

Abstract
We investigated the relative free energies of hapten binding to the germ line and mature forms of the 48G7 antibody Fab fragments by applying a continuum model to structures sampled from molecular dynamics simulations in explicit solvent. Reasonable absolute and very good relative free energies were obtained. As a result of nine somatic mutations that do not contact the hapten, the affinity-matured antibody binds the hapten >104 tighter than the germ line antibody. Energetic analysis reveals that van der Waals interactions and nonpolar contributions to solvation are similar and drive the formations of both the germ line and mature antibody–hapten complexes. Affinity maturation of the 48G7 antibody therefore appears to occur through reorganization of the combining site geometry in a manner that optimizes the balance of gaining favorable electrostatic interactions with the hapten and losing those with solvent during the binding process. As reflected by lower rms fluctuations in the antibody–hapten complex, the mature complex undergoes more restricted fluctuations than the germ line complex. The dramatically increased affinity of the 48G7 antibody over its germ line precursor is thus made possible by electrostatic optimization.
The immune system is capable of producing antibodies that can bind virtually any molecule with high affinity through the combinatorial association of variable, joining, and diversity genes with subsequent affinity maturation (1). Affinity maturation involves repeated cycles of somatic mutations in the variable regions, preferentially stimulating high-affinity antibodies in the resulting diverse pool of antibodies until an optimal affinity is achieved (2, 3). Indeed, nature has solved many of the complex problems associated with molecular recognition by generating and screening a large library of proteins over the course of a few weeks during an immune response. By unraveling the mechanism behind molecular recognition, we will gain a deeper understanding of the principles that lead to high-affinity and selective inhibitors for a target receptor in the area of drug design.
Over the past decade, chemists have tapped the enormous diversity of the immune system to obtain antibody catalysts for chemical reactions of interest by immunizing with transition-state analog haptens. Research involving catalytic antibodies has provided us with structural insights into the process of affinity maturation. Specifically, high-resolution x-ray crystal structures for the germ line and mature Fab fragments of the esterolytic antibody 48G7, by themselves and in complex with the p-nitrophenyl phosphonate hapten, have been attained (4, 5). As a result of nine somatic mutations that are distant from the combining site, the mature antibody has a >104 greater affinity for the hapten with a dissociation constant (Kd) of 10 nM, compared with a Kd of 135 μM for the germ line antibody (6). Conformational changes occur in the variable region of the germ line antibody in response to binding hapten; these changes become preorganized in the combining site of the mature antibody, which undergoes little change in structure on binding hapten. Structural aspects that lead to the dramatic difference in binding affinities between the germ line and mature antibody–hapten complexes are not well understood. The availability of high-resolution structural information on the 48G7 antibody presents the opportunity to perform a detailed energetic analysis of the system.
A practical approach to calculating free energies is one developed by Srinivasan et al. in 1998 (7), which involves applying a continuum model to solute configurations sampled as “snapshots” from a molecular dynamics (MD) simulation by using explicit solvent. For each solute configuration, a molecular mechanical energy is determined. Free energies of solvation are estimated by using finite-difference Poisson–Boltzmann (PB) calculations for the electrostatics contribution and a surface-area-dependent term for the nonelectrostatic contribution to solvation. Solute entropic contributions are estimated from a harmonic analysis. This approach has been used to investigate nucleic acid conformational problems, such as the relative stability of the “A” and “B” forms of nucleic acid duplexes (7, 8); it has also been used for a protein–RNA complex (C. Reyes and P.A.K., unpublished observations) and for different mutants of a peptide–protein complex (9). Here the approach of Srinivasan et al. (7) is applied to a small molecule–protein complex problem: the relative binding free energies of the 48G7 germ line and mature antibody–hapten complexes.
Methods
Preparation.
Coordinates were extracted from the x-ray crystal structures of the 48G7 germ line and mature Fab fragments complexed with the p-nitrophenyl phosphonate hapten (1aj7 and 1 gaf for the germ line and mature Fab–hapten complexes, respectively, in the Protein Data Bank) (4, 5). All crystallographic waters were included (151 waters for the mature Fab–hapten complex). Hydrogen atoms were added to the heavy-atom positions of the crystal structures with the Leap module of the amber 5.0 package (10). HisL189, HisL198, and HisL55 (in the mature Fab–hapten complex only) were protonated at the Δ-nitrogen; HisH35, HisH164, and HisH200 were protonated at the ɛ-nitrogen [light chain (L) and heavy chain (H)]. Charges for the hapten were obtained by restrained electrostatic potential fitting (11), and the electrostatic potentials were produced by single-point quantum mechanical calculations at the Hartree–Fock level with a 6–31G* basis set. The resulting partial atomic charges of the hapten are shown in supplemental Table 5 (see www.pnas.org). Bond, angle, and dihedral parameters for the hapten not included in the Cornell et al. force field (12) are listed in supplemental Table 6.
Simulation.
MD simulations were performed by using the Cornell et al. force field (12) and the amber 5.0 suite of programs (10). Long-range nonbonded interactions were truncated by using a 12-Å residue-based cutoff. The SHAKE algorithm was applied to constrain all bonds to their equilibrium values, thus removing high-frequency vibrations (13). Each system was solvated by placing a spherical cap of TIP3P water molecules (14) with a radius of 25 Å from the geometric center of the hapten. Unfavorable interactions within the structures were relieved with steepest descent followed by conjugate gradient energy minimization until the rms of the elements in the gradient vector was less than 10−4 kcal/(mol⋅Å). Solvent in the systems (including crystallographic waters) was then equilibrated for 25 ps while raising the temperature from 0 K to 300 K. A second equilibration phase of 100 ps at 300 K was applied with the “belly” option in which only those residues within 12 Å of the hapten geometric center were allowed to move. Constant temperature was maintained by the Berendsen coupling algorithm (15) with separate solute–solvent and solvent–solvent coupling. Sampling of reasonable configurations for the given stable states of the antibody–hapten complex structures was conducted by running a 500-ps belly simulation with a 2-fs time step at 300 K.
Energetic Analysis.
The general strategy used here involves calculating energies for “snapshot” configurations taken from the MD trajectories of the antibody–hapten complexes and then averaging the values (7). Unbound antibody and hapten snapshots were also taken from the antibody–hapten complex trajectories. Fifty snapshots taken at 10-ps intervals from each antibody–hapten complex trajectory were used for analysis. For consistency, all waters were removed from each snapshot before energy calculations were performed. Removal of crystallographic waters was not a concern, because none of the waters lie within the combining site.
Total molecular-mechanical energies (Egas) and internal energy (Eint where int = bonds, angles, and dihedrals), van der Waals (EvdW), and electrostatic (Eelec) components were determined by using the anal module of the amber 5.0 package (10) with the same force field (12) used in the MD simulations, with the inclusion of solute–solute pairwise interactions. No cutoff was used for the evaluation of nonbonded interactions.
Solute entropic contributions were estimated for the 50th snapshots taken from the trajectories by using the nmode module of the amber 5.0 package (10), which involves a harmonic approximation to the normal modes and standard (quantum) formulas. Residues lying outside of the belly region in the simulations were removed before minimizing and proceeding with normal mode analysis. All minimizations and normal mode calculations were performed with a distance-dependent dielectric (ɛ = 4 r, where r = interatomic distance in Å) to mimic solvent screening. Steepest descent followed by conjugate gradient minimizations was carried out with a nonbonded cutoff of 12 Å until the rms of the elements in the gradient vector was less than 10−4 kcal/(mol⋅Å). The structures were further minimized with no cutoff for nonbonded interactions by using conjugate gradient and then Newton–Raphson minimizations until the rms of the elements in the gradient vector was less than 10−5 kcal/(mol⋅Å). Normal mode calculations were then carried out with no cutoff for nonbonded interactions.
The electrostatic contribution to the solvation free energy (GPB) was determined with the PB approach (16). This approach involves using a continuum solvent model, which represents the solute as a low dielectric medium (ɛ = 1) with embedded charges and the solvent as a high dielectric medium (ɛ = 80) with no salt. Atomic charge values were taken from the Cornell et al. force field (12) as consistent with the molecular-mechanical energy calculations. However, atomic radii were taken from the PARSE parameter set (17) instead of the Cornell et al. force field (12) because of the small size of hydrogens in the latter. The dielectric boundary is the contact surface between the radii of the solute and the radius (1.4 Å) of a water probe molecule. The delphi 2.0 computer program package was used to numerically solve the linearized PB equation on a cubic lattice by using iterative finite-difference methods (16). One thousand iterations were performed for each calculation. The cubic lattice, which had a grid spacing of 0.5 Å, was scaled such that its dimensions were 80% larger than the longest dimension of the solute; the points on the boundary of the grid were set to the sum of Debye–Huckel potentials (18).
The nonpolar contribution to the solvation free energy (Gnp) was determined by using the solvent-accessible surface area algorithm (SASA) of Sanner (19) in a linear parameterization, where Gnp = γ (SASA) + β and the constants γ and β are 0.00542 kcal/Å2 and 0.92 kcal/mol, respectively, for use with PARSE atomic radii values (17). The solvent probe radius was set to 1.4 Å.
Computational Mutagenesis.
Desired alanine and glycine mutations were made by editing the snapshots from the trajectories before energetic analysis by using an approach developed by Massova and Kollman (9). In this approach, a residue is mutated to an alanine by truncating the residue at Cγ and replacing Cγ with a hydrogen atom at a 1.09-Å distance from Cβ along the Cγ–Cβ bond; likewise, a glycine mutation involves truncating the residue at Cβ and replacing Cβ with a hydrogen atom. The partial charges of the residue were then changed to those appropriate for alanine or glycine by simply changing the topology files. Energetic analysis of the mutants was performed as described above for the wild-type structures.
Results and Discussion
Structure and Dynamics of the Antibody–Hapten Complexes.
MD simulations of the 48G7 germ line and mature antibody–hapten complexes were performed with a spherical cap of explicit waters centered at the hapten and a “belly,” in which only atoms within a defined distance from the hapten were allowed to move during the simulation. Stable trajectories of the complexes resulted, as demonstrated by steady rms fluctuations (see Fig. 1) and rms deviations from their minimized crystal structures (data not shown). As shown in Fig. 1, the belly atoms in the germ line complex experienced greater rms fluctuations (0.4 to 0.6 Å) than those in the mature complex (0.3 to 0.5 Å). rms deviations of the belly atoms in the complexes from their minimized crystal structures were 1 Å and 0.7 Å, as averaged over the last 100 ps of the simulations for the germ line and mature complexes, respectively. These results suggest that the germ line complex has greater flexibility than the mature complex. The importance of this greater flexibility is underscored by two well-discussed notions of affinity maturation (4, 6): (i) the germ line antibody must readily adapt to a variety of antigens to efficiently protect the host from disease; and (ii) the plasticity of the germ line antibody facilitates its remodeling by somatic mutation into a “mature” conformation that has a tighter fit for the repeatedly introduced antigen.
Figure 1.

rms fluctuations of the germ line and mature antibody–hapten complexes. rms fluctuations are defined as rms deviations of the structure at a given time from the average structure of the MD simulation. Only mobile atoms of the simulation, or “belly” atoms (see Methods), were considered for the rms fluctuations.
Energetic Analysis of the Antibody–Hapten Complexes.
The results from energetic analysis of 50 equally spaced snapshots taken from each of the two MD simulations are summarized in Table 1. All energetic analysis was done for only a single MD trajectory of the desired antibody–hapten complex with unbound antibody and hapten snapshots taken from snapshots of that trajectory. The binding free energy (ΔGbind) for the complex was estimated by the following expression:
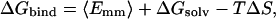 |
where 〈Emm〉 is the molecular-mechanical energy, ΔGsolv is the solvation energy, and −TΔS is the solute entropic contribution. The molecular-mechanical energy consists of internal (〈ΔEint〉), van der Waals (〈ΔEvdW〉), and electrostatic (〈ΔEelec〉) components. Note that, because the structures of each antibody in its unbound and bound states were the same, the internal component of the molecular-mechanical energy has zero contribution to the binding free energy (〈ΔEint〉 = 0). The solvation energy consists of an electrostatic contribution (ΔGPB) and a nonpolar contribution (ΔGnp). Solute entropic contributions were determined only for the last snapshots of the MD trajectories. The calculated free energies of germ line complex formation (ΔGbind = −9.1 kcal/mol) and of mature complex formation (ΔGbind = −15.7 kcal/mol) agree fairly well with those from experiment (−5.3 kcal/mol and −10.9 kcal/mol, respectively, for the germ line and mature complexes) (6). The favorable formations of these complexes are driven by the van der Waals contributions (〈ΔEvdW〉 values) and the nonpolar contributions to solvation (ΔGnp values). These nonelectrostatic components are very similar for both the germ line and mature complex formations (〈ΔEvdW〉 = −35.9 kcal/mol and ΔGnp = −4.4 kcal/mol for the germ line complex; 〈ΔEvdW〉 = −35.5 kcal/mol and ΔGnp 4.2 kcal/mol for the mature complex). The solute entropic contributions (−TΔS values) are similar as well for the germ line and mature complex formations (18.4 kcal/mol and 19.3 kcal/mol, respectively).
Table 1.
Energetic analysis of 48G7 antibody–hapten complex formations
| Germ line | Mature | |
|---|---|---|
| 〈ΔEelec〉 | −304.9 (2.5) | −349.1 (1.5) |
| 〈ΔEvdw〉 | −35.9 (0.5) | −35.5 (0.4) |
| 〈ΔEmm〉 | −340.8 (2.4) | −384.6 (1.4) |
| ΔGnp | −4.4 (0.0) | −4.2 (0.1) |
| ΔGPB | 317.6 (2.2) | 354.0 (1.2) |
| ΔGsolv | 313.3 (2.2) | 349.6 (1.2) |
| ΔGelec,tot | 12.7 (0.9) | 4.8 (0.5) |
| ΔGtot | −27.5 (0.6) | −35.0 (0.5) |
| −TΔS | 18.4 | 19.3 |
| ΔGbind | −9.1 | −15.7 |
Values in parentheses are SEM.
Results are mean energies (in kcal/mol) and SEM from 50 equally spaced “snapshot” configurations of a 500-ps MD simulation 〈ΔEmm〉. Each snapshot contains only the solute, which consists of all antibody and hapten atoms, including hydrogens. Definitions of the remaining energetic components are as follows: 〈ΔEelec〉 = electrostatic molecular-mechanical energy; 〈ΔEvdW〉 = van der Waals molecular-mechanical energy; 〈ΔEmm〉 = total molecular-mechanical energy (〈ΔEelec〉 + 〈ΔEvdW〉 + 〈ΔEint〉); ΔGnp = nonpolar contribution to the solvation energy; ΔGPB = electrostatic contribution to the solvation energy calculated by the PB approach; ΔGsolv = total solvation energy (ΔGPB + ΔGnp), ΔGelec,tot = total electrostatic energy (〈ΔEelec〉 + ΔGPB); ΔGtot = total energy without solute entropic contribution (〈ΔEmm〉 + ΔGsolv); − TΔS = solute entropic contribution, where T = temperature and S = sum of translational, rotational, and vibrational entropies; and ΔGbind = total energy with solute entropic contribution (ΔGtot − TΔS). Energetic contributions for the unbound antibody, unbound hapten, and antibody-hapten complex states in both the germ line and mature systems are shown in Tables 5 and 6, respectively, of supplementary material (see www.pnas.org).
It is important to consider the electrostatic component of the molecular-mechanical energy (〈ΔEelec〉) together with the electrostatic contribution to solvation (ΔGPB) when examining the role of electrostatics in the antibody–hapten complex formations. As demonstrated by numerous studies (20–26), electrostatics generally disfavor the docking of ligand and receptor molecules because the unfavorable change in the electrostatics of solvation is mostly, but not fully, compensated by the favorable electrostatics within the resulting ligand–receptor complex. Indeed, the total electrostatic energy contributions (ΔGelec,tot values) for both the germ line and mature antibody–hapten complex formations are unfavorable, with values of 12.7 kcal/mol and 4.8 kcal/mol, respectively. The mature complex formation is less unfavorable than the germ line complex formation because of a less positive total electrostatic term in which the penalty paid by the electrostatics of solvation is better compensated by favorable electrostatic interactions within the complex. Thus, even though electrostatics destabilize antibody–hapten complex formation, it is the optimized balance of opposing electrostatic contributions that leads to tighter binding in the mature complex.
On the affinity maturation of the 48G7 antibody, changes in either its conformation or amino acid sequence may account for the optimization of electrostatics. In other words, the nine somatic mutations may increase the binding affinity of the antibody for hapten directly through long-range electrostatic interactions or indirectly by inducing a conformational change that leads to improved electrostatic interactions within the immediate binding vicinity. To isolate the effects of conformational change from those of sequence change, we produced identical amino acid sequences for the germ line and mature antibodies while preserving their respective original conformations; this was done by replacing selected residues at the positions of the nine somatic mutations in one or both antibodies to alanines (or glycines, if the residues were glycines in either antibody) with no subsequent minimization. Details of this computational mutagenesis approach are described in Methods. We then proceeded to conduct an energetic analysis of the resulting “mutant” antibodies.
Results from our energetic analysis of the mutant germ line and mature antibodies are shown in Table 2. The total free energies (ΔGtot values) for the mutant germ line and mature antibody–hapten complex formations are different, with values of −27.7 kcal/mol and −35.2 kcal/mol, respectively. These values are essentially unchanged from those of the wild-type germ line and mature antibody–hapten complex formations, which are −27.5 kcal/mol and −35.0 kcal/mol, respectively (see Table 1). Conformational differences are therefore key to the affinity maturation of the 48G7 antibody.
Table 2.
Energetic analysis of mutant 48G7 antibody–complex formations
| kcal/mol
|
||
|---|---|---|
| Germ line mutant | Mature mutant | |
| 〈ΔEelec〉 | −374.0 (2.5) | −370.0 (1.3) |
| 〈ΔEvdw〉 | −35.8 (0.5) | −35.4 (0.4) |
| 〈Emm〉 | −409.8 (2.4) | −405.4 (1.2) |
| ΔGnp | −4.3 (0.0) | −4.4 (0.0) |
| ΔGPB | 386.5 (2.2) | 374.6 (1.0) |
| ΔGsolv | 382.1 (2.2) | 370.3 (1.0) |
| ΔGelec,tot | 12.5 (0.9) | 4.6 (0.5) |
| ΔGtot | −27.7 (0.7) | −35.2 (0.5) |
In order to produce mutant germ line and mature antibodies with identical amino acid sequences, the following mutations were made: SerL30, AspL55, GluH42, AsnH56, AsnH76 → Ala and SerL34 → Gly in the germ line antibody; AsnL30, HisL55, LysH42, AspH56, LysH76, ThrH78 → Ala and ValH55, AspH65 → Gly in the mature antibody. See Table 1 legend for definitions of the energy components.
This result is not too surprising, because the somatic mutations are distant from the hapten (the closest one being ≈6 Å away). Nonetheless, the similarity of the results for the wild-type and mutant antibody–hapten complexes suggests that long-range electrostatic interactions have little, if any, effect on the formation of the complexes. The difference in the binding affinities of the germ line and mature antibodies for the hapten must therefore lie in the short-range electrostatics, which can be largely influenced by reorganization of the combining site. Electrostatic interactions between the hapten and the residues in the germ line and mature antibodies were thus explored, dividing the hapten into three moieties: a nitroaryl “head” moiety, a phosphonate moiety, and an alkyl “tail” moiety.
As shown in Table 3, the phosphonate moiety of the hapten forms the bulk of the favorable electrostatic interactions between the hapten and the residues in both the germ line and mature antibodies. In fact, the net sum of electrostatic interactions between the antibody and hapten (〈ΔEelec〉) is more favorable for the mature complex (〈ΔEelec〉 = −349.1 kcal/mol) than for the germ line complex (〈ΔEelec〉 = −304.9 kcal/mol) largely because of a more negative electrostatic term for the phosphonate moiety of the hapten (−234.4 kcal/mol for the mature complex vs. −187.6 kcal/mol for the germ line complex). Note that the electrostatic interactions of the hapten tail are more favorable in the germ line complex (−135.8 kcal/mol) than in the mature complex (−117.5 kcal/mol). However, the differences in the electrostatic interactions of the tail are not as great as those of the phosphonate moiety. The phosphonate moiety is thus a major binding determinant, as suggested earlier by crystallographic studies based on the formation of hydrogen bonds between the phosphonyl oxygens and the combining site residues, ArgL96 and HisH35, in both the germ line and mature complexes; in the mature complex, the phosphonyl oxygens also form an additional hydrogen bond with TyrH33 (4, 5). These crystallographic studies also indicate that the tail of the hapten may be disordered and highly flexible because of its weak electron density relative to that of the phosphonate moiety. This flexibility may introduce error into our calculations. Nonetheless, our results are based on MD simulations that show low rms fluctuations (<1 Å) for the tail region of the hapten in both the germ line and mature complexes.
Table 3.
Electrostatic interactions between hapten and antibody

| kcal/mol
|
||
|---|---|---|
| Germ line | Mature | |
| Head (1) | 18.4 (0.3) | 2.8 (0.3) |
| Phosphonate (2) | −187.6 (0.9) | −234.4 (0.8) |
| Tail (3) | −135.8 (2.1) | −117.5 (1.4) |
| Total | −304.9 (2.5) | −349.1 (1.5) |
Results are mean interaction energies (in kcal/mol) and SEM from 50 configurations. Note that the values in the row labeled “totals” correspond to the 〈ΔEelec〉 and 〈ΔEvdW〉 values shown in Table 1.
The interaction between the phosphonate moiety of the hapten and TyrH33 is key to the more favorable network of electrostatic interactions between the phosphonate moiety of the hapten and the mature antibody. This interaction, which is worth about −17 kcal/mol, is much more attractive than it is in the germ line complex, where it amounts to about −5 kcal/mol. With such a favorable electrostatic interaction between just the TyrH33 and the phosphonate moiety of the hapten, the electrostatic interactions formed by TyrH33 may actually compensate, or more than compensate, for the penalty paid in the electrostatics of solvation on antibody–hapten complex formation.
These possibilities were addressed by developing a strategy for determining the total electrostatic contribution of an individual residue (i.e., TyrH33), which includes both its favorable electrostatic interactions with the hapten and its unfavorable loss of electrostatic interactions with solvent. This strategy involves creating a “discharged” version of the residue by setting all partial atomic charges within the residue to zero and using the resulting modified topology file for each snapshot taken from the MD simulation of the antibody–hapten complex. One can thus estimate the total electrostatic contribution of the residue relative to its discharged hydrophobic version. Electrostatic contributions were calculated for the resulting “mutants” as described in Methods for the wild-type antibody–hapten complexes.
Results for the discharged TyrH33 mutants of both the germ line and mature antibodies in complexes with the hapten are shown in Table 4 along with the corresponding values for the wild-type antibody–hapten complexes. As predicted, the total electrostatic contributions of TyrH33 to the germ line and mature complex formations are neutral, if not favorable, relative to the discharged version of the residue (ΔΔGelec,tot = −5.4 kcal/mol for the germ line complex formation and ΔΔGelec,tot = −8.1 kcal/mol for the mature complex formation). Interestingly, the net electrostatic interactions formed by TyrH33 in the complex are about the same in the germ line and mature complexes; however, they are distributed differently as a result of the different orientations of the hapten in the two complexes. As shown in Fig. 2, the phosphonate moiety of the hapten is located in similar positions for both complexes, whereas the head and the tail of the hapten are in rather different positions. In the germ line complex, the hydroxyl of TyrH33 forms a strong hydrogen bond (worth −13 kcal/mol) with the carboxyl end of the hapten. This hydrogen bond is absent in the mature complex, because the tail of the hapten has oriented itself away from TyrH33; however, this missing hydrogen bond is compensated by a strong hydrogen bond (worth −17 kcal/mol) between the hydroxyl of a newly oriented TyrH33 and the phosphonate moiety of the hapten. The net ΔΔEelec (Table 4) is actually about 1 kcal/mol more favorable in the germ line antibody, but this is compensated by ΔΔGPB, the solvation contribution, which is less favorable than that of the mature complex by about 4 kcal/mol. This more destabilizing electrostatic contribution to solvation is because of greater burial of TyrH33 on binding of the germ line antibody to hapten with a solvent-accessible surface area of 45.2 Å2 in the unbound state and 25.1 Å2 in the bound state, compared with the values for the mature antibody, which are 22.4 Å2 and 12.1 Å2 for the unbound and bound states, respectively.
Table 4.
Electrostatic analysis of TyrH33 in 48G7 antibody–hapten complex formations
| kcal/mol
|
||
|---|---|---|
| Germ line | Mature | |
| ΔΔGPB | 12.4 (0.8) | 8.5 (0.1) |
| 〈ΔΔEelec〉 | −17.7 (1.2) | −16.5 (0.3) |
| ΔΔGelec,tot | −5.3 (0.5) | −8.0 (0.3) |
“Mutant” corresponds to the antibody–hapten complex system with “discharged” TyrH33 (all partial atomic charges in TyrH33 set to zero). See Table 1 legend for definitions of electrostatic energetic terms.
Figure 2.

Orientations of TyrH33 and hapten in the 48G7 germ line and mature antibody–hapten complexes. Orientations are taken from representative structures of the appropriate antibody–hapten complex trajectories. Hydrogen bonds are indicated by dashed lines. (A) Germ line antibody–hapten complex. The carboxyl end of the hapten is ideally oriented to form a hydrogen bond with the hydroxyl of TyrH33. (B) Mature antibody–hapten complex. Hydrogen bonding is not possible between TyrH33 and the newly positioned carboxyl end of the hapten. However, TyrH33 has shifted toward the phosphonate moiety to enable the formation of a strong hydrogen bond between its hydroxyl and one of the phosphonyl oxygens. Produced with the midasplus graphics program (26).
Approximations and Future Directions.
Our approach involving the combination of MD simulations in explicit solvent with the use of continuum solvent models gives reasonable absolute and relative estimates of free energies for the formations of the 48G7 germ line and mature antibody–hapten complexes. These results encourage using this approach as an alternative to Aqvist’s linear interaction energy method (27, 28), which also calculates free energies of binding from MD simulations.
A few advantages exist in our approach vs. the linear interaction energy method. One advantage is that our approach requires only one MD simulation, which is for the ligand–receptor complex. The linear interaction energy method requires the generation of two simulations, one for the unbound ligand and one for the ligand–receptor complex. Second, our approach also has the advantage of using the PB treatment of electrostatics, which tends to be more accurate than the linear response approximation for electrostatics used in Aqvist’s method. Accurate treatment of electrostatics is particularly important for highly charged systems, such as the 48G7 germ line and mature antibody–hapten complexes, which involve a net charge of −2 on the hapten and net charges of +3 and +5 for the germ line and mature antibodies, respectively. Thirdly, our approach has the advantage of requiring no empirical parameters that are specific for a given system, whereas Aqvist’s method involves an empirical expression for nonpolar effects, which contains parameter values that vary between systems (29).
There are some approximations in our study. First, we have used the snapshots from the MD trajectory of each antibody–hapten complex as representative structures of the unbound antibody and hapten states. Approximating the unbound and bound states of the antibody or of the hapten as the same would be problematic if major conformational changes occur on complex formation. However, only small conformational changes in the binding regions take place in the 48G7 germ line and mature complexes (4). Furthermore, it has been shown for a peptide–protein complex (9) and a protein–RNA complex (C. Reyes and P.A.K., unpublished observations) that, if the true unbound states of the ligand and receptor of interest are used instead of being taken from the complex MD simulations, the resulting absolute binding free energies are of similar magnitudes as when all the free energies are extracted from the single antibody–hapten complex simulation.
Another uncertainty is the harmonic approximation to the solute entropy and its estimation by only a single normal mode calculation. The harmonic approximation involved in normal mode analysis is generally a crude one and, in addition, because of the very shallow potential surface with explicit water, we have removed the waters and done the normal mode analysis with a distance-dependent dielectric (ɛ = 4 r, where r = interatomic distance in Å). We used a distance-dependent dielectric, rather than setting the dielectric equal to one, because minimization with fully charged residues, in the latter case without the waters, is very unrealistic and is sure to move the structure far from the x-ray-determined position. We emphasize that we have used normal mode analysis only to determine approximate estimates of the change in solute entropy on antibody–hapten complex formation.
According to our results, the affinity maturation of the 48G7 antibody involves the optimization of electrostatics; van der Waals interactions appear optimal to begin with in the germ line antibody. These conclusions are consistent with the findings for the Diels–Alderase antibody 39-A11 (30), in which the mature form binds a hydrophobic hapten ≈40-fold tighter than the germ line form. In this situation, the increase in binding affinity is not as dramatic as that for the 48G7 antibody, because the germ line antibody has the ability to form nearly optimal interactions with the hapten. Thus, only two somatic mutations are necessary for the affinity maturation of 39-A11 (30).
As is evident in our energetic analysis of the germ line and mature antibody–hapten complexes, there is a delicate balance between the favorable gas-phase electrostatics term and the unfavorable change in electrostatic contribution to the solvation. In our electrostatic analysis of TyrH33, desolvation of the residue on binding of the antibody to the hapten plays a key role in determining the different extents of stabilization provided by the electrostatics of the residue toward complex formation. This type of analysis appears to be useful in isolating the electrostatic effects of individual residues and is likely to uncover more details about the optimization of the 48G7 antibody when applied to other residues in the combining site vicinity.
Conclusions
Using a combination of molecular mechanical energies derived from MD simulations in explicit solvent, solvation free energies derived from a continuum solvent model, and solute entropic contributions derived from normal mode analysis, we have obtained reasonable absolute and very good relative free energies for the 48G7 germ line and mature antibody–hapten complex formations. Energetic analysis reveals that van der Waals interactions and nonpolar contributions to solvation provide the basis for the favorable absolute free energies of the germ line and mature complexes and are optimal even before affinity maturation. The key to the >104 times tighter binding in the mature complex lies in the electrostatics. By counteracting the favorable electrostatic interactions that form between the antibody and the hapten, the desolvation of the antibody plays an important role in determining the effect of electrostatics, as a whole, on the formation of the antibody–hapten complex. Alanine/glycine scanning reveals that the interactions of the charged residues involved in somatic mutations are not changed by affinity maturation. Rather, the combining site geometry appears to be reorganized in a manner that optimizes the electrostatics. Increasing the rigidity of the antibody structure further optimizes the binding affinity of the antibody for the hapten. This increased rigidity is apparent in the lower rms fluctuations of the mature complex relative to the germ line complex.
The results of this study have provided us with insight into the process of affinity maturation. First, the immune system must be able to produce high-affinity and specific antibodies rapidly, before the antigen has time to damage the host. In the case of 48G7, this rapid response appears to be achieved through the likely ability of the germ line antibody to adapt and bind to a variety of antigens, because it is more flexible than the mature antibody. Through already optimal hydrophobic interactions but suboptimal electrostatic interactions, the germ line antibody may bind with reasonable affinity to each of these antigens. Greater affinity and specificity are then accomplished through the optimization of electrostatic interactions in the binding site and increased rigidity of the antibody during affinity maturation of the 48G7 antibody.
In the design of an ideal ligand for a given receptor, we must consider the electrostatics and van der Waals interactions as well as the specific arrangement of hydrogen bonds. Optimization, as has been done by the 48G7 antibody, can involve minimizing the desolvation penalty for favorable electrostatic interactions. Such an optimization strategy requires less precision and a more limited set of residues than fine-tuning the precise arrangement of hydrogen bonds and packing of van der Waals interactions. Thus, it is likely that molecular design efforts will benefit from effective strategies for optimizing electrostatics, including desolvation (31, 32), as well as from developing reliable methods for creating ideal hydrogen bonding or van der Waals interactions.
Supplementary Material
Acknowledgments
We thank P. Schultz and R. Stevens for providing the crystal structures of the 48G7 antibody system; B. Honig for the delphi program; M. Sanner for the solvent-accessible surface area algorithm; and members of our research group, particularly O. Donini, J. Pitera, and C. Reyes, for their helpful suggestions. This work was supported in part by a National Institutes of Health (NIH) Grant GM-29072 to P.A.K. and by a National Science Foundation Fellowship to L.T.C. Graphics were provided by the Computer Graphics Laboratory, University of California, San Francisco (T. Ferrin, prinicipal investigator, NIH P41 Grant RR-01081).
Abbreviations
- H
heavy chain of antibody
- L
light chain of antibody
- MD
molecular dynamics
- PB
Poisson–Boltzmann
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Tonegawa S. Nature (London) 1983;302:575–581. doi: 10.1038/302575a0. [DOI] [PubMed] [Google Scholar]
- 2.Kim S, Davis M, Sinn E, Patten P, Hood L. Cell. 1981;27:573–581. doi: 10.1016/0092-8674(81)90399-8. [DOI] [PubMed] [Google Scholar]
- 3.Siskind G W, Benacerraf B. Adv Immunol. 1969;10:1–50. doi: 10.1016/s0065-2776(08)60414-9. [DOI] [PubMed] [Google Scholar]
- 4.Wedemayer G J, Patten P A, Wang L H, Schultz P G, Stevens R C. Science. 1997;276:1665–1669. doi: 10.1126/science.276.5319.1665. [DOI] [PubMed] [Google Scholar]
- 5.Wedemayer G J, Wang L H, Patten P A, Schultz P G, Stevens R C. J Mol Biol. 1997;268:390–400. doi: 10.1006/jmbi.1997.0974. [DOI] [PubMed] [Google Scholar]
- 6.Patten P A, Gray N S, Yang P L, Marks C B, Wedemayer G J, Boniface J J, Stevens R C, Schultz P G. Science. 1996;271:1086–1091. doi: 10.1126/science.271.5252.1086. [DOI] [PubMed] [Google Scholar]
- 7.Srinivasan J, Cheatham T E, Cieplak P, Kollman P A, Case D A. J Am Chem Soc. 1998;120:9401–9409. [Google Scholar]
- 8.Cheatham T E, Srinivasan J, Case D A, Kollman P A. J Biomol Struct Dyn. 1998;16:265–280. doi: 10.1080/07391102.1998.10508245. [DOI] [PubMed] [Google Scholar]
- 9.Massova I, Kollman P A. J Am Chem Soc. 1999;121:8133–8143. [Google Scholar]
- 10.Case D A, Pearlman D A, Caldwell J W, Cheatham T E, III, Ross W S, Simmerling C L, Darden T A, Merz K M, Stanton R V, Cheng A L, et al. amber 5.0. San Francisco: University of California; 1997. [Google Scholar]
- 11.Bayly C I, Cieplak P, Cornell W D, Kollman P A. J Phys Chem. 1993;97:10269–10280. [Google Scholar]
- 12.Cornell W D, Cieplak P, Bayly C I, Gould I R, Merz K M, Ferguson D M, Spellmeyer D C, Fox T, Caldwell J W, Kollman P A. J Am Chem Soc. 1995;117:5179–5197. [Google Scholar]
- 13.Ryckaert J P, Ciccotti G, Berendsen H J C. J Comput Phys. 1977;23:327–341. [Google Scholar]
- 14.Jorgensen W L, Chandrasekhar J, Madura J D, Impey R W, Klein M L. J Comput Phys. 1983;79:926–935. [Google Scholar]
- 15.Berendsen H J C, Postma J P M, van Gunsteren W F, DiNola A, Haak J R. J Comput Phys. 1984;81:3684–3690. [Google Scholar]
- 16.Sharp K A, Honig B. Annu Rev Biophys Biophys Chem. 1990;19:301–332. doi: 10.1146/annurev.bb.19.060190.001505. [DOI] [PubMed] [Google Scholar]
- 17.Sitkoff D, Sharp K A, Honig B. J Phys Chem. 1994;98:1978–1988. [Google Scholar]
- 18.Gilson M K, Honig B H. Nature (London) 1987;330:84–86. doi: 10.1038/330084a0. [DOI] [PubMed] [Google Scholar]
- 19.Sanner M F, Olson A J, Spehner J C. Biopolymers. 1996;38:305–320. doi: 10.1002/(SICI)1097-0282(199603)38:3%3C305::AID-BIP4%3E3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 20.Novotny J, Sharp K. Prog Biophys Mol Biol. 1992;58:203–224. doi: 10.1016/0079-6107(92)90006-r. [DOI] [PubMed] [Google Scholar]
- 21.Novotny J, Bruccoleri R E, Davis M, Sharp K A. J Mol Biol. 1997;268:401–411. doi: 10.1006/jmbi.1997.0961. [DOI] [PubMed] [Google Scholar]
- 22.Misra V K, Sharp K A, Friedman R A, Honig B. J Mol Biol. 1994;238:245–263. doi: 10.1006/jmbi.1994.1285. [DOI] [PubMed] [Google Scholar]
- 23.Misra V K, Hecht J L, Sharp K A, Friedman R A, Honig B. J Mol Biol. 1994;238:264–280. doi: 10.1006/jmbi.1994.1286. [DOI] [PubMed] [Google Scholar]
- 24.Sharp K A. Biophys Chem. 1996;61:37–49. doi: 10.1016/0301-4622(96)00021-x. [DOI] [PubMed] [Google Scholar]
- 25.Shen J, Wendoloski J. J Comput Chem. 1996;17:350–357. [Google Scholar]
- 26.Bruccoleri R E, Novotny J, Davis M E. J Comput Chem. 1997;18:268–276. [Google Scholar]
- 27.Aqvist J, Medina C, Samuelsson J E. Prot Eng. 1994;7:385–391. doi: 10.1093/protein/7.3.385. [DOI] [PubMed] [Google Scholar]
- 28.Hansson T, Marelius J, Aqvist J. J Comput Aided Mol Des. 1998;12:27–35. doi: 10.1023/a:1007930623000. [DOI] [PubMed] [Google Scholar]
- 29.Wang W, Wang J, Kollman P A. Prot Struct Funct Genet. 1999;34:395–402. [PubMed] [Google Scholar]
- 30.Romesberg F E, Spiller B, Schultz P G, Stevens R C. Science. 1998;279:1929–1933. doi: 10.1126/science.279.5358.1929. [DOI] [PubMed] [Google Scholar]
- 31.Lee L P, Tidor B. J Chem Phys. 1997;106:8681–8690. [Google Scholar]
- 32.Kangas E, Tidor B. J Chem Phys. 1998;109:7522–7545. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}