

Abstract
The phosphatidylinositol 3-kinase (PI3K) signaling pathway is an important regulator of many cellular events, including apoptosis, proliferation, and motility. Enhanced activation of this pathway can occur through several mechanisms, such as inactivation of its negative regulator, phosphatase and tensin homolog deleted on chromosome ten (PTEN) and activating mutations and gene amplification of the gene encoding the catalytic subunit of PI3K (PIK3CA). These genetic abnormalities have been particularly associated with follicular thyroid neoplasia and anaplastic thyroid cancer, suggesting an important role for PI3K signaling in these disorders. In this review, the role of PI3K pathway activation in thyroid cancer will be discussed, with a focus on recent advances.
Keywords: thyroid carcinoma, anaplastic thyroid cancer, PIK3CA, AKT, PKB, PTEN, Cowden syndrome
Introduction
Activation of the phosphatidylinositol 3-kinase (PI3K) pathway is a common event in many cancers, including thyroid neoplasias. Class I PI3Ks are a family of proteins comprised of a regulatory and a catalytic subunit (p85 and p110 subunits, respectively) that are activated by receptor tyrosine kinases and other signaling molecules resulting in regulation of a wide variety of cellular functions 1. Of the subfamilies of Class I PI3Ks, PI3Kα is the best studied in thyroid cancer. Activation of PI3K results in the formation of phosphatidylinositol 3,4,5, triphosphate (PIP3) which subsequently recruits proteins with pleckstrin homology (PH) domains to the cytosolic membrane (Figure 1). Several key PH domain-containing proteins transduce signaling through multiple downstream signaling cascades, including phosphoinositide-dependent kinase -1 (PDK1) and AKT 2, 3. PDK1 has been shown to phosphorylate a large number of kinases, including AKT, serum and glucocorticoid-induced kinase (SGK), protein kinase A, several protein kinase C isoforms, and others that are known to regulate cell growth, metabolism, and apoptosis. In addition to PH domains, several other phosphoinositide-binding domains have been described supporting even broader consequences of PI3K signaling2, 3.
Figure 1. PI3K Signaling Cascade.
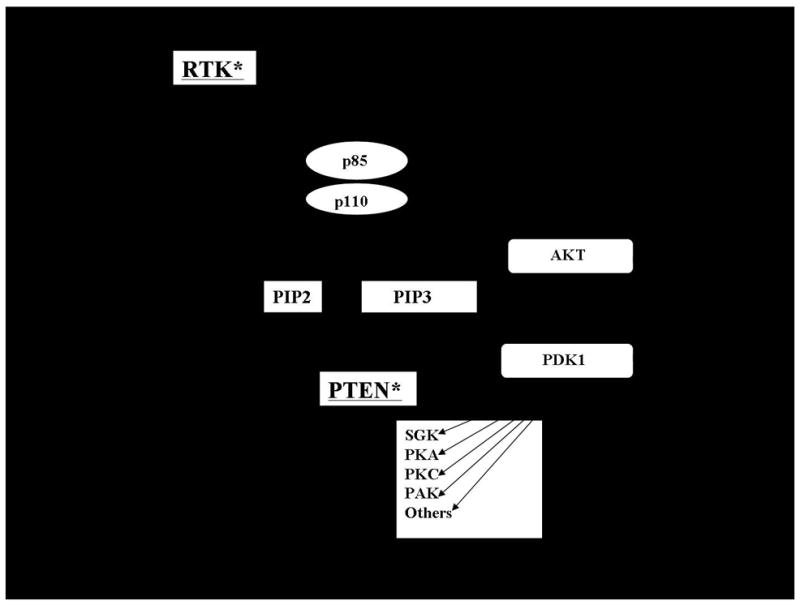
Upon activation by receptor tyrosine kinases (RTK), PI3 Kinase which is comprised of a p85 regulatory and p110 catalytic domain converts PIP2 to PIP3. PIP3 subsequently interacts with proteins containing PH domains, such as PDK1 and AKT, allowing for membrane recruitment (dark arrows). PDK1 phosphorylates AKT and other downstream targets including PKA, PKC isoforms, PAK, SGK, and other targets to regulate cell behavior. In the case of AKT, an additional phosphorylation by a PDK2 activity mediated by one of several kinases (see text) is required for full activation. PI3K signaling is inhibited by the lipid phosphatase activity of PTEN which converts PIP3 to PIP2. RTKs also activate the RAS/RAF/MEK signaling cascade which has important biological effects as well. RAS is also a site of cross-talk between these pathways through its ability to activate PI3K. Genetic alterations that result in constitutive signaling occur in thyroid cancer; the location of these alterations in these pathways are underlined and notified with an asterisk.
The best studied downstream kinase in the PI3K pathway in thyroid cancer is AKT. After its recruitment to the membrane, AKT is phosphorylated at two critical sites for activation of the protein, threonine 308 by PDK1 and serine 473 by PDK2, an activity that has been ascribed to several kinases, including the rapamycin insensitive companion of target of rapamycin/target of rapamycin (rictor/TOR) complex 4, DNA-dependent kinase5, PKCβII 6, intergin-linked kinase (ILK) 7, and AKT 8 (i.e. autophosphorylation). Phosphorylated AKT isoforms subsequently interact with effectors in both the cytosol and nucleus to regulate cell proliferation, apoptosis, metabolism, and motility. A detailed discussion of the frequency and potential functional role of AKT activation in thyroid cancer, and the potential importance of specific isoforms and subcellular localization in these effects has been recently published 9. While the importance of AKT as a key mediator of the downstream effects of PI3K has been demonstrated, other PI3K signaling pathways are also activated and regulate cell behavior 3.
One important negative regulator of this pathway is phosphatase and tensin homolog deleted on chromosome ten (PTEN). PTEN is a dual function lipid and protein phosphatase. Its lipid phosphatase function leads to the conversion of PIP3 to PIP2, thereby opposing the activity of PI3K, reducing membrane localization of PH domain-containing proteins such as PDK1 and AKT, and inhibiting subsequent signaling activity 3. The importance of the function of the PH domain in regulating AKT activity is highlighted by a recent report by Carpten et al. 10 in which a glutamic acid to lysine substitution at amino acid 17 (E17K) in the PH domain of AKT1 was identified in 8% of breast, 6% of colorectal, and 2% of ovarian cancers that were analyzed. The AKT1 E17K mutant was shown to display altered subcellular localization to the plasma membrane, exhibit enhanced levels of activity, and to transform fibroblasts and induce leukemia in a murine model.
In addition to upstream inhibition of the pathway by PTEN, downstream inhibition can also occur through complex crosstalk mechanisms, by the effects of protein phosphatases, or by disruption of protein stabilization. For example, AKT is stabilized by chaperone proteins, such as adaptor protein containing pleckstrin homology domain, phosphotyrosine-binding domain, leucine zipper region (APPL), Cdc37, and heat shock protein 90 (HSP90) 11, 12. While not specific for AKT inhibition, compounds that disrupt Hsp90 binding to client proteins have been developed for potential therapeutic benefit and exhibit anti-proliferative effects in thyroid cells 13. Inactivation of AKT activity also occurs via the activities of protein phosphatase 2A at Thr 308 and PHD leucine-rich repeat protein phosphatase (PHLPP) at Ser 473 14–16. A second isoform of PHLPP was recently defined by Brognard et, al., that not only inactivates AKT, but also inhibits cell-cycle progression, and promotes apoptosis through distinct AKT isoforms 17.
PTEN in Thyroid Cancer
PTEN in Cowden Syndrome
Perhaps the strongest evidence supporting a role for PI3K signaling in the development of thyroid neoplasia is Cowden syndrome (MIM 15830). Cowden syndrome is an autosomal dominant syndrome characterized by the development of benign hamartomas in multiple organ systems, such as the skin and gastrointestinal tract, as well as an increased risk of developing breast and thyroid cancer. Inactivating mutations of PTEN have been defined as the cause of Cowden syndrome 18 and can be identified in the majority of individuals meeting established diagnostic criteria 19. Thyroid neoplasia can be identified in approximately two-thirds of affected individuals. Thyroid cancer, most frequently follicular thyroid cancer (FTC), is identified in approximately 10% of individuals with Cowden’s syndrome. Thus, thyroid neoplasia is included as one of the major diagnostic criteria supporting a clinical diagnosis of Cowden syndrome 19. These genetic data provide strong evidence for a role of PI3K signaling in the development of FTC. Subsequently, several groups created murine models of generalized loss of Pten 20–22. In these models, homozygous deletion of the Pten gene resulted in embryonic lethality, while Pten+/− mice developed neoplasias in multiple organ systems resembling Cowden syndrome, including the thyroid tumors. In these tumors, loss of the remaining Pten allele was frequently identified, consistent with the role of Pten as a tumor suppressor gene that requires loss of expression of both alleles to result in tumor formation. Evidence of increased activation of PI3K signaling was also shown in the tumors and it was shown that the deficiency of Akt1 reduced the formation of tumors in the Pten +/− mice in additional cross-breeding experiments 23. These data confirm that loss of Pten expression can result in neoplastic transformation in vivo.
To analyze more specifically the ability of Pten loss to cause thyroid neoplasia, Yeager et al. developed a mouse strain (PtenL/L;TPO-Cre) using Cre-mediated recombination to delete Pten specifically in the thyroid 24. In these interesting studies, mice with complete loss of Pten expression in the thyroid gland developed hyperplastic euthyroid goiters in association with increased PI3K signaling. Over time, a number of mice developed nodular hyperplasia and benign follicular tumors, although cancers were not identified. It was particularly interesting that there were phenotypic differences between the female and male mice. The female mice were reported to have a higher proliferative index in the follicular cells at younger ages than the males and were more likely to develop follicular adenomas at 8–10 months of age. The authors subsequently identified an increase in expression and phosphorylation of estrogen receptor α, a downstream target of AKT, in the Pten null thyroid glands of the female mice. In addition to these experiments, the authors also compared the findings in the PtenL/L;TPO-Cre to their Pten +/− mice that developed hyperplastic nodules at a lower frequency. They identified loss of expression of the remaining wild type allele in the nodular tissue, further supporting the ability of increased PI3K signaling to induce thyroid nodules. Taken together, these data suggest that genetic loss of Pten is sufficient to induce thyroid neoplasia in vivo.
PTEN in Sporadic Thyroid Cancer
Several groups have assessed sporadic benign and malignant thyroid cancers for mutations, deletions, and changes in expression levels of PTEN. Nearly all of these studies demonstrate that mutations or deletions in PTEN are rare events in sporadic thyroid cancer, occurring at a low frequency in most of the studies, particularly in differentiated thyroid cancers [Table 1 and 25–28]. In addition to gene mutations, loss of heterozygosity (LOH) at one of the PTEN alleles also occurs in thyroid cancer 26, 29, 30. However, it has not been reported to occur in tumors with a mutation in the other allele; thus, this LOH would not be predicted to result in complete loss of PTEN gene expression as seems to be required for tumor formation (see above) unless other mechanisms were involved in loss of allelic expression. Indeed, it has been reported that reduced expression of PTEN protein occurs in a fairly high percentage of anaplastic thyroid cancers, and less frequently in FTC and PTC samples when analyzing tumors at the protein level by immunohistochemistry 30, 31. This has been correlated with LOH and with the identification of PTEN promoter hypermethylation. For example, Gimm, et al30 reported the common reduction of PTEN protein expression as well as differences in PTEN subcellular localization compared with normal tissue in sporadic thyroid cancers associated with PTEN LOH in one allele, suggesting that both epigenetic and post-translational regulation of PTEN might be involved in thyroid cancers.
Table 1.
Frequency of Gene Abnormalities in PTEN and PI3KCA in Sporadic Thyroid Cancers
| Gene Abnormality | FA | FTC | PTC | ATC | References |
|---|---|---|---|---|---|
| PTEN Mutation* | 0/183 (0%) | 10/147 (6.8%) | 3/163 (1.8%) | 8/59 (13.6%) | 25–28, 32 |
| PTEN Reduction** | 1/36 (2.7%) | 17/76 (22.4%) | 5/14 (35.7%) | 9/22 (40.9%) | 29, 39 |
| PIK3CA Mutation | 0/180 (0%) | 13/167 (7.8%) | 5/296 (1.7%) | 22/133 (16.5%) | 28, 32, 47, 48 |
| PIK3CA Amplification | 23/177 (13%) | 38/137 (27.7%) | 33/286 (11.5%) | 20/48 (42%) | 28, 32, 48 |
Does not include studies demonstrating loss of heterozygosity or hypermethylation. These are noted in the text.
Includes only data relating to mRNA levels. Western blot and IHC studies not included. They are noted in the text.
In some cases, downstream signaling activity has been assessed. These are described in the text.
As discussed in detail below, several recent studies in follicular thyroid neoplasias demonstrated that PTEN mutations rarely occur in tumors with other PI3K-activating genetic changes, such as mutations or amplification of PIK3CA 28, 32, providing important genetic evidence for a role for PI3K signaling in sporadic follicular tumor formation. It is also of interest that activation of RAS and loss of PPARγ function, which may be involved in the activity of PPARγ/PAX8, have been reported to activate PI3K signaling 33–35 and are associated with follicular thyroid tumor development and with AKT activation in human tumors 36. Thus, while PTEN mutations appear to be relatively uncommon in sporadic FTC, LOH, promoter methylation, and loss of protein expression may be more commonly identified. These data, in combination with the lack of genetic overlap between PTEN mutations and other FTC-related genetic alterations support a role for PI3K signaling in follicular tumorigenesis.
The frequency of PTEN mutations has also been reported in papillary and anaplastic thyroid cancers (PTC and ATC, respectively). In general, the mutation rate of PTEN in PTC is lower than FTC (see Table 1) 25–28, 32, 37, and while the frequency of PTCs with reduced PTEN mRNA expression appears to be higher than the number with mutations, it seems unlikely that PTEN loss itself plays a central role early in PTC tumorigenesis based on the well-defined role for BRAF and ERK signaling in these tumors. When taken together, mutations in PTEN or other genetic abnormalities predicted to increase AKT activity in PTC are also reported to occur at a less frequent rate than in FTC in general (Table 1) and they do not appear to be mutually exclusive of known PTC-inducing mutations such as activating mutations of BRAF 28. This is distinct from mutations that results in enhanced activation of the RAS/RAF/ERK pathway that appear to be mutually exclusive in PTCs 38. These data suggest that genetic changes leading to increased PI3K pathway signaling may represent a later-stage event in PTC in general. Consistent with this hypothesis is the observation that enhanced activation of AKT appears to occur most notably in the invasive fronts of PTCs 36 and the higher frequency of PI3K-related genetic changes in ATCs in comparison to well differentiated PTCs (Table 1 and see below). In addition, based on signaling data from human tumors, the degree of PI3K signaling may differ depending on the initiating oncogene in PTC 36.
In comparison to differentiated FTCs and PTCs, loss of PTEN expression and mutations of the PTEN gene are reported to occur at a higher frequency in ATCs 25, 28, 29, 39. This may not be surprising as these tumors harbor many genetic changes, suggesting overall genomic instability. The functional role for the loss of PTEN function and subsequent increased PI3K signaling in the development of ATC is uncertain, although in vitro data supports a role for this pathway in proliferation, survival, and motility in thyroid cancer cell lines, especially those with loss of PTEN 39. Thus, it appears that loss of PTEN expression and function may be important both in the development of FTCs and more broadly in the progression and dedifferentiation of thyroid cancers.
PI3K in Thyroid Cancer
PIK3CA Mutations and Amplification in Sporadic Thyroid Cancer
The PIK3CA gene encodes the catalytic subunit of class 1A PI3K (p110α). As described above, PI3K is comprised of a regulatory subunit (p85) which forms a heterodimer with the catalytic subunit (p110), leading to generation of PIP3 and initiation of its signaling cascade. Samuels, et al 40 and other groups identified cancer-related mutations in PIK3CA that largely reside in hot-spot regions of the gene that encode the helical and kinase domains of the protein 1. These mutations have been shown to be cause expression of constitutively active PI3Kα proteins that are capable of inducing malignant transformation in vitro and in vivo 41–43. In addition to activating mutations, PIK3CA gene amplification has also been implicated in cancer and has been associated with increased PI3K signaling 44–46. Over the past several years, the frequency of PIK3CA mutations and gene amplification has been analyzed in sporadic thyroid carcinomas.
In follicular thyroid cancer, the frequency of mutations in the “hot spot” domains of PIK3CA has ranged from 6 to 13% depending on the population 28, 32, 47, 48. By contrast, 24–28% of FTCs reported are described to have gene amplification of PIK3CA (defined as 4 or more copies) 28, 32, 48. Intriguingly, in studies where mutations and amplification of PIK3CA were studied in conjunction with PTEN and RAS mutations, the presence of these changes appeared to be mutually exclusive; however PPARγ/PAX8 analysis was not included in these studies. Overall, these studies support a potential genetic role for genetic abnormalities involving PIK3CA in follicular thyroid cancer.
In PTC, the frequency of mutations in PIK3CA appears to be lower than FTC in several studies, with occurrence rates ranging from 0–3% (see Table 1). Similarly, while gene amplification of PIK3CA is more common than mutations in PTC, its frequency is lower than in FTCs with occurrence rates ranging from 5–14% 28, 32, 47, 48. In addition, while these changes were rare and relatively exclusive from PTEN or RAS gene mutations, they were not found to be independent of activating mutations of BRAF, the most common pathogenic mutation in PTC.
Of significant interest has been the recent finding that anaplastic thyroid cancers frequently harbor mutations and gene amplification in PIK3CA. Garcia-Rosten, et al identified PIK3CA hot-spot gene mutations in 23% of seventy ATCs 47. Levels of phospho-AKT were scored as moderate or high in the majority of ATCs whether or not a PIK3CA mutation was present, suggesting multiple mechanisms for enhanced AKT signaling in these tumors. It was noted that tumors with moderate or high levels of pAKT statistically had higher levels of markers of proliferation. Immunoactive pAKT was detected in both the cytosol and nucleus in the majority of cases with higher levels of AKT overactivation. PIK3CA gene amplification and PTEN mutations and expression levels were not assessed in this study. A second study that included samples from fifty patients with ATC was recently published by Hou, et al 28. In this study, PIK3CA mutations were identified in 12% of cases, gene amplification was noted in 42% of cases, and PTEN mutations were identified in 16% of cases. Different from the well-differentiated thyroid cancers, these were not mutually exclusive in this group of ATCs. When considered as a group, 58% of ATCs harbored one of these three genetic abnormalities predicted to increase PI3K signaling. In comparison, 31% of benign follicular adenomas, 55% of FTCs and 24% of PTCs in this study had one of these abnormalities. The authors thus concluded that the data supported a role for PI3K signaling in thyroid tumorigenesis, particularly for follicular neoplasias, and in thyroid cancer progression toward ATC. Table 1 includes a summary of the mutation and published expression level data for PTEN and PIK3CA in human thyroid neoplasias.
Other Potential Mechanisms for PI3K Activation in Thyroid Cancer
Activation by Thyroid Oncogenes and Tyrosine Kinase Receptors
A variety of other mechanisms might also be responsible for activation of PI3K signaling in thyroid cancer, including signaling through oncogenes, such as RET/PTC or RAS, and signaling through other receptor tyrosine kinases known to be commonly overexpressed in thyroid cancers. An association between expression of both RET/PTC and constitutively active N-RAS and enhanced PI3K signaling has been reported in human thyroid cancers at the level of cell signaling studies 36 and gene expression 49. While the activation of downstream targets in the classical PI3K signaling cascade is known to be initiated by RET/PTC and other receptor tyrosine kinases, recent evidence suggests that RET/PTC3 may also be capable of activating PDK1 and AKT through tyrosine phosphorylation in a PI3K-independent manner 50, 51. In addition to RET/PTC, activated RAS is known to interact with and activate PI3K directly 33. In human tumors, RAS gene mutations have been associated with follicular neoplasias 52 and with the follicular variant of papillary cancer 53. RAS mutations have been shown to be associated with AKT activation 36 and to be largely mutually exclusive of PIK3CA and PTEN genetic abnormalities in well-differentiated thyroid cancers 28, 32, 47. While the focus of this manuscript is on PI3K signaling, it is important to recognize that the relative importance PI3K signaling versus other pathways in the action of the RET/PTC and activated RAS in thyroid cancer is not fully elucidated. The importance of RAF/ERK signaling in RET/PTC downstream effects has been demonstrated 54, and while there are distinct genetic signatures for thyroid cancer harboring specific oncogenes, there is overlap between the expression profiles for tumors with BRAF V600E mutations and RET/PTC rearrangements 49. Thus, it seems likely that both (and other) pathways are important in the oncogenic action of RET/PTC and activated RAS in thyroid cancer.
In addition to oncogenes, PI3K signaling may also be activated by other events that occur in thyroid cancer, such as overexpression of a variety of receptor tyrosine kinases, such as cMET, the FGF, IGF-1, and VEGF receptors, and others 55. Many of these receptors are known to regulate angiogenesis and/or cell proliferation and invasion through activation of PI3K and other pathways, including the RAS/RAF cascade, suggesting their potential suitability as therapeutic targets.
Activation of the PI3K regulatory domain
In addition to mutations or gene amplifications of PIK3CA, PI3K signaling can be increased through cell signaling at the level of the regulatory domain of PI3K (p85). Potentially relevant for thyroid cancer are in vitro studies that have demonstrated the ability of thyroid hormone to activate PI3 kinase through interactions between thyroid hormone receptors and p85 in the cytosol in several cell types 56, 57. In addition, a particular thyroid hormone receptor β mutant (TRβpv/pv) that is capable of inducing invasive and eventually dedifferentiated FTC in vivo in a knock-in model in association with PI3K activation in the setting of high TSH levels 58, 59, also appears to activate PI3K signaling via interactions with the p85 subunit 60. It has recently been reported that treating these thyroid cancer-prone mice with the PI3K inhibitor LY294002 retards growth of the primary tumors and inhibits tumor invasion and metastases 61. While the clinical relevance of this thyroid hormone receptor β mutant is uncertain, the data provide evidence that disruption of PI3K signaling is capable of reducing thyroid cancer progression in an endogenous model system of thyroid cancer characterized by enhanced PI3K signaling. Further studies in additional model systems are needed to determine if this is a more generalized finding.
Activation of Downstream Signaling Molecules in the PI3K Pathway
Finally, the role of non-AKT downstream effectors of PI3K in thyroid cancer is only now being elucidated. Important roles for mTOR activation in thyroid neoplasia that may be independent of AKT, for example, are being studied 62. Recent data also suggest an important role for p21 activated kinases, a family of proteins activated by PDK1, in thyroid cancer cell motility 63. Activation of additional effectors of PDK1 such as protein kinase A, have also been shown to be associated with the development of thyroid cancer 64. PKC signaling, which also can be activated through the activity of PDK1 65, has also been shown to be upregulated in some thyroid tumors as well 66, 67. Thus, it is likely that new information regarding the key upstream regulators and downstream targets of PI3K involved in thyroid tumorigenesis and progression will help define important potential therapeutic targets for thyroid cancer.
PI3K as a Target for Thyroid Cancer Therapy
PI3K itself has been suggested to be a target for novel cancer therapy, although it has not yet been studied in clinical trials in thyroid cancer 68. The recent development of PI3K isoform-specific inhibitors, particularly PI3Kα inhibitors, have further raised the possibility of targeting this pathway for treatment of patients with cancers harboring PIK3CA mutations or that display increased PI3K signaling through other mechanisms, including those detailed above 1, 69, 70. Because PI3K signaling has important metabolic and neurological effects, side effects such as insulin resistance or other off-target effects will need to be closely monitored and might impact the dosing strategies or the ultimately utility of this approach 2, 68. It may be reasonable to consider inhibition of PI3K as part of a combinatorial therapeutic strategy. Indeed, Berns, et al 71 recently reported that the PI3K pathway was crucial in predicting resistance to trastuzumab in breast cancer. Whether or not PI3K inhibition plays a similar role in the therapeutic resistance of poorly differentiated thyroid cancer remains to be determined. In addition to inhibiting PI3K directly, inhibitors of several downstream effectors, such as mammalian target of rapamycin (mTOR) 72 are also being developed and will be interesting to test in preclinical thyroid cancer systems.
Summary
Dysregulated PI3K signaling is a common event in thyroid cancer. Genetic evidence supporting the ability of constitutive PI3K signaling to cause follicular neoplasias derives from observations studying Cowden syndrome, a genetic syndrome caused by inactivation of PTEN, and by the mutually exclusive nature of loss of PTEN expression and activating mutations and amplifications of the PIK3CA gene in sporadic follicular thyroid cancers. Genetic alterations that result in activated PI3K signaling have also been identified in sporadic well differentiated papillary and anaplastic thyroid cancers, particularly in the latter, suggesting a role for PI3K signaling in dedifferentiated thyroid cancers. Finally, PI3K activation can also occur through the action of receptor tyrosine kinases and through cross-talk from signaling molecules known to be activated in thyroid cancer. Taken together, these data suggest that dysregulated PI3K signaling plays an important role in thyroid neoplasia.
Acknowledgments
This work was supported by a grant (5 R01 CA102572-02) from the NIH to MDR.
Footnotes
Disclosure: Dr. Ringel has received speaker honoraria from Genzyme Corporation and Abbott Laboratories.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Vogt PK, Kang S, Elsliger MA, Gymnopoulos M. Cancer-specific mutations in phosphatidylinositol 3-kinase. Trends Biochem Sci. 2007;32(7):342–349. doi: 10.1016/j.tibs.2007.05.005. [DOI] [PubMed] [Google Scholar]
- 2.Cully M, You H, Levine AJ, Mak TW. Beyond PTEN mutations: the PI3K pathway as an integrator of multiple inputs during tumorigenesis. Nat Rev Cancer. 2006;6(3):184–192. doi: 10.1038/nrc1819. [DOI] [PubMed] [Google Scholar]
- 3.Blanco-Aparicio C, Renner O, Leal JF, Carnero A. PTEN, more than the AKT pathway. Carcinogenesis. 2007;28(7):1379–1386. doi: 10.1093/carcin/bgm052. [DOI] [PubMed] [Google Scholar]
- 4.Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and Regulation of Akt/PKB by the Rictor-mTOR Complex. Science. 2005;307(5712):1098–1101. doi: 10.1126/science.1106148. [DOI] [PubMed] [Google Scholar]
- 5.Feng J, Park J, Cron P, Hess D, Hemmings BA. Identification of a PKB/Akt Hydrophobic Motif Ser-473 Kinase as DNA-dependent Protein Kinase. J Biol Chem. 2004;279(39):41189–41196. doi: 10.1074/jbc.M406731200. [DOI] [PubMed] [Google Scholar]
- 6.Kawakami Y, Nishimoto H, Kitaura J, et al. Protein kinase C betaII regulates Akt phosphorylation on Ser-473 in a cell type- and stimulus-specific fashion. J Biol Chem. 2004;279(46):47720–47725. doi: 10.1074/jbc.M408797200. [DOI] [PubMed] [Google Scholar]
- 7.Lynch DK, Ellis CA, Edwards PA, Hiles ID. Integrin-linked kinase regulates phosphorylation of serine 473 of protein kinase B by an indirect mechanism. Oncogene. 1999;18(56):8024–8032. doi: 10.1038/sj.onc.1203258. [DOI] [PubMed] [Google Scholar]
- 8.Toker A, Newton AC. Akt/Protein kinase B is regulated by autophosphorylation at the hypothetical PDK-2 site. J Biol Chem. 2000;275(12):8271–8274. doi: 10.1074/jbc.275.12.8271. [DOI] [PubMed] [Google Scholar]
- 9.Shinohara M, Chung YJ, Saji M, Ringel MD. AKT in Thyroid Tumorigenesis and Progression. Endocrinology 2007. 2007;148(3):942–947. doi: 10.1210/en.2006-0937. [DOI] [PubMed] [Google Scholar]
- 10.Carpten JD, Faber AL, Horn C, et al. A transforming mutation in the pleckstrin homology domain of AKT1 in cancer. Nature. 2007;448(7152):439–444. doi: 10.1038/nature05933. [DOI] [PubMed] [Google Scholar]
- 11.Basso AD, Solit DB, Chiosis G, Giri B, Tsichlis P, Rosen N. Akt forms an intracellular complex with Hsp90 and Cdc37 and is destabilized by inhibitors of Hsp90 function. J Biol Chem. 2002;277(42):39858–39866. doi: 10.1074/jbc.M206322200. [DOI] [PubMed] [Google Scholar]
- 12.Mitsuuchi Y, Johnson SW, Sonoda G, Tanno S, Golemis EA, Testa JR. Identification of a chromosome 3p14.3–1.1 gene, APPL, encoding an adaptor molecule that interacts with the oncoprotein-serine/threonine kinase AKT2. Oncogene. 1999;18(35):4891–4898. doi: 10.1038/sj.onc.1203080. [DOI] [PubMed] [Google Scholar]
- 13.Braga-Basaria M, Hardy E, Gottfried R, Burman KD, Saji M, Ringel MD. 17-Allylamino-17-Demethoxygeldanamycin Activity against Thyroid Cancer Cell Lines Correlates with Heat Shock Protein 90 Levels. J Clin Endocrinol Metab. 2004;89(6):2982–2988. doi: 10.1210/jc.2003-031767. [DOI] [PubMed] [Google Scholar]
- 14.Cantley LC, Neel BG. New insights into tumor suppression: PTEN suppresses tumor formation by restraining the phosphoinositide 3-kinase/AKT pathway. Proc Natl Acad Sci U S A. 1999;96(8):4240–4245. doi: 10.1073/pnas.96.8.4240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gao T, Furnari F, Newton AC. PHLPP: A Phosphatase that Directly Dephosphorylates Akt, Promotes Apoptosis, and Suppresses Tumor Growth. Mol Cell. 2005;18(1):13–24. doi: 10.1016/j.molcel.2005.03.008. [DOI] [PubMed] [Google Scholar]
- 16.Trotman LC, Alimonti A, Scaglioni PP, Koutcher JA, Cordon-Cardo C, Pandolfi PP. Identification of a tumour suppressor network opposing nuclear Akt function. Nature. 2006;441(7092):523–527. doi: 10.1038/nature04809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brognard J, Sierecki E, Gao T, Newton AC. PHLPP and a second isoform, PHLPP2, differentially attenuate the amplitude of Akt signaling by regulating distinct Akt isoforms. Mol Cell. 2007;25(6):917–931. doi: 10.1016/j.molcel.2007.02.017. [DOI] [PubMed] [Google Scholar]
- 18.Liaw D, Marsh DJ, Li J, et al. Germline mutations of the PTEN gene in Cowden disease, an inherited breast and thyroid cancer syndrome. Nat Genet. 1997;16(1):64–67. doi: 10.1038/ng0597-64. [DOI] [PubMed] [Google Scholar]
- 19.Pilarski R, Eng C. Will the real Cowden syndrome please stand up (again)? Expanding mutational and clinical spectra of the PTEN hamartoma tumour syndrome. J Med Genet. 2004;41(5):323–326. doi: 10.1136/jmg.2004.018036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Di Cristofano A, Pesce B, Cordon-Cardo C, Pandolfi PP. Pten is essential for embryonic development and tumour suppression. Nat Genet. 1998;19(4):348–355. doi: 10.1038/1235. [DOI] [PubMed] [Google Scholar]
- 21.Podsypanina K, Ellenson LH, Nemes A, et al. Mutation of Pten/Mmac1 in mice causes neoplasia in multiple organ systems. Proc Natl Acad Sci U S A. 1999;96(4):1563–1568. doi: 10.1073/pnas.96.4.1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stambolic V, Tsao MS, Macpherson D, Suzuki A, Chapman WB, Mak TW. High incidence of breast and endometrial neoplasia resembling human Cowden syndrome in pten+/− mice. Cancer Res. 2000;60(13):3605–3611. [PubMed] [Google Scholar]
- 23.Chen ML, Xu PZ, Peng XD, et al. The deficiency of Akt1 is sufficient to suppress tumor development in Pten+/− mice. Genes Dev. 2006;20(12):1569–1574. doi: 10.1101/gad.1395006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yeager N, Klein-Szanto A, Kimura S, Di Cristofano A. Pten Loss in the Mouse Thyroid Causes Goiter and Follicular Adenomas: Insights into Thyroid Function and Cowden Disease Pathogenesis. Cancer Res. 2007;67(3):959–966. doi: 10.1158/0008-5472.CAN-06-3524. [DOI] [PubMed] [Google Scholar]
- 25.Dahia PLM, Marsh DJ, Zheng Z, et al. Somatic deletions and mutations in the Cowden disease gene, PTEN, in sporadic thyroid tumors. Cancer Res. 1997;57:4710–4713. [PubMed] [Google Scholar]
- 26.Halachmi N, Halachmi S, Evron E, et al. Somatic mutatons of the PTEN/MMAC1 tumor suppressor gene in sporadic follicular thyroid tumors. Genes Chromosomes Cancer. 1998;23(3):239–243. doi: 10.1002/(sici)1098-2264(199811)23:3<239::aid-gcc5>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- 27.Hsieh MC, Lin SF, Shin SJ, Liu TC, Chang JG, Lee JP. Mutation analysis of PTEN/MMAC 1 in sporadic thyroid tumors. Kaohsiung J Med Sci. 2000;16(1):9–12. [PubMed] [Google Scholar]
- 28.Hou P, Liu D, Shan Y, et al. Genetic Alterations and Their Relationship in the Phosphatidylinositol 3-Kinase/Akt Pathway in Thyroid Cancer. Clin Cancer Res. 2007;13(4):1161–1170. doi: 10.1158/1078-0432.CCR-06-1125. [DOI] [PubMed] [Google Scholar]
- 29.Frisk T, Foukakis T, Dwight T, et al. Silencing of the PTEN tumor-suppressor gene in anaplastic thyroid cancer. Genes Chromosomes Cancer. 2002;35(1):74–80. doi: 10.1002/gcc.10098. [DOI] [PubMed] [Google Scholar]
- 30.Gimm O, Perren A, Weng LP, et al. Differential nuclear and cytoplasmic expression of PTEN in normal thyroid tissue, and benign and malignant epithelial thyroid tumors. Am J Pathol. 2000;156(5):1693–1700. doi: 10.1016/s0002-9440(10)65040-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Alvarez-Nunez F, Bussaglia E, Mauricio D, et al. PTEN promoter methylation in sporadic thyroid carcinomas. Thyroid. 2006;16(1):17–23. doi: 10.1089/thy.2006.16.17. [DOI] [PubMed] [Google Scholar]
- 32.Wang Y, Hou P, Yu H, et al. High prevalence and mutual exclusivity of genetic alterations in the phosphatidylinositol-3-kinase/akt pathway in thyroid tumors. J Clin Endocrinol Metab. 2007;92(6):2387–2390. doi: 10.1210/jc.2006-2019. [DOI] [PubMed] [Google Scholar]
- 33.Rodriguez-Viciana P, Warne PH, Dhand R, et al. Phosphatidylinositol-3-OH kinase as a direct target of Ras. Nature. 1994;370(6490):527–532. doi: 10.1038/370527a0. [DOI] [PubMed] [Google Scholar]
- 34.Farrow B, Evers BM. Activation of PPARgamma increases PTEN expression in pancreatic cancer cells. Biochem Biophys Res Commun. 2003;301(1):50–53. doi: 10.1016/s0006-291x(02)02983-2. [DOI] [PubMed] [Google Scholar]
- 35.Aiello A, Pandini G, Frasca F, et al. PPAR-γ agonists induce partial reversion of epithelial-mesenchymal transition in anaplastic thyroid cancer cells. Endocrinology. 2006;147(9):4463–4475. doi: 10.1210/en.2005-1610. [DOI] [PubMed] [Google Scholar]
- 36.Vasko V, Saji M, Hardy E, et al. Akt activation and localization correlate with tumor invasion and oncogene expression in thyroid cancer. J Mol Genet. 2004;41(3):161–170. doi: 10.1136/jmg.2003.015339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gimm O, Attie-Bitach T, Lees JA, Vekemans M, Eng C. Expression of the PTEN tumour suppressor protein during human development. Hum Mol Genet. 2000;9(11):1633–1639. doi: 10.1093/hmg/9.11.1633. [DOI] [PubMed] [Google Scholar]
- 38.Fagin JA. How thyroid tumors start and why it matters: kinase mutants as targets for solid cancer pharmacotherapy. J Endocrinol. 2004;183(2):249–256. doi: 10.1677/joe.1.05895. [DOI] [PubMed] [Google Scholar]
- 39.Bruni P, Boccia A, Baldassarre G, et al. PTEN expression is reduced in a subset of sporadic thyroid carcinomas: evidence that PTEN-growth suppressing activity in thyroid cancer cells mediated by p27kip1. Oncogene. 2000;19(28):3146–3155. doi: 10.1038/sj.onc.1203633. [DOI] [PubMed] [Google Scholar]
- 40.Samuels Y, Wang Z, Bardelli A, et al. High Frequency of Mutations of the PIK3CA Gene in Human Cancers. Science. 2004;304(5670):554. doi: 10.1126/science.1096502. [DOI] [PubMed] [Google Scholar]
- 41.Bader AG, Kang S, Vogt PK. Cancer-specific mutations in PIK3CA are oncogenic in vivo. Proc Natl Acad Sci U S A. 2006;103(5):1475–1479. doi: 10.1073/pnas.0510857103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gymnopoulos M, Elsliger MA, Vogt PK. Rare cancer-specific mutations in PIK3CA show gain of function. Proc Natl Acad Sci U S A. 2007;104(13):5569–5574. doi: 10.1073/pnas.0701005104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kang S, Bader AG, Vogt PK. Phosphatidylinositol 3-kinase mutations identified in human cancer are oncogenic. Proc Natl Acad Sci U S A. 2005;102(3):802–807. doi: 10.1073/pnas.0408864102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bertelsen BI, Steine SJ, Sandvei R, Molven A, Laerum OD. Molecular analysis of the PI3K-AKT pathway in uterine cervical neoplasia: frequent PIK3CA amplification and AKT phosphorylation. Int J Cancer. 2006;118(8):1877–1883. doi: 10.1002/ijc.21461. [DOI] [PubMed] [Google Scholar]
- 45.Byun DS, Cho K, Ryu BK, et al. Frequent monoallelic deletion of PTEN and its reciprocal associatioin with PIK3CA amplification in gastric carcinoma. Int J Cancer. 2003;104(3):318–327. doi: 10.1002/ijc.10962. [DOI] [PubMed] [Google Scholar]
- 46.Wu G, Xing M, Mambo E, et al. Somatic mutation and gain of copy number of PIK3CA in human breast cancer. Breast Cancer Res. 2005;7(5):R609–616. doi: 10.1186/bcr1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Garcia-Rostan G, Costa AM, Pereira-Castro I, et al. Mutation of the PIK3CA Gene in Anaplastic Thyroid Cancer. Cancer Res. 2005;65(22):10199–10207. doi: 10.1158/0008-5472.CAN-04-4259. [DOI] [PubMed] [Google Scholar]
- 48.Wu G, Mambo E, Guo Z, et al. Uncommon Mutation but Common Amplifications of the PIK3CA gene in Thyroid Tumors. J Clin Endocrinol Metab. 2005;90(8):4688–4693. doi: 10.1210/jc.2004-2281. [DOI] [PubMed] [Google Scholar]
- 49.Giordano TJ, Kuick R, Thomas DG, et al. Molecular classification of papillary thyroid carcinoma: distinct BRAF, RAS, and RET/PTC mutation-specific gene expression profiles discovered by DNA microarray analysis. Oncogene. 2005;24(44):6646–6656. doi: 10.1038/sj.onc.1208822. [DOI] [PubMed] [Google Scholar]
- 50.Kim DW, Hwang JH, Suh JM, et al. RET/PTC (rearranged in transformation/papillary thyroid carcinomas) tyrosine kinase phosphorylates and activates phosphoinositide-dependent kinase 1 (PDK1): an alternative phosphatidylinositol 3-kinase-independent pathway to activate PDK1. Mol Endocrinol. 2003;17(7):1382–1394. doi: 10.1210/me.2002-0402. [DOI] [PubMed] [Google Scholar]
- 51.Jung HS, Kim DW, Jo YS, et al. Regulation of PKB tyrosine phosphorylation by thyroid-specific oncogene Ret/PTC kinases. Mol Endocrinol. 2005;19(11):2748–2759. doi: 10.1210/me.2005-0122. [DOI] [PubMed] [Google Scholar]
- 52.Vasko V, Ferrand M, Di Cristofaro J, Carayon P, Henry JF, de Micco C. Specific Pattern of RAS Oncogene Mutations in Follicular Thyroid Tumors. J Clin Endocrinol Metab. 2003;88(6):2745–2752. doi: 10.1210/jc.2002-021186. [DOI] [PubMed] [Google Scholar]
- 53.Adeniran AJ, Zhu Z, Gandhi M, et al. Correlation between genetic alterations and microscopic features, clinical manifestations, and prognostic characteristics of thyroid papillary carcinomas. Am J Surg Pathol. 2006;30(2):216–222. doi: 10.1097/01.pas.0000176432.73455.1b. [DOI] [PubMed] [Google Scholar]
- 54.Mitsutake N, Miyagishi M, Mitsutake S, et al. BRAF mediates RET/PTC-induced MAPK activation in thyroid cells: functional support for requirement of the RET/PTC-RAS-BRAF pathway in papillary thyroid carcinogenesis. Endocrinology. 2005;147(2):1014–1019. doi: 10.1210/en.2005-0280. [DOI] [PubMed] [Google Scholar]
- 55.Kondo T, Ezzat S, Asa SL. Pathogenetic mechanisms in thyroid follicular-cell neoplasia. Nat Rev Cancer. 2006;6(4):292–306. doi: 10.1038/nrc1836. [DOI] [PubMed] [Google Scholar]
- 56.Cao X, Kambe F, Moeller LC, Refetoff S, Seo H. Thyroid hormone induces rapid activation of Akt/PKB-mTOR-p70S6K cascade through phosphatidylinositol 3-kinase in human fibroblasts. Mol Endocrinol. 2005;19:102–112. doi: 10.1210/me.2004-0093. [DOI] [PubMed] [Google Scholar]
- 57.Kenessey A, Ojamaa K. Thyroid hormone stimulates protein synthesis in the cardiomyocyte by activating the Akt-mTOR and p70S6K pathways. J Biol Chem. 2006;281(30):20666–20672. doi: 10.1074/jbc.M512671200. [DOI] [PubMed] [Google Scholar]
- 58.Suzuki H, Willingham MC, Cheng SY. Mice with a mutation in the thyroid hormone receptor beta gene spontaneously develop thyroid carcinoma: a mouse model of thyroid carcinogenesis. Thyroid. 2002;12(11):963–969. doi: 10.1089/105072502320908295. [DOI] [PubMed] [Google Scholar]
- 59.Kim CF, Vasko VV, Kato Y, et al. AKT Activation Promotes Metastasis in a Mouse Model of Follicular Thyroid Carcinoma. Endocrinology. 2005;146(10):4456–4463. doi: 10.1210/en.2005-0172. [DOI] [PubMed] [Google Scholar]
- 60.Furuya F, Hanover JA, Cheng S-y. Activation of phosphatidylinositol 3-kinase signaling by a mutant thyroid hormone beta receptor. Proc Natl Acad Sci USA. 2006;103(6):1780–1785. doi: 10.1073/pnas.0510849103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Furuya F, Lu C, Willingham MC, Cheng SY. Inhibition of phosphatidylinositol 3′ kinase delays tumor progression and blocks metastatic spread in a mouse model of thyroid cancer. Carcinogenesis. 2007;28(12):2451–2458. doi: 10.1093/carcin/bgm174. [DOI] [PubMed] [Google Scholar]
- 62.Brewer C, Yeager N, Di Cristofano A. Thyroid-stimulating hormone initiated proliferative signals converge in vivo on the mTOR kinase without activating AKT. Cancer Res. 2007;67(17):8002–8006. doi: 10.1158/0008-5472.CAN-07-2471. [DOI] [PubMed] [Google Scholar]
- 63.Porchia LM, Guerra M, Wang YC, et al. 2-Amino-N-{4-[5-(2-phenanthrenyl)-3-(trifluoromethyl)-1H-pyrazol-1-yl]-phe nyl} Acetamide (OSU-03012), a Celecoxib Derivative, Directly Targets p21-Activated Kinase. Mol Pharmacol. 2007;72(5):1124–1131. doi: 10.1124/mol.107.037556. [DOI] [PubMed] [Google Scholar]
- 64.Sandrini F, Matyakhina L, Sarlis NJ, et al. Regulatory subunit type I-alpha of protein kinase A (PRKAR1A): a tumor-suppressor gene for sporadic thyroid cancer. Genes Chromosomes Cancer. 2002;35(2):182–192. doi: 10.1002/gcc.10112. [DOI] [PubMed] [Google Scholar]
- 65.Dutil EM, Toker A, Newton AC. Regulation of conventional protein kinase C isozymes by phosphoinositide-dependent kinase 1 (PDK-1) Curr Biol. 1998;8(25):1366–1375. doi: 10.1016/s0960-9822(98)00017-7. [DOI] [PubMed] [Google Scholar]
- 66.Shimizu T, Usuda N, Sugenoya A, et al. Immunohistochemical evidence for the overexpression of protein kinase C in proliferative diseases of human thyroid. Cell Mol Biol. 1991;37(8):813–821. [PubMed] [Google Scholar]
- 67.Eszlinger M, Krohn K, Berger K, et al. Gene Expression Analysis Reveals Evidence for Increased Expression of Cell Cycle-Associated Genes and Gq-Protein-Protein Kinase C Signaling in Cold Thyroid Nodules. J Clin Endocrinol Metab. 2005;90(2):1163–1170. doi: 10.1210/jc.2004-1242. [DOI] [PubMed] [Google Scholar]
- 68.Hennessy BT, Smith DL, Ram PT, Lu Y, Mills GB. Exploiting the PI3K/AKT pathway for cancer drug discovery. Nat Rev Drug Discov. 2005;4(12):988–1004. doi: 10.1038/nrd1902. [DOI] [PubMed] [Google Scholar]
- 69.Hayakawa M, Kaizawa H, Moritomo H, et al. Synthesis and biological evaluation of 4-morpholino-2-phenylquinazolines and related derivatives as novel PI3 kinase p110alpha inhibitors. Bioorg Med Chem. 2006;14(20):6847–6858. doi: 10.1016/j.bmc.2006.06.046. [DOI] [PubMed] [Google Scholar]
- 70.Marion F, Williams DE, Patrick BO, et al. Liphagal, a Selective inhibitor of PI3 kinase alpha isolated from the sponge akacoralliphaga: structure elucidation and biomimetic synthesis. Org Lett. 2006;8(2):321–324. doi: 10.1021/ol052744t. [DOI] [PubMed] [Google Scholar]
- 71.Berns K, Horlings HM, Hennessy BT, et al. A functional genetic approach identifies the PI3K pathway as a major determinant of trastuzumab resistance in breast cancer. Cancer Cell. 2007;12(4):395–402. doi: 10.1016/j.ccr.2007.08.030. [DOI] [PubMed] [Google Scholar]
- 72.Abraham RT, Gibbons JJ. The Mammalian Target of Rapamycin Signaling Pathway: Twists and Turns in the Road to Cancer Therapy. Clin Cancer Res. 2007;13(11):3109–3114. doi: 10.1158/1078-0432.CCR-06-2798. [DOI] [PubMed] [Google Scholar]