

Abstract
Recent decades have witnessed tremendous advances in the neuroscience of emotion, learning and memory, and in animal models for understanding depression and anxiety. This review focuses on new rationally designed psychiatric treatments derived from preclinical human and animal studies. Nonpharmacological treatments that affect disrupted emotion circuits include vagal nerve stimulation, rapid transcranial magnetic stimulation and deep brain stimulation, all borrowed from neurological interventions that attempt to target known pathological foci. Other approaches include drugs that are given in relation to specific learning events to enhance or disrupt endogenous emotional learning processes. Imaging data suggest that common regions of brain activation are targeted with pharmacological and somatic treatments as well as with the emotional learning in psychotherapy. Although many of these approaches are experimental, the rapidly developing understanding of emotional circuit regulation is likely to provide exciting and powerful future treatments for debilitating mood and anxiety disorders.
Major depressive disorder (MDD) is the most common of all psychiatric disorders. MDD ranks among the top causes of worldwide disease burden and disability, with lifetime risk of 7–12% in men and 20–25% in women1. Although good treatments such as selective serotonin reuptake inhibitors (SSRIs) are effective and available, up to 20% of patients completely fail to respond to standard interventions and nearly 60% may not achieve adequate response2. The different anxiety disorders, including panic disorder, post-traumatic stress disorder (PTSD) and phobias, are also extremely common, with a combined lifetime prevalence of over 28%, and with a similar societal cost-burden to that of MDD3. Anxiety disorders can be extremely debilitating and, overall, have rates of failure to respond similar to those of MDD.
In this review, mood and anxiety disorders will be considered together for several reasons: (i) comorbidity between anxiety and depression is the rule and not the exception, with up to 90% of patients with anxiety disorders experiencing clinical depression at some point in their lifetime4; (ii) there is a significant problem of diagnostic classification, with highly overlapping symptom criteria; (iii) from a neuroimaging perspective the circuits involved in both sets of disorders can be difficult to distinguish; and (iv) the most powerful treatments for both disorders are the same, including antidepressants such as SSRIs and cognitive behavioral therapy (CBT). It is not that biologically meaningful subclassifications do not exist within the broad categories of emotional disorders, rather that the current clinical descriptions are probably not identifying the phenotypic clusters of disorders that may be most useful from a neurobiological and treatment perspective.
Several lines of evidence suggest that there are specific neural circuits within the limbic-cortical system that mediate stress-responsiveness, mood and emotional regulation. Disorders of mood and anxiety represent brain-based disorders that lead to dysregulation of these circuits. Traditional psychiatric medication, psychotherapy and somatic therapies converge in bringing homeostasis to these disrupted circuits. New neurostimulatory therapies based on progress in understanding emotion circuitry and new pharmacological therapies based on understanding emotional learning are likely to provide more rapid and robust methodologies for treating these debilitating and complex disorders.
Abnormal circuit modulation in mood and anxiety disorders
Human imaging studies using positron emission tomography (PET) and functional magnetic resonance imaging (fMRI) have examined differences in brain regional activation in depressed and anxious subjects relative to controls and in patients before and after treatment. This review focuses on components of depression such as sad or dysphoric affect, negative emotions, impaired cognition and anxiety-related symptoms. Many brain areas may underlie some of the different symptom clusters of depression5. In contrast to the brain regions that bring about the negative emotional components of depression, the nucleus accumbens, along with other areas involved in reward processing, are also likely to be involved in the anhedonic components of depression6–10. Some of these areas may be equally important in the circuitry of depression, but will not be examined here due to space constraints.
The areas most reproducibly found to be dysregulated in common emotional disorders are the prefrontal cortex (PFC) and subgenual cingulate cortex (Cg25), which seem to be involved in emotion experience and processing, as well as the subcortical hippocampus and amygdala, which are involved in emotional memory formation and memory retrieval5,11–14.
For the purposes of illustration, this review will focus primarily on data related to the role of Cg25 in emotion regulation and processing, and the role of the amygdala in emotional memory formation and expression. Cg25 is involved in the production of sad emotions and in antidepressant treatment response. It is activated during transient sadness, and after recovery from depression its activity is decreased compared with baseline after recovery from depression12 (Fig. 1a). Cg25 decreases in activity are seen in response to chronic fluoxetine treatment for MDD (Fig. 1b), as well as during recovery from depression related to Parkinson’s disease after chronic fluoxetine treatment (Fig. 1c). Interestingly, subjects who are randomized to placebo but show a natural recovery from symptoms of depression also have decreased activity in Cg25 from baseline (Fig. 1d). Activity in Cg25 before treatment predicts treatment response with CBT15 (Fig. 1e). Additionally, a response to CBT for social phobia is accompanied by decreases in Cg25 activity, and responders have greater decreases in Cg25 activity than do nonresponders16 (Fig. 1f).
Figure 1.
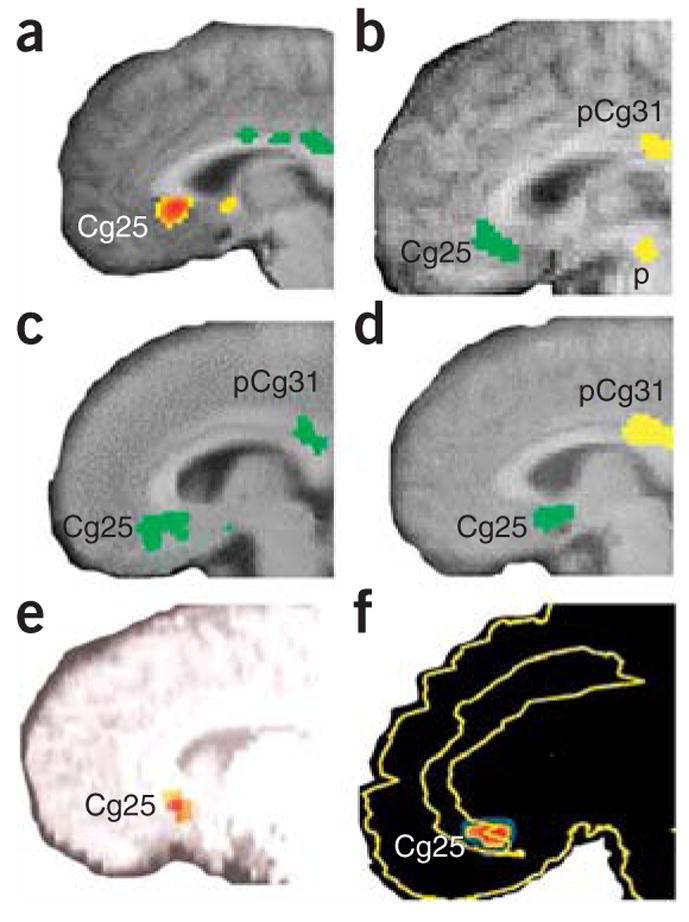
Subgenual cingulate cortex activation across studies. (a) Transient sadness in healthy volunteers increases activity (red) in Cg25 (arrow) measured with positron emission tomography (PET) (from ref. 12. Reprinted with permission from the American Journal of Psychiatry, copyright 1999, American Psychiatric Association.). (b) Decreased Cg25 activity (green) with chronic fluoxetine treatment for depression. (c) Cg25 decrease (green) in recovery with chronic fluoxetine from Parkinson’s disease–related depression. (d) Natural recovery with decreased Cg25 activity (green) in patients treated with placebo. Panels b–d reprinted from ref. 11 by permission of Oxford University Press. (e) Predictors of response in subjects responding to CBT for depression included low pretreatment Cg25 activity (red) (from ref. 15. Reprinted with permission from the American Journal of Psychiatry, copyright 1999, American Psychiatric Association.). (f) Subgenual cortical decreased activity (red) was common in responders compared with nonresponders for those responding both to citalopram and to CBT for social phobia (from ref. 16. Reprinted from Archives of General Psychiatry, copyright 2002, American Medical Association. All rights reserved.).
Overactivation of the amygdala is also implicated in depression and anxiety17 (Fig. 2). Amygdala activation decreases with recovery from mood symptoms. Studies that implicate Cg25 also find significant amygdala decreases with response to CBT treatment for social phobia16 (Fig. 2a) and report that sustained amygdala activity before treatment predicts antidepressant response to CBT15 (Fig. 2c).
Figure 2.
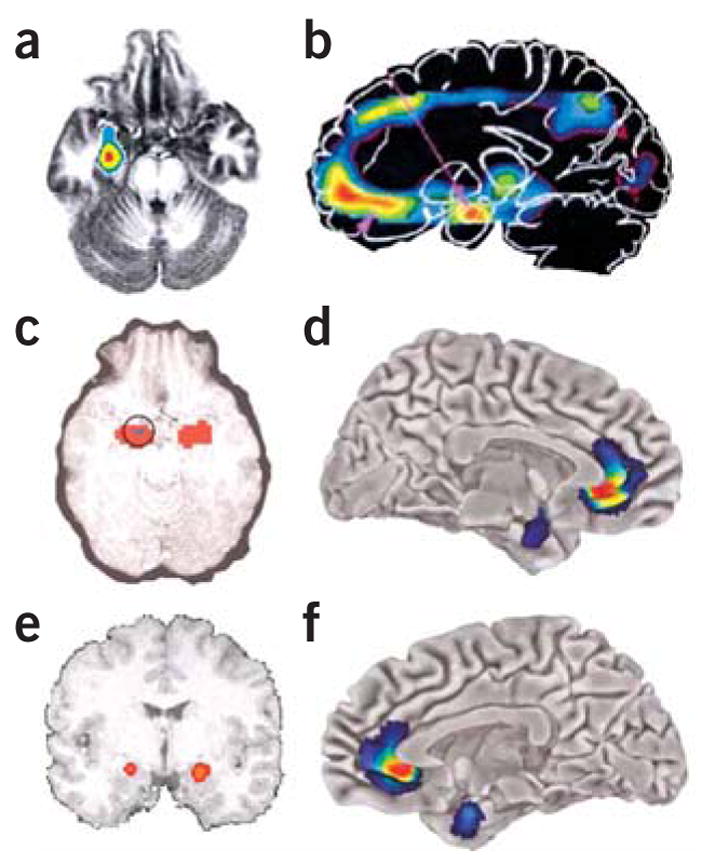
Amygdala activation across studies. (a) Responders compared with nonresponders showed greater decreases (red to blue indicates greatest to least decrease) in anxiety-induced amygdala activation in both citalopram and CBT treatment for social phobia (ref. 16. Reprinted from Archives of General Psychiatry, copyright 2002, American Medical Association. All rights reserved.). (b) In familial MDD, areas of abnormally increased cerebral blood flow (red) compared with that in controls include the amygdala (from ref. 17. Reprinted from Biological Psychiatry, copyright 2000, with permission from Elsevier.). (c) Predictors of response in subjects responding to CBT for depression included high sustained emotion-induced amygdala activity (circled; red indicates increased regional cerebral blood flow in response to negative words) (from ref. 15. Reprinted with permission from the American Journal of Psychiatry, copyright 1999, American Psychiatric Association.). (d–f) Left (d) and right (f) hemispheres showing that subgenual cingulate and amygdala show reductions in gray matter volume (red to blue indicates most to least volume decrease) in 5-HTTLPR high-risk short-allele carriers compared with homozygous long-allele genotypes (Reprinted by permission from Macmillan Publishers, Ltd.: Nature Neuroscience, ref. 19, copyright 2005.). Lorazepam, a benzodiazepine used for anxiety treatment, dose dependently attenuates the amygdata activation induced by emotional face viewing (e; red indicates greatest decrease in regional cerebral blood flow compared with placebo) (from ref. 99. Reprinted from Archives of General Psychiatry, copyright 2005, American Medical Association. All rights reserved.).
A genetic polymorphism has repeatedly been implicated in gene by environment interactions for disorders of emotional dysregulation: the serotonin promoter polymorphism 5-HTTLPR (see review by Canli and Lesch18 in this issue). Carriers of the risk-conferring 5-HTTLPR polymorphism have reduced gray matter volume in the perigenulate region surrounding Cg25 as well as in the amygdala19 (Fig. 2d,f). Additionally, these areas are tightly coupled with the processing of negative affect in long-allele carriers, whereas they are functionally uncoupled in the at-risk short-allele carriers. Together these data indicate that prefrontal-limbic circuits, the Cg25 and amygdala areas in particular, may be critically involved in emotional processing and regulation in mood and anxiety disorders.
Neurostimulation therapies modulate dysregulated circuits
Several somatic therapies, available or under investigation, may modulate this disrupted circuit activity. Vagus nerve stimulation therapy (VNS) is approved by the FDA for treatment of medication-resistant depression and was approved earlier for the treatment of epilepsy20. Although the approach seems to be tolerable for long-term use in patients, and relatively few patients have relapsed in the early depression trials, long-term efficacy in refractory patients still remains to be demonstrated.
The initial reasoning behind the use of VNS followed from its apparent effects of elevating mood in patients with epilepsy20, combined with evidence that VNS affects limbic activity in neuroimaging studies21. Furthermore, VNS alters concentrations of serotonin, norepinephrine, GABA and glutamate within the brain22–24, suggesting that VNS may help correct dysfunctional neurotransmitter modulatory circuits in patients with depression. In VNS, patients have a battery-powered generator implanted in the chest wall that connects to a wire wrapped around the left vagus nerve in the neck. This wire sends intermittent electrical pulses through the vagal afferent connections, which normally convey visceral and homeostatic information to the brainstem. Although several regions within the medulla receive vagal inputs, the nucleus of the tractus solitarius (NTS) receives the greatest number of vagal afferent synapses (Fig. 3a,b). These afferents carry visceral sensation from the pharynx, larynx, trachea and thoracoabdominal organs, somatic sensation from around the ear, and taste sensation. Related to its antidepressant and antiepileptic effects are the polysynaptic connections through the NTS to the noradrenergic locus coeruleus and to the serotonergic raphe nuclei25. Additionally, there are vagal disynaptic projections through the NTS to the thalamus, hypothalamus, central amygdala nucleus, bed nucleus of the stria terminalis and nucleus accumbens. Finally, the vagus directly innervates the reticular thalamic formation, which is involved in coordinating a variety of activities, including sleep-wake cycle, alertness and attention26,27, all behaviors that are frequently disrupted in depression. Patients notice improved alertness during VNS, and a study of patients without sleep disorders revealed improved diurnal alertness during VNS28.
Figure 3.
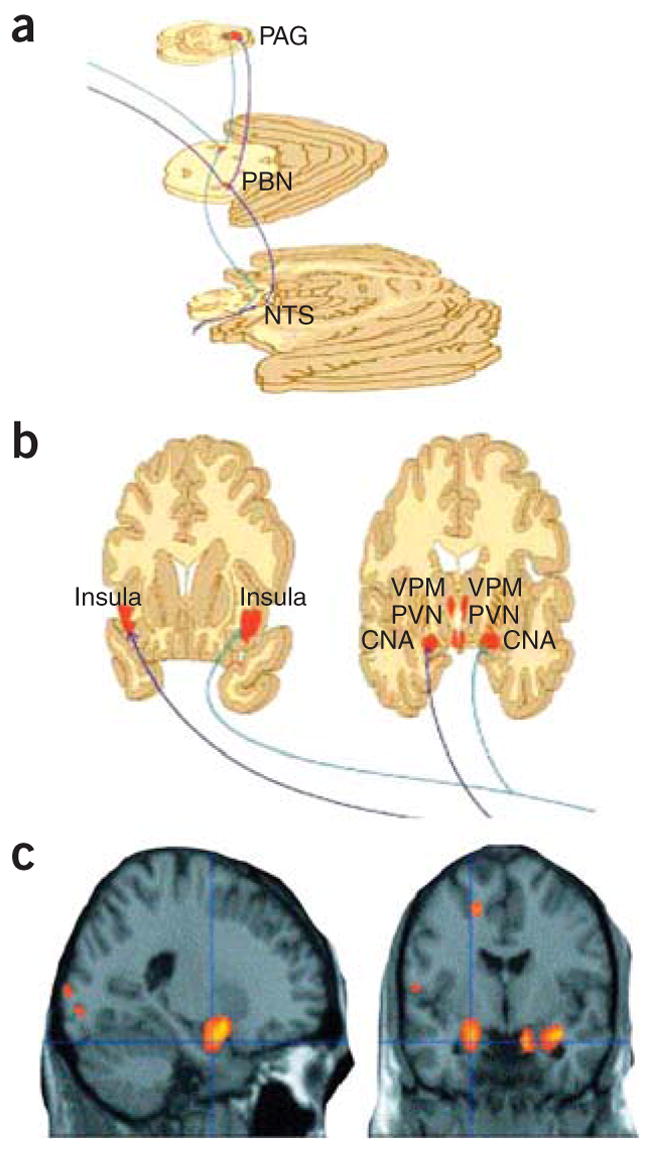
Ascending projections from vagus–nucleus solitarius pathways with VNS. (a) Brainstem view of ascending bilateral pathways of the central autonomic, reticular activating and limbic systems receiving afferent input from the vagus nerve. Vagal–bilateral NTS projections, through synapses in the parabrachial nuclei, project to more rostral and cortical regions. (b) Projections of NTS and parabrachial nucleus provide dense innervation of autonomic, reticular and limbic forebrain structures (from ref. 26. Reprinted from Neurology, by permission of Lippincott Williams & Wilkins.). NTS, nucleus of the tractus solitarius; PBN, parabrachial nucleus; PAG, periaqueductal gray matter; CNA, central nucleus of the amygdala; PVN, periventricular nucleus of the hypothalamus; VPM, ventral posteromedial nucleus of the thalamus. (c) Changes in amygdala and hippocampal regional cerebral blood flow by therapeutic vagus nerve stimulation after 4 weeks of VNS for depression (red-yellow represent areas of greatest decrease in regional cerebral blood flow relative to pretreatment values). Panels a and b reprinted from Psychiatry Research: ref. 29, copyright 2005, with permission from Elsevier.
Functional imaging suggests that VNS leads to acute changes in hypothalamus, orbitofrontal cortex, amygdala, hippocampus, insula, medial prefrontal cortex and cingulate29,30 (Fig. 3c). Chronic VNS treatment leads to significant ventromedial prefrontal cortex deactivation, similarly to other approaches to depression treatment31. Several studies of VNS use in depression demonstrate that it is reasonably well tolerated and that patients treated with VNS for depression maintain low rates of relapse32, so this approach may prove to be an important adjunct for refractory depression.
Transcranial magnetic stimulation
Repetitive transcranial magnetic stimulation (rTMS) and electroconvulsive therapy (ECT) are both somatic treatments relying upon altering local and distant neural circuits within the brain. ECT has been used since the 1930s as a treatment for depression, but was not approved by the FDA until 1979. ECT involves the application of brief electrical pulses to the scalp to induce depolarization of cortical neurons and resultant brain seizures. ECT is among the most effective treatments in psychiatry for refractory depression, yet its mechanisms of action remains unclear33. Furthermore, although it is now a largely safe and effective treatment approach, there are numerous disadvantages, including anesthesia exposure, postseizure amnesia and relapse of depression, which is common within 6 months after treatment34,35. rTMS is a relatively new technique in which focal magnetic stimulation is applied external to the scalp, inducing electrical stimulation in cortical tissue36–38. TMS occurs when a powerful, pulsed magnetic field is created with the pulsing of high-intensity current through an electromagnetic coil placed near the scalp. This technique was originally introduced as a tool to probe local cortical function through noninvasive stimulation and later was found to have antiepileptic activity37.
Because of the ability of rTMS to cause transient activity changes in focal brain regions, it was predicted that rTMS might have antidepressant activity similar to that of ECT, if the local prefrontal cortex depolarization effects of ECT are a primary contributing factor to its antidepressant function. Early trials of rTMS in patients with depression confirmed this prediction39,40. A meta-analytic review, which examined 33 clinical trials performed since 1996 on the efficacy of rTMS in depression41, concluded that rTMS is more effective in the treatment of depression than sham rTMS, with a large effect size of 0.71. In contrast, another review42 that arguably was too conservative in the trial inclusion criteria41 found a lack of efficacy of rTMS in a much smaller number of select trials. Another recent, multisite review is promising in both treatment efficacy and tolerability43. Unfortunately, there still remains limited consensus evidence regarding which are the most effective treatment parameter combinations. Thus although the data remain promising as a future treatment methodology, we need more knowledge regarding which patients are likely to receive optimal benefit and which treatment parameters are optimal, and we need larger, rigorously controlled studies.
rTMS is also potentially useful as a probe for understanding brain activity changes with response to treatment with rTMS and ECT. By using a modified TMS coil and a conventional MRI scanner, the magnetic field induced by a TMS coil can be visualized44. With this approach, one can noninvasively and precisely determine the locations of magnetic and electrical field induction. PET in combination with rTMS indicates that TMS at 1 Hz inhibits cortical excitability, whereas high-frequency TMS (20 Hz) enhances cortical excitability45,46, consistent with preclinical data showing that low-frequency TMS leads to cortical inhibition and quenching of seizure activity. These studies also suggest the intriguing idea that there is differential antidepressant response dependent on baseline level of cerebral glucose metabolism, with baseline hypometabolism responding better to high-frequency stimulation that seems to enhance excitability, and baseline hypermetabolism responding better to low-frequency stimulation that seems to dampen excitability45.
In preclinical studies, rTMS modulates neural circuits and neurotransmitter systems thought to be involved with mood regulation. rTMS acutely modulates dopamine and serotonin levels33. Chronic rTMS modulates cortical β-adrenergic receptors, decreases type 5-HT2 and increases type 5-HT1a serotonergic receptors in frontal cortex, and increases NMDA receptors in hypothalamus and basolateral amygdala. Furthermore, rTMS at 10 Hz in a rat model of depression leads to enhanced hippocampal plasticity after stress using in vitro LTP as a model47.
Magnetic seizure therapy
Magnetic seizure therapy (MST) involves the induction of relatively focal seizure activity through the transcranial induction of cortical depolarization using focused magnetic stimulation. MSTseems to offer greater control of intracerebral current intensity than is possible with ECT and thus may result in fewer cognitive side effects33. The use of therapeutic magnetic seizure induction began at University Hospital in Bern, Switzerland, in May 2000. The first randomized, within-subject trial comparing ECT and MST in ten patients indicated that MST seems to have fewer side effects, is associated with faster recovery of orientation and is superior to ECT on measures of attention, category fluency and retrograde amnesia48. Ongoing studies are more fully examining its efficacy in the treatment of depression. As with ECT, the specific neural circuitry being altered with MST remains unclear, but it is hoped that the ability to focally stimulate cortical areas will provide a new tool to dissect the critical circuitry involved in recovery from depression.
Deep brain stimulation
Deep brain stimulation (DBS) with implanted electrodes has been used with increasing frequency and success in the treatment of Parkinson’s disease49 (Fig. 4a,b). DBS may also modulate disorders of emotion, in addition to its known role in treating disorders of motion. An early report suggested that DBS could rapidly alter emotion in Parkinson’s patients. This study reported the immediate onset of severe, debilitating depressive symptoms in a subject receiving DBS just below the left subthalamic nucleus50. DBS targeting the caudate may lead to recovery from depressive symptoms10,51,52, but this work remains preliminary.
Figure 4.
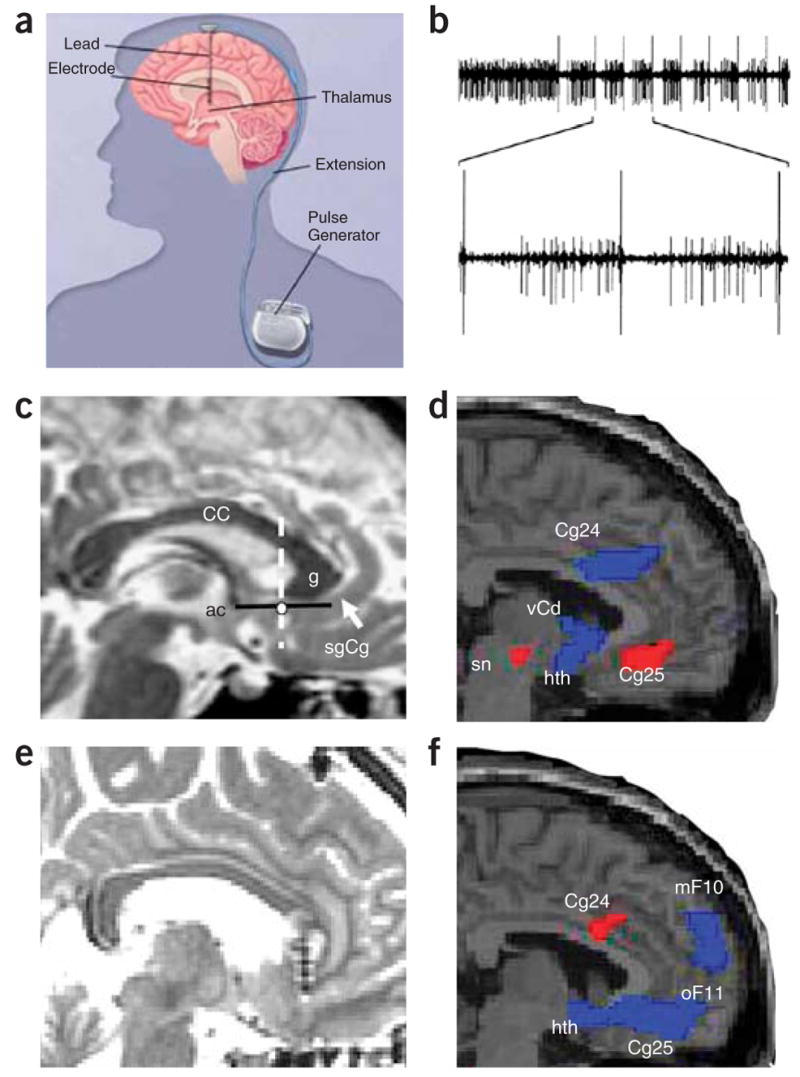
Deep brain stimulation. (a) Schematic of DBS used for Parkinson’s patients (modified from WebMD, http://www.medicinenet.com/deep_brain_stimulation/article.htm). (b) One possible mechanism of action of DBS-induced inhibition: recordings from a globus pallidus internus (GPi) high-frequency-discharge neuron showing inhibitory periods after each stimulus pulse (50 μA) (from ref. 100. Copyright 2000 by American Physiological Society. Reproduced with permission of American Physiological Society via Copyright Clearance Center.). (c) Preoperative MRI target localization for DBS for refractory depression. White dot, location of the sgCg white matter targeted for electrode placement in the DBS depression study, shown in a single subject; white arrow, sgCg gyrus; dotted line, anterior-posterior location of the electrode along the line (black) between anterior commissure and genu of the corpus callosum. (d) Baseline PET-derived blood flow: depressed patients show increased activity (red) in Cg25 compared with healthy controls. (e) Postoperative MRI in a single DBS patient showing the electrode tip in the sgCg white matter. (f) At 3 months, regions of blood flow change measured with PET in treatment responders show decreased activity (blue) compared with pretreatment. Panels c–f reprinted by permission from an article published in Neuron, ref. 58, copyright Elsevier 2005. CC, corpus callosum; ac, anterior commissure; g, genu of the corpus callosum; sgCg, subgenual cingulate; Cg24, cingulate area 24; sn, substantia nigra; hth, hypothalamus; mF10, medial frontal area 10; oF11, orbito-frontal area 11.
In treatment-refractory depressed patients, focusing DBS on another region, prefrontal cortex area Cg25, has been examined more extensively. Limbic-cortical pathways mediate depressed mood, according to functional neuroimaging studies11,53. Work from our group (H.S.M.) demonstrates that Cg25 is involved in acute sadness as well as in the effects of antidepressant treatment in depression12,54 (Fig. 1). Based on data that Cg25 is metabolically overactive in treatment-resistant depression and that activity in this area correlates with response to treatment55–57, we examined whether the application of chronic DBS to Cg25 would reduce depression symptoms in patients with severe refractory depression58. Chronic stimulation of white matter tracts adjacent to the subgenual gyrus (Fig. 4) was associated with a robust, sustained remission of depression in four of six previously refractory patients. Furthermore, the antidepressant action was associated with marked reduction in local cerebral blood flow as well as with changes in downstream limbic and cortical sites measured with PET (Fig. 4d,f). These data indicate that disrupting the pathological limbic-cortical circuit in depression with DBS may effectively reverse treatment-resistant depression in some patients.
The mechanisms that mediate responses to chronic treatment remain incompletely understood, but it is clear that DBS produces both local and distant brain activity effects59. The behavioral and neuroimaging changes in the above study are consistent with a DBS effect of reducing elevated baseline Cg25 activity. DBS-induced activation of inhibitory GABAergic afferents and/or synaptic or metabolic failure induced by high-frequency activity are some of the more likely mechanisms of action currently under study (Fig. 4b)60,61. Other distal effects on remote cortical and brainstem areas could occur both through indirect trans-synaptic effects, as a direct result from Cg25 decreased activity, or through direct anterograde or retrograde activation of white matter projections that pass through the field of stimulation.
In addition to these chronic effects, some subjects noted a sudden mood change with stimulation within the operating room. This is consistent with an acute effect of deactivating area Cg25, which dynamically correlates with changes in mood (that is, activity increases with sadness and decreases with alleviation of dysphoria in depression)12.
DBS is also being examined to treat refractory obsessive-compulsive disorder, a severely disabling disorder in which patients are overwhelmed with obsessional and repetitive thoughts and often cannot inhibit disruptive compulsive behavior. In contrast to MDD, OCD is thought to involve overactivation of cortico-striatal-limbic circuits, and there are several theoretical connections between OCD and Parkinson’s disease implying that they may involve different components of the striatal-cortical systems62. DBS focusing on the internal capsule extending into the ventral striatum shows promise for treatment of OCD63. A long-term outcome, open-label study using 36-month chronic stimulation suggests promising long-term effects of DBS in highly treatment-resistant OCD64. The subjects demonstrated significant decreases in OCD symptoms and improvements in overall functioning. Notably, concomitant changes in depression symptoms were also described, facilitating pilot studies of anterior internal capsule stimulation for depression.
Together, the early data on DBS suggest that it might be a powerful tool to specifically target dysregulated neural circuits. As the circuitry underlying emotional regulation continues to be worked out, DBS may provide an important approach to the treatment of refractory mood and anxiety disorders.
Pharmacological therapies to modulate emotional learning
Emotional circuits show consistent, robust and significant changes in activity when mood and anxiety disorders are treated with certain forms of talk therapy, which are often similar to changes seen with pharmacological and somatic treatments (Figs. 1 and 2)15,16,53. These findings suggest that with emotional learning, the brain can use endogenous homeostatic mechanisms to correct dysregulated patterns of activity. Standard pharmacological treatments tend to target symptom clusters but not necessarily the root cause of the disorder. Thus, an alternative approach is the possibility of enhancing the specific new learning that occurs with psychotherapy. An even more exciting, yet still very exploratory, idea is the possibility that certain disorders, such as post-traumatic stress disorder (PTSD), could be prevented before they ever develop by preventing the fear ‘overlearning’ that is thought to occur during memory consolidation after trauma65.
The pathways mediating this learning are very complex. However, specific learning processes involved in the extinction of fear (new emotional learning that competes with or inhibits aversive memories) are well understood (Fig. 5). Disorders of aversive emotion respond to behavioral therapy, and this process, which uses extinction mechanisms, may be enhanced with specific pharmacotherapy. It is also theoretically possible that the initial consolidation of a fear memory may be disrupted before the overlearning of a trauma and subsequent anxiety disorder develop. Finally, animal studies indicate that already-formed memories may remain labile when reactivated and thus may be subject to the same sort of disruption as consolidation of new memories. Together, these approaches offer tantalizing future ways to modulate fear, aversive and traumatic memories to alleviate suffering due to negative memories, as we discuss in more detail below.
Figure 5.
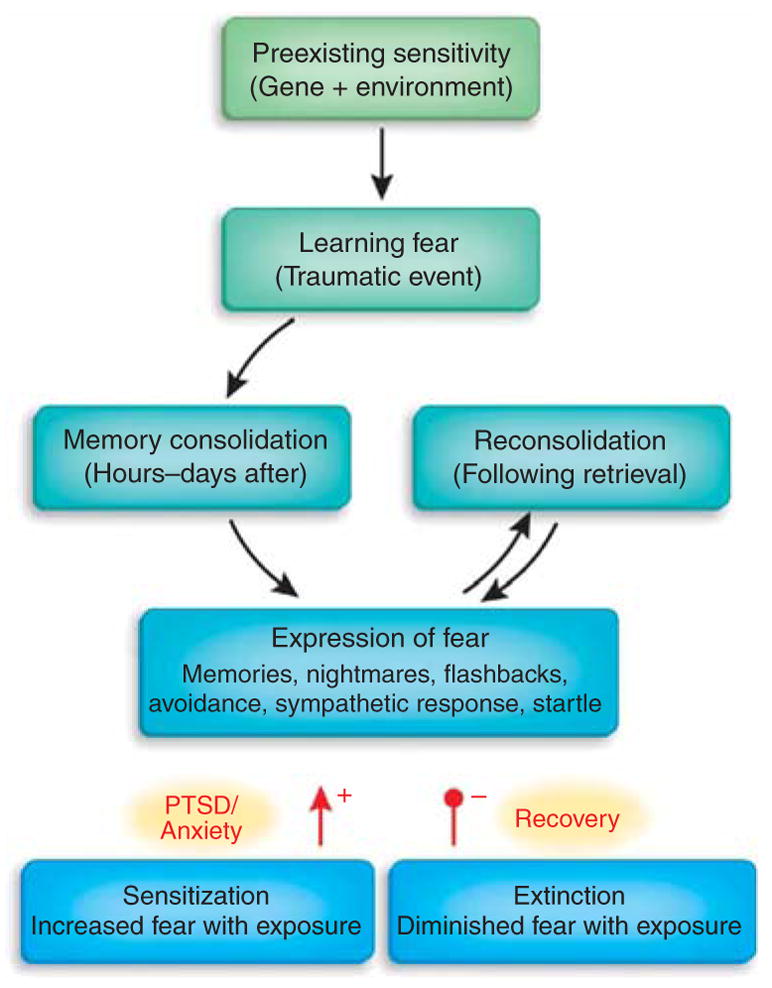
Emotional learning processes related to fear. The strength and regulation of emotional memories is affected by many factors both before and after the traumatic or fearful event occurs. Genetic heritability comprises up to ~40% of the risk for both depression and PTSD, and early childhood abuse is a very strong risk factor for all mood and anxiety disorders. Memories are not permanent at the time of the trauma, rather they undergo a period of consolidation in which they shift from a labile state to a more permanent state. Some evidence, at least for relatively recent memories, indicates that even after memories become consolidated, they become labile again when recalled—a process known as reconsolidation. The expression of traumatic memories, which can be the source of many symptoms in fear-related disorders, is diminished by the process of extinction, when repeated therapeutic exposures to the fear-related cues reduces or inhibits the fear memories over time. In contrast, there is some evidence that in those who develop PTSD and other pathology, a combination of avoidance of sufficient exposure with intrusive and uncontrollable memories leads to sensitization of the fear response. Arrowhead (+), sensitization increases expression of fear; filled circle (−), extinction decreases expression.
Enhancing extinction of fear
The inhibition of fear acquired by associative learning has been studied in both animals and humans. After the pairing of an aversive unconditioned stimulus (UCS) to a neutral conditioned stimulus, a conditioned fear response is established. If the neutral conditioned stimulus is then repeatedly presented in the absence of the UCS, a procedure known as extinction training, the result is an inhibition of the conditioned fear response to the neutral conditioned stimulus. From an operational perspective, extinction may thus be defined as “a reduction in the strength or probability of a conditioned fear response as a consequence of repeated presentation of the conditioned stimulus in the absence of the UCS”66.
A variety of behavioral observations support the hypothesis that extinction is a form of learning and not ‘unlearning’ or the forgetting of a conditioned association (reviewed in ref. 67). Thus, enhancing neurotransmission at NMDA receptors, which are involved in new learning processes, might facilitate extinction68. D-Cycloserine (DCS) is a partial NMDA agonist, acting at the strychnine-insensitive glycine recognition site of the NMDA receptor complex to enhance NMDA receptor activity69. Both systemic and amygdala-specific administration of DCS dose-dependently enhance extinction of previously conditioned fear but do not influence fear in rats that have not received extinction training68. These findings have been replicated in studies measuring extinction of fear with startle and freezing and with extinction of appetitive cues, such as cocaine-conditioned place preference70–73. Collectively, data from rodent studies indicate that DCS, a drug already used in humans for treating tuberculosis, may have potential use in facilitating extinction-based therapies for human anxiety disorders.
Behavioral therapies for anxiety disorders generally involve some form of extinction training66. This involves graded exposure to the feared object or event in the absence of any likely actual harm. This exposure may occur in the imagination, as when a narrative is read or listened to by the patient, or the patient may directly encounter the feared stimulus. Considering the similarity between extinction training in rodents and exposure therapy for anxiety disorders in humans, we were interested in finding ways to integrate pharmacotherapy with psychotherapy, which have not generally been combined into a treatment more effective than either alone. Sometimes combining pharmacotherapy with psychotherapy can even make a bad situation worse74. However, extinction-based therapies for anxiety may be an exception to this trend.
To determine whether DCS would improve extinction of fear in human patients with fear-related disorders, we ran a double-blind, placebo-controlled trial in a controlled exposure protocol75. Twenty-eight subjects with fear of heights (acrophobia) were treated with two sessions of behavioral therapy using virtual reality exposure to heights within a glass elevator. Single doses of placebo or DCS were taken before each therapy session. Exposure therapy combined with DCS resulted in significantly larger reductions of acrophobia symptoms on all main outcome measures, including fear within the virtual environment, acrophobia symptoms outside of the environment and height-related anxiety. Subjects receiving DCS also showed significantly greater decreases in a physiological measure of anxiety during the test exposure. These pilot data provided initial support for the use of acute dosing of DCS as an adjunct to exposure-based psychotherapy to accelerate the associative learning processes that contribute to correcting psychopathology.
DCS has also been used in a double-blind, placebo-controlled trial for the treatment of social anxiety disorder by exposure therapy. In this study, 27 subjects received four exposure therapy sessions combined with DCS or placebo. Those receiving DCS in addition to exposure therapy reported significantly less social anxiety compared with patients receiving exposure therapy plus placebo. Controlled effect sizes were in the medium to large range76. Additionally, DCS has been used to accelerate reduction of obsession-related distress in patients with OCD undergoing extinction-based exposure therapy. DCS given before therapy decreased both the number of exposure sessions required to achieve clinical milestones and the rate of therapy dropout. After four exposure sessions, patients in the DCS group reported significantly greater decreases in obsession-related distress compared with the placebo group77. There are two negative findings of DCS use in the augmentation of extinction in humans, one using exposure therapy for the treatment of spider phobia78 and the second using extinction for a conditioned cue in healthy human subjects79. However, in both studies, the placebo-controlled groups had good recovery as well. This suggests, as in the animal studies, that if a ‘floor effect’ of complete extinction is accomplished, then DCS may not augment extinction any further. DCS may be most useful in disorders that require many therapy sessions for a response or that are refractory to normal exposure-based therapies. Studies are underway on the use of DCS to augment exposure-based CBT for PTSD, panic disorder, social anxiety, OCD and depression. Based on the data that DCS enhances extinction of place preference conditioned to cocaine in rats71, at least two human studies are also examining DCS effects in exposure-based treatment of cravings in disorders of addiction.
As the neuroscience of extinction is advancing, other molecular pathways that might be amenable to augmentation have been identified. In the acquisition and consolidation processes mediating extinction of fear, these include the endogenous cannabinoid system, dopamine pathways, adrenergic pathways and brain-derived neurotrophic factor, among others. We hope that ongoing advances in the neurobiology and neuropharmacology of extinction of fear will lead to new types of pharmacotherapeutic approaches for refractory mood and anxiety disorders.
Preventing consolidation of traumatic memories
With certain disorders, notably PTSD, the time of the insult that created the disorder is known—it is when the initiating trauma occurred. New preclinical approaches are examining mechanisms of consolidation of learning and how this consolidation process could be interrupted to prevent the development of trauma-related disorders.
Decades of preclinical research demonstrate that memories do not immediately become permanent at the time of the initial experience. Instead they exist in a labile state for at least hours and possibly days, during which they become consolidated into a more permanent memory (Fig. 5). There is a host of in vitro and in vivo data in animals outlining the molecular, synaptic, neurotransmitter and systems-level changes that may occur during this consolidation process80. This literature suggests that during the immediate period after fear training in the animal model and after trauma in the human patient, it may be possible to disrupt the consolidation of new fear memories that are in the process of being formed65.
Although there are philosophical and ethical questions raised by a potential amnestic agent that would ‘erase’ a memory fully, it is increasingly clear that differential memory systems are encoding different aspects of a memory in parallel. In other words, declarative or explicit memory systems involving hippocampal-cortical pathways are likely to be encoding the ‘what, when and where’ of an event, while in parallel, amygdala-cortical pathways are encoding the emotional salience and aversiveness of the memory. Therefore, agents that block the ‘emotional overconsolidation’ of a traumatic memory perhaps could preserve the declarative aspects of the same memory. This idealized approach would not render the patient fully amnestic, but would potentially prevent PTSD.
Many of the ideas on interruption of consolidation stem from a protocol called ‘avoidance learning’, in which an animal is conditioned in a box with two compartments. When it steps from one compartment to the next, it receives a mild footshock80. After this training, the animal then avoids spending time in the compartment in which it was shocked, even if there are no more reinforcing shocks. Consolidation of contextual fear memory in this protocol is disrupted after training by a variety of agents, including dopaminergic, cholinergic and other modulatory drugs, but most notably those involving the adrenal stress system—epinephrine and glucocorticoids81,82. Activation of the endogenous fight or flight adrenergic and glucocorticoid responses seems to be involved in strengthening these memories after the event. Notably, some of these manipulations do not impair consolidation of different types of memory, such as pavlovian fear conditioning, in contrast to their effects on consolidation of inhibitory avoidance learning. This difference in the animal models may have to do with the different neural circuits involved in cued fear conditioning and inhibitory avoidance learning.
Fear consolidation (at least with certain forms of fear training such as inhibitory avoidance learning) is blocked after training by an antagonist of noradrenergic activation. Propranolol, a common β-blocker used for hypertension, can block central β1 and β2 adrenergic receptors. Clinical trials are testing the use of propranolol to prevent the formation of PTSD after trauma. In one study, subjects who presented to an emergency room immediately after a traumatic event (primarily motor vehicle accidents)83 randomly received either propranolol or placebo, orally four times daily for 10 days, followed by a taper period. The first dose of medicine was administered, on average, 4 hours after the traumatic event. One month after the trauma, PTSD measures trended lower in the 11 completers who took propranolol compared to the 20 completers who did not. Also when exposed to script-driven mental imagery of the trauma, a significant number of those who took placebo had ongoing physiological symptoms of PTSD, whereas none of the subjects who took propranolol did. Given the length of time that subjects took propranolol, and given that only 1 or 2 doses would have occurred during the expected period of early memory consolidation, it is unclear whether these effects are due to ‘blockade of consolidation’ versus ‘prevention of stress sensitization’—another hypothesis of PTSD development—but the pilot data are intriguing.
A second open-label study examined 19 completers, 11 of whom took propranolol three times daily beginning 2–20 h after the trauma for 7 days followed by a taper period84. At 2 months after trauma, levels of PTSD symptoms were significantly different in the subjects treated with propranolol. These studies were not blinded and had small numbers of subjects. Furthermore, given that propranolol is used frequently to treat hypertension and social anxiety disorder, significant amnestic properties might have been expected to be clinically obvious by now. Larger randomized clinical trials are ongoing to examine whether these promising pilot findings are robust and reproducible in a larger controlled and blinded design.
Another consolidation-blockade approach is based on the findings that lower cortisol levels after trauma predict subsequent PTSD85,86. Combined with a large body of data that glucocorticoids are involved in emotional memory encoding and retrieval and that high doses of systemic glucocorticoids decrease the memory-enhancing effects of catecholamines87, this finding indicates that glucocorticoids given after trauma may enhance normal stress responsiveness and prevent overconsolidation of trauma memories. Pilot data in humans support these hypotheses. Exogenously administered stress doses of cortisol reduced the development of subsequent PTSD in medical-surgical patients after septic shock88. A randomized, double-blinded study replicated the finding with 11 patients that received placebo and 9 that received hydrocortisone for 6 days after septic shock. Thirty-one months later, significantly more subjects who had received placebo compared with hydrocortisone had PTSD symptoms from their intensive-care hospital experience88. These studies also have small numbers of subjects, yet provide intriguing early data.
Giving corticosterone to rats following a reminder trial 24 h after fear conditioning89 indicates that enhanced glucocorticoid modulation may enhance the extinction component of fear, similarly to the mechanisms described above. However, the inverted ‘U’ relationship of the glucocorticoid system and the complexities of consolidation, extinction and reconsolidation make the story complicated. In a similar protocol, inactivating glucocorticoid receptors with an antagonist led to a disruption of memory retrieval when tested later90. Although these results at first seem contradictory, they are consistent with the increasing data implying that every memory recall event is accompanied by competition between molecular events that strengthen the original memory (reconsolidation) and those that inhibit that memory (extinction)70. By differentially enhancing or inhibiting these processes, opposing effects on the state of the existing fear memories may be obtained.
Other neuromodulators91, including dopamine92 and glutamate-NMDA receptor activation93, are implicated in the consolidation of fear memories. This work remains in its early stages, but there is room for optimism that a better understanding of the mechanisms of fear consolidation will lead to direct translational interventions that may one day prevent the development of anxiety disorders in acutely traumatized subjects and that psychopharmacological interventions in emergency rooms, on the battlefield or after a disaster may be as important in later PTSD prevention as early routine medical interventions after stroke and myocardial infarction are today.
Preventing reconsolidation of traumatic memories
As with consolidation, memories remain labile and associative when they are recalled. Reactivated memories are also sensitive to pharmacological disruption70,94–96. Local infusion of protein synthesis inhibitors into discrete brain regions seems to prevent the ‘reconsolidation’ of the memory after it is made labile again through reactivation. Protein synthesis inhibitors do not easily cross the blood-brain barrier and can be toxic, so they are unlikely to be used in studies in humans. However, other, more benign drugs, given in animals acutely with recall of fear memories, may also act to reduce later memory expression, possibly through inhibiting reconsolidation mechanisms. For example, NMDA receptor agonists96, the β-adrenergic antagonist timolol95 or propranolol97 block reconsolidation, and thus may serve as useful agents in future human studies of reconsolidation.
Reconsolidation occurs for recently learned memories but not distant memories98. It is curious that propranolol has not been reported to block reconsolidation in humans, as the initial report in animals was almost a decade ago and such an experiment would be relatively easy and safe. One possibility is that the human pilot studies of propranolol after trauma in the emergency room (above) were actually not targeting consolidation, but were instead targeting the early reactivation, and thus reconsolidation, process in the days and weeks after trauma. Together, these data indicate that treatment with a β-adrenergic antagonist in the weeks after trauma might be effective in preventing later PTSD formation, possibly through impairing reconsolidation and sensitization processes early, but that disrupting the remote memories from chronic PTSD may not be possible using reconsolidation-impairing approaches. If that turns out to be the case, we can hope that the mechanisms discussed above to enhance extinction of fear may eventually be optimal in more chronic cases.
Conclusions
This is a very exciting time in the fields of learning and memory and psychiatry, because the growing literature on the differential processes involved in memory formation are now increasingly amenable to translational human studies. This review has focused on recent developments in understanding the neural circuit processes underlying mood and anxiety disorders. It is hoped that recent progress in understanding the neurobiology of emotional regulation and dysregulation—when these emotion states cannot be ‘turned off’—will lead to powerful and exciting new treatments for mood and anxiety disorders based on neuroscientifically informed rational approaches.
We have focused on an incomplete, but illustrative, set of brain areas that are thought to be involved in these disorders of emotion. New experimental treatments may alleviate symptoms through the correction of dysregulated circuit activity as well as prevent overlearning or enhance extinction learning in circuits mediating negative emotional memories. Combining the neurobiology of emotional learning and memory with translational studies in human disease provides for an exciting set of opportunities for the treatment of these often treatment-refractory disorders.
Acknowledgments
This authors are supported by grants from the US National Institutes of Mental Health (K.J.R., MH071537 and MH069884; H.S.M., P50 MH58922, P50 MH077083 and 1R01MH073719), US National Institute on Drug Abuse (K.J.R., DA-019624), NARSAD, Burroughs Wellcome Fund, Stanley Medical Research Foundation, Woodruff Fund and The Dana Foundation.
Footnotes
COMPETING INTERESTS STATEMENT
The authors declare competing financial interests: details accompany the full-text HTML version of the paper at http://www.nature.com/natureneuroscience/.
References
- 1.Kessler RC, Chiu WT, Demler O, Merikangas KR, Walters EE. Prevalence, severity, and comorbidity of 12-month DSM-IV disorders in the National Comorbidity Survey Replication. Arch Gen Psychiatry. 2005;62:617–627. doi: 10.1001/archpsyc.62.6.617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Keller MB, et al. Time to recovery, chronicity, and levels of psychopathology in major depression. A 5-year prospective follow-up of 431 subjects. Arch Gen Psychiatry. 1992;49:809–816. doi: 10.1001/archpsyc.1992.01820100053010. [DOI] [PubMed] [Google Scholar]
- 3.Greenberg P, et al. The economic burden of anxiety disorders in the 1990s. J Clin Psychiatry. 1999;60:427–435. doi: 10.4088/jcp.v60n0702. [DOI] [PubMed] [Google Scholar]
- 4.Gorman JM. Comorbid depression and anxiety spectrum disorders. Depress Anxiety. 1996;4:160–168. doi: 10.1002/(SICI)1520-6394(1996)4:4<160::AID-DA2>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 5.Ressler KJ, Nemeroff CB. Role of serotonergic and noradrenergic systems in the pathophysiology of depression and anxiety disorders. Depress Anxiety. 2000;12:2–19. doi: 10.1002/1520-6394(2000)12:1+<2::AID-DA2>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 6.Berton O, et al. Essential role of BDNF in the mesolimbic dopamine pathway in social defeat stress. Science. 2006;311:864–868. doi: 10.1126/science.1120972. [DOI] [PubMed] [Google Scholar]
- 7.Bornstein SR, Schuppenies A, Wong ML, Licinio J. Approaching the shared biology of obesity and depression: the stress axis as the locus of gene-environment interactions. Mol Psychiatry. 2006;11:892–902. doi: 10.1038/sj.mp.4001873. [DOI] [PubMed] [Google Scholar]
- 8.Nestler EJ, Carlezon WA., Jr The mesolimbic dopamine reward circuit in depression. Biol Psychiatry. 2006;59:1151–1159. doi: 10.1016/j.biopsych.2005.09.018. [DOI] [PubMed] [Google Scholar]
- 9.Tremblay LK, et al. Functional neuroanatomical substrates of altered reward processing in major depressive disorder revealed by a dopaminergic probe. Arch Gen Psychiatry. 2005;62:1228–1236. doi: 10.1001/archpsyc.62.11.1228. [DOI] [PubMed] [Google Scholar]
- 10.Schlaepfer TE, et al. Deep brain stimulation to reward circuitry alleviates anhedonia in refractory major depression. Neuropsychopharmacology. doi: 10.1038/sj.npp.1301408. advance online publication 11 April 2007. [DOI] [PubMed] [Google Scholar]
- 11.Mayberg HS. Modulating dysfunctional limbic-cortical circuits in depression: towards development of brain-based algorithms for diagnosis and optimised treatment. Br Med Bull. 2003;65:193–207. doi: 10.1093/bmb/65.1.193. [DOI] [PubMed] [Google Scholar]
- 12.Mayberg HS, et al. Reciprocal limbic-cortical function and negative mood: converging PET findings in depression and normal sadness. Am J Psychiatry. 1999;156:675–682. doi: 10.1176/ajp.156.5.675. [DOI] [PubMed] [Google Scholar]
- 13.Drevets WC. Functional anatomical abnormalities in limbic and prefrontal cortical structures in major depression. Prog Brain Res. 2000;126:413–431. doi: 10.1016/S0079-6123(00)26027-5. [DOI] [PubMed] [Google Scholar]
- 14.Milad MR, et al. Recall of fear extinction in humans activates the ventromedial prefrontal cortex and hippocampus in concert. Biol Psychiatry. doi: 10.1016/j.biopsych.2006.10.011. published online 9 January 2007. [DOI] [PubMed] [Google Scholar]
- 15.Siegle GJ, Carter CS, Thase ME. Use of FMRI to predict recovery from unipolar depression with cognitive behavior therapy. Am J Psychiatry. 2006;163:735–738. doi: 10.1176/ajp.2006.163.4.735. [DOI] [PubMed] [Google Scholar]
- 16.Furmark T, et al. Common changes in cerebral blood flow in patients with social phobia treated with citalopram or cognitive-behavioral therapy. Arch Gen Psychiatry. 2002;59:425–433. doi: 10.1001/archpsyc.59.5.425. [DOI] [PubMed] [Google Scholar]
- 17.Drevets WC. Neuroimaging studies of mood disorders. Biol Psychiatry. 2000;48:813–829. doi: 10.1016/s0006-3223(00)01020-9. [DOI] [PubMed] [Google Scholar]
- 18.Canli T, Lesch KP. Long story short: the serotonin transporter in emotion regulation and social cognition. Nat Neurosci. 2007;10:1103–1109. doi: 10.1038/nn1964. [DOI] [PubMed] [Google Scholar]
- 19.Pezawas L, et al. 5-HTTLPR polymorphism impacts human cingulate-amygdala interactions: a genetic susceptibility mechanism for depression. Nat Neurosci. 2005;8:828–834. doi: 10.1038/nn1463. [DOI] [PubMed] [Google Scholar]
- 20.George MS, et al. Vagus nerve stimulation for the treatment of depression and other neuropsychiatric disorders. Expert Rev Neurother. 2007;7:63–74. doi: 10.1586/14737175.7.1.63. [DOI] [PubMed] [Google Scholar]
- 21.Henry TR, et al. Brain blood flow alterations induced by therapeutic vagus nerve stimulation in partial epilepsy: I Acute effects at high and low levels of stimulation. Epilepsia. 1998;39:983–990. doi: 10.1111/j.1528-1157.1998.tb01448.x. [DOI] [PubMed] [Google Scholar]
- 22.Ben-Menachem E, et al. Effects of vagus nerve stimulation on amino acids and other metabolites in the CSF of patients with partial seizures. Epilepsy Res. 1995;20:221–227. doi: 10.1016/0920-1211(94)00083-9. [DOI] [PubMed] [Google Scholar]
- 23.Krahl SE, Senanayake SS, Handforth A. Seizure suppression by systemic epinephrine is mediated by the vagus nerve. Epilepsy Res. 2000;38:171–175. doi: 10.1016/s0920-1211(99)00089-3. [DOI] [PubMed] [Google Scholar]
- 24.Walker BR, Easton A, Gale K. Regulation of limbic motor seizures by GABA and glutamate transmission in nucleus tractus solitarius. Epilepsia. 1999;40:1051–1057. doi: 10.1111/j.1528-1157.1999.tb00818.x. [DOI] [PubMed] [Google Scholar]
- 25.Craig AD. How do you feel? Interoception: the sense of the physiological condition of the body. Nat Rev Neurosci. 2002;3:655–666. doi: 10.1038/nrn894. [DOI] [PubMed] [Google Scholar]
- 26.Henry TR. Therapeutic mechanisms of vagus nerve stimulation. Neurology. 2002;59:S3–S14. doi: 10.1212/wnl.59.6_suppl_4.s3. [DOI] [PubMed] [Google Scholar]
- 27.Cox CL, Huguenard JR, Prince DA. Nucleus reticularis neurons mediate diverse inhibitory effects in thalamus. Proc Natl Acad Sci USA. 1997;94:8854–8859. doi: 10.1073/pnas.94.16.8854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Malow BA, et al. Vagus nerve stimulation reduces daytime sleepiness in epilepsy patients. Neurology. 2001;57:879–884. doi: 10.1212/wnl.57.5.879. [DOI] [PubMed] [Google Scholar]
- 29.Zobel A, et al. Changes in regional cerebral blood flow by therapeutic vagus nerve stimulation in depression: an exploratory approach. Psychiatry Res. 2005;139:165–179. doi: 10.1016/j.pscychresns.2005.02.010. [DOI] [PubMed] [Google Scholar]
- 30.Bohning DE, et al. Feasibility of vagus nerve stimulation-synchronized blood oxygenation level-dependent functional MRI. Invest Radiol. 2001;36:470–479. doi: 10.1097/00004424-200108000-00006. [DOI] [PubMed] [Google Scholar]
- 31.Nahas Z, et al. Serial vagus nerve stimulation functional MRI in treatment-resistant depression. Neuropsychopharmacology. 2007;32:1649–1660. doi: 10.1038/sj.npp.1301288. [DOI] [PubMed] [Google Scholar]
- 32.Sackeim HA, et al. Durability of antidepressant response to vagus nerve stimulation (VNSTM) Int J Neuropsychopharmacol. doi: 10.1017/S1461145706007425. published online 9 February 2007. [DOI] [PubMed] [Google Scholar]
- 33.Lisanby SH, et al. New developments in electroconvulsive therapy and magnetic seizure therapy. CNS Spectr. 2003;8:529–536. doi: 10.1017/s1092852900019003. [DOI] [PubMed] [Google Scholar]
- 34.Sackeim HA, et al. Cognitive consequences of low-dosage electroconvulsive therapy. Ann NY Acad Sci. 1986;462:326–340. doi: 10.1111/j.1749-6632.1986.tb51267.x. [DOI] [PubMed] [Google Scholar]
- 35.Carpenter LL. Neurostimulation in resistant depression. J Psychopharmacol. 2006;20:35–40. doi: 10.1177/1359786806064327. [DOI] [PubMed] [Google Scholar]
- 36.Epstein CM, Schwartzberg DG, Davey KR, Sudderth DB. Localizing the site of magnetic brain stimulation in humans. Neurology. 1990;40:666–670. doi: 10.1212/wnl.40.4.666. [DOI] [PubMed] [Google Scholar]
- 37.Barker AT, Jalinous R, Freeston IL. Non-invasive magnetic stimulation of human motor cortex. Lancet. 1985;1:1106–1107. doi: 10.1016/s0140-6736(85)92413-4. [DOI] [PubMed] [Google Scholar]
- 38.Paus T, Barrett J. Transcranial magnetic stimulation (TMS) of the human frontal cortex: implications for repetitive TMS treatment of depression. J Psychiatry Neurosci. 2004;29:268–279. [PMC free article] [PubMed] [Google Scholar]
- 39.Pascual-Leone A, Rubio B, Pallardo F, Catala MD. Rapid-rate transcranial magnetic stimulation of left dorsolateral prefrontal cortex in drug-resistant depression. Lancet. 1996;348:233–237. doi: 10.1016/s0140-6736(96)01219-6. [DOI] [PubMed] [Google Scholar]
- 40.George MS, et al. Mood improvement following daily left prefrontal repetitive transcranial magnetic stimulation in patients with depression: a placebo-controlled crossover trial. Am J Psychiatry. 1997;154:1752–1756. doi: 10.1176/ajp.154.12.1752. [DOI] [PubMed] [Google Scholar]
- 41.Herrmann LL, Ebmeier KP. Factors modifying the efficacy of transcranial magnetic stimulation in the treatment of depression: a review. J Clin Psychiatry. 2006;67:1870–1876. doi: 10.4088/jcp.v67n1206. [DOI] [PubMed] [Google Scholar]
- 42.Couturier JL. Efficacy of rapid-rate repetitive transcranial magnetic stimulation in the treatment of depression: a systematic review and meta-analysis. J Psychiatry Neurosci. 2005;30:83–90. [PMC free article] [PubMed] [Google Scholar]
- 43.O’Reardon JP, et al. Efficacy and safety of transcranial magnetic stimulation in the acute treatment of major depression: a multisite randomized controlled trial. Biol Psychiatry. doi: 10.1016/j.biopsych.2007.01.018. published online 14 June 2007. [DOI] [PubMed] [Google Scholar]
- 44.Bohning DE, et al. Mapping transcranial magnetic stimulation (TMS) fields in vivo with MRI. Neuroreport. 1997;8:2535–2538. doi: 10.1097/00001756-199707280-00023. [DOI] [PubMed] [Google Scholar]
- 45.Kimbrell TA, et al. Frequency dependence of antidepressant response to left prefrontal repetitive transcranial magnetic stimulation (rTMS) as a function of baseline cerebral glucose metabolism. Biol Psychiatry. 1999;46:1603–1613. doi: 10.1016/s0006-3223(99)00195-x. [DOI] [PubMed] [Google Scholar]
- 46.Speer AM, et al. Opposite effects of high and low frequency rTMS on regional brain activity in depressed patients. Biol Psychiatry. 2000;48:1133–1141. doi: 10.1016/s0006-3223(00)01065-9. [DOI] [PubMed] [Google Scholar]
- 47.Kim EJ, et al. Repetitive transcranial magnetic stimulation protects hippocampal plasticity in an animal model of depression. Neurosci Lett. 2006;405:79–83. doi: 10.1016/j.neulet.2006.06.023. [DOI] [PubMed] [Google Scholar]
- 48.Kosel M, Frick C, Lisanby SH, Fisch HU, Schlaepfer TE. Magnetic seizure therapy improves mood in refractory major depression. Neuropsychopharmacology. 2003;28:2045–2048. doi: 10.1038/sj.npp.1300293. [DOI] [PubMed] [Google Scholar]
- 49.Benabid AL. Deep brain stimulation for Parkinson’s disease. Curr Opin Neurobiol. 2003;13:696–706. doi: 10.1016/j.conb.2003.11.001. [DOI] [PubMed] [Google Scholar]
- 50.Bejjani BP, et al. Transient acute depression induced by high-frequency deep-brain stimulation. N Engl J Med. 1999;340:1476–1480. doi: 10.1056/NEJM199905133401905. [DOI] [PubMed] [Google Scholar]
- 51.Kopell BH, Greenberg B, Rezai AR. Deep brain stimulation for psychiatric disorders. J Clin Neurophysiol. 2004;21:51–67. doi: 10.1097/00004691-200401000-00007. [DOI] [PubMed] [Google Scholar]
- 52.Aouizerate B, et al. Deep brain stimulation of the ventral caudate nucleus in the treatment of obsessive-compulsive disorder and major depression. Case report. J Neurosurg. 2004;101:682–686. doi: 10.3171/jns.2004.101.4.0682. [DOI] [PubMed] [Google Scholar]
- 53.Brody AL, et al. Regional brain metabolic changes in patients with major depression treated with either paroxetine or interpersonal therapy: preliminary findings. Arch Gen Psychiatry. 2001;58:631–640. doi: 10.1001/archpsyc.58.7.631. [DOI] [PubMed] [Google Scholar]
- 54.Seminowicz DA, et al. Limbic-frontal circuitry in major depression: a path modeling metanalysis. Neuroimage. 2004;22:409–418. doi: 10.1016/j.neuroimage.2004.01.015. [DOI] [PubMed] [Google Scholar]
- 55.Dougherty DD, et al. Cerebral metabolic correlates as potential predictors of response to anterior cingulotomy for treatment of major depression. J Neurosurg. 2003;99:1010–1017. doi: 10.3171/jns.2003.99.6.1010. [DOI] [PubMed] [Google Scholar]
- 56.Goldapple K, et al. Modulation of cortical-limbic pathways in major depression: treatment-specific effects of cognitive behavior therapy. Arch Gen Psychiatry. 2004;61:34–41. doi: 10.1001/archpsyc.61.1.34. [DOI] [PubMed] [Google Scholar]
- 57.Nobler MS, et al. Structural and functional neuroimaging of electroconvulsive therapy and transcranial magnetic stimulation. Depress Anxiety. 2000;12:144–156. doi: 10.1002/1520-6394(2000)12:3<144::AID-DA6>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 58.Mayberg HS, et al. Deep brain stimulation for treatment-resistant depression. Neuron. 2005;45:651–660. doi: 10.1016/j.neuron.2005.02.014. [DOI] [PubMed] [Google Scholar]
- 59.Davis KD, et al. Globus pallidus stimulation activates the cortical motor system during alleviation of parkinsonian symptoms. Nat Med. 1997;3:671–674. doi: 10.1038/nm0697-671. [DOI] [PubMed] [Google Scholar]
- 60.Lozano AM, Dostrovsky J, Chen R, Ashby P. Deep brain stimulation for Parkinson’s disease: disrupting the disruption. Lancet Neurol. 2002;1:225–231. doi: 10.1016/s1474-4422(02)00101-1. [DOI] [PubMed] [Google Scholar]
- 61.McIntyre CC, Savasta M, Kerkerian-Le Goff L, Vitek JL. Uncovering the mechanism(s) of action of deep brain stimulation: activation, inhibition, or both. Clin Neurophysiol. 2004;115:1239–1248. doi: 10.1016/j.clinph.2003.12.024. [DOI] [PubMed] [Google Scholar]
- 62.Harbishettar V, Pal PK, Janardhan Reddy YC, Thennarasu K. Is there a relationship between Parkinson’s disease and obsessive-compulsive disorder? Parkinsonism Relat Disord. 2005;11:85–88. doi: 10.1016/j.parkreldis.2004.07.006. [DOI] [PubMed] [Google Scholar]
- 63.Nuttin B, Cosyns P, Demeulemeester H, Gybels J, Meyerson B. Electrical stimulation in anterior limbs of internal capsules in patients with obsessive-compulsive disorder. Lancet. 1999;354:1526. doi: 10.1016/S0140-6736(99)02376-4. [DOI] [PubMed] [Google Scholar]
- 64.Greenberg BD, et al. Three-year outcomes in deep brain stimulation for highly resistant obsessive-compulsive disorder. Neuropsychopharmacology. 2006;31:2384–2393. doi: 10.1038/sj.npp.1301165. [DOI] [PubMed] [Google Scholar]
- 65.Pitman RK, Delahanty DL. Conceptually driven pharmacologic approaches to acute trauma. CNS Spectr. 2005;10:99–106. doi: 10.1017/s109285290001943x. [DOI] [PubMed] [Google Scholar]
- 66.Rothbaum BO, Davis M. Applying learning principles to the treatment of posttrauma reactions. Ann NY Acad Sci. 2003;1008:112–121. doi: 10.1196/annals.1301.012. [DOI] [PubMed] [Google Scholar]
- 67.Myers KM, Davis M. Behavioral and neural analysis of extinction. Neuron. 2002;36:567–584. doi: 10.1016/s0896-6273(02)01064-4. [DOI] [PubMed] [Google Scholar]
- 68.Walker DL, Ressler KJ, Lu KT, Davis M. Facilitation of conditioned fear extinction by systemic administration or intra-amygdala infusions of D-cycloserine as assessed with fear- potentiated startle in rats. J Neurosci. 2002;22:2343–2351. doi: 10.1523/JNEUROSCI.22-06-02343.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Monahan JB, Handelmann GE, Hood WF, Cordi AA. D-cycloserine, a positive modulator of the N-methyl-D-aspartate receptor, enhances performance of learning tasks in rats. Pharmacol Biochem Behav. 1989;34:649–653. doi: 10.1016/0091-3057(89)90571-6. [DOI] [PubMed] [Google Scholar]
- 70.Lee JL, Milton AL, Everitt BJ. Reconsolidation and extinction of conditioned fear: inhibition and potentiation. J Neurosci. 2006;26:10051–10056. doi: 10.1523/JNEUROSCI.2466-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Botreau F, Paolone G, Stewart J. D-Cycloserine facilitates extinction of a cocaine-induced conditioned place preference. Behav Brain Res. 2006;172:173–178. doi: 10.1016/j.bbr.2006.05.012. [DOI] [PubMed] [Google Scholar]
- 72.Ledgerwood L, Richardson R, Cranney J. Effects of D-cycloserine on extinction of conditioned freezing. Behav Neurosci. 2003;117:341–349. doi: 10.1037/0735-7044.117.2.341. [DOI] [PubMed] [Google Scholar]
- 73.Yang YL, Lu KT. Facilitation of conditioned fear extinction by D-cycloserine is mediated by mitogen-activated protein kinase and phosphatidylinositol 3-kinase cascades and requires de novo protein synthesis in basolateral nucleus of amygdala. Neuroscience. 2005;134:247–260. doi: 10.1016/j.neuroscience.2005.04.003. [DOI] [PubMed] [Google Scholar]
- 74.Otto M. Learning and “unlearning” fears: preparedness, neural pathways, and patients. Biol Psychiatry. 2002;52:917–920. doi: 10.1016/s0006-3223(02)01719-5. [DOI] [PubMed] [Google Scholar]
- 75.Ressler KJ, et al. Cognitive enhancers as adjuncts to psychotherapy: use of D-cycloserine in phobic individuals to facilitate extinction of fear. Arch Gen Psychiatry. 2004;61:1136–1144. doi: 10.1001/archpsyc.61.11.1136. [DOI] [PubMed] [Google Scholar]
- 76.Hofmann SG, et al. Augmentation of exposure therapy with D-cycloserine for social anxiety disorder. Arch Gen Psychiatry. 2006;63:298–304. doi: 10.1001/archpsyc.63.3.298. [DOI] [PubMed] [Google Scholar]
- 77.Kushner MG, et al. D-Cycloserine augmented exposure therapy for obsessive compulsive disorder. Biol Psychiatry. doi: 10.1016/j. biopsych.2006.12.020. published online 22 June 2007. [DOI] [PubMed] [Google Scholar]
- 78.Guastella AJ, Dadds MR, Lovibond PF, Mitchell P, Richardson R. A randomized controlled trial of the effect of D-cycloserine on exposure therapy for spider fear. J Psychiatr Res. 2007;41:466–471. doi: 10.1016/j.jpsychires.2006.05.006. [DOI] [PubMed] [Google Scholar]
- 79.Guastella AJ, Lovibond PF, Dadds MR, Mitchell P, Richardson R. A randomized controlled trial of the effect of D-cycloserine on extinction and fear conditioning in humans. Behav Res Ther. 2007;45:663–672. doi: 10.1016/j.brat.2006.07.005. [DOI] [PubMed] [Google Scholar]
- 80.McGaugh JL. Memory–a century of consolidation. Science. 2000;287:248–251. doi: 10.1126/science.287.5451.248. [DOI] [PubMed] [Google Scholar]
- 81.Ferry B, Roozendaal B, McGaugh JL. Basolateral amygdala noradrenergic influences on memory storage are mediated by an interaction between β- and α1-adrenoceptors. J Neurosci. 1999;19:5119–5123. doi: 10.1523/JNEUROSCI.19-12-05119.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Quirarte GL, Roozendaal B, McGaugh JL. Glucocorticoid enhancement of memory storage involves noradrenergic activation in the basolateral amygdala. Proc Natl Acad Sci USA. 1997;94:14048–14053. doi: 10.1073/pnas.94.25.14048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Pitman RK, et al. Pilot study of secondary prevention of posttraumatic stress disorder with propranolol. Biol Psychiatry. 2002;51:189–192. doi: 10.1016/s0006-3223(01)01279-3. [DOI] [PubMed] [Google Scholar]
- 84.Vaiva G, et al. Immediate treatment with propranolol decreases posttraumatic stress disorder two months after trauma. Biol Psychiatry. 2003;54:947–949. doi: 10.1016/s0006-3223(03)00412-8. [DOI] [PubMed] [Google Scholar]
- 85.Yehuda R, et al. Low urinary cortisol excretion in Holocaust survivors with posttraumatic stress disorder. Am J Psychiatry. 1995;152:982–986. doi: 10.1176/ajp.152.7.982. [DOI] [PubMed] [Google Scholar]
- 86.Yehuda R, Golier JA, Yang RK, Tischler L. Enhanced sensitivity to glucocorticoids in peripheral mononuclear leukocytes in posttraumatic stress disorder. Biol Psychiatry. 2004;55:1110–1116. doi: 10.1016/j.biopsych.2004.02.010. [DOI] [PubMed] [Google Scholar]
- 87.Borrell J, De Kloet ER, Versteeg DH, Bohus B. Inhibitory avoidance deficit following short-term adrenalectomy in the rat: the role of adrenal catecholamines. Behav Neural Biol. 1983;39:241–258. doi: 10.1016/s0163-1047(83)90910-x. [DOI] [PubMed] [Google Scholar]
- 88.Schelling G, et al. The effect of stress doses of hydrocortisone during septic shock on posttraumatic stress disorder in survivors. Biol Psychiatry. 2001;50:978–985. doi: 10.1016/s0006-3223(01)01270-7. [DOI] [PubMed] [Google Scholar]
- 89.Cai WH, Blundell J, Han J, Greene RW, Powell CM. Postreactivation glucocorticoids impair recall of established fear memory. J Neurosci. 2006;26:9560–9566. doi: 10.1523/JNEUROSCI.2397-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Tronel S, Alberini CM. Persistent disruption of a traumatic memory by postretrieval inactivation of glucocorticoid receptors in the amygdala. Biol Psychiatry. 2007;62:33–39. doi: 10.1016/j.biopsych.2006.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Schafe GE, Nader K, Blair HT, LeDoux JE. Memory consolidation of Pavlovian fear conditioning: a cellular and molecular perspective. Trends Neurosci. 2001;24:540–546. doi: 10.1016/s0166-2236(00)01969-x. [DOI] [PubMed] [Google Scholar]
- 92.Guarraci FA, Frohardt RJ, Falls WA, Kapp BS. The effects of intra-amygdaloid infusions of a D2 dopamine receptor antagonist on Pavlovian fear conditioning. Behav Neurosci. 2000;114:647–651. doi: 10.1037//0735-7044.114.3.647. [DOI] [PubMed] [Google Scholar]
- 93.Shimizu E, Tang YP, Rampon C, Tsien JZ. NMDA receptor-dependent synaptic reinforcement as a crucial process for memory consolidation. Science. 2000;290:1170–1174. doi: 10.1126/science.290.5494.1170. [DOI] [PubMed] [Google Scholar]
- 94.Nader K, Schafe GE, Le Doux JE. Fear memories require protein synthesis in the amygdala for reconsolidation after retrieval. Nature. 2000;406:722–726. doi: 10.1038/35021052. [DOI] [PubMed] [Google Scholar]
- 95.Przybyslawski J, Roullet P, Sara SJ. Attenuation of emotional and nonemotional memories after their reactivation: role of beta adrenergic receptors. J Neurosci. 1999;19:6623–6628. doi: 10.1523/JNEUROSCI.19-15-06623.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Przybyslawski J, Sara SJ. Reconsolidation of memory after its reactivation. Behav Brain Res. 1997;84:241–246. doi: 10.1016/s0166-4328(96)00153-2. [DOI] [PubMed] [Google Scholar]
- 97.Debiec J, Ledoux JE. Disruption of reconsolidation but not consolidation of auditory fear conditioning by noradrenergic blockade in the amygdala. Neuroscience. 2004;129:267–272. doi: 10.1016/j.neuroscience.2004.08.018. [DOI] [PubMed] [Google Scholar]
- 98.Eisenberg M, Dudai Y. Reconsolidation of fresh, remote, and extinguished fear memory in medaka: old fears don’t die. Eur J Neurosci. 2004;20:3397–3403. doi: 10.1111/j.1460-9568.2004.03818.x. [DOI] [PubMed] [Google Scholar]
- 99.Paulus MP, Feinstein JS, Castillo G, Simmons AN, Stein MB. Dose-dependent decrease of activation in bilateral amygdala and insula by lorazepam during emotion processing. Arch Gen Psychiatry. 2005;62:282–288. doi: 10.1001/archpsyc.62.3.282. [DOI] [PubMed] [Google Scholar]
- 100.Dostrovsky JO, et al. Microstimulation-induced inhibition of neuronal firing in human globus pallidus. J Neurophysiol. 2000;84:570–574. doi: 10.1152/jn.2000.84.1.570. [DOI] [PubMed] [Google Scholar]