

Abstract
Tyrosyl-DNA phosphodiesterase 1 (Tdp1) is a recently discovered enzyme that catalyzes the hydrolysis of 3′-phosphotyrosyl bonds. Such linkages form in vivo following the DNA processing activity of topoisomerase I (Top1). For this reason, Tdp1 has been implicated in the repair of irreversible Top1-DNA covalent complexes, which can be generated by either exogenous or endogenous factors. Tdp1 has been regarded as a potential therapeutic co-target of Top1 in that it seemingly counteracts the effects of Top1 inhibitors, such as camptothecin and its clinically used derivatives. Thus, by reducing the repair of Top1-DNA lesions, Tdp1 inhibitors have the potential to augment the anticancer activity of Top1 inhibitors provided there is a presence of genetic abnormalities related to DNA checkpoint and repair pathways. Human Tdp1 can also hydrolyze other 3′-end DNA alterations including 3′-phosphoglycolates and 3′-abasic sites indicating it may function as a general 3′-DNA phosphodiesterase and repair enzyme. The importance of Tdp1 in humans is highlighted by the observation that a recessive mutation in the human TDP1 gene is responsible for the inherited disorder, spinocerebellar ataxia with axonal neuropathy (SCAN1). This review provides a summary of the biochemical and cellular processes performed by Tdp1 as well as the rationale behind the development of Tdp1 inhibitors for anticancer therapy.
1. INTRODUCTION
Cancer is generally characterized by the presence of endogenous DNA damage. However, while DNA damage underlies carcinogenesis, it has also been exploited as a means to treat cancer. For example, the cytotoxic effects of the two main therapeutic modalities that are clinically used to treat malignances (i.e. chemotherapy or radiation) are directly related to their propensity to generate DNA damage. Moreover, DNA damage is also responsible for many of the side effects of these therapies, such as bone marrow suppression, gastrointestinal toxicities, and hair loss. The sensitivity of cancer cells to DNA damaging agents is probably related to intrinsic deficiencies in DNA repair and checkpoint mechanisms. However, the capacity of cancer cells to recognize DNA damage and initiate DNA repair is a key mechanism for therapeutic resistance to chemotherapy. Therefore, the targeting of DNA repair enzymes for anticancer therapeutic intervention can be used as a strategy to potentiate the cytotoxicity of the currently available DNA damaging agents toward cancer cells. In this review we will summarize the enzymatic activities, the biological and cellular functions of tyrosyl-DNA phosphodiesterase 1 (Tdp1), as well as discuss the rationale for targeting Tdp1 for cancer therapy.
2. TOPOISOMERASE I
2.1 Biological Functions and Catalytic Mechanism
DNA topoisomerase IB (Top1) is a ubiquitous enzyme that regulates DNA topology by relaxing positive and negative supercoiling in the DNA. These changes in the DNA topological state permit efficient processing of the genetic material during fundamental cellular events such as replication, transcription, and chromatin remodeling [1-4]. More importantly, Top1 has been identified as a therapeutic target in cancer chemotherapy following the discovery that camptothecin (CPT) is a specific Top1 inhibitor [5] and the FDA approval of the two CPT derivatives, topotecan and irinotecan, for clinical use [6].
The catalytic mechanism of Top1 involves a reversible transesterification reaction, as shown in Fig. 1A. In brief, following DNA binding the Top1 catalytic cycle is initiated by the nucleophilic attack of the phenolic hydroxyl group of the active site tyrosine (Tyr723 for human Top1) on the scissile phosphate, which generates a covalent complex between the protein and the 3′-phosphate of the DNA via a phosphotyrosyl bond. This transient DNA single-strand break (i.e. cleavage complex) subsequently allows controlled rotation of the cleaved DNA strand around its intact complement, effectively removing any local helical tension. Once the DNA is relaxed, the covalent intermediate is reversed via a second transesterification reaction, wherein the free DNA 5′-hydroxyl acts as a nucleophile to attack the 3′-phosphotyrosyl linkage; thus restoring the continuity of the original DNA duplex. For more extensive reviews covering the biochemistry and biology of DNA topoisomerase I, see [1-4].
Fig. (1).

A) Human Top1-mediated DNA cleavage and religation mechanisms. Tyr723 is the active site tyrosine involved in the transesterification reaction. The bases flanking the Top1 cleavage site are referred to as -1 and +1 for the bases at the 3′ and 5′ DNA termini, respectively. B) Representative circumstances resulting in the formation of trapped Top1 cleavage complexes (i.e. (i) Top1 inhibitors or preexisting DNA lesions, such as (ii) DNA strand breaks or (iii) nucleotide base damage).
2.2 The Formation of Irreversible Top1 Cleavage Complexes
Under normal circumstances, the rate of religation is much faster than the rate of cleavage, which allows the Top1-DNA cleavage complexes to be a transitory intermediate event of the Top1 catalytic cycle [1]. However, a variety of conditions (briefly schematized in Fig. 1B), have been shown to increase the frequency of Top1-DNA cleavage complexes by reducing or inhibiting the rate of the religation reaction (for reviews see [7, 8]). For example, Top1 inhibitors, such as camptothecin (CPT) and its clinically used derivatives, as well as several non-CPT Top1 inhibitors including the indenoisoquinolines and the indolocarbazoles, selectively and reversibly bind to the Top1-DNA interface [6, 9] and slow the rate of Top1-mediated DNA religation. This is especially the case when Top1 relaxes positive supercoiling [10]. Failure to reseal this normally transient break in the DNA results in the generation of a Top1-linked DNA single-strand break that can be transformed into a prolonged double-stranded break following collision of replication forks with the drug-stabilized cleavage complex [11-14]. In a similar manner, a number of naturally occurring DNA lesions, such as strand breaks [15], abasic sites, base mismatches, and certain oxidized or modified bases [16], have also been shown in vitro to physically block the Top1 religation reaction through the misalignment the 5′-hydroxyl with the tyrosyl-DNA phosphodiester backbone. In cells, both UV irradiation- [17] and hydrogen peroxide-induced [18] DNA damage have also been shown to trap Top1 cleavage complexes. Repair of these irreversible Top1-DNA cleavage complexes has been suggested as an important part of DNA metabolism given that mutants defective in specific DNA repair pathways demonstrate increased sensitivity to Top1 inhibitors. However, all repair pathways require DNA termini consisting of a 5′-phosphate and 3′-hydroxyl for appropriate processing by DNA ligase. Thus, Top1-DNA lesions present a unique challenge for the DNA repair machinery given that the DNA strand break is encumbered with a protein adduct, which must be removed before the DNA can be religated.
3. TYROSYL-DNA PHOSPHODIESTERASE
3.1 Biological Functions and Catalytic Mechanism
In 1996, Nash and coworkers discovered an enzymatic activity from yeast S.cerevisiae that specifically hydrolyzes the covalent bond between a tyrosine residue and a DNA 3′-phosphate [19]. Because Top1 is the only known enzyme to generate 3′-phosphotyrosine linkages in vivo, the observed enzymatic activity was proposed to be associated with the repair of DNA lesions, which develop from irreversible Top1-DNA cleavage complexes [19, 20]. The yeast gene encoding this activity was subsequently cloned and appropriately named tyrosyl-DNA phosphodiesterase 1 (Tdp1). All eukaryotic species examined thus far contain a well-conserved homologue of Tdp1. Specifically, the human genome encodes a single copy of TDP1 consisting of 17 exons: two 5′ non-coding exons and 15 coding exons [21], and has been shown to possess a similar 3′phosphotyrosyl-processing activity to that of the originally identified yeast enzyme [22], while having only minimal sequence identity (∼15%) [23]. The greatest sequence variance resides in the N-terminal domain of Tdp1, which is poorly conserved or absent in most Tdp1 homologues of lower eukaryotes. The N-terminus (1-148) of human Tdp1 has been shown to be dispensable for enzymatic activity [22], but seems to have evolved to play a regulatory function. For example, the N-terminal domain has been shown to be necessary for specific protein-protein interactions (i.e. DNA ligase III) (discussed below) [24, 25]. In addition, an in silico search for potential nuclear localization signals (NLS) in the human Tdp1 protein sequence reveals two putative NLSs in the N-terminal domain, one of which is located in the nonessential region (Fig. 2A). This is in agreement with studies using fluorescence microscopy demonstrating localization of Tdp1 to the nucleus in human cells [26].
Fig. (2).

A) Schematic representation of the human Tdp1 domain structure. The N-terminal and C-terminal domains correspond to residues 1-350 and 351-608, respectively. Positions of the “HKN” motifs and NLS (nuclear localization signals) motifs are shown in black and white, respectively. Arrows indicate active site residues. B) Ribbon diagram of Tdp1 structure (Δ148). The domain colors correspond to those shown in (A). The active site residues (H263, K265, H493, and K495) are shown as stick structures in blue (Figure modified from [40]). C) Structure of the Tdp1-vanadate-peptide-DNA complex (1NOP). The orientation and domain colors of Tdp1 are identical as in (B). Tdp1 is shown as a molecular surface and the vanadate-peptide-DNA substrate mimic is shown as ball-and-stick structures with the DNA colored in green, the vanadate colored in red, and the Top1-derived peptide colored in white (Figure modified from [41]).
Tdp1 is physiologically important in humans given that a homozygous mutation in the TDP1 gene (A1478G), which results in a histidine to arginine mutation (H493R, shown in italics in Fig. 2A) in the active site of Tdp1, is responsible for the rare autosomal recessive neurodegenerative disease, spinocerebellar ataxia with axonal neuropathy (SCAN1) [21]. Individuals harboring the SCAN1 mutation develop symptoms that are restricted to the nervous system during their teenage years [21]. In contrast to other diseases associated with defects in DNA repair proteins, SCAN1 patients do not have an increase in cancer susceptibility. Initially, it was proposed that the SCAN1 mutation disrupted the Tdp1 active site symmetry leading to a loss-of-function [21, 24, 27], yet recent studies have suggested an additional hypothesis wherein the accumulation of covalent Tdp1-DNA intermediates might be responsible for the DNA damage leading to the SCAN1 phenotype (see Fig. 3, legend) [28].
Fig. (3).

Proposed reaction mechanism for human Tdp1. A) The first step of the reaction involves the nucleophilic attack of the phosphodiester-containing substrate by the imidazole Nε2 atom of His263. H493 donates a proton to the tyrosyl moiety of the leaving group. The phosphohistidine intermediate is shown in (B). Interactions between the non-bridging oxygens and the active site lysine residues (Lys265 and Lys495) serve to stabilize the transition intermediate. C) The next step of the reaction involves second a nucleophilic attack by a water molecule activated by H493, generating final 3′-phosphate product and free Tdp1 (D) (A-D modified from [40]). (E) The SCAN-1 mutations (H493R) leads to an accumulation of the Tdp1-DNA intermediate and a defect in Tdp1 turnover rate.
Tdp1 is a member of the phospholipase D superfamily [22], which encompasses a diverse group of enzymes that catalyze phosphodiester bond cleavage on substrates ranging from phospholipids to DNA, or in the case of Tdp1, protein-DNA complexes. The majority of the sequence identity between Tdp1 and the members of the PLD family is restricted to two copies of an active site signature motif H(X)K(X4)D, also referred to as an HKD motif [29], which has been implicated in the catalytic mechanism of these enzymes [30, 31]. Two such motifs are present in the N- and C-terminal domains of human Tdp1, both of which contain the conserved histidine and lysine residues (263HTK265 and 495HIK497) but lack the aspartate residues that exist in other HKD motifs (Fig. 2A). Thus, it has been suggested that Tdp1 represents a distinct subclass of the PLD superfamily based on its unique “HKN” motifs. The importance of the two signature HKN motifs for Tdp1 enzymatic activity has been established by site-directed mutagenesis. Indeed, mutation of H263 produces a catalytically inactive enzyme, whereas mutating H495, K265, or K497 results in impaired enzymatic activity [22, 32]. These results are consistent with studies performed on other members of the PLD family [33-37].
The affiliation of Tdp1 with the PLD superfamily has been further established through comparative analysis of the three-dimensional structure of the catalytically active N-terminal truncated human Tdp1 (Δ1-148) (Fig. 2A) [38] with the known crystal structures of two members of the PLD superfamily, a PLD from Streptomyces [30] and a bacterial nuclease from Salmonella tryphimurium [39]. Albeit the low sequence identity amid these three enzymes, they share a strikingly similar tertiary structure. In addition, the superimposition of the active sites supports the notion that all three enzymes participate in analogous reaction chemistry. The crystallographic studies of human Tdp1 reveals a monomeric structure comprised of two topologically similar α-β-α domains. Each domain contributes a histidine and lysine residue to form the active site, which is centrally located at the pseudo-twofold axis of symmetry between the two domains (Fig. 2B). The Tdp1 crystal structure also reveals that four additional residues, which are conserved in all Tdp1 orthologs, N283, Q294, N516, and E538, are also positioned near the active site [38].
In order to gain further insight into the substrate binding properties and catalytic mechanism of Tdp1, co-crystal structures were solved of human Tdp1 bound to vanadate [40, 41], an oxyanion form of pentavalent vanadium. Unlike the phosphate ion, vanadate can form five-coordinate species allowing it to serve as a transition state analogue and eliminate enzyme turnover. The molecular assembly of vanadate, Tdp1, a Top1-derived peptide containing the active site tyrosine, and a single-stranded DNA oligonucleotide into a quaternary structure is shown in Fig. 2C. Initial examination of the quaternary structure revealed an asymmetrical substrate-binding channel that extended in opposite directions from the active site across the entire surface of Tdp1. The DNA moiety bound to one side of the active site in a relatively narrow cleft that is predominantly positively charged (Fig. 2C, shown behind the DNA (green)). Conversely, the peptide moiety bound to the other side of the active site in a relatively large, more open cleft that contains a mixed charge distribution (Fig. 2C, shown behind the peptide (white)) [38, 40, 41]. Interestingly, the DNA-binding groove is highly conserved among Tdp1 orthologues, while the peptide-binding pocket shows much less sequence conservation between different species [41, 42].
A closer look into the active site of the quaternary structure [40, 41] shows that the vanadate ion exhibits trigonal bipyramidal geometry, which is consistent with a transition state of an SN2 nucleophilic attack on phosphate. The apical coordination to the vanadate molecule is filled by the imidazole Nε2 atom of the active site H263 residue of Tdp1 and the oxygen atom of the tyrosine side chain of the Top1-derived peptide, whereas the 3′-hydroxyl of the DNA oligonucleotide occupies one of the three equatorial positions. The remaining two equatorial oxygens, which represent the nonbridging oxygens of the phosphodiester substrate, form hydrogen bonds with the amino groups of K265 and K495. It is well-recognized that the incoming and outgoing groups in an SN2 reaction occupy the two axial positions of a trigonal bipyramid [43]. Therefore, it is evident from the Tdp1-vanadate structure that the first step in the Tdp1 catalytic mechanism involves the nucleophilic attack of the phosphotyrosyl bond by the H263 residue from the N-terminal HKN motif. Additionally, the H493 residue from the C-terminal HKN motif acts as a general acid and donates a proton to the apical tyrosine-containing peptide-leaving group (Fig. 3A). This results in the formation of a transient covalent phosphoamide bond between the Nε2 atom of H263 and the 3′-end of the DNA (Fig. 3B). Hydrolysis of this covalent intermediate is proposed to be carried out by a water molecule that is activated by the H493 residue acting as a general base (Fig. 3C) [40]. The second SN2 reaction step in the Tdp1 catalytic mechanism is supported by in vitro biochemical studies with the SCAN1 H493R mutant, which leads to an accumulation of Tdp1-DNA covalent intermediate [28]. Overall, the final product released after Tdp1 catalysis is a DNA molecule with a 3′-phosphate end (Fig. 3D).
3.2 Tdp1 Substrates
A stalled Top1-DNA complex is an unmanageable substrate for Tdp1 because of the limited access of the scissile phosphotyrosyl bond, which is internally located within the Top1-DNA complex [44]. This statement is corroborated by the poor processing by Tdp1 of a suicide complex made of full-length human Top1 covalently linked to a DNA oligonucleotide (Fig. 4F (iii)) [45]. Prior denaturation of the Top1-DNA complex, which presumably results in enhanced steric access of the phosphotyrosyl bond, is required for Tdp1 enzymatic activity toward a full-length Top1-DNA covalent complex [19]. Thus, it has been suggested that the Top1 must undergo proteolysis in order for efficient Tdp1 activity [19, 41, 45], which is in agreement with studies demonstrating that the effectiveness of Tdp1 processing decreases as the length of the Top1 polypeptide is extended [45]. Accordingly, several studies have shown that Top1 is degraded in some cells lines following CPT treatment, suggesting that Top1 ubiquitination and degradation contribute to CPT resistance [46-49]. In addition, prevention of Top1 degradation by the proteasome inhibitors, MG132 [47] or PS-341 (clinically used) [50], results in increased sensitivity to CPT. These studies imply that Top1 degradation via a ubiquitin-proteasome pathway may play a role in the repair of Top1-mediated damage, wherein Top1 degradation serves to prepare the Top1 cleavage complexes for hydrolysis by Tdp1.
Fig. (4).

Outline of the substrate binding pocket and the different substrates processed by Tdp1. A) Molecular surface of Tdp1 in an orientation identical to that shown in Fig. 2C. The active site region is shown in white. The black dotted lines define the substrate binding pocket (Figure modified from [40]). B) Substrate orientation in the Tdp1 binding pocket. The R-group refers to the structures shown in (C) and (D). The arrow indicates the Tdp1 cleavage site. C) and D) illustrate the physiological and non-physiological substrates of Tdp1 (Figure modified and updated from [61]). E) and F) show both favorable and unfavorable the Tdp1 substrates. The black circle represents a 3′-end phosphotyrosine. E) (i) long single-strand, (ii) tailed duplex, (iii) gapped duplex. F) (i) short single-strand, (ii) nicked duplex, (iii) full-length Top1.
In addition to a Top1-derived peptide and a single tyrosine residue, a variety of other moieties that block to the 3′-end of the DNA from being substrates for DNA polymerases or ligases are hydrolyzed by Tdp1 (Fig. 4C and 4D), although less effectively than a 3′-phosphotyrosyl end [19]. The broad substrate specificity of Tdp1 is consistent with analysis of the human Tdp1 structure bound to a Top1-derived peptide, which demonstrates that there are no specific contacts between the tyrosine and the Tdp1 polypeptide [41]. One physiologically relevant Tdp1 substrate is the 3′-phosphoglycolate (Fig. 4C), which can be generated by oxidative DNA damage [51]. In addition, wild-type Tdp1 has been shown to hydrolyze the covalent phosphoamide intermediate proposed to form in SCAN1 cells between the mutant Tdp1 and the DNA (Fig. 3E) [52]. Other naturally occurring substrates for Tdp1 include 3′-abasic sites, 3′-nucleosides, and 3′-ribonucleosides (Fig. 4C) [52]. However, it is interesting to note that the Tdp1 is only able to remove a single terminal 3′-mononucleoside and is incapable of processing a DNA end containing a 3′-phosphate [52], which demonstrates that Tdp1 cannot act as a 3′-exonuclease. Yeast Tdp1 has also been shown to process a topoisomerase II (Top2) cleavage complex or a 5′-phosphotyrosyl bond [53]. Yet, the effects of Top2 inhibitors on TDP1-deficient yeast cells have been contradictory [53, 54]. Moreover, a single study in mammalian cells has demonstrated that overexpression of an active Tdp1 significantly reduces the DNA damage caused by Top2 inhibitors [26].
Besides the physiological substrates, numerous synthetic Tdp1 substrates have been identified (Fig. 4D). Specifically, phosphotyrosine analogues, such as 3′-(4-nitro)phenol and 3′(4-methyl)phenol, have been used in kinetic analysis of the Tdp1 catalytic mechanism [32]. Other artificial substrates include a 3′-linker-biotin [52] and a fluorescent 3′-4-methylumbelliferone [55], which could potentially be employed in high-throughput screens for Tdp1 inhibitors, similar to how the 3′-BV tag has been used (discussed below).
The second component of the Tdp1 substrate is the DNA. Indeed, we previously showed that the efficiency of Tdp1 to hydrolyze peptide-DNA substrates decreases when the length of the oligonucleotide is shortened from 13 to 4 nucleotides [45]. Several structural variations of DNA that mimic substrates Tdp1 may encounter in vivo have also been examined, such as single-strand, tailed duplex, gapped duplex, and nicked duplex. Tdp1 has been shown to cleave all the substrates listed to a similar degree, with the exception of the nicked duplex (Fig. 4E and 4F) [19, 54, 56]. These observations suggest that Tdp1 acts after the 5′-end of the broken DNA has been digested or displaced to provide access to the phosphotyrosyl bond. However, the lack of preference between single-strand and duplex DNA substrates is in disagreement with the proposed binding mode of DNA to the Tdp1 enzyme, which has resulted in an alternative model for binding of duplex DNA to Tdp1 [56]. In general, both the structure of the DNA segment bound to Top1 and the length of the Top1 polypeptide chain determine the biochemical activity of Tdp1 [45].
3.3 Tdp1 Repair Pathways
The 3′-phosphate-end generated by Tdp1 must be hydrolyzed to a 3′-hydroxyl in order for further DNA repair to occur. Polynucleotide kinase phosphatase (PNKP), a bifunctional enzyme with 5′-kinase and 3′-phosphatase activities, has been suggested as a reasonable candidate in human cells for the repair of these 3′-phosphate lesions. In vitro studies have shown that T4 kinase, the bacteriophage equivalent of PNKP, can hydrolyze the resulting 3′-phosphate produced by Tdp1 [19]. It has also been shown that human A549 cells that are defective in PNKP accrue similar levels of CPT-induced strand breaks in comparison to SCAN1 cells [24], which suggested that Tdp1 and PNKP function in the same repair pathway in human cells. Furthermore, PNKP is known to interact with the XRCC1 protein, which together with the other base excision repair (BER) proteins, such as DNA polymerase β, DNA ligase III, and poly(ADP-ribose)polymerase 1(PARP-1) forms a multiprotein DNA repair complex [57]. Recently, Tdp1 has also been shown to be a member of the BER repair complex through interactions with DNA ligase III [24] and XRCC1 [25]. XRCC1-deficient cells have been discovered to be hypersensitive to CPT and defective in Tdp1 activity, which is in agreement with a direct role of Tdp1 in association with the BER complex for the removal and repair of Top1-mediated DNA damage [25]. In addition, the interaction of XRCC1 with PNKP has been shown to stimulate both the 5′-kinase and 3′-phosphatase activities of this enzyme [57]. Similarly, XRCC1 interacts with DNA ligase III and increases the intracellular stability of the ligase [58, 59]. The association PARP-1 with the XRCC1 complex has also been observed. However, its function in the repair of Top1-mediated DNA damage remains to be determined. PARP-1 is a nuclear enzyme often termed a molecular nick sensor, which recognizes and binds to DNA single or double strand breaks [60]. Indeed, the hypersensitivity of PARP-1-deficient cells to CPT has been shown to be directly related to a functional defect in Tdp1 [61], which suggests that PARP-1 may be involved in the targeting of a BER complex to sites of Top1-mediated DNA damage. The effects on Tdp1 activity after the association with the XRCC1 complex are still unresolved. However, based on these observations and several others, a scheme for the role of the XRCC1 repair complexes in the repair of Top1-mediated DNA lesions has been proposed (Fig. 5) [8]. Following degradation or denaturation of Top1 to expose the phosphotyrosyl linkage, Tdp1 hydrolyzes the Top1-DNA bond. Next, PNKP hydrolyzes the resulting 3′-phosphate end and catalyzes the phosphorylation of the 5′-end of the DNA. Lastly, DNA polymerase β replaces the missing DNA segment and DNA ligase III reseals the DNA.
Fig. (5).

Proposed repair of Top1 cleavage complexes by the XRCC-1-dependent pathway. The XRCC1 complex including the associated repair enzymes is shown in the center of the figure. Tdp1 hydrolyzes the Top1 phosphotyrosyl bond. PNKP hydrolyzes the resulting 3′-phosphate and phosphorylates the 5′-hydroxyl. DNA polymerase β fills the gap and DNA ligase III reseals the DNA. PARP-1 may play a role in the initial recognition of the Top1-mediated DNA damage. (Figure modified from [8, 19, 76])
3.4 Rationale for Targeting Tdp1 for Cancer Therapy
The initial understanding of the role of Tdp1 in DNA repair and the rationale for targeting Tdp1 for anticancer therapy in combination with CPT came about by taking advantage of the genetic manipulations possible in yeast. Tdp1 is one of several potentially redundant pathways involved in the repair of Top-mediated damage in yeast [54, 61-65]. This has been demonstrated through the observation that significant increases in sensitivity to CPT in TDP1-defective yeast are conditional upon at least one additional mutation in other DNA repair and checkpoint genes. For instance, hypersensitivity to CPT is observed when TDP1 and the checkpoint gene Rad9 are simultaneously inactivated [54]. In addition, a second group of conditional genes are the three sets of genes implicated in the structure-specific endonuclease repair pathways. Rad1/Rad10 (XPF/ERCC1) functions primarily in the nucleotide excision repair pathway, where it cleaves on the 5′-side of the repair bubble formed around bulky DNA lesions [65-67]. Mus81/Mms4 (Mus81/Eme1) and Mre11/Rad50/Xrs2 (Mre11/Rad50/Nbs1) cleave a 3′-flap upstream of a branch point [68, 69] and function independently from the Tdp1 pathway (Fig. 6A) [62, 63]. Mutations in each of these genes render TDP1-deficient yeast cells highly sensitive to CPT. Specific homologues for each of the genes mentioned above are present in humans (shown in parentheses), with the exception of Rad9.
Fig. (6).
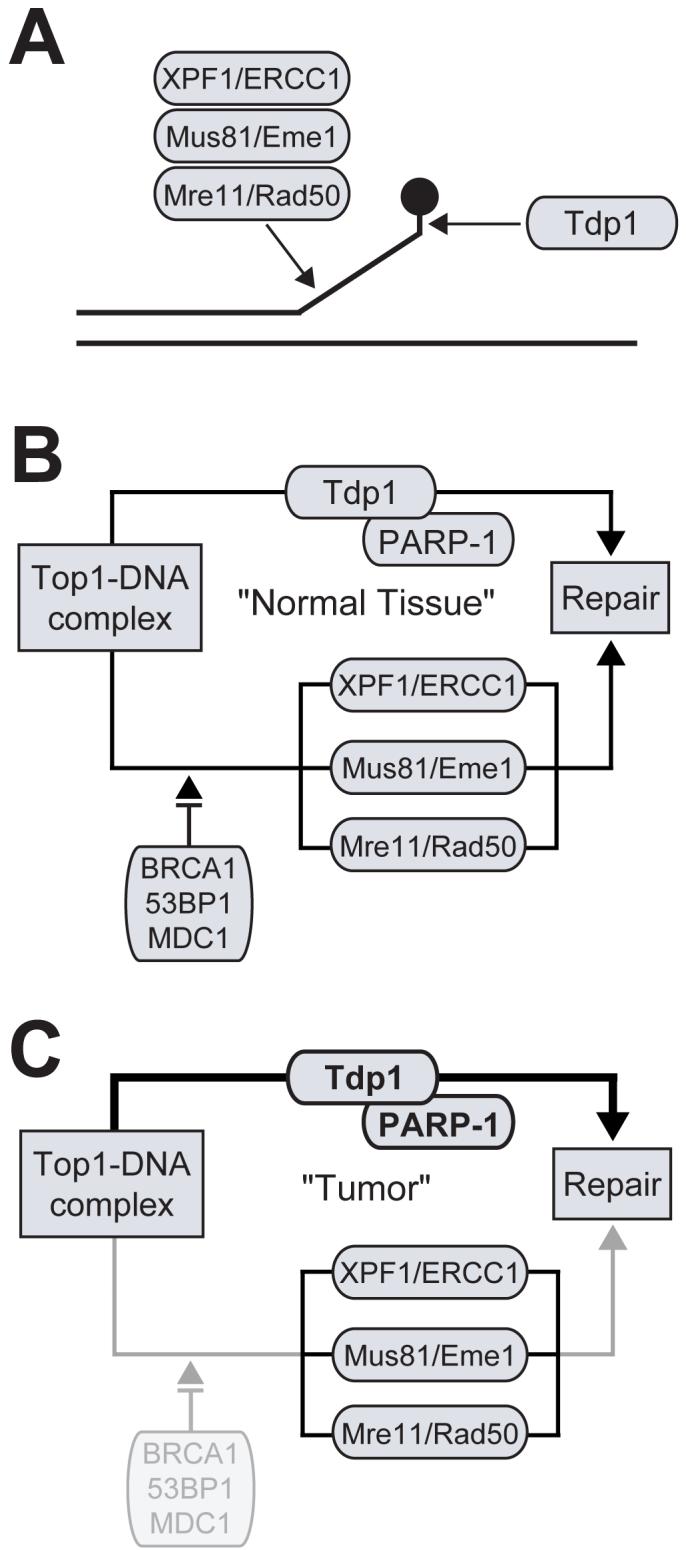
Redundant pathways involved the repair of Top1 cleavage complexes in mammalian cells. A) The processing of a Top1 cleavage complex by Tdp1 and three different structure-specific 3′-endonucleases. B) Schematic representation of the Tdp1-dependent pathway and checkpoint-dependent 3′-endonuclease pathway, which together are implicated in the repair of Top1 cleavage complexes. B) DNA repair and checkpoint deficiencies, such as in cancer cells, results in enhanced dependency for the repair of Top1 cleavage complexes via the Tdp1-dependent pathway.
In humans, there is no single Rad9 orthologue readily identifiable, but rather a family of tandem BRCT domain-containing proteins that act in the checkpoint response. Some of the Rad9 human orthologues include BRCA1, 53BP1, and MDC1, which are commonly referred to as checkpoint mediators [70]. Based on the insight gained from yeast, it has been proposed that there are two redundant pathways involved in repairing Top1-mediated DNA damage in humans (Fig. 6A). The first is a Tdp1-dependent pathway involving collaboration with the previously mentioned XRCC1 complex. The second is a Tdp1-independent, checkpoint-dependent pathway involving the Rad9 human homologues in the checkpoint response and the removal of damage by the three parallel functioning endonucleases. Even though there is redundancy in the repair of Top1-mediated DNA damage, it may not be critical to the clinical relevance of Tdp1 inhibition, since it is well established that deficiencies in checkpoint mechanisms are a common feature in cancer cells [71]. For example, if cancer-related checkpoint inactivation arises, then Tdp1 becomes the principle source for the removal of Top1-mediated DNA damage (Fig. 6B).
Studies performed in SCAN1 cells have further established proof of principle for the development of Tdp1 inhibitors in combination with CPT in anticancer drug therapy. Specifically, homozygous mutant SCAN1 cells have been shown to accumulate more total DNA strand breaks than normal lymphoblastoid cells after treatment with CPT [24, 72]. SCAN1 cells have also demonstrated enhanced sensitivity to the killing effects of CPT [28, 72]. Furthermore, complementary studies have shown that overexpression of wild-type Tdp1 protects cells against CPT-induced cell death [26, 73], whereas the inactive mutant Tdp1H263A does not [26]. Taken together, these data implicate Tdp1 in the repair of Top1-mediated DNA damage and demonstrate that a single defect in Tdp1 activity in human cells is adequate for CPT hypersensitivity, which is in contrast to the conditional mutations required in TDP1-deficient yeast cells. Thus, inhibition of Tdp1 can be envisioned to potentiate or synergize the cytotoxic effects of the clinically used Top1 inhibitors.
3.5 Tdp1 Inhibitors
Both vanadate and tungstate inhibit Tdp1 at millimolar concentrations because of their ability to mimic the transition state of Tdp1 (Fig. 7) [40]. Vanadate and tungstate have been shown to be useful in co-crystallization studies, such as in the case of Tdp1 [40, 41]. However, they cannot be used as pharmacological inhibitors because of their broad activity against phosphoryl transfer reactions. Another inhibitor of Tdp1 is the aminoglycoside neomycin (IC50, 8 mM) (Fig. 7) [74], which was originally tested based on its reported ability to inhibit PLD [75].
Fig. (7).

Chemical structures of Tdp1 inhibitors reported to date.
These are the only known inhibitors of Tdp1, therefore, a search for novel, more potent Tdp1 inhibitors is warranted. To assist in screening process for additional Tdp1 inhibitors, we have developed a high-throughput assay that uses electrochemiluminesent (ECL) substrates [77]. The DNA substrates have ruthenium labels (BV-TAG™) designed to emit light when stimulated (Fig. 4). In the presence of recombinant Tdp1, the ECL substrate is processed resulting in loss of the ECL signal. Thus, an inhibitor of Tdp1 would prevent this loss in signal. We have screened the “Diversity Set” that consists of 1981 compounds from the Developmental Therapeutics Program of the National Cancer Institute against human Tdp1. These compounds are representative of the chemical and structural diversity of the available repository of more that 140,000 chemicals. Several inhibitors of Tdp1 were identified. Fig. 7 shows the chemical structures of NSC 305831, NSC 118695, and NSC 88915, which were discovered using our high-throughput screen and confirmed by biochemical assays to be potent inhibitors of Tdp1 with IC50s in the micromolar range ([77]; Dexheimer et. al., in preparation). Additional studies are needed to gain insights into their mechanism of action and their cellular pharmacology.
4. SUMMARY AND PERSPECTIVES
The discovery of novel Tdp1 inhibitors is now possible as Tdp1 can be expressed as a biological active recombinant protein and high throughput assays are relatively easy to set up. In addition to biochemical screening, Tdp1 crystal structures could provide a means for in silico screening of chemical libraries. Lead optimization can also benefit from existing crystal structures and from the generation of new co-crystal structures with Tdp1 inhibitors. Biochemical inhibitors of Tdp1 should enhance the cytotoxicity of camptothecins and delay the removal of Top1 cleavage complexes in cell culture, thereby providing a cellular step for compound selection. The overall therapeutic goal is the development of new drugs that would selectively enhance the activity of Top1 inhibitors in tumors with preexisting DNA repair and cell cycle checkpoint deficiencies.
REFERENCES
- [1].Champoux JJ. Annu. Rev. Biochem. 2001;70:369. doi: 10.1146/annurev.biochem.70.1.369. [DOI] [PubMed] [Google Scholar]
- [2].Keck JL, Berger JM. Nat. Struct. Biol. 1999;6:900. doi: 10.1038/13260. [DOI] [PubMed] [Google Scholar]
- [3].Pommier Y, Pourquier P, Fan Y, Strumberg D. Biochim. Biophys. Acta. 1998;1400:83. doi: 10.1016/s0167-4781(98)00129-8. [DOI] [PubMed] [Google Scholar]
- [4].Wang JC. Nat. Rev. Mol. Cell Biol. 2002;3:430. doi: 10.1038/nrm831. [DOI] [PubMed] [Google Scholar]
- [5].Hsiang YH, Hertzberg R, Hecht S, Liu LF. J. Biol. Chem. 1985;260:14873. [PubMed] [Google Scholar]
- [6].Pommier Y. Nat. Rev. Cancer. 2006;6:789. doi: 10.1038/nrc1977. [DOI] [PubMed] [Google Scholar]
- [7].Pourquier P, Pommier Y. Adv. Cancer Res. 2001;80:189. doi: 10.1016/s0065-230x(01)80016-6. [DOI] [PubMed] [Google Scholar]
- [8].Pommier Y, Redon C, Rao VA, Seiler JA, Sordet O, Takemura H, Antony S, Meng L, Liao Z, Kohlhagen G, Zhang H, Kohn KW. Mutat. Res. 2003;532:173. doi: 10.1016/j.mrfmmm.2003.08.016. [DOI] [PubMed] [Google Scholar]
- [9].Marchand C, Antony S, Kohn KW, Cushman M, Ioanoviciu A, Staker BL, Burgin AB, Stewart L, Pommier Y. Mol. Cancer Ther. 2006;5:287. doi: 10.1158/1535-7163.MCT-05-0456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].McClendon AK, Osheroff N. Biochemistry. 2006;45:3040. doi: 10.1021/bi051987q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Chen AY, Liu LF. Annu. Rev. Pharmacol. Toxicol. 1994;34:191. doi: 10.1146/annurev.pa.34.040194.001203. [DOI] [PubMed] [Google Scholar]
- [12].Gupta M, Fujimori A, Pommier Y. Biochim. Biophys. Acta. 1995;1262:1. doi: 10.1016/0167-4781(95)00029-g. [DOI] [PubMed] [Google Scholar]
- [13].Pommier Y, Pourquier P, Urasaki Y, Wu J, Laco GS. Drug Resist. Updat. 1999;2:307. doi: 10.1054/drup.1999.0102. [DOI] [PubMed] [Google Scholar]
- [14].Strumberg D, Pilon AA, Smith M, Hickey R, Malkas L, Pommier Y. Mol. Cell Biol. 2000;20:3977. doi: 10.1128/mcb.20.11.3977-3987.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Pourquier P, Pilon AA, Kohlhagen G, Mazumder A, Sharma A, Pommier Y. J. Biol. Chem. 1997;272:26441. doi: 10.1074/jbc.272.42.26441. [DOI] [PubMed] [Google Scholar]
- [16].Pourquier P, Ueng LM, Kohlhagen G, Mazumder A, Gupta M, Kohn KW, Pommier Y. J. Biol. Chem. 1997;272:7792. doi: 10.1074/jbc.272.12.7792. [DOI] [PubMed] [Google Scholar]
- [17].Lanza A, Tornaletti S, Rodolfo C, Scanavini MC, Pedrini AM. J. Biol. Chem. 1996;271:6978. doi: 10.1074/jbc.271.12.6978. [DOI] [PubMed] [Google Scholar]
- [18].Daroui P, Desai SD, Li TK, Liu AA, Liu LF. J. Biol. Chem. 2004;279:14587. doi: 10.1074/jbc.M311370200. [DOI] [PubMed] [Google Scholar]
- [19].Yang SW, Burgin AB, Jr., Huizenga BN, Robertson CA, Yao KC, Nash HA. Proc. Natl. Acad. Sci. USA. 1996;93:11534. doi: 10.1073/pnas.93.21.11534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Pouliot JJ, Yao KC, Robertson CA, Nash HA. Science. 1999;286:552. doi: 10.1126/science.286.5439.552. [DOI] [PubMed] [Google Scholar]
- [21].Takashima H, Boerkoel CF, John J, Saifi GM, Salih MA, Armstrong D, Mao Y, Quiocho FA, Roa BB, Nakagawa M, Stockton DW, Lupski JR. Nat. Genet. 2002;32:267. doi: 10.1038/ng987. [DOI] [PubMed] [Google Scholar]
- [22].Interthal H, Pouliot JJ, Champoux JJ. Proc. Natl. Acad. Sci. USA. 2001;98:12009. doi: 10.1073/pnas.211429198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Cheng TJ, Rey PG, Poon T, Kan CC. Eur. J. Biochem. 2002;269:3697. doi: 10.1046/j.1432-1033.2002.03059.x. [DOI] [PubMed] [Google Scholar]
- [24].El-Khamisy SF, Saifi GM, Weinfeld M, Johansson F, Helleday T, Lupski JR, Caldecott KW. Nature. 2005;434:108. doi: 10.1038/nature03314. [DOI] [PubMed] [Google Scholar]
- [25].Plo I, Liao ZY, Barcelo JM, Kohlhagen G, Caldecott KW, Weinfeld M, Pommier Y. DNA Repair. (Amst) 2003;2:1087. doi: 10.1016/s1568-7864(03)00116-2. [DOI] [PubMed] [Google Scholar]
- [26].Barthelmes HU, Habermeyer M, Christensen MO, Mielke C, Interthal H, Pouliot JJ, Boege F, Marko D. J. Biol. Chem. 2004;279:55618. doi: 10.1074/jbc.M405042200. [DOI] [PubMed] [Google Scholar]
- [27].Zhou T, Lee JW, Tatavarthi H, Lupski JR, Valerie K, Povirk LF. Nucleic Acids Res. 2005;33:289. doi: 10.1093/nar/gki170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Interthal H, Chen HJ, Kehl-Fie TE, Zotzmann J, Leppard JB, Champoux JJ. EMBO J. 2005;24:2224. doi: 10.1038/sj.emboj.7600694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Waite M. Biochim. Biophys. Acta. 1999;1439:187. doi: 10.1016/s1388-1981(99)00094-3. [DOI] [PubMed] [Google Scholar]
- [30].Stuckey JA, Dixon JE. Nat. Struct. Biol. 1999;6:278. doi: 10.1038/6716. [DOI] [PubMed] [Google Scholar]
- [31].Ponting CP, Kerr ID. Protein Sci. 1996;5:914. doi: 10.1002/pro.5560050513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Raymond AC, Rideout MC, Staker B, Hjerrild K, Burgin AB., Jr. J. Mol. Biol. 2004;338:895. doi: 10.1016/j.jmb.2004.03.013. [DOI] [PubMed] [Google Scholar]
- [33].Gottlin EB, Rudolph AE, Zhao Y, Matthews HR, Dixon JE. Proc. Natl. Acad. Sci. USA. 1998;95:9202. doi: 10.1073/pnas.95.16.9202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Iwasaki Y, Horiike S, Matsushima K, Yamane T. Eur. J. Biochem. 1999;264:577. doi: 10.1046/j.1432-1327.1999.00669.x. [DOI] [PubMed] [Google Scholar]
- [35].Rudolph AE, Stuckey JA, Zhao Y, Matthews HR, Patton WA, Moss J, Dixon JE. J. Biol. Chem. 1999;274:11824. doi: 10.1074/jbc.274.17.11824. [DOI] [PubMed] [Google Scholar]
- [36].Sung TC, Roper RL, Zhang Y, Rudge SA, Temel R, Hammond SM, Morris AJ, Moss B, Engebrecht J, Frohman MA. EMBO J. 1997;16:4519. doi: 10.1093/emboj/16.15.4519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Xie Z, Ho WT, Exton JH. J. Biol. Chem. 2000;275:24962. doi: 10.1074/jbc.M909745199. [DOI] [PubMed] [Google Scholar]
- [38].Davies DR, Interthal H, Champoux JJ, Hol WG. Structure. 2002;10:237. doi: 10.1016/s0969-2126(02)00707-4. [DOI] [PubMed] [Google Scholar]
- [39].Leiros I, Secundo F, Zambonelli C, Servi S, Hough E. Structure. 2000;8:655. doi: 10.1016/s0969-2126(00)00150-7. [DOI] [PubMed] [Google Scholar]
- [40].Davies DR, Interthal H, Champoux JJ, Hol WG. J. Mol. Biol. 2002;324:917. doi: 10.1016/s0022-2836(02)01154-3. [DOI] [PubMed] [Google Scholar]
- [41].Davies DR, Interthal H, Champoux JJ, Hol WG. Chem. Biol. 2003;10:139. doi: 10.1016/s1074-5521(03)00021-8. [DOI] [PubMed] [Google Scholar]
- [42].Davies DR, Interthal H, Champoux JJ, Hol WG. J. Med. Chem. 2004;47:829. doi: 10.1021/jm030487x. [DOI] [PubMed] [Google Scholar]
- [43].Lowe G, Cullis PM, Jarvest RL, Potter BV, Sproat BS. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 1981;293:75. doi: 10.1098/rstb.1981.0062. [DOI] [PubMed] [Google Scholar]
- [44].Redinbo MR, Stewart L, Kuhn P, Champoux JJ, Hol WG. Science. 1998;279:1504. doi: 10.1126/science.279.5356.1504. [DOI] [PubMed] [Google Scholar]
- [45].Debethune L, Kohlhagen G, Grandas A, Pommier Y. Nucleic Acids Res. 2002;30:1198. doi: 10.1093/nar/30.5.1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Beidler DR, Cheng YC. Mol. Pharmacol. 1995;47:907. [PubMed] [Google Scholar]
- [47].Desai SD, Li TK, Rodriguez-Bauman A, Rubin EH, Liu LF. Cancer Res. 2001;61:5926. [PubMed] [Google Scholar]
- [48].Desai SD, Liu LF, Vazquez-Abad D, D’Arpa P. J. Biol. Chem. 1997;272:24159. doi: 10.1074/jbc.272.39.24159. [DOI] [PubMed] [Google Scholar]
- [49].Zhang HF, Tomida A, Koshimizu R, Ogiso Y, Lei S, Tsuruo T. Cancer Res. 2004;64:1114. doi: 10.1158/0008-5472.can-03-2858. [DOI] [PubMed] [Google Scholar]
- [50].Cusack JC, Jr., Liu R, Houston M, Abendroth K, Elliott PJ, Adams J, Baldwin AS., Jr. Cancer Res. 2001;61:3535. [PubMed] [Google Scholar]
- [51].Inamdar KV, Pouliot JJ, Zhou T, Lees-Miller SP, Rasouli-Nia A, Povirk LF. J. Biol. Chem. 2002;277:27162. doi: 10.1074/jbc.M204688200. [DOI] [PubMed] [Google Scholar]
- [52].Interthal H, Chen HJ, Champoux JJ. J. Biol. Chem. 2005;280:36518. doi: 10.1074/jbc.M508898200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Nitiss KC, Malik M, He X, White SW, Nitiss JL. Proc. Natl. Acad. Sci. USA. 2006;103:8953. doi: 10.1073/pnas.0603455103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Pouliot JJ, Robertson CA, Nash HA. Genes Cells. 2001;6:677. doi: 10.1046/j.1365-2443.2001.00452.x. [DOI] [PubMed] [Google Scholar]
- [55].Rideout MC, Raymond AC, Burgin AB., Jr. Nucleic Acids Res. 2004;32:4657. doi: 10.1093/nar/gkh796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Raymond AC, Staker BL, Burgin AB., Jr. J. Biol. Chem. 2005;280:22029. doi: 10.1074/jbc.M502148200. [DOI] [PubMed] [Google Scholar]
- [57].Whitehouse CJ, Taylor RM, Thistlethwaite A, Zhang H, Karimi-Busheri F, Lasko DD, Weinfeld M, Caldecott KW. Cell. 2001;104:107. doi: 10.1016/s0092-8674(01)00195-7. [DOI] [PubMed] [Google Scholar]
- [58].Caldecott KW, Tucker JD, Stanker LH, Thompson LH. Nucleic Acids Res. 1995;23:4836. doi: 10.1093/nar/23.23.4836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Taylor RM, Wickstead B, Cronin S, Caldecott KW. Curr. Biol. 1998;8:877. doi: 10.1016/s0960-9822(07)00350-8. [DOI] [PubMed] [Google Scholar]
- [60].de Murcia G, Menissier de Murcia J. Trends Biochem. Sci. 1994;19:172. doi: 10.1016/0968-0004(94)90280-1. [DOI] [PubMed] [Google Scholar]
- [61].Pommier Y, Barcelo JM, Rao VA, Sordet O, Jobson AG, Thibaut L, Miao ZH, Seiler JA, Zhang H, Marchand C, Agama K, Nitiss JL, Redon C. Prog. Nucleic Acid Res. Mol. Biol. 2006;81:179. doi: 10.1016/S0079-6603(06)81005-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Deng C, Brown JA, You D, Brown JM. Genetics. 2005;170:591. doi: 10.1534/genetics.104.028795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Liu C, Pouliot JJ, Nash HA. Proc. Natl. Acad. Sci. USA. 2002;99:14970. doi: 10.1073/pnas.182557199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Liu C, Pouliot JJ, Nash HA. DNA Repair. (Amst) 2004;3:593. doi: 10.1016/j.dnarep.2004.03.030. [DOI] [PubMed] [Google Scholar]
- [65].Vance JR, Wilson TE. Proc. Natl. Acad. Sci. USA. 2002;99:13669. doi: 10.1073/pnas.202242599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Roberts JA, White MF. Nucleic Acids Res. 2005;33:6662. doi: 10.1093/nar/gki974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Bardwell AJ, Bardwell L, Tomkinson AE, Friedberg EC. Science. 1994;265:2082. doi: 10.1126/science.8091230. [DOI] [PubMed] [Google Scholar]
- [68].Bastin-Shanower SA, Fricke WM, Mullen JR, Brill SJ. Mol. Cell Biol. 2003;23:3487. doi: 10.1128/MCB.23.10.3487-3496.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].D’Amours D, Jackson SP. Nat. Rev. Mol. Cell Biol. 2002;3:317. doi: 10.1038/nrm805. [DOI] [PubMed] [Google Scholar]
- [70].Zhou BB, Elledge SJ. Nature. 2000;408:433. doi: 10.1038/35044005. [DOI] [PubMed] [Google Scholar]
- [71].Dasika GK, Lin SC, Zhao S, Sung P, Tomkinson A, Lee EY. Oncogene. 1999;18:7883. doi: 10.1038/sj.onc.1203283. [DOI] [PubMed] [Google Scholar]
- [72].Miao ZH, Agama K, Sordet O, Povirk L, Kohn KW, Pommier Y. DNA Repair. (Amst) 2006 doi: 10.1016/j.dnarep.2006.07.004. [DOI] [PubMed] [Google Scholar]
- [73].Nivens MC, Felder T, Galloway AH, Pena MM, Pouliot JJ, Spencer HT. Cancer Chemother. Pharmacol. 2004;53:107. doi: 10.1007/s00280-003-0717-6. [DOI] [PubMed] [Google Scholar]
- [74].Liao Z, Thibaut L, Jobson A, Pommier Y. Mol. Pharmacol. 2006;70:366. doi: 10.1124/mol.105.021865. [DOI] [PubMed] [Google Scholar]
- [75].Huang Y, Qureshi IA. Chen, H. Mol. Cell Biochem. 1999;197:195. doi: 10.1023/a:1006930706311. [DOI] [PubMed] [Google Scholar]
- [76].Caldecott KW. Cell. 2003;112:7. doi: 10.1016/s0092-8674(02)01247-3. [DOI] [PubMed] [Google Scholar]
- [77].Antony S, Marchand C, Stephen AG, Thibaut L, Agama KK, Fisher RJ, Pommier Y. Nucleic Acids Res. 2007;35:4474. doi: 10.1093/nar/gkm463. [DOI] [PMC free article] [PubMed] [Google Scholar]