

Abstract
Engineering the permanent formation of a receptor-ligand complex has a number of promising applications in chemistry, biology, and medicine. Antibodies and other proteins can be excellent receptors for synthetic ligands such as probes or drugs. Because proteins possess an array of nucleophilic sites, the placement of an electrophile on the synthetic ligand to react with a nucleophile on the macromolecule is a standard practice. Previously, we have used the site-directed incorporation of cysteine nucleophiles at the periphery of an antibody’s binding site, paired with the chemical design of weakly electrophilic ligands, to produce receptor-ligand pairs that conjugate specifically and permanently (Corneillie et al. (2004) Bioconjugate Chem.15, 1392-1402 and references therein). After protein expression in Drosophila S2 cells we found, as is frequently observed, that the engineered cysteine was reversibly blocked by disulfide linkage to a cysteine monomer (cysteinylated). Removal of the cysteine monomer requires some care because of the need to preserve other disulfide linkages in the protein. Here we report that cysteinylation can be used to advantage by treating the cysteine monomer as a leaving group and the protein disulfide as an electrophile with special affinity for thiols. Two ligands bearing thiol side chains were synthesized and incubated with the cysteinylated antibody Fab fragment 2D12.5 G54C, with the finding that both ligands become covalently attached within a few minutes under physiological conditions. The attachment is robust even in the presence of excess thiol reagents. This rapid, specific conjugation is particularly interesting for biomedical applications.
Introduction
According to Means and Feeney (1), site-specific derivatization of proteins (affinity labeling) began with the attachment of haloacetyl reagents to enzyme active sites, resulting in irreversible inhibition (e.g., 2, 3), and the diazonium coupling of a hapten to an antibody’s binding site (4). The derivatizing reagents were prepared by synthesizing a substrate or ligand for the macromolecular target, and incorporating an electrophilic reactive group. The ligand’s strong affinity for a particular site concentrates the reactive group there, where its reaction with a nearby amino acid side chain is promoted by mutual proximity. This approach has been widely employed in biomedical research, from protein structural studies to the development of new pharmaceuticals (e.g., 5). Notably, even the antibiotic penicillin may be regarded as an affinity label (6).
Recently, an enhanced form of affinity labeling has been introduced which is aimed at the development of macromolecules that permanently capture small probe molecules (7, 8, 9, 10, 11, 12). Here the macromolecular target site is genetically modified to make a reactive partner for the affinity label, thus permitting rapid attachment by modestly reactive synthetic ligands that bind near the mutated residue. A related strategy, used for drug discovery screening, is tethering library ligands to a cysteine-mutant receptor by disulfide formation (13, 14, 15). Here the small molecules are in disulfide form, to react with free cysteine on the protein; a reducing buffer containing a thiol provides selective pressure so that only the ligands with highest affinity remain bound in vitro.
Because proteins and other biological molecules commonly possess an array of nucleophilic sites but rarely contain electrophilic groups, the placement of an electrophile on the synthetic ligand to react with a nucleophile on the macromolecule seems to be a universal practice (e.g., 13, 14, 15, 16, 17, 18, 19, 20). A potential weakness of this strategy is that the electrophilic reagent may react with nucleophiles other than the target because there are so many. This is manifested by the occasional development of allergic reactions to drugs such as penicillin (21).
For the capture of probe molecules, it is attractive to use an antibody’s binding site and engineer the antibody to display an appropriately located cysteine thiol side chain because of its distinctive nucleophilicity (12). However, in practice it is frequently observed that such Cys-mutant proteins have their solvent-accessible cysteines reversibly blocked by disulfide linkage to a cysteine monomer, or sometimes another natural thiol such as glutathione or coenzyme A (22, 23).
Reductive removal of this naturally occurring protecting group without reducing the natural disulfide bonds of an antibody is difficult to achieve quantitatively (10, 24). While seeking improved strategies for this step, we conjectured that rather than removing the cysteine monomer during purification of the protein, it might be possible to use protein cysteinylation as an advantage by thinking of the disulfide as a specialized electrophile and the cysteine monomer as a leaving group. An affinity label bearing a nucleophilic thiol group might bind and displace the cysteine, forming a new disulfide bond as illustrated in Figure 1. This hypothesis was plausible because we had engineered the antibody such that this cysteine would lie to the side of the antibody’s binding site, not likely to interfere with normal binding of the synthetic ligand. Here we describe experimental proof that this reaction does indeed take place, with a rate and efficiency that point to further development.
Figure 1.
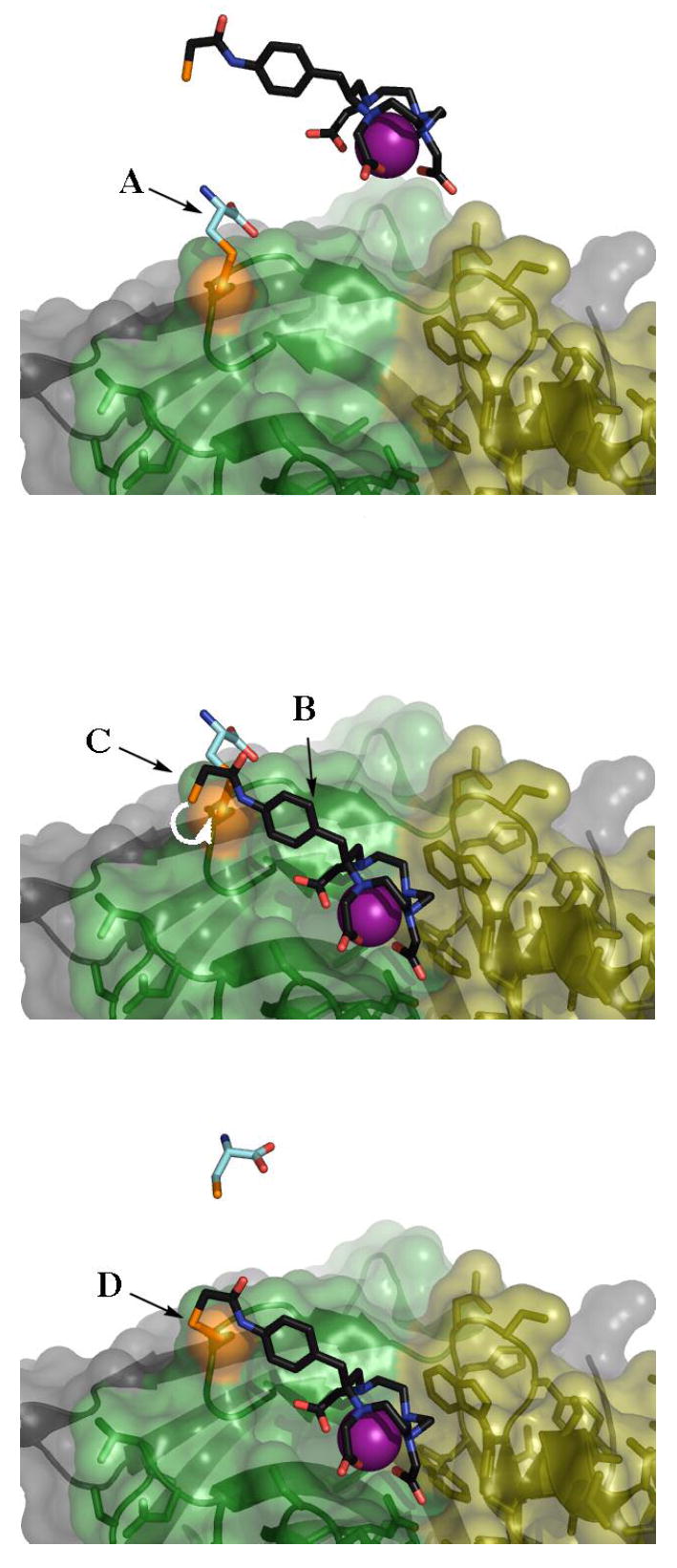
Infinite affinity binding of ligand 1 with cysteinylated 2D12.5 G54C Fab. Top: a cysteine monomer (A), is in disulfide linkage with the G54C when the protein is first expressed. The binding pocket of the 2D12.5 G54C Fab is pictured with CDR heavy chain regions shown in green, CDR light chain regions shown in yellow and constant regions shown in gray. Ligand 1 is rendered in cpk colors, with the metal in purple. All sulfur atoms involved in the bond formation are colored orange. Middle: association of chelate 1 with the binding pocket of the Fab (B) allows the free thiol group on the ligand to displace the cysteine monomer (C). Bottom: the disulfide bond (D) between the 2D12.5 G54C Fab and chelate 1 prevents dissociation of the ligand from the binding pocket.
We expressed the cysteinylated anti-DOTA1 antibody Fab fragment 2D12.5 G54C in Drosophila S2 cells as described (10). After DTT reduction, we used amino-acid analysis to confirm the release of cysteine monomer from the Fab.2 As test affinity labels we synthesized nucleophilic compounds 1 and 2 to compare with previously reported electrophiles 3 and 4 (Chart 1). The method of Weiss (25) was used to prepare 1 from thiocyanate, bromoacetate, and aminobenzyl-DOTA; Traut’s reagent was used to prepare 2.2
Chart 1.

M = yttrium(III).
Determination of Covalent Bond Formation between the Cysteinylated 2D12.5 G54C Fab Protein and 90Y-1 or 90Y-2
As suggested by Figure 1, each chelate was added to cysteinylated Fab to allow the formation of a new disulfide linkage. To assure that the measured rates of covalent attachment would reflect the reaction in the binding site rather than the rate of association, the final concentration of the 90Y-chelate was 10 μM and the protein was 1 μM in PBS buffer, pH 7.4. This solution was allowed to incubate at 37 °C for periods up to 160 min.2
In each case the half-lives for covalent attachment were a few minutes at 37 °C, pH 7.4, and the attached ligands migrated with the protein during denaturing (non-reducing) gel electrophoresis. The results for ligands 1 and 2, shown in Figure 2, are similar to the previous results for electrophiles 3 and 4 reacting with the free sulfhydryl form of this antibody (10).
Figure 2.
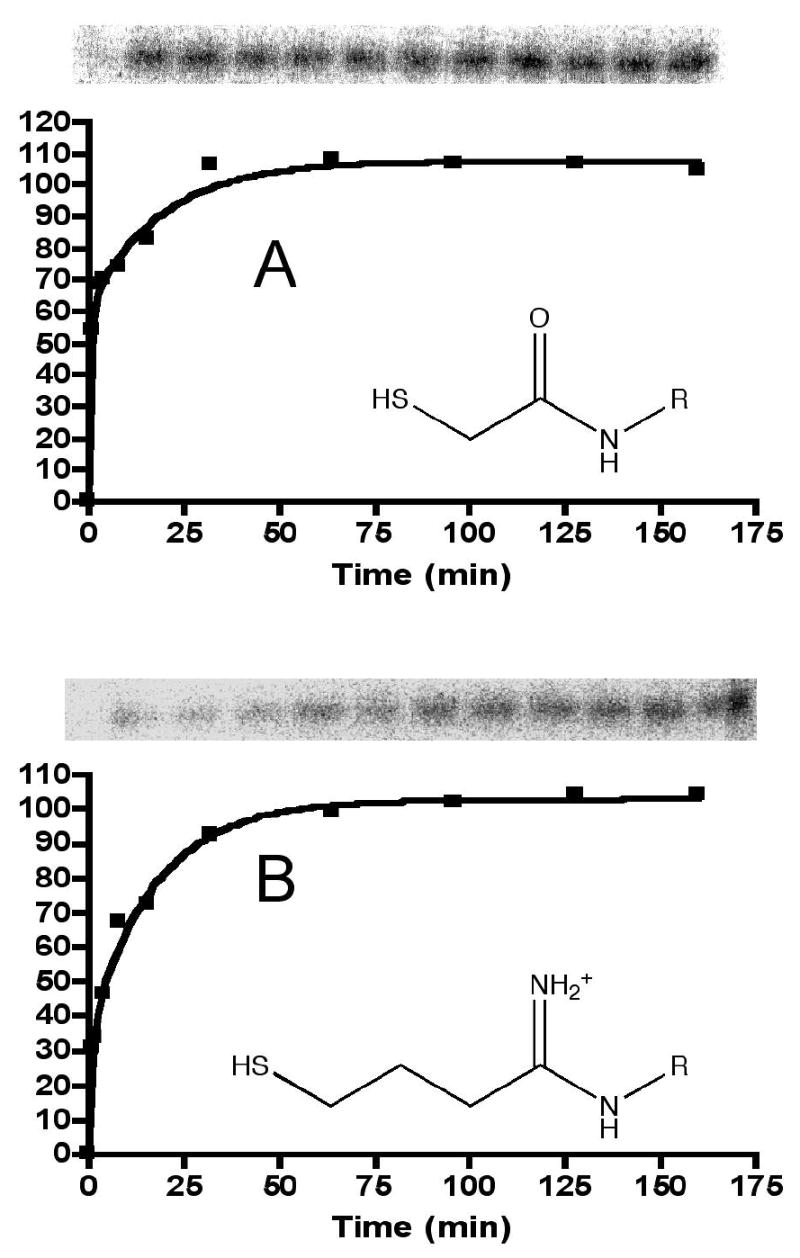
Panel A: covalent attachment of 90Y-1 to 2D12.5 G54C Fab as a function of time at 37 °C, pH 7.4, showing gel phosphorimages and plot of density. Panel B: covalent attachment of 90Y-2 to 2D12.5 G54C Fab.
Quantitation of Covalent Bond Formation between the Cysteinylated 2D12.5 G54C Fab and 90Y-1 or 90Y-3 Before and After Mild Reduction
As will be shown below, the protein as expressed contains a small fraction of heavy chain cysteine-54 with a free thiol side chain (~7%), and a larger fraction of cysteinylated cysteine-54. Normally before the 2D12.5 G54C Fab protein can react to a satisfactory degree with either 3 or 4 it must be reduced in order to increase the proportion of free thiol side chain at Cys-54, because the disulfide is unreactive with electrophiles. This must be a mild treatment in order to avoid reduction of the structurally important disulfides, such as the interchain disulfide between the light and heavy chains of the Fab protein.
A sample of 2D12.5 G54C was treated with 25 μM DTT1 followed by overnight incubation at 4 °C (10). Samples of 2D12.5 Fab G54C with or without DTT treatment were incubated with excess 90Y-1 or 90Y-3, followed by gel filtration to remove excess chelate not associated with the Fab. The resulting samples of Fab contained both reversibly bound ligand and covalently bound ligand. To determine the percentage of chelate covalently bound to the protein, reversibly bound chelate was separated from the protein on a denaturing (nonreducing) polyacrylamide gel.2
The results for the four samples, reduced or non-reduced 2D12.5 G54C Fab reacting with either 90Y-1 or 90Y-3, are shown in Table 1. For the naturally expressed protein without DTT treatment, covalent bond formation with thiol 90Y-1 was approximately 11× more likely than reaction with acrylamide 90Y-3. Mild reduction of the cysteinylated Fab increased covalent attachment of electrophile 90Y-3 approximately 6×, but had little effect on the covalent attachment of 90Y-1. The reason for 90Y-1 having such a high efficiency here is that it becomes partially oxidized during metallation: for this experiment 29% was in the disulfide form,2 which can react with the reduced Cys-54.
Table 1.
Covalent attachment, as a percentage of total bound ligand, of 90Y-1 and 90Y-3 to 2D12.5 G54C Fab with and without DTT treatment.2
| 2D12.5 G54C | 2D12.5 G54C w/DTT | |
|---|---|---|
| 90Y-1* | 83 ± 4 % | 77 ± 5 % |
| 90Y-3 | 7.6 ± 0.1 % | 47 ± 1 % |
90Y-1 had a notably high efficiency of binding to DTT-treated G54C, which is only partially reduced to the sulfhydryl form, because the 90Y-1 sample contained both the free sulfhydryl (71%) and the disulfide form (29%) of 90Y-1.
Having formed the disulfide, we sought an indication of how stable the bond is. Because the binding site has high affinity for the DOTA chelate, the loss of the chelate after disulfide reduction should be disfavored. This may be envisioned as similar to the situation of the light and heavy chains of the Fab, which are held together by strong van der Waals interactions in addition to the interchain disulfide bond. We treated samples of the Y-2-Fab disulfide with increasing amounts of BME.1 We analyzed the samples by denaturing, non-reducing gel electrophoresis immediately after removing the BME by a desalting column (Figure 3), finding that there is neither significant reduction of the Fab interchain disulfide nor loss of Y-2 until the concentration of free BME exceeds 1 mM, where a small amount of interchain disulfide reduction becomes detectable. At 10 mM BME, the interchain disulfide is half reduced, whereas the Y-2-Fab disulfide is only approximately 20% reduced.2 It is also notable that a significant portion of the 25kD disulfide-linked Y-2-heavy chain is produced at higher BME concentrations, confirming that the interchain disulfide is reduced while the Y-2 disulfide remains intact. Since the Y-2-Fab disulfide bond is more stable than the interchain disulfide, Y-2-Fab should remain intact under physiological conditions.
Figure 3.
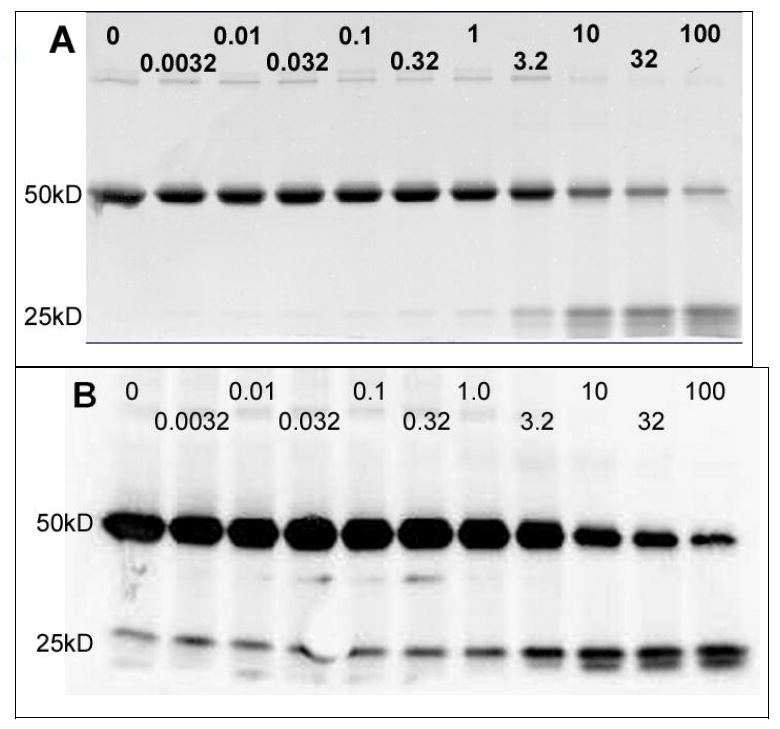
Sypro total protein stained gel (A) and anti-DOTA western blot (B) analysis of samples of 2D12.5 G54C Fab conjugated to Y-2 by disulfide bond formation. From left to right in each panel, the samples were treated with increasing concentrations of β-mercaptoethanol; numbers shown are BME concentration, mM.
While all four ligands in Chart 1 were designed to be unreactive in ordinary circumstances but to react quickly upon binding to the specific antibody, the thiol-disulfide reaction is particularly mild. The properties of the disulfide bond are of special interest for probe capture applications, because under physiological conditions the only likely reaction is thiol-disulfide interchange. In the human circulatory system, thiol-disulfide interchange reactions are possible since both thiols and disulfides are present. Normally there are micromolar concentrations of small thiol-containing molecules in human plasma. However, studies of the relative concentrations of cysteine (≈ 9 μM) and its oxidation partner cystine (≈ 47 μM), compared to glutathione (≈ 2 μM) and oxidized glutathione (≈ 0.05 μM), show that the partners are not in equilibrium (26). Thus interchange reactions do not occur efficiently in the absence of a special circumstance such as proximity in a binding site.
Another interesting feature of this system is that it may be possible to displace the cysteine monomer in vitro with other thiols, either natural or synthetic, before use with the affinity label. In this way we might adjust the reactivity of the disulfide, or perhaps attach a drug or probe molecule to be released upon ligand binding.
Supporting Information Available
Experimental details and spectra for chelate synthesis, determination of the Cys-blocking residue, proof of the specificity of the disulfide that is formed, and comparison of disulfide stability (24 pages, PDF). See any current masthead page for ordering information and Web access instructions.
Acknowledgments
We thank other members of the Meares lab for their contributions and helpful discussions, and NIH Research Grants CA016861 and CA098207 (C.F.M.) for support.
Footnotes
Abbreviations: BME, β-mercaptoethanol; DOTA, 1,4,7,10-tetraazacyclododecane- N,N′,N″,N′″-tetraacetic acid; DTT, dithiothreitol; Fab, antigen-binding fragment of an antibody.
Experimental details are provided in Supporting Information.
References
- 1.Means GE, Feeney RE. Chemical Modifications of Proteins: History and Applications. Bioconjugate Chem. 1990;1:2–12. doi: 10.1021/bc00001a001. [DOI] [PubMed] [Google Scholar]
- 2.Baker BR, Lee WW, Tong E, Ross LO. Potential Anticancer Agents. LXVI. Non-classical Antimetabolites. III. 4-(Iodoacetamido)-Salicylic Acid, an Exo Alkylating Irreversible Inhibitor of Glutamic Dehydrogenase. J Am Chem Soc. 1961;83:3713–3714. [Google Scholar]
- 3.Schoellmann G, Shaw E. Direct evidence for the presence of histidine in the active center of chymotrypsin. Biochemistry. 1963;2:252–255. doi: 10.1021/bi00902a008. [DOI] [PubMed] [Google Scholar]
- 4.Wofsy L, Metzger H, Singer SJ. Affinity labeling-A general method for labeling the active sites of antibody and enzyme molecules. Biochemistry. 1962;1:1031–1039. doi: 10.1021/bi00912a013. [DOI] [PubMed] [Google Scholar]
- 5.Fry DW, Bridges AJ, Denny WA, Doherty A, Greis KD, Hicks JL, Hook KE, Keller PR, Leopold WR, Loo JA, McNamara DJ, Nelson JM, Sherwood V, Smaill JB, Trumpp-Kallmeyer S, Dobrusin EM. Specific, irreversible inactivation of the epidermal growth factor receptor and erbB2, by a new class of tyrosine kinase inhibitor. Proc Natl Acad Sci USA. 1998;95:12022–12027. doi: 10.1073/pnas.95.20.12022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yocum RR, Waxman DJ, Rasmussen JR, Strominger JL. Mechanism of penicillin action: penicillin and substrate bind covalently to the same active site serine in two bacterial D-alanine carboxypeptidases. Proc Natl Acad Sci USA. 1979;76:2730–2734. doi: 10.1073/pnas.76.6.2730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chmura AJ, Orton MS, Meares CF. Antibodies with infinite affinity. Proc Natl Acad Sci USA. 2001;98:8480–8484. doi: 10.1073/pnas.151260298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chmura AJ, Schmidt BD, Corson DT, Traviglia ST, Meares CF. Electrophilic chelating agents for binding of metal chelates to engineered antibodies. J Controlled Release. 2002;78:249–258. doi: 10.1016/s0168-3659(01)00485-0. [DOI] [PubMed] [Google Scholar]
- 9.Corneillie TM, Whetstone PA, Lee KC, Wong JP, Meares CF. Converting weak binders into infinite binders. Bioconjugate Chem. 2004;15:1389–1391. doi: 10.1021/bc049825e. [DOI] [PubMed] [Google Scholar]
- 10.Corneillie TM, Lee KC, Whetstone PA, Wong JP, Meares CF. Irreversible Engineering of the Multielement-Binding Antibody 2D12. 5 and Its Complementary Ligands. Bioconjugate Chem. 2004;15:1392–1402. doi: 10.1021/bc049824m. [DOI] [PubMed] [Google Scholar]
- 11.Corneillie TM, Whetstone PA, Meares CF. Irreversibly binding anti-metal chelate antibodies: Artificial receptors for pretargeting. J Inorg Biochem. 2006;100:882–890. doi: 10.1016/j.jinorgbio.2006.01.004. [DOI] [PubMed] [Google Scholar]
- 12.Butlin NG, Meares CF. Antibodies with infinite affinity: origins and applications. Accts Chem Res. 2006;39:780–787. doi: 10.1021/ar020275e. [DOI] [PubMed] [Google Scholar]
- 13.Erlanson DA, Braisted AC, Raphael DR, Randal M, Stroud RM, Gordon EM, Wells JA. Site-directed ligand discovery. Proc Natl Acad Sci USA. 2000;97:9367–9372. doi: 10.1073/pnas.97.17.9367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Arkin MR, Randal M, DeLano WL, Hyde J, Luong TN, Oslob JD, Raphael DR, Taylor L, Wang J, McDowell RS, Wells JA, Braisted AC. Binding of small molecules to an adaptive protein-protein interface. Proc Natl Acad Sci USA. 2003;100:1603–1608. doi: 10.1073/pnas.252756299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Buck E, Wells JA. Disulfide trapping to localize small-molecule agonists and antagonists for a G protein-coupled receptor. Proc Natl Acad Sci USA. 2005;102:2719–2724. doi: 10.1073/pnas.0500016102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pollack SJ, Nakayama GR, Schultz PG. Science. Vol. 242. Washington, DC: 1988. Introduction of nucleophiles and spectroscopic probes into antibody combining sites; pp. 1038–1040. [DOI] [PubMed] [Google Scholar]
- 17.Salerno A, Lawrence DS. Covalent modification with concomitant inactivation of the cAMP-dependent protein kinase by affinity labels containing only L-amino acids. J Biol Chem. 1993;268:13043–13049. [PubMed] [Google Scholar]
- 18.Yan XW, Corbin JD, Francis SH, Lawrence DS. Precision targeting of protein kinases - An affinity label that inactivates the cGMP- but not the cAMP-dependent protein kinase. J Biol Chem. 1996;271:1845–1848. doi: 10.1074/jbc.271.4.1845. [DOI] [PubMed] [Google Scholar]
- 19.Levitsky K, Ciolli CJ, Belshaw PJ. Selective inhibition of engineered receptors via proximity-accelerated alkylation. Org Lett. 2003;5:693–696. doi: 10.1021/ol027448k. [DOI] [PubMed] [Google Scholar]
- 20.Chen G, Heim A, Riether D, Yee D, Milgrom Y, Gawinowicz MA, Sames D. Reactivity of functional groups on the protein surface: development of epoxide probes for protein labeling. J Am Chem Soc. 2003;125:8130–8133. doi: 10.1021/ja034287m. [DOI] [PubMed] [Google Scholar]
- 21.Blanca M, Cornejo-Garcia JA, Torres MJ, Mayorga C. Specificities of B cell reactions to drugs. The penicillin model. Toxicology. 2005;209:181–184. doi: 10.1016/j.tox.2004.12.018. [DOI] [PubMed] [Google Scholar]
- 22.Gadgil HS, Bondarenko PV, Pipes GD, Dillon TM, Banks D, Abel J, Kleemann GR, Treuheit MJ. Identification of cysteinylation of a free cysteine in the Fab region of a recombinant monoclonal IgG1 antibody using Lys-C limited proteolysis coupled with LC/MS analysis. Anal Biochem. 2006;355:165–174. doi: 10.1016/j.ab.2006.05.037. [DOI] [PubMed] [Google Scholar]
- 23.Odaka M, Kiribuchi K, Allison WS, Yoshida M. In vivo affinity label of a protein expressed in Escherichia coli. Coenzyme A occupied the AT(D)P binding site of the mutant F1-ATPase beta subunit (Y307C) through a disulfide bond. FEBS Lett. 1993;336:231–235. doi: 10.1016/0014-5793(93)80809-9. [DOI] [PubMed] [Google Scholar]
- 24.Stimmel JB, Merrill BM, Kuyper LF, Moxham CP, Hutchins JT, Fling ME, Kull FC., Jr Site-specific conjugation on serine right-arrow cysteine variant monoclonal antibodies. J Biol Chem. 2000;275:30445–30450. doi: 10.1074/jbc.M001672200. [DOI] [PubMed] [Google Scholar]
- 25.Weiss U. N-Arylamides of Mercaptoacetic Acid. I. Analogs of a Carbamylmercaptoacetanilide. Am Chem Soc. 1947;69:2682–2684. [Google Scholar]
- 26.Jones DP, Mody VC, Jr, Carlson JL, Lynn MJ, Sternberg P., Jr Redox analysis of human plasma allows separation of pro-oxidant events of aging from decline in antioxidant defenses. Free Radic Biol Med. 2002;33:1290–1300. doi: 10.1016/s0891-5849(02)01040-7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Experimental details and spectra for chelate synthesis, determination of the Cys-blocking residue, proof of the specificity of the disulfide that is formed, and comparison of disulfide stability (24 pages, PDF). See any current masthead page for ordering information and Web access instructions.