

Abstract
Mammalian soluble thiamine triphosphatase (ThTPase) is a 25-kDa cytosolic enzyme that specifically catalyzes the conversion of thiamine triphosphate (ThTP) to thiamine diphosphate and has an absolute requirement for divalent cations. We have investigated the kinetic properties of recombinant mouse thiamine triphosphatase (mThTPase) and determined its solution structure by NMR spectroscopy. Residues responsible for binding Mg2+ and ThTP were determined from NMR titration experiments. The binding of Mg2+ induced only a minor local conformational change, whereas ThTP binding was found to cause a more global conformational change. We derived a structural model for the mThTPase·ThTP·Mg2+ ternary complex and concluded from this that whereas free mThTPase has an open cleft fold, the enzyme in the ternary complex adopts a tunnel fold. Our results provide a functional rationale for a number of conserved residues and suggest an essential role for Mg2+ in catalysis. We propose a mechanism underlying the high substrate specificity of mThTPase and discuss the possible role of water molecules in enzymatic catalysis.
Two classes of enzymes are known to convert thiamine triphosphate (ThTP)3 to thiamine diphosphate in animal tissues: a membrane-associated enzyme that is yet to be characterized and a soluble thiamine triphosphatase (ThTPase). ThT-Pase was first identified in rat brain (1) and has been found in most mammalian tissues studied to date (2). Biochemical studies of the purified, homogeneous ThTPase further revealed that the enzyme is a 24-25-kDa monomer with virtually absolute specificity for ThTP (3, 4). The enzyme has an alkaline pH optimum, and its catalytic activity depends critically on a divalent metal cofactor, such as Mg2+.
ThTPase is thought to regulate the intracellular concentration of ThTP, which is found in most animal cells but at a much lower level than other forms of thiamine (5). Several lines of biochemical evidence have shown that ThTP can phosphorylate proteins in the electric organ of Torpedo marmorata and in rodent brain (6). In addition, ThTP is believed to act as one of the messengers in cell signaling in response to cellular stress (7). Despite the relatively high turnover rate of ThTP in neural cells (8), its level is strictly maintained at a low concentration in vivo, presumably through the activity of ThTPase.
The sequence of ThTPase does not closely resemble that of any other protein identified in the mammalian genomes. However, recent bioinformatic analysis has suggested that mammalian 25-kDa ThTPase and adenylate cyclase CyaB from Aeromonas hydrophila define an ancient superfamily of domains, called the CYTH domains, which play a role at the interface of polyphosphate and nucleotide metabolism (9). The structures of several CYTH domains have been determined, including class IV adenylyl cyclase from Yersinia pestis (YpAC4, Protein Data Bank (PDB) 2FJT) (10) and proteins with unknown functions from Pyrococcus furiosus (PDB accession numbers 1YEM and 2DC4), Vibrio parahemeolyticus (PDB 2ACA), and Nitrosomonas europaea (PDB 2FBL). With the exception of the hypothetical protein NE1496 from N. europaea, these structures are characterized by a closed tunnel fold resembling that initially described for the RNA triphosphatase from Saccharomyces cerevisiae (Cet1, PDB 1D8H) (11). Given the fact that Cet1 and the two proposed founding members of CYTH family, ThTPase and CyaB (9), are all metal-dependent enzymes that act on the triphosphate-containing substrates, it has been proposed that the tunnel fold is characteristic of a larger enzyme superfamily, namely the “triphosphate tunnel metalloenzyme” (TTM) superfamily (12). Despite its more open fold, Keppetipola et al. (13) classified the hypothetical protein NE1496 from N. europaea as a member of the TTM superfamily and gave it the name “NeuTTM,” which we use here.
The enzymatic mechanism of TTM proteins has remained elusive. Sequence analysis and recent mutational studies have identified a few conserved residues, arrayed inside the tunnel, as essential for catalysis (12, 13). However, how these residues participate in enzyme catalysis is unclear.
We present here the solution structure of mouse ThTPase (mThTPase) determined by NMR spectroscopy. Unlike most of the TTM proteins studied so far, the enzyme has an open cleft like NeuTTM. We used NMR titration experiments to identify residues responsible for Mg2+ and substrate (ThTP) binding. These titration results, together with the intermolecular NOEs, enabled us to build a structural model of the ground state ternary complex, mThTPase·ThTP·Mg2+, in which mThTPase adopts a tunnel fold characteristic of a TTM protein. Our results provide a framework for further characterization of ThTPase and shed light on its catalytic mechanism.
EXPERIMENTAL PROCEDURES
Informatics—We used the Sesame laboratory information management system (14) developed at the Center for Eukaryotic Structural Genomics to select this target and to track all subsequent steps in this study.
Cloning and Protein Production—The gene encoding mTh-TPase (BC025562) was purchased from Open Biosystems. 15N/15N,13C-labeled proteins were expressed and purified following the standard Center for Eukaryotic Structural Genomics pipeline protocols for plasmid construction (15), protein production (16), and protein purification (17).
ThTPase Enzyme Assay—The ThTPase assay was carried out as previously described (18, 19). The standard reaction medium contained 50 mm Na-TAPS (pH 8.3), 10 mm MgCl2, 0.5 mm ThTP (synthesized as described (20)), and the enzyme preparation at the appropriate dilution. The total volume was 0.1 ml. After incubation (20 min, 37 °C), the reaction was stopped by the addition of 1 ml of phosphate reagent (21).
NMR Spectroscopy—All NMR spectra were recorded at the National Magnetic Resonance Facility at Madison (NMRFAM) using 600- and 900-MHz Varian Inova and 750-MHz Bruker spectrometers equipped with 1H, 15N, 13C triple-resonance cryogenic probes. Unless indicated otherwise, the sample temperature was controlled at 25 °C.
The sample used for determining the structure of mThTPase contained 1.4 mm [U-13C, U-15N]mThTPase, 10 mm MOPS, 100 mm NaCl, 5 mm dithiothreitol, in 90% H2O, 10% D2OatpH 7.0. A suite of three-dimensional heteronuclear NMR experiments, including HNCACB, CBCA(CO)NH, HNCO, HBHA(CO)NH, C(CO)NH, and HCCH total correlation spectroscopy were acquired for sequential backbone and non-aromatic side chain assignments of free mThTPase. Three-dimensional 15N-edited NOESY-HSQC (τmix = 100 ms), three-dimensional aliphatic 13C-edited NOESY-HSQC (τmix = 100 ms), and three-dimensional aromatic 13C-edited NOESY-HSQC (τmix = 100 ms) data sets were acquired to measure NOEs used for additional assignments (side chain amide groups and aromatic groups) and distance constraints.
The sample used in determining the structure of the mThTPase·ThTP complex (used in modeling the ternary complex) contained 1.4 mm [U-13C, U-15N]mThTPase, 2 mm unlabeled ThTP, 10 mm MOPS, 100 mm NaCl, and 5 mm dithiothreitol in 90% H2O, 10% D2O at pH 7.0. Data sets acquired with this sample included three-dimensional HNCA and three-dimensional HN(CO)CA for sequence specific assignments and three-dimensional 15N-edited NOESY-HSQC (τmix = 100 ms) and three-dimensional 13C-edited NOESY-HSQC (τmix = 100 ms) for NOE assignments. In addition, a 13C,15N-filtered, 13C-edited NOESY (τmix = 150 ms) (22) spectrum was collected at 30 °C to detect intermolecular NOEs between mThTPase and ThTP.
Two-dimensional 1H,15N HSQC spectra were acquired to monitor the titration of Mg2+ or ThTP to 0.5 mm U-15N-labeled mThTPase, with ThTP at 0, 0.05, 0.1, 0.2, 0.3, 0.5, 1.0 mm and Mg2+ at 0, 1, 2, 4, 8 mm, respectively. The spectra were processed and analyzed, respectively, with the nmrPipe (23) and Sparky software packages. The raw, time-domain NMR data sets and chemical shift assignments have been deposited in the BioMagResBank (BMRB) data base under accession number 15063.
Structure Calculations—Interproton distance constraints for free mThTPase were derived from a three-dimensional 15N-edited NOESY-HSQC spectrum and a three-dimensional 13C-edited NOESY-HSQC spectrum. Backbone ϕ and ψ angles were derived from TALOS-based analysis of backbone chemical shifts (24). The automated iterative refinement module (25) in the CYANA software package (26) was used to generate the initial NOE assignments and the initial set of structural models. These NOE assignments were subsequently corrected and extended through iterative analysis of NMR spectra and structural calculation. In the final round of CYANA refinement, 4698 NOE restraints and 289 dihedral angle restraints were used. In addition, 176 hydrogen bond restraints, generated from analysis of the secondary chemical shifts and from observed NOEs characteristic for α-helices and β-sheets, were also added to the set of restraints. Of the 100 final structures calculated by CYANA, the 20 structures with the lowest target functions were chosen for further refinement in explicit water solvent by Xplor-NIH (27) followed by validation by Procheck-NMR (28). The statistics for the 20 final structures are listed in Table 1. The coordinates for these structural models, along with the restraints employed, have been deposited in the PDB under accession number 2JMU.
TABLE 1.
Statistics for the 20 energy-minimized conformers of free mThTPase and the mThTPase:ThTP:Mg2+ ternary complex
| Distance constraints | mThTPase | mThTPase:ThTP:Mg2+ |
|---|---|---|
| Long [(i - j) ≥ 5] (intramolecular) | 1455 | 1384 |
| Medium [1 ≤ (i - j) < 5] | 683 | 671 |
| Sequential [(i - j) = 1] | 921 | 912 |
| Intraresidue [i = j] | 1639 | 1636 |
| Intermolecular | ||
| Unambiguous | 21 | |
| Ambiguous | 18 | |
| Dihedral angle constraints (ϕ and ψ) | 289 | 289 |
| Hydrogen bond constraints | 176 | 174 |
| Average pairwise r.m.s.d. to the mean structure (Å) | ||
| Residues 5-213 | ||
| Backbone (Cα, C′, N, O) | 1.06 ± 0.17 | 1.11 ± 0.17 |
| Heavy atoms | 1.43 ± 0.15 | 1.43 ± 0.14 |
| Regular secondary structural elements | ||
| Backbone (Cα, C′, N, O) | 0.73 ± 0.13 | 0.64 ± 0.10 |
| Heavy atoms | 1.06 ± 0.12 | 1.01 ± 0.07 |
| Deviations from idealized covalent geometry | ||
| Bond (Å) | 0.019 ± 0.001 | 0.002 ± 0.001 |
| Angles (°) | 1.218 ± 0.024 | 0.477 ± 0.006 |
| Impropers (°) | 1.455 ± 0.046 | 0.561 ± 0.006 |
| r.m.s.d. | ||
| r.m.s.d. from experimental distance restraints (Å) | 0.018 ± 0.001 | 0.007 ± 0.001 |
| r.m.s.d. from experimental dihedral restraints (°) | 0.585 ± 0.044 | 0.251 ± 0.055 |
| Ramachandran statistics (% of all residues) | ||
| Most favored | 86.7 | 81.2 |
| Additionally allowed | 11.2 | 13.9 |
| Generously allowed | 1.2 | 3.1 |
| Disallowed | 0.9 | 1.8 |
Based on chemical shift and NOE analysis, most constraints used for structure calculation of free mThTPase were retained in calculating the structure of mThTPase in complex with ThTP and Mg2+. However, a number of NOE differences were also identified, which led to the removal of 112 NOEs specific for free mThTPase and the addition of 29 NOEs specific for complexed mThTPase. In addition, 16 intermolecular NOEs between mThTPase and ThTP were derived from a three-dimensional 13C-edited NOESY-HSQC spectrum and a 13C,15N-filtered, 13C-edited NOESY-HSQC spectrum, and 20 ambiguous distance restraints were derived from NMR titration and mutational studies. The structure of the ternary complex was calculated through standard annealing and torsion angle dynamics using the program CNS 1.1 (29).
RESULTS
Enzyme Activity of mThTPase—The ThTPases from rat (rThTPase) (1), bovine (bThTPase) (3), human (hThTPase) (18), and pig (pThTPase) (19) have been shown to exhibit a large variation of catalytic efficiency. Analysis of the effect of ThTP concentration on the activity of recombinant mThTPase indicated non-Michaelis-Menten kinetics, with inhibition by excess substrate at [ThTP] > 0.5 mm (Fig. 1A). Thus, it was not possible to determine accurate Km and kcat values for this enzyme. To explain substrate inhibition, it might be assumed that the enzyme is able to catalyze hydrolysis of the Mg-ThTP complex but not free ThTP. At constant Mg2+ concentration, increasing substrate concentration would result in an accumulation of free ThTP, which might act as a competitive inhibitor. If this explanation were valid, substrate inhibition should be strong at low Mg2+ concentration and should be reversed by excess Mg2+. Fig. 1A clearly shows that this is not the case; substrate inhibition is not reversed in the presence of high [Mg2+] (10 mm) when compared with a lower concentration (0.2 mm). It is therefore hard to explain substrate inhibition if we assume that the substrate can only bind to the active site. It seems more likely that ThTP also binds to some peripheral non-catalytic site with lower affinity. Substrate inhibition was observed previously for rThTPase (1) but at a much higher substrate concentration, [ThTP] > 3 mm, than for mThTPase. By contrast, no substrate inhibition was observed for bThTpase (3), hThTPase (18), or pThTPase (19). Our results also suggest that the activating effect of Mg2+ reflects cation binding to an activation site on the protein rather than complexation with the substrate. Indeed, the apparent Ka for Mg2+ is independent of ThTP concentration (0.50 ± 0.09 and 0.45 ± 0.06 mm, respectively, at 0.2 and 0.5 mm ThTP), suggesting that it reflects the Kdiss between Mg2+ and the activation site of the enzyme (Fig. 1B). As has been found for all other ThTPases, the enzymatic activity of mThTPase is pH-dependent (Fig. 1C). The optimal pH for mThTPase was found to be 8.5, in agreement with that for hThTPase (18).
FIGURE 1.

Kinetic properties of recombinant mThTPase. A, ThTP hydrolysis by recombinant mThTPase as a function of substrate concentration. The incubation medium contained 10 (filled circle) or 0.2 mm (open circle) MgCl2 and 50 mm Na-TAPS, pH 8.3. B, effect of Mg2+ concentration on the specific activity of mThTPase in the presence of 0.5 (filled circle) or 0.2 mm (open circle) ThTP and 50 mm Na-TAPS, pH 8.3. The concentration of free Mg2+ was calculated from total [MgCl2] and [ThTP] using the value of 65 μm for the Kd of the Mg-ThTP complex (32). C, effect of pH on the specific activity of mThTPase in the presence of 10 mm MgCl2 and 0.5 mm ThTP.
Three-dimensional Structure of Free mThTPase—The 20 conformers of mThTPase that represent its solution structure, superimposed by the backbone atoms of their structured regions, are shown in Fig. 2A. These structures overlay the average structure (residues 5-213) with a backbone atom r.m.s.d. of 1.06 Å and an all heavy atom r.m.s.d. of 1.43 Å. Analysis of the backbone ϕ and ψ dihedral angles showed that 99.1% of these angles fall within the allowed region in the Ramachandran plot (Table 1). The outliers correspond to residues in the poorly defined N and C termini and loop regions. The structure shows that mThTPase in solution is monomeric; this result is in agreement with observed 15N R1/R2 relaxation rates (data not shown).
FIGURE 2.

NMR solution structure of mThTPase. A, stereoscopic view of the backbone trace of the final family of 20 conformers representing the structure with helical, sheet, and loop residues colored red, green, and gray, respectively. B, ribbon diagram of the mThTPase structure, with individual elements of secondary structure labeled. For clarity, the disordered residues 1-3 and 215-224 are not shown. C, schematic of the secondary structure topology. The color schemes in B and C are the same as in A.
The overall structure of mThTPase is dominated by an antiparallel β-sheet that is folded in half to form a cup-shaped shallow cleft (Fig. 2B). The β-sheet consists of nine strands with order 9 1 8 7 6 2 3 4 5 (Fig. 2C). The two longest strands are β2 (28-41) and β6 (116-131), which extend from one rim of the cleft to the other and form the core of the cleft. On the β6 side of the central β2-β6 sheet, the wall of the cleft is formed by four half-length or shorter strands (β7, 141-149; β8, 153-162; β1, 6-14; β9, 183-185). On the β2 side of the central β2-β6 sheet, the opposite wall of the cleft is formed by additional strands (β3, 53-57; β4, 61-66; β5, 80-82). This architecture, reinforced by the natural twist of the β-strands, causes the strands at the two edges of the β-sheet (β1 and β5) to curl toward each other. Further closure of the cleft is blocked by the two C-terminal antiparallel α-helices (α7, 193-201; α8, 203-212) situated over one end of the cleft. The α7- and α8-helices tightly associate with each other through coil-coil interactions and are anchored to the β-sheet by two hydrophobic clusters: a cluster of residues (Tyr-39, Trp-53, Leu-194, Leu-198, Leu-209) on one side of the two helices and a cluster of residues (Phe-121, Phe-151, Tyr-153, Ala-119, Lys-193, Tyr-197) on the other side of the two helices. Several helices are attached to the outside face of the β-sheet. Three helices (α1, 18-25; α5, 165-167; α6, 168-182) cover the outer faces of strands β1, β2, β6, β7, and β8 so as to enclose a hydrophobic cluster composed of side chains from residues Val-8, Phe-12, Leu-22, Leu-29, Trp-128, Ile-144, Leu-146, Val-162, Lys-165, and Leu-182. Three additional helices (α2, 46-49; α3, 85-96; α4, 106-113) are attached to the outer faces of strands β2 and β3 so as to enclose another hydrophobic cluster composed of side chains from residues Leu-47, Leu-54, Trp-62, Ile-88, Leu-92, Phe-93, Leu-95, Val-106, Leu-110, Leu-113, and Leu-115.
The mThTPase structure also contains poorly defined regions. The most prominent of these is the loop that connects β4 and β5 (lβ4,β5, 67-79). This 13-residue loop, which protrudes from one rim of the cleft, constitutes the major dynamic element at the top of the cleft. Residues at both the N terminus (1-4) and C terminus (214-224) are structurally disordered as evidenced by chemical shift and NOE analysis.
NMR Titrations—To map the Mg2+ and ThTP binding sites in mThTPase, we carried out separate titrations of the enzyme with Mg2+ or ThTP by adding increasing amounts of Mg2+ or ThTP to an NMR sample containing 0.5 mm mThTPase. At each titration point, a 1H,15N HSQC spectrum was collected to monitor changes in chemical shifts (Fig. 3, A and B). Changes in 1HN and 15N chemical shifts between the free state of mThT-Pase and the Mg2+ or ThTP-bound state were calculated according to the formula, Δδave(HN) = [(ΔδHN2 + (ΔδN/5)2)/2]½, where ΔδHN and ΔδN correspond, respectively, to the chemical shift differences in 1HN and 15N. In the absence of Mg2+, we saw no evidence for hydrolysis of the added ThTP.
FIGURE 3.

Interaction of mThTPase with Mg2+ and ThTP. A, overlaid two-dimensional 1H,15N HSQC spectra of 0.5 mm [U-15N]mThTPase in the presence of various concentrations of Mg2+ highlighting the progressive chemical shift changes of selected residues (bars). B, overlaid two-dimensional 1H,15N HSQC spectra of 0.5 mm [U-15N]mThTPase in the presence of various concentrations of ThTP illustrating the discontinuous chemical shift changes between free mThTPase and the mThTPase·ThTP complex. C, surface representation of the solution structure of mThTPase showing in red the residues that display the largest chemical shift changes (Δδave > 0.05 ppm) between 0 and 8 mm Mg2+. D, surface representation of the solution structure of mThTPase showing the residues that display large chemical shift changes between 0 and 1 mm added ThTP (red, Δδave > 0.4 ppm; yellow, 0.4 ppm >Δδave > 0.3 ppm). The side chain 13Cβ,ε signals from residues Asp-145, Asp-147, and Met-195 (also shown in red) displayed chemical shifts changes > 0.9 ppm.
In the titration with Mg2+, progressive shifting of NMR peaks from their positions in free mThTPase after each addition (Fig. 3A) indicated that free mThTPase is capable of binding Mg2+. Furthermore, all the chemical shift changes fell into the fast exchange limit on the NMR time scale, indicative of weak binding. The peaks that moved upon Mg2+ addition correspond to a small number of residues in mThTPase (supplemental Fig. S1), suggesting that Mg2+ binding leads to a local change in the conformation of mThTPase. Residues that displayed the largest chemical shift changes were identified as residues 7-11, 142-149, 157-160, 190, and 193. This result is consistent with the earlier suggestion, from studies on Cet1 and CthTTM (a homologue of NeuTTM from Clostridium thermocellum) (11, 13), that residues equivalent to Glu-7, Glu-9, Asp-145, Glu-157, and Glu-159 in mThTPase are responsible for metal binding. Indeed, when mapped onto the structure of mThTPase, these most perturbed residues are all located inside the cleft (Fig. 3C) and appear to define a single Mg2+ binding site. On the basis of the Mg2+-dependent chemical shift changes and the assumption of 1:1 stoichiometry, a dissociation constant (Kd) of 1.1 mm was estimated for the mThTPase·Mg2+ complex. Residues Ala-190 and Lys-193, along with those residues exhibiting minor titration shifts (32-34, 55-64, 120-125) (supplemental Fig. S1), are located peripheral to this site. Thus, their chemical shift changes presumably arise from a secondary or indirect binding effect.
The titration of mThTPase with ThTP (Fig. 3B) led to spectral changes consistent with a slow process on the chemical shift timescale (indicative of tight binding with Kd ≤ 1 μm). The addition of ThTP led to decreased intensities of peaks from free mTh-TPase and the appearance of new peaks corresponding to the mThTPase·ThTP complex. The effects were found to be widespread and included residues on all nine β-strands and in the region between α6 and α8 (supplemental Fig. S1). Those residues with the largest backbone titration shifts Δδave(HN) included Glu-9, His-31, Glu-81, Thr-123, Arg-125, Glu-157, Glu-189, Ala-190, Lys-193, and to a lesser extent, Arg-10, Lys-65, Val-33, and Leu-194. Furthermore, large side chain chemical shift changes were observed for a few residues exhibiting only moderate backbone chemical shift perturbations. These residues included Asp-145 (Δδ(Cβ) =-2.2 ppm), Asp-147 (Δδ(Cβ) = 1.4 ppm), Ala-192 (Δδ(Cβ) =-1.0 ppm), and Met-195 (Δδ(Cε) = 0.9 ppm). When mapped onto the structure of mThTPase, the residues with the largest chemical shift perturbations were located in the interior of the cleft (Fig. 3D). The presence of ThTP also induced severe broadening of signals from residues in this region: 6-11, 77-79, 125-128, 157-160, 188-189, and 191-193. Analysis of the ThTP titration was consistent with a mThTPase·ThTP stoichiometry of 1:1. Although no significant spectral changes occurred upon increasing the ThTP concentration from 1 to 2 mm, when the ThTP concentration was increased to 4 mm or higher, signals from a number of residues exhibited additional broadening and chemical shift changes; these included the indole group of Trp-53 and the backbone amides of Tyr-79 and Glu-81. It remains to be determined whether this is the result of binding to the secondary site, whose existence was suggested by the kinetic data (Fig. 1A).
To further establish the role of Mg2+ in mThTPase function, we also examined changes in the spectrum of the mThTPase·ThTP complex upon Ca2+ addition (supplemental Fig. S2). Ca2+ has been shown to inhibit hThTPase activity through competition with Mg2+ (1, 18). Our results showed that the addition of Ca2+ to the enzyme-substrate complex led to the progressive shifting of a small number of peaks and to the reappearance of some mThTPase peaks that had disappeared in the spectrum of the ThTPase·ThTP complex. The intensities of these signals were consistently enhanced in the presence of Ca2+. Most of the affected peaks, such as those from Glu-7, Arg-125, and Glu-159, mapped to the Mg2+ or substrate binding sites (supplemental Fig. S2).
Structural Model of the mThTPase·ThTP·Mg2+ Ternary Complex—The NMR titrations described above provided insight into the Mg2+ and ThTP binding sites of mThTPase. In deriving a structural model for the mThTPase·ThTP·Mg2+ ternary complex, we introduced distance constraints between Mg2+ and the side chain carboxylates of Glu-7, Glu-9, Asp-145, Glu-157, and Glu-159 and ambiguous distance constraints between Mg2+ and the β-, γ-phosphate groups of ThTP. On the basis of the NMR titration results with added ThTP and the effects of ThTPase mutations on catalysis (12, 13), we introduced ambiguous distance constraints between ThTP and Lys-11, Arg-55, Arg-57, Lys-65, Glu-81, Arg-125, Asp-147, and Lys-193, Met-195 of mThTPase. These inferred constraints, along with experimentally derived dihedral angle and NOE constraints (Table 2), served as input for calculating the structure of the mThTPase·ThTP·Mg2+ complex (Fig. 4A).
TABLE 2.
Intermolecular 1H-1H NOEs between ThTP and mThTPase
| ThTP | mThTPase |
|---|---|
| C6′-H | Ala-192 (HN, Hα) |
| C2-H | Lys-65 (Hδ2,3), Ala-192 (Hβ), Met-195 (Hγ2, H∈) |
| C3,5′-H2 | Ala-192 (Hβ), Met-195 (Hγ2/3, H∈) |
| 5-CH2 | Lys-65 (H δ2,3) |
| 2′-CH3 | Met-195 (H∈), Leu-210 (Hα, Hβ2/3, Hδ1,2), Ala-213 (Hβ) |
| 4′-CH3 | Lys-65 (Hδ2,3), Ala-192 (Hβ) |
FIGURE 4.

Structural model of the ternary complex. A, stereoscopic
view of the backbones of the family of 20 conformers representing the ternary
complex of mThTPase (magenta), Mg2+ (gold), and
ThTP (green). B, ribbon diagram of structure of the ternary
complex. C, ribbon diagram of structure of hThTPase bound to citric
acid (CIT) and 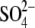 .
D, stereoscopic view of overlaid structures of free mThTPase
(blue) and complexed mThTase (magenta).
.
D, stereoscopic view of overlaid structures of free mThTPase
(blue) and complexed mThTase (magenta).
The 20 conformers representing the structure of the mThTPase·ThTP·Mg2+ complex (Fig. 4A) loosely define the positions of Mg2+ and ThTP. In the complex, mThTPase adopts a tunnel fold, formed by joining of the N-terminal ends of β1 and β5. The substrate assumes a “C”-shaped conformation that aligns along the groove (Fig. 4B). The phosphate groups of the substrate are anchored near the Mg2+, and the aminopyrimidine and thiazolium rings lie in a more hydrophobic site at the far end of the cleft.
Subsequent to the deposition and release of our structure of mThTPase, an
x-ray crystal structure of hThTPase, bound to a citric acid molecule and a
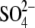 ion, became available from the
Protein Data Bank (accession number 3BHD)
(Fig. 4C).
Interestingly, the structure of hThTPase exhibits a tunnel fold that
superimposes well with our model of mThTPase in the ternary complex. The
backbone r.m.s.d. between the hThTPase structure and the lowest energy
conformer of the mThTPase·ThTP·Mg2+ complex (over the
structured region) was 2.6 Å. By contrast, the backbone r.m.s.d. between
corresponding regions of the lowest energy conformer of free mThTPase and the
hThTPase structure was 8.0 Å. Further analysis of the hThTPase structure
revealed that the citrate molecule, which has abundant negative charges,
occupies a position that is structurally equivalent to that of the
triphosphate group of ThTP in the
mThTPase·ThTP·Mg2+ternary complex. The residues of
hThTPase that make contacts with the citric acid (Glu-7, Glu-9, Lys-11,
Trp-53, Arg-55, Lys-65, His-76, Tyr-79, Glu-157, Ala-192, and Lys-193) are the
same ones involved in binding phosphates in the
mThTPase·ThTP·Mg2+ternary complex. In addition, the
ion, became available from the
Protein Data Bank (accession number 3BHD)
(Fig. 4C).
Interestingly, the structure of hThTPase exhibits a tunnel fold that
superimposes well with our model of mThTPase in the ternary complex. The
backbone r.m.s.d. between the hThTPase structure and the lowest energy
conformer of the mThTPase·ThTP·Mg2+ complex (over the
structured region) was 2.6 Å. By contrast, the backbone r.m.s.d. between
corresponding regions of the lowest energy conformer of free mThTPase and the
hThTPase structure was 8.0 Å. Further analysis of the hThTPase structure
revealed that the citrate molecule, which has abundant negative charges,
occupies a position that is structurally equivalent to that of the
triphosphate group of ThTP in the
mThTPase·ThTP·Mg2+ternary complex. The residues of
hThTPase that make contacts with the citric acid (Glu-7, Glu-9, Lys-11,
Trp-53, Arg-55, Lys-65, His-76, Tyr-79, Glu-157, Ala-192, and Lys-193) are the
same ones involved in binding phosphates in the
mThTPase·ThTP·Mg2+ternary complex. In addition, the
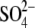 ion in the hThTPase structure,
which is positioned similarly to the α-phosphate of ThTP in the
mThTPase·ThTP·Mg2+ complex, helps to close the tunnel
through interactions with residues Arg-55, Arg-57, and Lys-65. We conclude
that the x-ray structure of the (citric acid,
ion in the hThTPase structure,
which is positioned similarly to the α-phosphate of ThTP in the
mThTPase·ThTP·Mg2+ complex, helps to close the tunnel
through interactions with residues Arg-55, Arg-57, and Lys-65. We conclude
that the x-ray structure of the (citric acid,
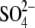 )-bound hThTPase, which adopts a
tunnel structure, is a better model of the substrate complex than of the free
enzyme. Thus it is possible that free hThT-Pase may adopt an open structure of
the type observed here for mThTPase.
)-bound hThTPase, which adopts a
tunnel structure, is a better model of the substrate complex than of the free
enzyme. Thus it is possible that free hThT-Pase may adopt an open structure of
the type observed here for mThTPase.
Comparison of the structural models for free mThTPase and its ternary complex (mThTPase·ThTP·Mg2+) shows that the individual secondary structural elements are conserved but that the complex manifests a closed tunnel fold, distinct from the open cleft assumed by free mThTPase (Fig. 4D). This change is a consequence of inward bending of strands β1, β3, β4, and β5 from the two sides of cleft and the resulting antiparallel pairing of β1 with β5. Because most residues making contact with Mg2+ and the phosphate groups of ThTP are located in these strands, it appears that electrostatic attractions involving mThTPase, Mg2+, and the substrate are the driving forces for closing the cleft. Closure of the cleft may lead to proper alignment of the active site residues for catalysis and thus supports the hypothesis that the cleft fold needs to transit toward a tunnel conformation in order for catalysis to occur (13).
Structural analysis of the ternary complex revealed a number of contacts between the enzyme and the substrate. Several basic residues, including Lys-11, Arg-55, Arg-57, Lys-65, Arg-125, and Lys-193, stabilize the three phosphate groups through electrostatic contacts (Fig. 5A). Specifically, Lys-11 interacts with the α- and β-phosphate groups; Arg-55, Arg-57, and Lys-193 surround the γ-phosphate; and Lys-65 and Arg-125 are in close proximity with all the three phosphate groups. Through interactions with distinct phosphates, these residues presumably serve to align the phosphate groups relative to Mg2+ and may facilitate product release. Also, note that each of these basic residues forms an ion pair with an acidic residue in mThTPase. Lys-11 makes ionic contacts with both Glu-157 and Glu-188; Arg-55 and Arg-57 engage in a bidentate salt bridge with Glu-63; Lys-65 makes an ionic interaction with Glu-81; and Arg-125 forms a salt bridge with Asp-147. These ion pairs, most of which are conserved in NeuTTM and other TTMs, likely are required to properly orient the basic residues for efficient catalysis. Indeed, biochemical studies on CthTTM have indicated that the disruption of any of these ion pairs reduces the Mg2+-dependent ATPase activity of this enzyme (13).
FIGURE 5.

Close-up views of substrate binding to mThTPase. A, interaction of mThTPase with Mg2+ and substrate phosphate groups. B, interaction of mThTPase with the thiamine moiety. The color scheme is the same as in Fig. 4.
The aminopyrimidine and thiazolium rings of ThTP are accommodated in a hydrophobic pocket formed by residues Tyr-79, Ala-192, Met-195, Leu-210, and Ala-213 (Fig. 5B). This region of mThTPase undergoes a major reorganization upon complex formation. Residues Ala-190, Pro-191, and Val-196 move toward residues Tyr-153 and Val-155 and constitute a new hydrophobic cluster in the complex. This cluster is stabilized by the formation of a hydrogen bond between the hydroxyl group of Tyr-153 and the backbone oxygen of Pro-191. On the other side of α7, complex formation leads to participation of the side chain of Met-195 in the hydrophobic cluster enclosed by β2, α2, β3, α7, and α8.
DISCUSSION
On the basis of sequence comparisons, ThTPase was proposed as a founding member of the CYTH domain family (9) and, more recently, as a member of the TTM superfamily (12). As the name implies, most TTM domains contain a tunnel fold, as first described for Cet1 (11) (Fig. 6A), with highly conserved active site residues pointing into the tunnel (10-13). Although free mThTPase has an open cleft, it adopts the tunnel fold in the presence of substrate. This further establishes ThTPase as a member of the TTM family. One other protein in the family has been shown to have an open cleft similar to that of free mThTPase: the hypothetical protein NE1486 from N. europaea (PDB 2FBL) solved by the Midwest Center for Structural Genomics and recently assigned to the TTM family and renamed NeuTTM (13). Superposition of the backbone Cα atoms of the structures of NeuTTM and free mThTPase yielded an r.m.s.d. of 4.2 Å over 134 amino acids. An important open question is whether the substrate-bound form of NeuTTM adopts a tunnel fold.
FIGURE 6.

Comparison of mThTPase and selected members of the TTM superfamily. A, comparison of the structure of mouse thiamine triphosphatase (mThTPase, PDB 2JMU) determined here with those of selected members of the TTM superfamily: YpAC4, the class IV adenylyl cyclase from Y. pestis (PDB 2FJT); Cet1, the RNA triphosphatase from S. cerevisiae (PDB 1D8H); PF0863, the TTM protein from P. furiosus (PDB 1YEM), and NeuTTM, a hypothetical protein from N. europaea (PDB 2FBL). B, structure-based sequence alignment of TTM family members. Identical residues are colored in yellow and highlighted in blue; similar residues are highlighted in green; completely conserved residues are colored in red and highlighted in blue.
ThTPase sequences are highly conserved across various mammalian species (19). The 74% sequence identity between human hThTPase and mThTPase (Fig. 6A) suggests that they share a conserved molecular mechanism. By contrast, the sequence identity among members of the CYTH superfamily is considerably lower (Fig. 6A). Structure-based sequence alignment of mThTPase with other TTM domains indicated that highly conserved residues are located in nearly all the β-strands (except β5) and in some α-helices (α1, α4, α5/6, α7). These structural elements constitute the backbone of the cleft/tunnel-fold architecture. Of particular note, the β1-strand contains four completely conserved residues (Glu-7, Glu-9, Lys-11, Phe-12). Other completely conserved residues in TTM proteins include residues equivalent to Lys-65 and Arg-125 of mThTPase. The sequence comparison presented here, together with NMR titration results described above and the effects of previous mutational studies, enabled us to model the active site inside the hydrophilic groove of mThTPase. The nine highly conserved charged residues (Glu-7, Glu-9, Lys-11, Arg-55, Lys-65, Arg-125, Asp-145, Glu-157, Glu-159), which have been hypothesized to bind the substrate phosphates and divalent cations (10-13), are all recapitulated in mThTPase. The postulated phosphate binding residues (Lys-11, Arg-55, Lys-65, Arg-125) are splayed apart in free mThTPase but come close and point toward the tunnel center in the ternary complex. These observations not only provide an intimate link between ThTPase and other reported TTM proteins but also suggest a unique enzyme activation mechanism for ThTPase.
The largest sequence variations among members of the TTM family are found in the secondary structural elements corresponding to β5, α2, α3, α8, lβ4,β5, and lβ6,β7 of mThTPase. These variable regions include putative active site features of mThTPase, such as the mobile loop lβ4,β5 surrounding the cleft and the hydrophobic platform formed by one face of α7 and α8 (Fig. 5C). Other differences may explain the unique structural characteristics of mThTPase. For example, all other TTM proteins have been reported to be homodimers resulting from the pairing of strands α4-α4′ plus peripheral hydrophobic contacts. In mThTPase, which is a monomer, the potential dimer interface on α4 is blocked by an extra helix (α3). In addition, another extra helix (α8), present in mThTPase but not in most tunnel TTM proteins, forms a barrier that prevents closure of the cleft in free mThTPase. In the ternary complex, α8 must be pushed outward to allow the formation of the tunnel.
Despite their highly conserved active sites, TTM domains act on a diverse array of substrates. A characteristic feature of mThTPase is its specificity for ThTP. Results from previous studies (18), as well as this work (data not shown), indicate that the affinity of ThTPase for ATP is very low (Kd > 1 mm) when compared with that for ThTP (Kd ≤ 1 μm), although the recognition of their phosphate groups appears to involve the same set of active site residues (data not shown). The structure of mTh-TPase suggests that its high substrate specificity may derive from the residues in the C-terminal helix α8 that recognize the aminopyrimidine and thiazolium moieties of ThTP.
The activity of ThTPase is critically dependent on the proper divalent cation. Although the enzyme is activated by Mg2+, Ca2+ inhibits its activity by competition with Mg2+ (18). An investigation of the role of metal ion on the phosphatase activity of Cet1 (30) revealed that the binding of Mn2+ did not trigger a major conformational rearrangement. Instead, it presumably perturbed the alignment of the active site residues that ultimately influence substrate catalysis. In this study, we observed that the addition of Mg2+ or ThTP caused distinct conformational changes in mThTPase. Binding of ThTP was found to be tight and induced an extensive conformational change. By contrast, Mg2+ binding was weak, and residues whose chemical shifts were affected by added Mg2+ mapped to a localized site. The addition of the inhibitor Ca2+ to the ThTPase·ThTP complex stabilized active site residues that exhibited ms-scale dynamics (NMR exchange broadening) in the binary complex. Taken together, these results suggest that Mg2+, rather than assisting substrate binding, is essential for the precise alignment of active site residues with the substrate phosphates for catalysis. Although Ca2+ stabilizes active site residues, it may inhibit the activity by failing to achieve the proper alignment.
The optimal pH for the enzymatic catalysis of mThTPase occurs at pH 8.0. A plot of the log specific activity versus pH (Fig. 1C) indicates that the enzyme is activated by a single deprotonation step with pKa ≈ 7.5 and becomes less active above pH 10. The lower deprotonation could correspond to a general base that removes the proton from water when it attacks the γ phosphate, or it could be a group whose deprotonation activates the enzyme by altering the charge in the active site. The lower activity at high pH could be the result of any of a number of possibilities, including destabilization of the protein structure.
The active site conservation among TTM domains implies that the family utilizes a common catalytic mechanism. The NMR titrations, along with the structural models of mThTPase and its complexes presented here, support the hypothesis that residues Glu-7, Glu-9, Arg-55, Arg-125, Asp-145, Glu-157, and Glu-159 are essential for binding divalent metals and substrate phosphates. In particular, it appears that the water bridge that mediates the coordination of divalent metals with active site residues (e.g. Asp-145 in mThTPase) is also highly conserved (10-13). Given the conservation of the water bridge and the catalytic role of the water molecule in ATPase catalysis (31), it is intriguingly possible that a water bridge may serve as a nucleophilic agent to attack the substrates of TTM domains. Further mutational studies and biochemical characterization will be required to test this hypothesis.
Supplementary Material
Acknowledgments
We thank the many team members from the Center for Eukaryotic Structural Genomics who provided the infrastructure for this work and Dr. W. W. Cleland for helpful comments.
The atomic coordinates and structure factors (code 2JMU) have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ (http://www.rcsb.org/).
This work was supported by grants from the National Institutes of Health, Protein Structure Initiative through Grants P50 GM64598 and U54 GM074901 (to J. L. M.) and the Fonds de la Recherche Scientifique FNRS (F.R.S.-FNRS) through Grant 2.4.558.04.F (to L. B.). NMR data were collected at the National Magnetic Resonance Facility at Madison, which is supported in part by National Institutes of Health Grants P41 RR02301 and P41 GM66326 (to J. L. M.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1-S2.
Footnotes
The abbreviations used are: ThTP, thiamine triphosphate; ThTPase, thiamine triphosphatase; bThTPase, bovine thiamine triphosphatase; hThTPase, human thiamine triphosphatase; mThTPase, mouse thiamine triphosphatase; pThTPase, pig thiamine triphosphatase; rThTPase, rat thiamine triphosphatase; TTM, triphosphate tunnel metalloenzyme; NeuTTM, the TTM protein from N. europaea with known structure (PDB 2FBL); PDB, Protein Data Bank; HSQC, heteronuclear single quantum correlation; NOE, nuclear Overhauser effect; NOESY, NOE spectroscopy; MOPS, 3-(N-morpholino)-propanesulfonic acid; r.m.s.d., root-mean-square-deviation; TAPS; N-Tris(hydroxymethyl)methyl-3-aminopropanesulfonic acid.
References
- 1.Hashitani, Y., and Cooper, J. R. (1972) J. Biol. Chem. 247 2117-2119 [PubMed] [Google Scholar]
- 2.Penttinen, H. K., and Uotila, L. (1981) Med. Biol. 59 177-184 [PubMed] [Google Scholar]
- 3.Makarchikov, A. F., and Chernikevich, I. P. (1992) Biochim. Biophys. Acta 1117 326-332 [DOI] [PubMed] [Google Scholar]
- 4.Lakaye, B., Makarchikov, A. F., Antunes, A. F., Zorzi, W., Coumans, B., De Pauw, E., Wins, P., Grisar, T., and Bettendorff, L. (2002) J. Biol. Chem. 277 13771-13777 [DOI] [PubMed] [Google Scholar]
- 5.Iwata, H., Yabushita, Y., Doi, T., and Matsuda, T. (1985) Neurochem. Res. 10 779-787 [DOI] [PubMed] [Google Scholar]
- 6.Nghiem, H. O., Bettendorff, L., and Changeux, J. P. (2000) FASEB J. 14 543-554 [DOI] [PubMed] [Google Scholar]
- 7.Lakaye, B., Wirtzfeld, B., Wins, P., Grisar, T., and Bettendorff, L. (2004) J. Biol. Chem. 279 17142-17147 [DOI] [PubMed] [Google Scholar]
- 8.Bettendorff, L., Peeters, M., Wins, P., and Schoffeniels, E. (1993) J. Neurochem. 60 423-434 [DOI] [PubMed] [Google Scholar]
- 9.Iyer, L. M., and Aravind, L. (2002) BMC Genomics 3 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gallagher, D. T., Smith, N. N., Kim, S. K., Heroux, A., Robinson, H., and Reddy, P. T. (2006) J. Mol. Biol. 362 114-122 [DOI] [PubMed] [Google Scholar]
- 11.Lima, C. D., Wang, L. K., and Shuman, S. (1999) Cell 99 533-543 [DOI] [PubMed] [Google Scholar]
- 12.Gong, C., Smith, P., and Shuman, S. (2006) RNA (Cold Spring Harbor) 12 1468-1474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Keppetipola, N., Jain, R., and Shuman, S. (2007) J. Biol. Chem. 282 11941-11949 [DOI] [PubMed] [Google Scholar]
- 14.Zolnai, Z., Lee, P. T., Li, J., Chapman, M. R., Newman, C. S., Phillips, G. N., Jr., Rayment, I., Ulrich, E. L., Volkman, B. F., and Markley, J. L. (2003) J. Struct. Funct. Genomics 4 11-23 [DOI] [PubMed] [Google Scholar]
- 15.Thao, S., Zhao, Q., Kimball, T., Steffen, E., Blommel, P. G., Riters, M., Newman, C. S., Fox, B. G., and Wrobel, R. L. (2004) J. Struct. Funct. Genomics 5 267-276 [DOI] [PubMed] [Google Scholar]
- 16.Sreenath, H. K., Bingman, C. A., Buchan, B. W., Seder, K. D., Burns, B. T., Geetha, H. V., Jeon, W. B., Vojtik, F. C., Aceti, D. J., Frederick, R. O., Phillips, G. N., Jr., and Fox, B. G. (2005) Protein Expression Purif. 40 256-267 [DOI] [PubMed] [Google Scholar]
- 17.Jeon, W. B., Aceti, D. J., Bingman, C. A., Vojtik, F. C., Olson, A. C., Ellefson, J. M., McCombs, J. E., Sreenath, H. K., Blommel, P. G., Seder, K. D., Burns, B. T., Geetha, H. V., Harms, A. C., Sabat, G., Sussman, M. R., Fox, B. G., and Phillips, G. N., Jr. (2005) J. Struct. Funct. Genomics 6 143-147 [DOI] [PubMed] [Google Scholar]
- 18.Lakaye, B., Makarchikov, A. F., Wins, P., Margineanu, I., Roland, S., Lins, L., Aichour, R., Lebeau, L., El Moualij, B., Zorzi, W., Coumans, B., Grisar, T., and Bettendorff, L. (2004) Int. J. Biochem. Cell Biol. 36 1348-1364 [DOI] [PubMed] [Google Scholar]
- 19.Szyniarowski, P., Lakaye, B., Czerniecki, J., Makarchikov, A. F., Wins, P., Margineanu, I., Coumans, B., Grisar, T., and Bettendorff, L. (2005) Biochim. Biophys. Acta 1725 93-102 [DOI] [PubMed] [Google Scholar]
- 20.Bettendorff, L., Nghiem, H. O., Wins, P., and Lakaye, B. (2003) Anal. Biochem. 322 190-197 [DOI] [PubMed] [Google Scholar]
- 21.Lanzetta, P. A., Alvarez, L. J., Reinach, P. S., and Candia, O. A. (1979) Anal. Biochem. 100 95-97 [DOI] [PubMed] [Google Scholar]
- 22.Zwahlen, C., Legault, P., Vincent, S. J. F., Greenblatt, J., Konrat, R., and Kay, L. E. (1997) J. Am. Chem. Soc. 119 6711-6721 [Google Scholar]
- 23.Delaglio, F., Grzesiek, S., Vuister, G. W., Zhu, G., Pfeifer, J., and Bax, A. (1995) J. Biomol. NMR 6 277-293 [DOI] [PubMed] [Google Scholar]
- 24.Cornilescu, G., Delaglio, F., and Bax, A. (1999) J. Biomol. NMR 13 289-302 [DOI] [PubMed] [Google Scholar]
- 25.Güntert, P., Mumenthaler, C., and Wüthrich, K. (1997) J. Mol. Biol. 273 283-298 [DOI] [PubMed] [Google Scholar]
- 26.Herrmann, T., Güntert, P., and Wüthrich, K. (2002) J. Mol. Biol. 319 209-227 [DOI] [PubMed] [Google Scholar]
- 27.Schwieters, C. D., Kuszewski, J. J., Tjandra, N., and Marius, C. G. (2003) J. Magn. Reson. 160 65-73 [DOI] [PubMed] [Google Scholar]
- 28.Laskowski, R. A., Rullmann, J. A. C., MacArthur, M. W., Kaptein, R., and Thornton, J. M. (1996) J. Biomol. NMR 8 477-486 [DOI] [PubMed] [Google Scholar]
- 29.Brünger, A. T., Adams, P. D., Clore, G. M., Delano, W. L., Gros, P., Grossekunstleve, R. W., Jiang, J. S., Kuszewski, J., Nilges, M., Pannu, N. S., Read, R. J., Rice, L. M., Simonson, T., and Warren, G. L. (1998) Acta Crystallogr. Sect. D Biol. Crystallogr. 54 (Part 5), 905-921 [DOI] [PubMed] [Google Scholar]
- 30.Bisaillon, M., and Bougie, I. (2003) J. Biol. Chem. 278 33963-33971 [DOI] [PubMed] [Google Scholar]
- 31.Sousa, M. C., and McKay, D. B. (1998) Biochemistry 37 15392-15399 [DOI] [PubMed] [Google Scholar]
- 32.Barchi, R. L., and Viale, R. O. (1976) J. Biol. Chem. 251 193-197 [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.