rohrer B, Lu K, Lohr HR, Crouch RK, Ma JX. Downregulation of cone-specific gene expression and degeneration of cone photoreceptors in the Rpe65−/− mouse at early ages. Invest Ophthalmol Vis Sci. 2005;46:1473–79. doi: 10.1167/iovs.04-0653. [DOI] [PubMed] [Google Scholar]
- 120.Cornwall MC, Fain GL. Bleached pigment activates transduction in isolated rods of the salamander retina. J Physiol. 1994;480(Pt 2):261–79. doi: 10.1113/jphysiol.1994.sp020358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Fan J, Woodruff ML, Cilluffo MC, Crouch RK, Fain GL. Opsin activation of transduction in the rods of dark-reared Rpe65 knockout mice. J Physiol. 2005;568:83–95. doi: 10.1113/jphysiol.2005.091942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Fan J, Rohrer B, Moiseyev G, Ma JX, Crouch RK. Isorhodopsin rather than rhodopsin mediates rod function in rpe65 knock-out mice. Proc Natl Acad Sci USA. 2003;100:13662–67. doi: 10.1073/pnas.2234461100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Pang JJ, Chang B, Hawes NL, Hurd RE, Davisson MT, et al. Retinal degeneration 12 (rd12): a new, spontaneously arising mouse model for human Leber congenital amaurosis (LCA) Mol Vis. 2005;11:152–62. [PubMed] [Google Scholar]
- 124.Acland GM, Aguirre GD, Ray J, Zhang Q, Aleman TS, et al. Gene therapy restores vision in a canine model of childhood blindness. Nat Genet. 2001;28:92–95. doi: 10.1038/ng0501-92. [DOI] [PubMed] [Google Scholar]
- 125.Pang JJ, Chang B, Kumar A, Nusinowitz S, Noorwez SM, et al. Gene therapy restores vision-dependent behavior as well as retinal structure and function in a mouse model of RPE65 Leber congenital amaurosis. Mol Ther. 2006;13:565–72. doi: 10.1016/j.ymthe.2005.09.001. [DOI] [PubMed] [Google Scholar]
- 126.Acland GM, Aguirre GD, Bennett J, Aleman TS, Cideciyan AV, et al. Long-term restoration of rod and cone vision by single dose rAAV-mediated gene transfer to the retina in a canine model of childhood blindness. Mol Ther. 2005;12:1072–82. doi: 10.1016/j.ymthe.2005.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Jiang M, Pandey S, Fong HK. An opsin homologue in the retina and pigment epithelium. Invest Ophthalmol Vis Sci. 1993;34:3669–78. [PubMed] [Google Scholar]
- 128.Pandey S, Blanks JC, Spee C, Jiang M, Fong HK. Cytoplasmic retinal localization of an evolutionary homolog of the visual pigments. Exp Eye Res. 1994;58:605–13. doi: 10.1006/exer.1994.1055. [DOI] [PubMed] [Google Scholar]
- 129.Hao WS, Fong HKW. The endogenous chromophore of retinal G protein-coupled receptor opsin from the pigment epithelium. J Biol Chem. 1999;274:6085–90. doi: 10.1074/jbc.274.10.6085. [DOI] [PubMed] [Google Scholar]
- 130.Hara-Nishimura I, Matsumoto T, Mori H, Nishimura M, Hara R, Hara T. Cloning and nucleotide sequence of cDNA for retinochrome, retinal photoisomerase from the squid retina. FEBS Lett. 1990;271:106–10. doi: 10.1016/0014-5793(90)80383-t. [DOI] [PubMed] [Google Scholar]
- 131.Wenzel A, Oberhauser V, Pugh ENJ, Lamb TD, Grimm C, et al. The retinal G protein-coupled receptor (RGR) enhances isomerohydrolase activity independent of light. J Biol Chem. 2005;280:29874–84. doi: 10.1074/jbc.M503603200. [DOI] [PubMed] [Google Scholar]
- 132.Bernal S, Calaf M, Garcia-Hoyos M, Garcia-Sandoval B, Rosell J, et al. Study of the involvement of the RGR, CRPB1, and CRB1 genes in the pathogenesis of autosomal recessive retinitis pigmentosa. J Med Genet. 2003;40:e89. doi: 10.1136/jmg.40.7.e89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Rosenfeld PJ, Cowley GS, McGee TL, Sandberg MA, Berson EL, Dryja TP. A null mutation in the rhodopsin gene causes rod photoreceptor dysfunction and autosomal recessive retinitis pigmentosa. Nat Genet. 1992;1:209–13. doi: 10.1038/ng0692-209. [DOI] [PubMed] [Google Scholar]
- 134.Kumaramanickavel G, Maw M, Denton MJ, John S, Srikumari CR, et al. Missense rhodopsin mutation in a family with recessive RP. Nat Genet. 1994;8:10–11. doi: 10.1038/ng0994-10. [DOI] [PubMed] [Google Scholar]
- 135.Maeda A, Maeda T, Imanishi Y, Golczak M, Moise AR, Palczewski K. Aberrant metabolites in mouse models of congenital blinding diseases: formation and storage of retinyl esters. Biochemistry. 2006;45:4210–19. doi: 10.1021/bi052382x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Morimura H, Berson EL, Dryja TP. Recessive mutations in the RLBP1 gene encoding cellular retinaldehyde-binding protein in a form of retinitis punctata albescens. Invest Ophthalmol Vis Sci. 1999;40:1000–4. [PubMed] [Google Scholar]
- 137.Maw MA, Kennedy B, Knight A, Bridges R, Roth KE, et al. Mutation of the gene encoding cellular retinaldehyde-binding protein in autosomal recessive retinitis pigmentosa. Nat Genet. 1997;17:198–200. doi: 10.1038/ng1097-198. [DOI] [PubMed] [Google Scholar]
- 138.Burstedt MS, Forsman-Semb K, Golovleva I, Janunger T, Wachtmeister L, Sandgren O. Ocular phenotype of bothnia dystrophy, an autosomal recessive retinitis pigmentosa associated with an R234W mutation in the RLBP1 gene. Arch Ophthalmol. 2001;119:260–67. [PubMed] [Google Scholar]
- 139.Granse L, Abrahamson M, Ponjavic V, Andreasson S. Electrophysiological findings in two young patients with Bothnia dystrophy and a mutation in the RLBP1 gene. Ophthalmic Genet. 2001;22:97–105. doi: 10.1076/opge.22.2.97.2231. [DOI] [PubMed] [Google Scholar]
- 140.Saari JC, Nawrot M, Kennedy BN, Garwin GG, Hurley JB, et al. Visual cycle impairment in cellular retinaldehyde binding protein (CRALBP) knockout mice results in delayed dark adaptation. Neuron. 2001;29:739–48. doi: 10.1016/s0896-6273(01)00248-3. [DOI] [PubMed] [Google Scholar]
- 141.Yamamoto H, Simon A, Eriksson U, Harris E, Berson EL, Dryja TP. Mutations in the gene encoding 11-cis retinol dehydrogenase cause delayed dark adaptation and fundus albipunctatus. Nat Genet. 1999;22:188–91. doi: 10.1038/9707. [DOI] [PubMed] [Google Scholar]
- 142.Cideciyan AV, Haeseleer F, Fariss RN, Aleman TS, Jang GF, et al. Rod and cone visual cycle consequences of a null mutation in the 11-cis-retinol dehydrogenase gene in man. Vis Neurosci. 2000;17:667–78. doi: 10.1017/s0952523800175029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Shang E, Lai K, Packer AI, Paik J, Blaner WS, et al. Targeted disruption of the mouse cis-retinol dehydrogenase gene: visual and nonvisual functions. J Lipid Res. 2002;43:590–97. [PubMed] [Google Scholar]
- 144.Jang GF, Van Hooser JP, Kuksa V, McBee JK, He YG, et al. Characterization of a dehydrogenase activity responsible for oxidation of 11-cis-retinol in the retinal pigment epithelium of mice with a disrupted RDH5 gene. A model for the human hereditary disease fundus albipunctatus. J Biol Chem. 2001;276:32456–65. doi: 10.1074/jbc.M104949200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Kootstra NA, Verma IM. Gene therapy with viral vectors. Annu Rev Pharmacol Toxicol. 2003;43:413–39. doi: 10.1146/annurev.pharmtox.43.100901.140257. [DOI] [PubMed] [Google Scholar]
- 146.Verma IM, Weitzman MD. Gene therapy: twenty-first century medicine. Annu Rev Biochem. 2004;74:711–38. doi: 10.1146/annurev.biochem.74.050304.091637. [DOI] [PubMed] [Google Scholar]
- 147.Peden CS, Burger C, Muzyczka N, Mandel RJ. Circulating antiwild-type adeno-associated virus type 2 (AAV2) antibodies inhibit recombinant AAV2 (rAAV2)-mediated, but not rAAV5-mediated, gene transfer in the brain. J Virol. 2004;78:6344–59. doi: 10.1128/JVI.78.12.6344-6359.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Chirmule N, Xiao W, Truneh A, Schnell MA, Hughes JV, et al. Humoral immunity to adeno-associated virus type 2 vectors following administration to murine and nonhuman primate muscle. J Virol. 2000;74:2420–25. doi: 10.1128/jvi.74.5.2420-2425.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Lewin AS, Drenser KA, Hauswirth WW, Nishikawa S, Yasumura D, et al. Ribozyme rescue of photoreceptor cells in a transgenic rat model of autosomal dominant retinitis pigmentosa. Nat Med. 1998;4:967–71. doi: 10.1038/nm0898-967. [DOI] [PubMed] [Google Scholar]
- 150.LaVail MM, Yasumura D, Matthes MT, Drenser KA, Flannery JG, et al. Ribozyme rescue of photoreceptor cells in P23H transgenic rats: long-term survival and late-stage therapy. Proc Natl Acad Sci USA. 2000;97:11488–93. doi: 10.1073/pnas.210319397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Narfstrom K, Katz ML, Bragadottir R, Seeliger M, Boulanger A, et al. Functional and structural recovery of the retina after gene therapy in the RPE65 null mutation dog. Invest Ophthalmol Vis Sci. 2003;44:1663–72. doi: 10.1167/iovs.02-0595. [DOI] [PubMed] [Google Scholar]
- 152.Narfstrom K, Katz ML, Ford M, Redmond TM, Rakoczy E, Bragadottir R. In vivo gene therapy in young and adult RPE65−/− dogs produces long-term visual improvement. J Hered. 2003;94:31–37. doi: 10.1093/jhered/esg015. [DOI] [PubMed] [Google Scholar]
- 153.Hauswirth WW. The consortium project to treat Rpe65 deficiency in humans. Retina. 2005;25:S60. doi: 10.1097/00006982-200512001-00027. [DOI] [PubMed] [Google Scholar]
- 154.Lai CM, Yu MJ, Brankov M, Barnett NL, Zhou X, et al. Recombinant adeno-associated virus type 2-mediated gene delivery into the Rpe65−/− knockout mouse eye results in limited rescue. Genet Vaccines Ther. 2004;2:3. doi: 10.1186/1479-0556-2-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Batten ML, Imanishi Y, Tu DC, Doan T, Zhu L, et al. Pharmacological and rAAV gene therapy rescue of visual functions in a blind mouse model of Leber congenital amaurosis. PLoS Med. 2005;2:e333. doi: 10.1371/journal.pmed.0020333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.van Hooser JP, Aleman TS, He YG, Cideciyan AV, Kuksa V, et al. Rapid restoration of visual pigment and function with oral retinoid in a mouse model of childhood blindness. Proc Natl Acad Sci USA. 2000;97:8623–28. doi: 10.1073/pnas.150236297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Van Hooser JP, Liang Y, Maeda T, Kuksa V, Jang GF, et al. Recovery of visual functions in a mouse model of Leber congenital amaurosis. J Biol Chem. 2002;277:19173–82. doi: 10.1074/jbc.M112384200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Yoshizawa T, Wald G. Photochemistry of lodopsin. Nature. 1967;214:566–71. doi: 10.1038/214566a0. [DOI] [PubMed] [Google Scholar]
- 159.Ablonczy Z, Crouch RK, Goletz PW, Redmond TM, Knapp DR, et al. 11-cis-retinal reduces constitutive opsin phosphorylation and improves quantum catch in retinoid-deficient mouse rod photoreceptors. J Biol Chem. 2002;277:40491–98. doi: 10.1074/jbc.M205507200. [DOI] [PubMed] [Google Scholar]
- 160.Collins MD, Mao GE. Teratology of retinoids. Annu Rev Pharmacol Toxicol. 1999;39:399–430. doi: 10.1146/annurev.pharmtox.39.1.399. [DOI] [PubMed] [Google Scholar]
- 161.Zhao D, McCaffery P, Ivins KJ, Neve RL, Hogan P, et al. Molecular identification of a major retinoic-acid-synthesizing enzyme, a retinaldehyde-specific dehydrogenase. Eur J Biochem. 1996;240:15–22. doi: 10.1111/j.1432-1033.1996.0015h.x. [DOI] [PubMed] [Google Scholar]
- 162.Chambon P. A decade of molecular biology of retinoic acid receptors. FASEB J. 1996;10:940–54. [PubMed] [Google Scholar]
- 163.Delori FC, Staurenghi G, Arend O, Dorey CK, Goger DG, Weiter JJ. In vivo measurement of lipofuscin in Stargardt’s disease–Fundus flavimaculatus. Invest Ophthalmol Vis Sci. 1995;36:2327–31. [PubMed] [Google Scholar]
- 164.Cubeddu R, Taroni P, Hu DN, Sakai N, Nakanishi K, Roberts JE. Photophysical studies of A2-E, putative precursor of lipofuscin, in human retinal pigment epithelial cells. Photochem Photobiol. 1999;70:172–75. [PubMed] [Google Scholar]
- 165.Sparrow JR, Nakanishi K, Parish CA. The lipofuscin fluorophore A2E mediates blue light-induced damage to retinal pigmented epithelial cells. Investigative Ophthal Vis Sci. 2000;41:1981–89. [PubMed] [Google Scholar]
- 166.Schutt F, Davies S, Kopitz J, Holz FG, Boulton ME. Photodamage to human RPE cells by A2-E, a retinoid component of lipofuscin. Invest Ophthalmol Vis Sci. 2000;41:2303–8. [PubMed] [Google Scholar]
- 167.Finnemann SC, Leung LW, Rodriguez-Boulan E. The lipofuscin component A2E selectively inhibits phagolysosomal degradation of photoreceptor phospholipid by the retinal pigment epithelium. Proc Natl Acad Sci USA. 2002;99:3842–47. doi: 10.1073/pnas.052025899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Suter M, Reme C, Grimm C, Wenzel A, Jaattela M, et al. Age-related macular degeneration—the lipofuscin component N-retinyl-N-retinylidene ethanolamine detaches proapoptotic proteins from mitochondria and induces apoptosis in mammalian retinal pigment epithelial cells. J Biol Chem. 2000;275:39625–30. doi: 10.1074/jbc.M007049200. [DOI] [PubMed] [Google Scholar]
- 169.Sparrow JR, Cai B. Blue light-induced apoptosis of A2E-containing RPE: involvement of caspase-3 and protection by Bcl-2. Invest Ophthalmol Vis Sci. 2001;42:1356–62. [PubMed] [Google Scholar]
- 170.Eldred GE, Lasky MR. Retinal age pigments generated by self-assembling lysosomotropic detergents. Nature. 1993;361:724–26. doi: 10.1038/361724a0. [DOI] [PubMed] [Google Scholar]
- 171.Sparrow JR, Parish CA, Hashimoto M, Nakanishi K. A2E, a lipofuscin fluorophore, in human retinal pigmented epithelial cells in culture. Invest Ophthalmol Vis Sci. 1999;40:2988–95. [PubMed] [Google Scholar]
- 172.De S, Sakmar TP. Interaction of A2E with model membranes. Implications to the pathogenesis of age-related macular degeneration. J Gen Physiol. 2002;120:147–57. doi: 10.1085/jgp.20028566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Sparrow JR, Vollmer-Snarr HR, Zhou J, Jang YP, Jockusch S, et al. A2E-epoxides damage DNA in retinal pigment epithelial cells. Vitamin E and other antioxidants inhibit A2E-epoxide formation. J Biol Chem. 2003;278:18207–13. doi: 10.1074/jbc.M300457200. [DOI] [PubMed] [Google Scholar]
- 174.Sparrow JR, Fishkin N, Zhou J, Cai B, Jang YP, et al. A2E, a byproduct of the visual cycle. Vis Res. 2003;43:2983–90. doi: 10.1016/s0042-6989(03)00475-9. [DOI] [PubMed] [Google Scholar]
- 175.Cideciyan AV, Aleman TS, Swider M, Schwartz SB, Steinberg JD, et al. Mutations in ABCA4 result in accumulation of lipofuscin before slowing of the retinoid cycle: a reappraisal of the human disease sequence. Hum Mol Genet. 2004;13:525–34. doi: 10.1093/hmg/ddh048. [DOI] [PubMed] [Google Scholar]
- 176.Steinberg RH. Interactions between the retinal pigment epithelium and the neural retina. Doc Ophthalmol. 1985;60:327–46. doi: 10.1007/BF00158922. [DOI] [PubMed] [Google Scholar]
- 177.Jonas JB, Schneider U, Naumann GO. Count and density of human retinal photoreceptors. Graefes Arch Clin Exp Ophthalmol. 1992;230:505–10. doi: 10.1007/BF00181769. [DOI] [PubMed] [Google Scholar]
- 178.Lois N, Owens SL, Coco R, Hopkins J, Fitzke FW, Bird AC. Fundus autofluorescence in patients with age-related macular degeneration and high risk of visual loss. Am J Ophthalmol. 2002;133:341–49. doi: 10.1016/s0002-9394(01)01404-0. [DOI] [PubMed] [Google Scholar]
- 179.Einbock W, Moessner A, Schnurrbusch UE, Holz FG, Wolf S. Changes in fundus autofluorescence in patients with age-related maculopathy. Correlation to visual function: a prospective study. Graefes Arch Clin Exp Ophthalmol. 2005;243:300–5. doi: 10.1007/s00417-004-1027-3. [DOI] [PubMed] [Google Scholar]
- 180.Bindewald A, Bird AC, Dandekar SS, Dolar-Szczasny J, Dreyhaupt J, et al. Classification of fundus autofluorescence patterns in early age-related macular disease. Invest Ophthalmol Vis Sci. 2005;46:3309–14. doi: 10.1167/iovs.04-0430. [DOI] [PubMed] [Google Scholar]
- 181.Delori FC, Goger DG, Dorey CK. Age-related accumulation and spatial distribution of lipofuscin in RPE of normal subjects. Invest Ophthalmol Vis Sci. 2001;42:1855–66. [PubMed] [Google Scholar]
- 182.Wabbels B, Demmler A, Paunescu K, Wegscheider E, Preising MN, Lorenz B. Fundus autofluorescence in children and teenagers with hereditary retinal diseases. Graefes Arch Clin Exp Ophthalmol. 2006;244:36–45. doi: 10.1007/s00417-005-0043-2. [DOI] [PubMed] [Google Scholar]
- 183.Zhang K, Kniazeva M, Han M, Li W, Yu Z, et al. A 5-bp deletion in ELOVL4 is associated with two related forms of autosomal dominant macular dystrophy. Nat Genet. 2001;27:89–93. doi: 10.1038/83817. [DOI] [PubMed] [Google Scholar]
- 184.Karan G, Lillo C, Yang Z, Cameron DJ, Locke KG, et al. Lipofuscin accumulation, abnormal electrophysiology, and photoreceptor degeneration in mutant ELOVL4 transgenic mice: a model for macular degeneration. Proc Natl Acad Sci USA. 2005;102:4164–69. doi: 10.1073/pnas.0407698102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Lorenz B, Wabbels B, Wegscheider E, Hamel CP, Drexler W, Preising MN. Lack of fundus autofluorescence to 488 nanometers from childhood on in patients with early-onset severe retinal dystrophy associated with mutations in RPE65. Ophthalmology. 2004;111:1585–94. doi: 10.1016/j.ophtha.2004.01.033. [DOI] [PubMed] [Google Scholar]
- 186.D’Cruz PM, Yasumura D, Weir J, Matthes MT, Abderrahim H, et al. Mutation of the receptor tyrosine kinase gene Mertk in the retinal dystrophic RCS rat. Hum Mol Genet. 2000;9:645–51. doi: 10.1093/hmg/9.4.645. [DOI] [PubMed] [Google Scholar]
- 187.Katz ML, Eldred GE, Robison WGJ. Lipofuscin autofluorescence: evidence for vitamin A involvement in the retina. Mech Ageing Dev. 1987;39:81–90. doi: 10.1016/0047-6374(87)90088-1. [DOI] [PubMed] [Google Scholar]
- 188.Liu JH, Itagaki Y, Ben-Shabat S, Nakanishi K, Sparrow JR. The biosynthesis of A2E, a fluorophore of aging retina, involves the formation of the precursor, A2-PE, in the photoreceptor outer segment membrane. J Biol Chem. 2000;275:29354–60. doi: 10.1074/jbc.M910191199. [DOI] [PubMed] [Google Scholar]
- 189.Gal A, Li Y, Thompson DA, Weir J, Orth U, et al. Mutations in MERTK, the human orthologue of the RCS rat retinal dystrophy gene, cause retinitis pigmentosa. Nat Genet. 2000;26:270–71. doi: 10.1038/81555. [DOI] [PubMed] [Google Scholar]
- 190.Rakoczy PE, Zhang D, Robertson T, Barnett NL, Papadimitriou J, et al. Progressive age-related changes similar to age-related macular degeneration in a transgenic mouse model. Am J Pathol. 2002;161:1515–24. doi: 10.1016/S0002-9440(10)64427-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Zhang D, Brankov M, Makhija MT, Robertson T, Helmerhorst E, et al. Correlation between inactive cathepsin D expression and retinal changes in mcd2/mcd2 transgenic mice. Invest Ophthalmol Vis Sci. 2005;46:3031–38. doi: 10.1167/iovs.04-1510. [DOI] [PubMed] [Google Scholar]
- 192.Ambati J, Anand A, Fernandez S, Sakurai E, Lynn BC, et al. An animal model of age-related macular degeneration in senescent Ccl-2- or Ccr-2-deficient mice. Nat Med. 2003;9:1390–97. doi: 10.1038/nm950. [DOI] [PubMed] [Google Scholar]
- 193.Parish CA, Hashimoto M, Nakanishi K, Dillon J, Sparrow J. Isolation and one-step preparation of A2E and iso-A2E, fluorophores from human retinal pigment epithelium. Proc Nat Acad Sci USA. 1998;95:14609–13. doi: 10.1073/pnas.95.25.14609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Qu Z, Fischmeister R, Hartzell C. Mouse bestrophin-2 is a bona fide Cl(−) channel: identification of a residue important in anion binding and conduction. J Gen Physiol. 2004;123:327–40. doi: 10.1085/jgp.200409031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Palczewski K, van Hooser JP, Garwin GG, Chen J, Liou GI, Saari JC. Kinetics of visual pigment regeneration in excised mouse eyes and in mice with a targeted disruption of the gene encoding interphotoreceptor retinoid-binding protein or arrestin. Biochemistry. 1999;38:12012–19. doi: 10.1021/bi990504d. [DOI] [PubMed] [Google Scholar]
- 196.Weleber RG, Denman ST, Hanifin JM, Cunningham WJ. Abnormal retinal function associated with isotretinoin therapy for acne. Arch Ophthalmol. 1986;104:831–37. doi: 10.1001/archopht.1986.01050180065031. [DOI] [PubMed] [Google Scholar]
- 197.Law WC, Rando RR. The molecular basis of retinoic acid induced night blindness. Biochem Biophys Res Commun. 1989;161:825–29. doi: 10.1016/0006-291x(89)92674-0. [DOI] [PubMed] [Google Scholar]
- 198.Gamble MV, Mata NL, Tsin AT, Mertz JR, Blaner WS. Substrate specificities and 13-cis-retinoic acid inhibition of human, mouse and bovine cis-retinol dehydrogenases. Biochim Biophys Acta. 2000;1476:3–8. doi: 10.1016/s0167-4838(99)00232-0. [DOI] [PubMed] [Google Scholar]
- 199.Gollapalli DR, Rando RR. The specific binding of retinoic acid to Rpe65 and approaches to the treatment of macular degeneration. Proc Natl Acad Sci USA. 2004;101:10030–35. doi: 10.1073/pnas.0401936101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Radu RA, Mata NL, Nusinowitz S, Liu X, Sieving PA, Travis GH. Treatment with isotretinoin inhibits lipofuscin accumulation in a mouse model of recessive Stargardt’s macular degeneration. Proc Natl Acad Sci USA. 2003;100:4742–47. doi: 10.1073/pnas.0737855100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Berni R, Formelli F. In vitro interaction of fenretinide with plasma retinol-binding protein and its functional consequences. FEBS Lett. 1992;308:43–45. doi: 10.1016/0014-5793(92)81046-o. [DOI] [PubMed] [Google Scholar]
- 202.Noy N, Xu ZJ. Interactions of retinol with binding proteins: implications for the mechanism of uptake by cells. Biochemistry. 1990;29:3878–83. doi: 10.1021/bi00468a012. [DOI] [PubMed] [Google Scholar]
- 203.Vogel S, Piantedosi R, O’Byrne SM, Kako Y, Quadro L, et al. Retinol-binding protein-deficient mice: biochemical basis for impaired vision. Biochemistry. 2002;41:15360–68. doi: 10.1021/bi0268551. [DOI] [PubMed] [Google Scholar]
- 204.Radu RA, Han Y, Bui TV, Nusinowitz S, Bok D, et al. Reductions in serum vitamin A arrest accumulation of toxic retinal fluorophores: a potential therapy for treatment of lipofuscin-based retinal diseases. Invest Ophthalmol Vis Sci. 2005;46:4393–401. doi: 10.1167/iovs.05-0820. [DOI] [PubMed] [Google Scholar]
- 205.Naik HR, Kalemkerian G, Pienta KJ. 4-Hydroxyphenylretinamide in the chemoprevention of cancer. Adv Pharmacol. 1995;33:315–47. doi: 10.1016/s1054-3589(08)60673-0. [DOI] [PubMed] [Google Scholar]
- 206.Quadro L, Blaner WS, Salchow DJ, Vogel S, Piantedosi R, et al. Impaired retinal function and vitamin A availability in mice lacking retinol-binding protein. EMBO J. 1999;18:4633–44. doi: 10.1093/emboj/18.17.4633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Quadro L, Blaner WS, Hamberger L, Novikoff PM, Vogel S, et al. The role of extrahepatic retinol binding protein in the mobilization of retinoid stores. J Lipid Res. 2004;45:1975–82. doi: 10.1194/jlr.M400137-JLR200. [DOI] [PubMed] [Google Scholar]
- 208.Golczak M, Kuksa V, Maeda T, Moise AR, Palczewski K. Positively charged retinoids are potent and selective inhibitors of the trans-cis isomerization in the retinoid (visual) cycle. Proc Natl Acad Sci USA. 2005;102:8162–67. doi: 10.1073/pnas.0503318102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Golczak M, Imanishi Y, Kuksa V, Maeda T, Kubota R, Palczewski K. Lecithin:retinol acyltransferase is responsible for amidation of retinylamine, a potent inhibitor of the retinoid cycle. J Biol Chem. 2005;280:42263–73. doi: 10.1074/jbc.M509351200. [DOI] [PubMed] [Google Scholar]
- 210.Maiti P, Kong J, Kim SR, Sparrow JR, Allikmets R, Rando RR. Small molecule Rpe65 antagonists limit the visual cycle and prevent lipofuscin formation. Biochemistry. 2006;45:852–60. doi: 10.1021/bi0518545. [DOI] [PubMed] [Google Scholar]
- 211.Sieving PA, Chaudhry P, Kondo M, Provenzano M, Wu D, et al. Inhibition of the visual cycle in vivo by 13-cis retinoic acid protects from light damage and provides a mechanism for night blindness in isotretinoin therapy. PNAS. 2001;98:1835–40. doi: 10.1073/pnas.041606498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Imanishi Y, Batten ML, Piston DW, Baehr W, Palczewski K. Noninvasive two-photon imaging reveals retinyl ester storage structures in the eye. J Cell Biol. 2004;164:373–83. doi: 10.1083/jcb.200311079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Organisciak DT, Darrow RM, Barsalou L, Kutty RK, Wiggert B. Circadian-dependent retinal light damage in rats. Invest Ophthalmol Vis Sci. 2000;41:3694–701. [PubMed] [Google Scholar]
- 214.Gu X, Meer SG, Miyagi M, Rayborn ME, Hollyfield JG, et al. Carboxyethylpyrrole protein adducts and autoantibodies, biomarkers for age-related macular degeneration. J Biol Chem. 2003;278:42027–35. doi: 10.1074/jbc.M305460200. [DOI] [PubMed] [Google Scholar]
- 215.Crabb JW, Miyagi M, Gu X, Shadrach K, West KA, et al. Drusen proteome analysis: an approach to the etiology of age-related macular degeneration. Proc Natl Acad Sci USA. 2002;99:14682–87. doi: 10.1073/pnas.222551899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.AREDS. The Age-Related Eye Disease Study (AREDS): design implications. AREDS report no 1. Cont Clin Tri. 1999;20:573–600. doi: 10.1016/s0197-2456(99)00031-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Allikmets R, Shroyer NF, Singh N, Seddon JM, Lewis RA, et al. Mutation of the Stargardt disease gene (ABCR) in age-related macular degeneration. Science. 1997;277:1805–7. doi: 10.1126/science.277.5333.1805. [DOI] [PubMed] [Google Scholar]
- 218.Maugeri A, Klevering BJ, Rohrschneider K, Blankenagel A, Brunner HG, et al. Mutations in the ABCA4 (ABCR) gene are the major cause of autosomal recessive cone-rod dystrophy. Am J Hum Genet. 2000;67:960–66. doi: 10.1086/303079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Rozet JM, Gerber S, Ghazi I, Perrault I, Ducroq D, et al. Mutations of the retinal specific ATP binding transporter gene (ABCR) in a single family segregating both autosomal recessive retinitis pigmentosa RP19 and Stargardt disease: evidence of clinical heterogeneity at this locus. J Med Genet. 1999;36:447–51. [PMC free article] [PubMed] [Google Scholar]
- 220.Morimura H, Fishman GA, Grover SA, Fulton AB, Berson EL, Dryja TP. Mutations in the RPE65 gene in patients with autosomal recessive retinitis pigmentosa or leber congenital amaurosis. Proc Natl Acad Sci USA. 1998;95:3088–93. doi: 10.1073/pnas.95.6.3088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Liu L, Gudas LJ. Disruption of the lecithin:retinol acyltransferase gene makes mice more susceptible to vitamin A deficiency. J Biol Chem. 2005;280:40226–34. doi: 10.1074/jbc.M509643200. [DOI] [PubMed] [Google Scholar]
- 222.Katz ML, Redmond TM. Effect of Rpe65 knockout on accumulation of lipofuscin fluorophores in the retinal pigment epithelium. Invest Ophthalmol Vis Sci. 2001;42:3023–30. [PubMed] [Google Scholar]
RELATED REVIEWS
- Mark M, Ghyselinck NB, Chambon P. Function of retinoid nuclear receptors: lessons from genetic and pharmacological dissections of the retinoic acid signaling pathway during mouse embryogenesis. Annu Rev Pharmacol Toxicol. 2006;46:451–80. doi: 10.1146/annurev.pharmtox.46.120604.141156. [DOI] [PubMed] [Google Scholar]
- Chytil F. Rough and rocky road to the retinoid revolution. Annu Rev Nutr. 2003;23:1–16. doi: 10.1146/annurev.nutr.23.011702.073155. [DOI] [PubMed] [Google Scholar]
- Palczewski K. G protein-coupled receptor rhodopsin. Annu Rev Biochem. 2006;75:743–67. doi: 10.1146/annurev.biochem.75.103004.142743. [DOI] [PMC free article] [PubMed] [Google Scholar]