

Introduction
Both aerobic and anaerobic metabolism are accompanied by the production of reactive oxygen and/or nitrogen species, and organisms ranging from prokaryotes to mammals have evolved an elaborate and redundant complement of defenses to confer protection against oxidative and nitrosative insults. Compelling data also indicates that oxidants are produced and employed in physiological settings as signaling molecules in control of cell and tissue homeostasis, cell division, migration, contraction, and mediator production [1–4]. Collectively, these signaling oxidants--which include nitric oxide (NO), S-nitrosothiols (SNO, in particular S-nitrosoglutathione; GSNO) and hydrogen peroxide (H2O2)-- are produced mainly by NADPH-dependent enzymes (NO synthases and NADPH oxidases) whose expression is tightly controlled, compartmentalized and tissue-specific. Signaling by these reactive molecules is mainly carried out by targeted modifications of cysteine residues in proteins, including S-nitrosylation and S-oxidations. This manuscript reviews recent findings--made possible by methodological and technical advances that have allowed detection of oxidation events in proteins--and illuminates their significance. We also attempt to highlight some of the current pitfalls, and approaches needed to advance this important area of biochemical and biomedical research.
Oxidants as modulators of signal transduction
The specificity and selectivity of oxidant molecules is dictated by their chemical reactivity. Signal transduction cascades are rapidly and reversibly activated by specific agonists and stimuli, that transmit precisely regulated, compartmentalized signals, to produce a distinct molecular or biochemical outcome. Oxidants employed in signal transduction in physiological settings must exhibit substrate specificity and produce reversible oxidations. Highly reactive oxidant species such as ozone, hypochlorous acid, peroxynitrite, nitrogen dioxide and the hydroxyl radical, oxidize biomolecules without preference or specificity. Most oxidative modifications caused by these highly reactive oxidants, such as the 3-nitrotyrosine and protein carbonyls are also not easily reduced, and are thus unlikely to represent signaling events. Protein modifications by the aforementioned species may lead to the aberrent activation of signal transduction cascades, often resulting in pathophysiology. By contrast, physiological oxidants –principally NO/SNO, and O2.−/H2O2, have been implicated in reversible Cys-based modifications that underlie homeostatic control and diverse biological responses.
The role of NO in smooth muscle biology through its regulatory role in the activation of guanylate cyclase via binding to heme Iron and resultant production of cGMP is well established. However, even in vascular smooth muscle, a significant component of the relaxation response may be cGMP independent. In some vessels and species, the response may even be completely independent of cGMP [5–8], and it has been shown that animals deficient in a SNO-metabolizing enzyme have low vascular resistance ([9], JSS unpublished). By analogy, stringent genetic evidence for a role of NO (per se) in blood pressure control has not been provided. SNOs can signal via cGMP or S-nitrosylation. Taken together, these observations suggest that SNOs play a physiological role in regulation of vessel tone.
A cardinal role for S-nitrosylation in signaling that is independent of cGMP was pointed to early on [10, 11]. S-nitrosylation of specific cysteine residues (which will be discussed further below) has been detected in well over 100 proteins of all classes and is arguably the principal mechanism by which NO signals [1]. Much like phosphorylation by kinases, S-nitrosylation by NOSs influences protein activity, protein-protein interactions and protein location. S-nitrosylation thus serves as the prototypic redox-based signal. It is important to note that NO signalling by guanylate cyclase and S-nitrosylation are not necessarily mutually exclusive [12, 13]. S-nitrosylation has been implicated in transmitting signals downstream of all classes of receptors, including GPCRs, RTKs, TNF, Toll, glutaminergic and other [1, 14–16]--acting locally within subcellular signaling domains as well conveying signals from the cell surface to intracellular compartments, including the mitrochondria and the nucleus (e.g. by modification of transcription factors).
S-nitrosylation exhibits a number of essential features that are prototypic of biological signals and may be useful when considering the possibility that additional reactive oxidants subserve signaling roles: I) temporal regulation on physiological timescales (S-nitrosylation is stimulus-coupled and rapid), II) the existence of motifs within proteins that facilitate S-nitrosylation and/or provide specificity [1, 17], III) co-localization of target proteins with a source of NO (e.g. NOS enzyme) [18], IV) reversibility; and V) enzymatic control (e.g. S-nitrosoglutathione; SNO) reductase ([1, 19]) controls S-nitrosothiol tone [9, 20]). In addition, dysregulation of protein S-nitrosylation is associated with a growing list of diseases, including cystic fibrosis, asthma, heart disease, and neurodegenerative diseases. Therefore, understanding of the regulatory events that control S-nitrosothiol biology has the potential to lead to novel therapeutic opportunities [1].
An accumulating literature is making the case for signaling by H2O2. Stimulation of growth factor receptors, toll like receptors (TLR), and cytokine receptors, also leads to the activation of signaling cascades that are regulated by hydrogen peroxide (H2O2) as a result of activation of non-phagocytic oxidases (NOX), or dual oxidase (DUOX) (See Table 1) [18, 19, 21–24]. The expression of multiple isoforms of these enzymes in a variety of tissues provides evidence that the deliberate production of low levels of oxidants is a feature of many cells [25]. H2O2, a small and non-charged molecule that is readily produced and removed following these physiological stimuli, exhibits properties that are ideal for signal transduction [2]. Although it was initially believed that H2O2 can freely diffuse across membranes, recent genetic evidence suggests that some membranes are poorly permeable to H2O2, and that its transport may be regulated by aquaporin channel proteins [26, 27], which also regulates transport of NO [28]. H2O2 exerts its functions in signaling by causing reversible amino-acid oxidations, as will be discussed below.
Table 1.
Enzymes controlling reversible cysteine oxidations
| Family | Target oxidation | Designation | Reference |
|---|---|---|---|
| Thioredoxin | Disulfides | SS | [83] |
| Glutaredoxin | S-glutathionylated cysteines | PSSG | [47] |
| Bacterial glutaredoxin*1 | Disulfides | SS | [83, 99] |
| Protein disulfide isomerase | Disulfides | SS | [156] |
| Sulfiredoxin | Peroxiredoxin sulfinic acids | Prx-SO2H | [65] |
| GSNO reductase | S-nitrosothiols*2 | SNO | [20] |
In addition to the monothiol reduction of glutathionyl disulfides, bacterial Grx also reduces low molecular weight disulfides through a dithiol mechanism [83, 99]. However mammalian GRX displays marked substrate specificity towards S-glutathionylated proteins [100, 101]. Thus, the major target for Grx in mammalian cells in physiological setting is S-glutathione mixed disulfide (PSSG).
GSNO reductase decomposes low molecular weight S-nitrosothiols, thereby indirectly affecting S-nitrosylated protein content.
Oxidizable amino acids: Reactive cysteines as redox targets
Amino acids that are targets for reversible oxidation include cysteines with a low pKa sulhydryl group (4–5), which causes unique susceptibility to oxidation compared to the typical pKa of non-reactive cysteines (8.5) [29]. In addition, methionine, tryptophan, and tyrosine residues are also prone to oxidative modification, although the functional impact of those events in physiological signal transduction remains to be established. Regulation of cell signaling via the cysteine group is best established [30, 31], and will therefore be the focus of further discussion. Numerous classes of proteins contain free cysteine residues that are highly conserved across species, suggesting regulatory possibilities, beyond structural roles and metal ion coordination. In addition, cysteine residues can be modified through alternative redox-based modifications (SNO, SOH, SSG, S-S) that may enable differential effects on protein function [32–35]. In other words, distinct reversible modifications of cysteines (Figure 1) may lead to unique functional outcomes (Figure 2) as well as provide a mechanism through which differential responsitivity can be achieved. For example, sulfenic acid modification (SOH) (hydroxylation) of a single allosteric cysteine has been shown to elicit effects on structure and function that are distinct from those elicited by mixed disulfide (glutathionylation)[32]. Cysteines are thus believed to serve as molecular switches, capable of processing different redox-based signals into distinct functional responses [36–38].
Figure 1.
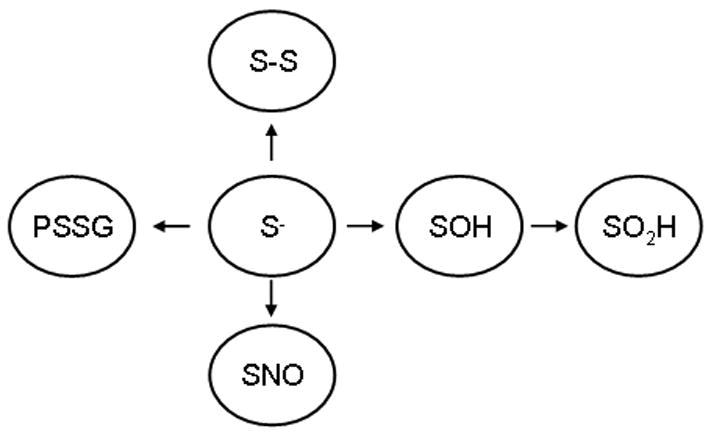
Schematic overview of various reversible modifications of reactive cysteines (S−). S-S; disulfide, PSSG; glutathionylation, SNO; nitrosylation, SOH; sulfenic acid, SO2H; sulfinic acid. Note that the arrows are strictly included for an illustrative purpose, and do not indicate directionality.
Figure 2.
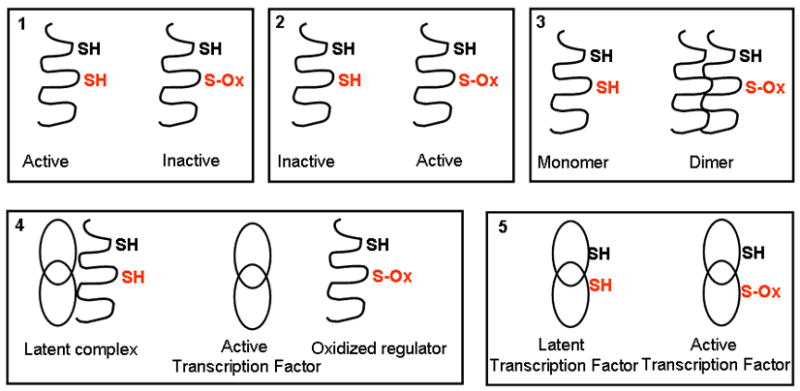
Schematic representation of mechanisms of redox regulation.
Well described is the scenario where cysteine oxidation inactivates a signaling protein, such as a protein tyrosine phospatase (box 1) or caspases. Additionally, a protein with a signaling function can also be directly activated by oxidation of a critical cysteine residue (box 2). Examples of this scenario are Ras, ryanodine receptor, dynamin, SERCA, and Src tyrosine kinase ([50], for review). Another mechanism whereby cysteine oxidation elicits a signal is through control of multimerization (box 3), such as the dimerization of various heat shock or chaperone proteins or self-assembly of dynamin. Box 4 visualizes the situation wherein cysteine oxidation of a regulatory protein causes its dissociation of its partner, thereby activating the function of the partner. An example of this represents the dissociation of oxidized, thioredoxin from Ask1, leading to Ask1 activation, the dissociation from oxidized Keap-1 from NRF-2, leading the activation of NRF-2 as a transcription factor, or the dissociation of nNOS from glucokinase, leading to activation of the kinase [243]. Direct oxidation of transcription factors, may also be sufficient to mediate their activation or inhibiton through altered interactions with DNA (box 5). Examples include the activation of prokaryotic OxyR, which shifts from a “dimeric” to “tetrameric” interaction with DNA upon oxidation, and NF-κB (p50, p65) or cJun whose binding with DNA is inhibited by S-oxidation.
Recent studies have shown that the sulfinic acid form of 2-cys peroxiredoxins can be reduced back to the sulfhydryl in an ATP-dependent enzyme-(sulfiredoxin, Srx) catalyzed reaction [39], and a reversible thioesterifcation has been reported to alter transcriptional activity [40], raising the possibility that additional Cys modifications may be found to have regulatory roles. Cysteines also are uniquely targeted by alkylating species (such as cyclopentenone prostaglandins, acrolein), hydroxynonenal and avicins (which can be formed endogenously, or encountered through environmental insults [41, 42]), and by acylation (which has important roles in localizing proteins to the membranes [43]). While a protein can have many cysteine residues, few are reactive (typically those in the thiolate, S− form). The pKa of a cysteine is dependent on the charge of adjacent amino acids within the three dimensional conformation or quaternary state. Furthermore, the susceptibility to oxidative modification may be governed by the reactivity of individual oxidants, which can be influenced by structural features of the protein (adjacent amino acids, or by coordination of metal ions). As an example, in hydrophobic regions, the reaction between NO and oxygen is accelerated, leading to enhanced formation of S-nitrosylating species [44]. Protein thiols found in consensus motifs wherein the Cys is adjacent to basic and acid residues or aromatic residues often serve as sites of nitrosylation [1]. In addition, transition metals may catalyze S-nitrosylation [1, 31].
S-nitrosylation may be mediated by NO, SNO, or several higher oxides [45]. Forman and colleagues [31] and Tannenbaum [46] emphasize that S-nitrosylation may involve nitrosative chemistry (nitrosation refers to chemical addition of NO+), but is not restricted to reactions of NO+. The term nitrosyl, which is mechanism neutral, aptly describes attachment of the NO moiety to a nucleophilic group or transition metal. S-nitrosylation is therefore preferred terminology in biological systems where chemical mechanism is rarely known. Needless to say, “-ylation” [31] is standard biochemical nomenclature for posttranslational modifications of proteins. Notably, searches of Pubmed with “S-nitrosation” as the key term yielded a small number of hits, whereas almost 100 papers on “S-nitrosylation”--covering major advances in the field—have been published this year alone. Few redox-based signaling functions or fundamental advances were identified using chemical nomenclature for NO+/nitrosation. Given these considerations, we refer to the NO-associated posttranslational modification of cysteine thiol as S-nitrosylation.
Like NO and SNO, H2O2 also oxidizes reactive cysteines, leading to the formation of sulfenic acids (SOH) which can be further oxidized to sulfinic (SO2H) and sulfonic acids (SO3H) [47–49]. Unlike NO/SNO, H2O2 cannot readily access hydrophobic compartments. Sulfenic acids also are byproducts of S-nitrosylation and targets of S-glutathionylation (PSSG). In particular, S-glutathionylation, the formation of a disulfide between the cysteine of glutathione and a cysteine moiety of a protein (also known as a protein mixed disulfide, or PSSG), is believed to be a protective mechanism that prevents further oxidation to sulfinic and sulfonic acids, which are not readily regenerated [47]. PSSG formation is an inevitable consequence of denitrosylation by GSNO reductase, but constitutes a minor pathway.
Relative importance of NO- or H2O2-dependent cysteine oxidations in redox signaling
The relative importance of NO/SNO- or H2O2-dependent oxidative modifications in cellular physiology vs. pathophysiology and the interplay of these regulatory oxidations is often denated, but prematurely so. Many years of work have established firmly the role of oxidants in pathophysiology and NO’s broad-based signaling function. These are the cornerstones of the field. Moreover, NO biology has enjoyed the use of highly specific inhibitors of enzymes that have uncovered its many physiological roles, whereas specific inhibitors of NADPH oxidases are not yet available. Thusfar, the signaling function of H2O2 –physiological vs. stress/adaptive --has been far more difficult to establish, important advances notwithstanding. One plausible reason for this lag may relate to the lack of commonly available technology to detect H2O2–based reversible cysteine oxidations methodology with sufficient specificity. The instability, or rapid reversal of these redox changes forms an argument for a signaling role, but of course poses practical problems in detecting them. Therefore, it is much easier to list physiological events regulated by NOSs than by NAPDH oxidases at this time, although evidence to support a role for multiple members of the NOX/Duox family in migration, cell differentiation, and mitogenesis is becoming increasingly apparent [25]. At the present time however, oxidative stress is more often linked to disease than nitrosative stress. The extent to which roles may be reversed is an area of active study. In this regard, both the NO and H2O2 systems rely on the principle of Cys-based posttranslational modifications. It is therefore of some surprise that while many reviews on the topic of redox signaling have been published [1, 3, 31, 50, 51] most highlight either H2O2 or NO selectively; co-existence of both oxidants and their respective chemical reactivities within the same cell is rarely considered.
A general classification scheme has been constructed for cysteine oxidations (including those induced by both NO and H2O2) which highlights that S-nitrosylation may be important in signaling functions relevant to cellular physiology and homeostasis, while the formation of sulfenic acids, and S-glutathionylation has been linked to cellular stress and adaptation [1, 52]. Indeed, a number of targets such as caspases, receptors (e.g. alpha-amino-3-hydroxy-5-methylisoxasole-4-propionic acid (AMPA) and NMDA), kinases, G proteins and ion channels have been demonstrated to be S-nitrosylated under physiological conditions [15, 53, 54]. In diverse cell types, S-nitrosylation can be detected in situ under both non-stimulated conditions and following physiological activation (e.g stimulation of GPCRs to dilate blood vessels or stimulate cardiac contractility [55, 56]). These findings suggest a role for S-nitrosylation in homeostatic control. Whether H2O2-based oxidative modifications (sulfenic acids and glutathionylation) will be afforded the same status is unclear, although high concentrations of GSH would seem to make it a likely substrate [47]. It remains to fully unraveled, for example, whether incorporation of GSH is an activating mechanism or part of a deactivation cycle (a route to reduce the protein) or both.
It seems likely that in physiological settings different cysteines will be targeted by different oxidative effectors; aforementioned regulation via compartmentalization of either NOX or NOS enzymes and various structural features (hydrophobicity, accessibility of GSH etc), will dictate which oxidative events prevail. Furthermore, as will be described below, regulatory enzymes important in redox control, such as thioredoxin, glutaredoxin, peroxiredoxin, and protein disulfide isomerase, are themselves affected and regulated by both H2O2 and NO-based cysteine oxidations. It is of interest to note that S-nitrosylation may be criticaly sensitive to oxygen tension, often being potentiated by hypoxia [15, 54, 57]. Further, it has been shown that oxidation/reduction (of a group) of thiols may regulate S-nitrosylation by an allosteric mechanism [57]. In aggregate, these observations highlight a need for additional studies to formally test the interplay between H2O2 and NO-dependent cysteine oxidations.
Control of S-nitrosothiol and H2O2 tone
Control of S-nitrosothiol tone
S-nitrosylation, as mentioned above, has emerged as a ubiquitous posttranslational modification, playing an essential role in NO biology. The evidence that S-nitrosylation serves as a physiological mode of signal transduction stems from a number of key observations. First and foremost, S-nitrosylation of proteins is demonstrably coupled to diverse physiological stimuli. Secondly, S-nitrosylation meets the requirements of a regulatory posttranslational modification, including specificity (it tends to target one or few thiols often found within consensus motifs), reversibility and and enzymatic control. [1, 58]. Numerous reaction channels may support S-nitrosylation chemistry in vivo, including the reaction of NO with thiols or thiyl radicals, dimerization of NO (on aromatic residues) and oxidation of NO to a molecule with NO+ character (followed by a reaction with thiols). Oxidation of NO may be catalyzed by transition metals, oxygen or NO itself; NO and O2 concentrate in hydrophobic compartments, which enables micellar catalysis and protects the SNO from solvent. Acidic conditions, which occur in microdomains of proteins, lysosomes or the mitochondrial inner space may also promote SNO formation from NO or nitrite (as reviewed in [1, 19]). Enzymes other than NOS have been shown to promote S-nitrosylation, employing some of the above-described chemistry, and include ceruloplasmin, membrane-associated mammalian hemoglobin and Ascaris hemoglobin. GSNO reductase, (discussed in detail in the next paragraph), which denitrosylates proteins in equilibrium with GSNO, is a major determinant of steady state levels of S-nitrosylated proteins. S-nitrosylation can also be reversed by specific metal ions, such as Cu+, the low MW antioxidant, ascorbate (see below) and thioredoxin (Trx) [59, 60]; the physiological relevance of the latter is still unclear.
Control of H2O2 tone
Peroxiredoxins (Prx) have emerged as critical regulatory systems that quench H2O2, and act along catalase and glutathione peroxidase. At least six mammalian Prx have been identified, subdivided into 2-Cys, atypical 2-Cys, and 1-Cys Prx, which exist as homodimers in different subcellular compartments ([50] for review). Prx are highly abundant and are believed to control the H2O2 tone. Their over-expression lowers intracellular levels of H2O2, and leads to inhibition of signaling induced by PDGF, TNFα, or ceramide, suggesting that Prx enzymes contribute to signaling by removing H2O2 [3, 50].
The catalytic mechanism of 2-Cys Prxs is relatively well understood. In the fully reduced state, the peroxidatic cysteine of one subunit of a head-to-tail homodimer is attacked by H2O2 to generate sulfenic acid (− SOH), and a conformational change facilitates attack of the resolving cysteine, resulting in an intersubunit disulfide bond. The disulfide reductase, thioredoxin (Trx), then reduces the intersubunit disulfide bond, regenerating reduced dimer. In eukaryotic Prxs, a C-terminal domain stabilizes the –SP-OH moiety, thereby promoting further oxidation of the peroxidatic cysteine to sulfinic acid (-SP-O2H). This process, which is commonly referred to as “over-oxidation” or “hyper-oxidation”, inactivates the normal catalytic cycle because disulfide bond formation with the resolving cysteine is abolished. Over-oxidation of Prx is believed to occur when H2O2 concentrations exceed the capacity of Prx regeneration by the Trx system, and originally was proposed to act as a “floodgate” to promote H2O2 signaling [61]. While a small portion of PrxI and II have been reported to be hyper-oxidized in response to physiological stimuli, PrxII dampens signaling through interactions with the PDGF receptor in the absence of hyper-oxidation [62].
Although sulfinic acid residues (SO2H) were originally thought to represent irreversible cysteine oxidations, Sulfiredoxins (Srx) have been identified as cysteine sulfinyl reductases that can repair the SO2H group of peroxiredoxin, regenerating active enzyme. Repair of the sulfinyl group supports the concept that the Prx inactivation-reactivation cycle represents a cell signaling loop [50, 63–66] (Table 1). This notion is supported by recent studies that show that cell cycle arrest is associated with recruitment of hyper-oxidized PrxII to high molecular weight oligomers, and that recovery is correlated with regeneration of reduced enzyme [67]. It is of significance to note that formation of SO2H forms are not limited to Prx, and that reactive cysteines in many other proteins can also be targets to “over-oxidation,” which may contribute to the regulation of their function.
The molecular regulation of Prx represents an intriguing area of investigation. Originally these enzymes were thought to be relatively weak scavengers of H2O2, but recent work shows that are as active as peroxidases as either catalase or glutathione peroxidase. What distinguishes peroxiredoxins from other peroxidases is their interaction with numerous regulatory proteins, including c-Abl, c-Myc, JNK, and the PDGF receptor [62, 68–71]. Little is known about how the oxidation state of Prxs influences protein-protein interactions, but one intriguing possibility is that oxidation state of peroxiredoxins may influence the subcellular location or activity of interacting factors. Linking protein-protein interactions to oxidation state, particularly for those proteins that interact with the C-terminal domains that promote hyper-oxidation, would permit peroxiredoxins to function as peroxide dosimeters in a wide variety of redox-dependent signaling events.
Peroxiredoxins have also have been shown to stack in complex 3-dimensional structures [72]. Prx-1 appears to have a function as a molecular chaperone, which is controlled by its phosphorylation state [73]. Phosphorylation of PrxI and II by cdc2/cyclin B complexes regulates oligomeric transitions and peroxidase activity, providing yet another mechanism to control the activity and structural state of these enzymes during the cell cycle. Changes in Prx mRNA and protein expression levels occur in many disease states, including cancer [74–77], suggesting that control of Prx function is regulated at both transcriptional and post-translational levels. While these observations show that the biological functions of individual Prxs exceed the mere reduction of H2O2, additional investigation is needed to define the array of protein complexes that include Prxs in mammalian cells, and to understand how changes in oxidation state and structural transitions of Prxs contribute to signal transduction in specific regulatory networks.
Biochemical and genetic regulation of reversible cysteine oxidations
Reversible cysteine oxidations potentially relevant to signal transduction include disulfides (S-S), sulfenic acid (SOH), glutathionylated cysteines (PSSG), nitrosylated cysteines (SNO) (Figure 1, Table 1). It is important to recognize that steady state levels of these oxidized moieties appear to be controlled directly or indirectly by a number of enzyme systems that control unique redox modules within cells (Table 1). It is notable that enzymes specifically metabolize superoxide (O2−), H2O2, and GSNO. No mammalian enzyme has been shown to metabolize NO.
GSNO reductase, also known as glutathione-dependent formaldehyde dehydrogenase, breaks down S-nitrosoglutathione. GSNO is converted to GSNHOH, ultimately forming ammonia (NH3) and glutathione disulfide (GSSG) [78]. GSNO-reductase is highly specific for GSNO and thus regulates the steady state levels of S-nitrosylated proteins that are in equilibrium with GSH [78, 79]. Mice deficient in GSNOR have low vascular and airway resistance, and increased cardiac ouput. Intriguingly, the activity of GSNO reductase had been demonstrated to by upregulated in a mouse model of asthma, mediating decreases in lung S-nitrosothiol levels, and increases in airway hyperresponsiveness (AHR), a cardinal feature of asthma. GSNO reductase knock-out mice had elevated levels of lung S-nitrosothiols, which act as endogenous bronchodilators and protected against increases in AHR or tachyphylaxis [20]. GSNO reductase knock-out mice were also hypotensive under anesthesia and showed enhanced susceptibility to mortality from endotoxin or septic shock [9]. GSNO reductase activity also has been linked to β-adrenergic receptor signaling via its regulation of S-nitrosylation state of G-protein coupled receptor kinase-2 [80], a central mediator of GPCR densensitization. Intriguingly, polymorphisms in the GSNO reductase gene were recently identified and demonstrated correlations with asthma susceptibility [81]. Of great interest, the knockout mice also exhibit insulin resistance [82] and show a unique signature of S-nitrosylated proteins: GSK-3-β, AKT and IRS are excessively S-nitrosylated well explaining the phenotype. In aggregate these findings illuminate the functional significance of the GSNO reductase system in general and protein S-nitrosylation by GSNO in particular, and they establish dispositively the role of S-nitrosothiols in the control of cardiac, metabolic, vascular, and respiratory function.
The disulfide reductases, thioredoxin (Trx) and glutaredoxin (Grx) play a major regulatory role in controlling redox homeostasis (Table 1). Both Trx and Grx contain a Trx fold and a common dithiol/disulfide active site motif (Cys-X-X-Cys, [83], for review). Trx catalyze the reversible reduction of disulfides, resulting in a reduced pool of dithiol target protein, and a disulfide in the active form of Trx which is then reduced by thioredoxin reductase, using electrons from NADPH. Two mammalian Trx systems exist: the cytosolic Trx1/TrxR1, and mitochondrial Trx2/TrxR2. A number of proteins have been identified to be redox regulated by Trx and include Nuclear Factor kappa B, (NF-κB), Activator Protein-1 (AP-1), p53, glucocorticoid and estrogen receptors, and HIF1α [84–89]. Trx1 is ubiquitously expressed and is upregulated under inflammatory conditions [83]. Intriguingly, following certain stimuli, Trx1 can also be translocated to the nucleus [90], or to the extracellular compartment following leaderless export [91]. Circulating levels of Trx were reported to be increased in patients with heart disease, rheumatoid arthritis, acute lung injury, or HIV infection, and showed various associations with severity of disease [92–95]. Extracellular Trx can act as a unique chemoattractant for neutrophils, monocytes, and T cells [96]. A 10-kDa C-terminally truncated variant of Trx (termed Trx80) is also secreted and present in plasma, and acts as a potent mitogenic cytokine that stimulates growth of resting human peripheral blood mononuclear cells [95], and differentiation of human CD14(+) monocytes into a novel phenotype in association with activation of mitogen activated protein kinases [97]. The use of a kinetic trapping technique to identify targets that bind extracellular Trx1, has unraveled a highly specific redox-based interaction between Trx1 and tumor necrosis factor receptor superfamily member 8 (also known as CD30), and that this interaction is important in the regulation of lymphocyte effector function [98].
Mammalian cells also contain glutaredoxins (Grx, also known as thiol transferases), which under physiological conditions act to reverse S-glutathionylated cysteines, restoring the protein cysteine to the sulfhydryl group ([47, 83, 99], for reviews). The Grx system consists of Grx enzymes, GSH, glutathione reductase, and NADPH. Grx carry out de-glutationylation reactions, also known as the reduction of protein-mixed disulfides (PSSG, glutathionylated cysteines) which requires the N-terminally located active site cysteine in Grx. The reduction of PSSG by Grx results in a mixed disulfide between GSH and the N-terminal active site cysteine (glutathionylation of Grx) which is then reduced by a second GSH molecule. Glutathione disulfide (GSSG) is finally regenerated by glutathione reductase at the expense of NADPH [47, 83, 99]. Although the major enzymatic action of mammalian Grx, under physiological conditions has been linked to de-glutathionylation of protein targets [47, 100], it should be highlighted that under conditions of oxidative stress or damage, where GSSG levels are increased, Grx causes increases in protein S-glutathionylation, instead of decreases [83]. In addition to the monothiol reduction of glutathionyl disulfides, bacterial Grx also reduce low molecular weight disulfides, or disulfides in ribonucleotide reductase through a dithiol mechanism [83, 99]. However for mammalian Grx, substrate specificity towards S-glutathionylated proteins has been demonstrated [101]. It is of interest to note that sulfiredoxin also was recently demonstrated to de-glutathionylate protein targets such as actin and protein tyrosine phosphatase 1B [102]. Multiple Grx genes are known in plants and in pro- and eukaryotes; in mammalian cells, Grx1 has been described and localized in the cytoplasm. In addition, Grx2 which has 1.5–3 fold higher catalytic efficiency than Grx1, is localized in mitochondria [47]. Steady state levels of Grx itself appear to be dynamically regulated, and increases in Grx occur in cancer and cardiovascular disease [83, 103]. Elevated levels of S-glutathionylated proteins in serum have been suggested to represent a risk-marker for arteriosclerosis obliterans [104]. Recent studies from our laboratory (Y J-H) have demonstrated that multiple stimuli, such as the TLR ligands, lipopolysaccharide (LPS) and CpG DNA, interleukin-4, and transforming growth factor-beta 1 (TGF-β1) result in decreases in Grx activity in primary lung epithelial cells, in association with decreases in Grx1 mRNA, in response to TGF-β1 [105]. In contrast, increases in Grx activity occurred in cells exposed to Interferon-gamma (IFN-γ), in association with increases in Grx1 mRNA, and increases in Grx activity and Grx1 mRNA were observed in mice with allergic airways disease [105].
It is of interest to note that both Grx1 and 2 can be S-nitrosylated, in association with the formation of one disulfide bridge in the active site, and nitrosylation of three structural cysteines. S-nitrosylation resulted in decreased activity of Grx [106]. Furthermore, Trx [107, 108] and protein disulfide isomerase (PDI) (see below) are also targets for S-nitrosylation. Intriguingly, in addition to Grx itself, Trx and PDI are targets for S-glutathionylation [109, 110]. S-glutathionylation of Trx at cysteine 72, abolished the disulfide reductase activity of Trx [109], whereas S-nitrosylation of cysteine 63 was reported to increase Trx activity [107]. S-nitrosylation of cysteine 72 has been recently implicated in transnitrosation of caspases, but did not seem to affect Trx activity [111, 112]. Interestingly Trx may also impact S-nitrosothiol homeostasis via its ability to cleave GSNO and protein SNO [60, 113]. These collective observations point to important cross regulation of the various redox systems which may have impact on steady state levels of various cysteine oxidation states controlled by these systems.
The aforementioned enzyme systems, operate together with classical antioxidant defenses consisting of superoxide dismutases, catalase or glutathione peroxidases to control steady state levels and/or molecular actions of superoxide anion (O2·), H2O2, NO, and SNO. Thus the notion is emerging that cells have evolved with multiple and independently regulated systems, consisting of cysteine/cystine, GSH/GSSG, and Trx-SH/Trx-SS to control the redox tone [114], a concept that also warrants inclusion of measurements of NO/SNO homeostasis (nitroso-redox homeostasis). Furthermore, the existence of multiple reversible cysteine modifications in proteins warrants a redefinition of the general concept of “oxidative and nitrosative stresses” to include the identity of particular oxidative event (culprit protein, Cys identity, oxidative or nitrosative modification), and its contribution to cellular or tissue (patho-) physiology. Because of the existence of independent regulatory systems, it has become apparent that concentrations of SNOs or H2O2 are kept under tight control to permit NO- or H2O2-induced signaling events in spatially and temporally controlled settings.
The impact of cysteine oxidations on cell signaling
A number of exciting cysteine targets in proteins have been identified and numerous reports have revealed that cysteine oxidation events can have important functional consequences for an array of signal transduction cascades. It has become apparent that multiple classes of regulatory proteins are reversibly oxidized by H2O2 or NO/SNO, and that these oxidation events also have broad implications. The full breadth of S-nitrosylation is beyond the scope of the current review; excellent examples exist for every major class of protein. In many of these cases, the evidence for physiological relevance includes, dependence (of S-nitrosylation-regulated activity) on NOSs, identification of SNO-protein in vivo, recapitulation of SNO effects with NO donors in vitro, and dominant-negative effects of Cys mutation (e.g. see [115]). Evidence of this sort has not yet emerged generally for H2O2 signaling. However, a few good examples may be paradigmatic of what the future holds. The occurance of cysteine modifications in multiple classes of proteins with signaling roles, and their functional impact will be discussed in the following paragraphs. It should be noted that many of these proteins have also been shown to be S-nitrosylated.
Regulation of tyrosine phosphatases
Analogies between phosphorylation and S-nitrosylation were made in the early 1990s [10, 11]. The first studies to broaden this idea beyond S-nitroylation to include other Cys oxidations centered around protein tyrosine phosphatases (PTPs, as reviewed in [17, 50]). The first demonstration that PTP is reversibly oxidized by H2O2 produced in response to EGF was reported in 1998 [116]. These enzymes which catalyze the dephosphorylation events on tyrosine residues are unique in that they possess a reactive cysteine (pKa= 4.7–5.4) in the catalytic domain which is required for enzymatic activity. Oxidation of this reactive cysteine leads to inactivation of the phosphatase (Figure 2, box 1), and results in an overall increase in tyrosine phosphorylation events, within a specific signaling module. Inactivation of tyrosine phosphatases by H2O2 has been demonstrated in a number of settings, including physiological scenarios, such as stimulation of cells with the growth factor, epidermal growth factor, (EGF), or PDGF [17]. Reversible inactivation of the tyrosine phosphatases, PTP-1B [116], PTEN [117], and SHP-2 [29] also have been reported, and a formation of a sulfenic acid intermediate was reported as the oxidative event, although the formation of sulfenyl amide species (Cys-S-N-R) and glutathionylated species also have been shown [118, 119]. Elegant biochemical analyses were done to demonstrate the sulfenic and sulfenyl amide forms of PTP1B in vitro, and the glutathionylated form of PTP1B has been shown in intact cells. Indeed, in stimulated macrophages that produce H2O2 on the outside of the cell, H2O2-dependent PTP1B glutathionylation occurs, although the rate constant for formation of the sulfenic acid intermediate suggests that it would not be the intermediate [120, 121]. PTP1B, PTP, PTEN and SHP2 are also substrates for inhibitory S-nitrosylation [1, 122–127], and singlet oxygen also inactivates protein tyrosine phosphatase-1B by oxidation of the active site cysteine [128], demonstrating that this class of proteases is a target for inhibition via multiple cysteine oxidations.
Oxidative inactivation of PTPs, coupled to the observations that tyrosine phosphorylation requires H2O2 production, suggests that activation of receptor tyrosine kinases per se following binding of growth factor may not be sufficient to increase steady state levels of protein tyrosine phosphorylation, and that concurrent inactivation of PTPs by H2O2 is needed [50]. Mitogen activated protein kinase phosphatases (MKP) also have a reactive cysteine, required for catalytic activity, and oxidation of the cysteine in MKPs result in inactivation of the phosphatase activity, leading to enhanced activation of MAPKs, including JNK and ERK [129–131]. The enhanced activation of protein kinases as a result of inactivation of the phosphatases represents one mechanism whereby H2O2-associated oxidations enhance signaling by RTKs. In contrast to earlier reports, the stress-inducible MKP, Sdp1 in yeast was recently demonstrated to acquire enhanced catalytic activity under oxidizing conditions via the formation of an intramolecular disulfide bridge which in coordination with an invariant histidine side chain facilitates recognition of tyrosine-phosphorylated MAPK, raising the potential that this pathway may be critical in adaptive responses to oxidative stress [132]. Whether similar MKPs that can be activated through oxidative mechanisms also exist in mammalian cells remains to be identified.
Regulation of proteases
Proteases such as caspases and matrix metalloproteinases are another example of enzyme-regulation by oxidative events. Caspases contain an active site cysteine and a role for NO in preventing caspase activation has been established based upon studies demonstrating that mitochondrial caspase 3 and 9 are S-nitrosylated under basal conditions. In response to a pro-apoptotic stimulus, such as Fas ligand of tumor necrosis factor-α (TNFα), caspase denitrosylation occurs in association with activation and subsequent execution of the apoptotic pathway [53, 133](Figure 2, box 1). Intriguingly, it has been suggested that Trx (Cys 73-SNO) may exert an antiapoptotic effect by trans-nitrosylation of pro-caspase 3 [111, 112]. In addition, H2O2 has been shown to inhibit the activation of caspases [134]. A recent study demonstrated that caspase-3 activation during TNFα-induced apoptosis was negatively regulated by S-glutathionylation, and could be reversed through the thiol transferase activity of Grx [135]. Not suprisingly, caspase activation can also be negatively regulated by direct cysteine alkylation with biologically generated electrophiles, such as acrolein or hydroxynonenal [42, 136].
In contrast to the oxidant sensitivity of critical cysteines in the family of caspases, the metzincin metalloproteinase superfamily, which include matrix metalloproteinases (MMPs) and a disintegrin and metalloproteases (ADAMs), contain an invariant cysteine residue in their prodomain region, which maintains latency by chelating the active site zinc center, and activation of these proteases requires disruption of this Cys-Zn interaction (the cysteine switch). Various studies have suggested that MMPs and ADAMs can be activated by oxidants or by NO/SNO, which is suggested to involve oxidation and/or S-nitrosylation of the prodomain cysteine residue [137–139], although this is generally supported by indirect evidence. Biochemical analysis of oxidative activation of purified MMPs has shown that NO or H2O2 are ineffective at directly activating these proteases, but they can be activated by more potent oxidants such as HOCl or ONOO− [140, 141]. However, since these oxidants also have the capacity to inactivate MMPs [140, 142, 143], and MMP activation is thought to occur primarily extracellularly after secretion, it is still unclear to what extent redox signaling events by NOX or NOS regulate MMP/ADAM activation.
Regulation of molecular adaptors and chaperones
Sophisticated systems of molecular chaperones have evolved that ascertain correct folding of newly synthesized proteins and prevent misfolding or protein aggregation. These protein systems consisting of the family of heat shock proteins (Hsp) and protein disulfide isomerase (PDI) are operative within the cytoplasm and endoplasmic reticulum, respectively [144]. Hsp and PDI also play a role in physiological signal transduction as they are part of signaling scaffolds, and control transport of S-nitrosothiols, respectively (see below). For instance, Hsp90 is essential for the activation of NF-κB, and is a part of the NF-κB signalsome [145, 146]. Because the molecular actions of Hsp and PDI rely on reactive cysteines, they are emerging as potential regulators of redox signaling. The cytosol and the endoplasmic reticulum (ER) have unique redox and ionic milieus, the cytosol (reducing) and the ER (more oxidizing), requiring distinct chaperone networks [36]. Studies initially conducted with Hsp33, which is present in prokaryotes and some eukaryotes, revealed that activation of some Hsp is linked to redox changes. Hsp33 in its inactive conformation has four reactive cysteine residues in the thiolate anion form that bind Zn2+. Under conditions of oxidative stress, the four thiolates form two intramolecular disulfide bonds which cause release of Zn, and subsequent dimerization of oxidized monomers, which effectuates full activation of Hsp33’s function as a chaperone [147–149]. Similarly, mammalian Hsp25, 60, 70, and 90 also have redox active cysteine residues, and their oxidation has been linked to the modification of various chaperone functions in conditions of oxidative stress [150–153]. Various cysteine oxidations have been linked to Hsp activation that include nitrosylation, glutathionylation, and disulfide formation [151, 152, 154, 155] (Figure 2, box 3). Hsp90 is, of note, regulated by S-nitrosylation [155].
PDI is a multi-domain, multi-functional member of the thioredoxin superfamily that contains two thioredoxin-like catalytic domains. PDI can catalyze thiol-disulfide oxidation, reduction and isomerization [156]. The PDI family comprises PDI and PDI-like proteins of which more than a dozen human members have been characterized [156]. PDI is mainly is present in the lumen of the endoplasmic reticulum ER of eukaryotic cells, where many folding factors and protein chaperones are co-located. PDI is oxidized by the ER oxidoreduction proteins Ero1-Lα and Ero1-Lβ in mammalian cells. Oxidized PDI, with active-site cysteines in the disulfide form, can act as an electron acceptor to form disulfide bonds in sulfhydryl-containing substrate proteins [157]. PDI is believed to act as a redox driven chaperone [158], although other reports have questioned the general concept of redox-regulated chaperone activity of PDI [159]. PDI is also surface associated, where it may play a role in transport of NO from S-nitrosothiols across membranes (whether directly or by maintaining surfact thiols in the reduced state is not clear) [160, 161]. Interestingly, S-nitrosylation of PDI [162, 163] has been linked to neurodegenerative disease [162]. Interestingly, PDI itself can mediate its own denitrosylation [163].
The transduction of molecular signals from receptors that themselves do not have kinase activity relies on the assembly of adaptor proteins in molecular scaffolds to recruit downstream kinases. The carefully timed assembly of these specialized “signalsomes” in precise cellular compartments ascertains the timing and specificity of downsteam responses [164, 165]. As an example, stimulation of tumor necrosis factor receptor-1 (TNF-R1) can lead to multiple outcomes, which depends on the differential presence or recruitment of adaptor proteins into the signalsome. While TRAF-2 is required for the activation of JNK, FADD is required for caspase activation, and RIP and Hsp90 are important for the activation of NF-κB. TRAF2 can bind to and activate apoptosis signal-regulating kinase (ASK1), an important regulator of apoptosis [166, 167]. Through a genetic screen, Trx was identified as an interacting partner of ASK1, and was demonstrated to regulate ASK1 function via a redox mechanism. The interaction between Trx and ASK1 was found to be highly dependent on the redox status of Trx. Reduced Trx binds ASK1 and keeps the enzyme inactive, suggesting that Trx is a physiological inhibitor of ASK1. Oxidation of Trx1, involving the formation of an intramolecular disulfide between cysteines 32 and 35 resulted in release of Trx1 from ASK1 which results in activation of ASK1 [167]. This redox dependent pathway from Trx to JNK has been demonstrated to occur in macrophages that were stimulated to produce H2O2 [168]. However, others reported redox independent regulation of ASK1 by Trx [169]. Trx is subject to S-nitrosylation at multiple sites. S-nitrosylation of cysteine 69 that lies outside the active site occurs under basal conditions, facilitates oxidoreductase activity, and is associated with protection of apoptosis [107]. Additional reports that S-nitrosylated Trx specifically transnitrosylates pro-caspase 3 [111, 112] underscore the functional importance of Trx in protection against apoptosis. In contrast, S-nitrosylation of active site site cysteines (32 and/or 35) promotes disruption of ASK1 and is associated with enhanced apoptosis [108]. In lung epithelial cells, exposure to H2O2, or expression of NOX1 disrupted the signaling through TNF-R1, leading to preferential and prolonged activation of JNK, while preventing the activation of IKK, and NF-κB. These events were associated with degradation and cleavage of RIP and Hsp90, repectively, two adaptor proteins required for the activation of IKK [170]. Further, H2O2 resulted in recruitment of TRAF2 and ASK1 to TNF-R1, which were functionally important in the activation of JNK and subsequently cell death. Collectively, these data confirm that oxidant-based signals utilize a TNF-R1-ASK-1JNK signaling axis to trigger cell death, and prevent activation of NF-κB [171].
The Nrf2-KEAP system provides another example of the functional significance of redox regulation of cellular adaptors. Nrf2-KEAP acts as a regulatory system that protects cells from an array of stresses. When cells are exposed to oxidative stress, electrophiles, or chemopreventive agents, the transcription factor Nrf2 activates antioxidant responsive element (ARE)-dependent gene expression to maintain cellular redox homeostasis. Because of its role in activation of genes with diverse functions, such as antioxidant and phase II enzymes, Nrf2 is now recognized as a key regulatory factor that controls expression of an array of genes important in the defense against diverse environmental insults, a topic that has been extensively reviewed, including in this journal [172, 173]. Nrf2 activation is prevented by binding to KEAP-1. Oxidative insults target KEAP-1 by targeting cysteines 273 and 288, which cause dissociation of KEAP-1 from Nrf2, allowing accumulation of Nrf2 in the nucleus and activation of target genes [174, 175]. Investigations into the mechanisms whereby KEAP-1 promotes degradation of Nrf2 have unraveled that KEAP-1 acts as a redox sensitive adaptor for a Cul3-based E3 ligase [176]. Thus, under homeostatic conditions, the complex of KEAP-1, Nrf2 and Cul3 ascertains rapid degradation of Nrf2 via the ubiquitin proteasome system, while under conditions of oxidative stress the associations between KEAP-1 Nrf2 and Cul2 are no longer intact, thereby preventing degradation of Nrf2 [177]. Importantly a somatic mutation of the KEAP-1 gene has been identified in lung cancer leading to a glycine to cysteine substitution in the DGR domain, which reduces KEAP-1’s affinity for Nrf2, providing a structural basis for the loss of KEAP-1 function and gain of Nrf2 function [178, 179]. These findings convincingly illustrate the importance of oxidation events in adaptor proteins in controlling redox signaling, and also that a disturbance in these interactions is associated with human disease.
Genetic screening for protein-protein interactions has identified that S-nitrosylation regulates protein-protein interactions, which include interactions of NOS with various substrates for S-nitrosylation [180]. These findings highlight the compartmentalized and targeted actions of NOS enzymes, and their specific S-nitrosylation of effector proteins that include NMDA and ryanodine receptor, G-protein coupled receptor kinase 2, caspases, GAPDH, NSF, dynamin, cycooxygenase 2 [1, 58, 80], and many other protein targets. S-nitrosylation also is known to affect the function of molecular chaperones, and this paradigm plays a central role in the regulation of apoptosis. Although the previous paragraph highlighted the significance of caspase S-nitrosylation in the prevention of apoptosis, a wealth of information has also accumulated in support of a pro-apoptotic role in settings where high levels of NO are present. This role of NO in cell death had been associated with formation of highly reactive oxidants, such as peroxynitrite (ONOO−) [181, 182]. However elegant recent studies demonstrate that in settings of high NO output, S-nitrosylation in fact plays a central role in apoptosis [183]. In the latter scenario, glyceraldehyde-3-dehydrogenase (GADPH) is the target of S-nitrosylation of catalytic site cysteine 150, which in turn causes binding of the E3 ligase, Siah1 to GAPDH, and stabilizes Siah1, which is otherwise rapidly turned over. The SNO-GAPDH-Siah1 complex can then translocate to the nucleus, where Siah1 targets nuclear proteins for degradation [184, 185].
Redox control through functional dimerization of redox enzymes with signaling intermediates
A number of studies recently emerged to support the notion that enzymes involved in redox homeostasis that include Trx, and its homologs, glutathione peroxidase, and glutathione S-transferase (GST), play a role in redox signaling through redox dependent interaction with a protein with signaling function. Perhaps the best known example of this scenario represents the interaction of Trx with its target proteins, including (ASK-1, Trx binding protein-2, and Trx interacting protein [83]. Oxidation of Trx results in a conformational change that changes interaction with the partner protein, leading to a signaling event (Figure 2, Box 4). Well described is the Trx1-oxidation dependent dissociation of Trx1 from ASK-1 which is critical to the activation of JNK [167] that was described earlier.
A recent study identified nucleoredoxin (Nrx), a member of the Trx family, as a critical regulator of Wnt/β-catenin, signaling pathway evolutionary conserved from nematodes to mammals. Specifically the authors demonstrated that Nrx suppresses Wnt-β-catenin, by redox-dependent binding to Dishevelled, and masking its basic PZD domain, which prevents activation of the pathway. Upon oxidation of reactive cysteines of Nrx1, its association with Dishevelled is lost which promotes stabilization of β-catenin and subsequent activation of the transcription factor T cell factor (TCF). In support of the redox dependence of the Dishevelled-Nrx1 interaction, exposure to H2O2 also resulted in disruption of the complex, leading to subsequent accumulation of β-catenin and activation of TCF [186]. It is worthwhile to mention that these conclusions were reached based upon mutagenesis experiments that encompassed mutations of cysteines 205 and 208 of Nrx, and that direct evidence for oxidation of those sites was not provided.
The Yap1 transcription factor in yeast S. cerevisiae regulates hydroperoxide homeostasis, and its activity is increased under conditions of oxidative stress. Intriguingly, redox control of Yap1 is not exerted through oxidation of Yap1 itself, but via glutathione peroxidase (Gpx)-like enzyme, Gpx3, which acts as the sensor and tranducer of the redox signals. Oxidation of cysteine 36 of Gpx3 resulted in an intermolecular disulfide bond with cysteine 598 of Yap-1, and the resolution of this intermolecular disulfide bond into a Yap1 intramolecular disulfide bond, causes activation of the regulator [187].
Glutathione S-transferase pi (GSTp) has emerged as a critical regulator of the JNK pathway. GSTp was demonstrated to be directly associated with JNK [188] and its monomeric form was shown to inhibit JNK activation in a manner that was independent on MEKK1-MKK4, the physiological upstream activators of JNK. In response to an oxidant stress, GSTp oligomerizes, and dissociates from JNK1, promoting its activation [189] (Figure 2, box 4). Importantly, mouse embryo fibroblasts from GSTp-null mice exhibited a high basal level of JNK activity, [189] and GSTp-null mice showed a phenotype of increased myeloproliferation, in association with activation of JNK and the JAK/STAT pathway [190], corroborating the functional significance of GSTp in the regulation of JNK, and potential other pathways in vivo. GSTp also is an important regulator of peroxiredoxin 6 (1-cysPrx), via a mechanism that involves heterodimerization of the oxidized catalytic cysteine of 1-cysPrx with pi GST followed by its GSH-mediated reduction and enzyme activation [191].
Redox regulation of transcription factors
In addition to redox control through oxidation of kinases and phosphatases, molecular adaptors, or chaperones, that can in turn regulate the activity of transcription factors, transcription factors themselves can also be direct targets of redox control, a topic that has been extensively reviewed [192]. Perhaps the first evidence for this was based upon the work on the bacterial transcription factor OxyR which was demonstrated to be directly activated by oxidants via the formation of a reversible disulfide bond [193–195] (Figure 2, box 5). Subsequent work has shown that S-nitrosylation, S-glutathionylation, and S-hydroxylation can also activate OxyR [32, 196]. This paradigm has been expanded; multiple families of transcription factors in mammalian cells that include NF-κB, AP-1, p53, HIF1α, and HSF are known to be modulated directly through oxidative changes. In many cases, critical reactive cysteines have been identified, which upon oxidation elicit a functional change either manifested as increased transactivation or inhibition. Examples of transcription factors that are inhibited following cysteine oxidation include the p50 and RelA (p65) subunits of NF-κB, and the c-Jun member of the AP-1 family. Further, both S-nitrosylation and S-glutathionylation of those proteins has been shown to interfere with their DNA binding [197–201], and S-nitrosylation is coupled to iNOS function in vivo. Multiple cysteines in murine p53 were found to be functionally important, and their selective mutation afftected various aspects of its function [202]. S-glutathionylation of cysteine 82 of p53 has been linked to decreased binding to DNA in association with the formation of monomers and high MW oligomers [203]. Interestingly, the redox state of cysteine 277 of human p53 has been linked to its ability to bind to bind p53 response elements in a sequence specific manner [204]. Hypoxia-inducible factor (HIF)1α also is regulated in a redox dependent manner, and its degradation and activation are regulated via cysteine oxidation. S-nitrosylation of HIF1α promotes its stabilization [205], and S-nitrosylation of Cys-800 activates HIF1α-p300 interaction and stimulates protein transactivation [206].
The importance of oxidation of Hsp in redox signaling was discussed in the previous paragraph. Interestingly, mammalian heat shock factor 1 (HSF1), a transcription factor critical in the induction of stress-inducible Hsp gene expression is directly activated by oxidant stress. Activation of mammalian Hsp gene expression by HSF1 requires conversion of its monomeric state to the DNA-binding competent homotrimer. The sensing of both stresses requires two cysteine residues within the HSF1 DNA-binding domain that are engaged in redox-sensitive disulfide bonds. HSF1 derivatives in which either or both cysteines were mutated are defective in stress-inducible trimerization and DNA binding, stress-inducible nuclear translocation, and Hsp gene trans-activation, and in the protection of mouse cells from stress-induced apoptosis [207].
An emerging body of evidence is accumulating to support the concept that alterations in the activities of transcription factors may not limited to their direct oxidative modification. Indeed chromatin remodeling factors, such as histone acyl transferases (HAT), or histone deacetylases (HDAC) also can sense redox changes, and thereby lead to changes in transcription. The sirtuin (sirt) family of nicotinamide adenine dinucleotide-dependant (NAD) deacetylases plays an important role in aging and metabolic regulation via control of the FOXO family of Forkhead transcription factors [208]. In response to oxidative stress, sirt1 forms a complex with FOXO3, leading to sirt1-dependent deacetylation of FOXO3, which mediates cell cycle arrest, resistance to oxidative stress, and inhibition of cell death [208]. Other reports corroborate a link between oxidative stress and sirt-dependent deacetylation of FOXO, subsequent enhanced activity of FOXO as a transcription factor, and cellular protection against oxidative stress, due to transcriptional upregulation of protective genes such as manganese superoxide dismutase and growth arrest and DNA damage inducible (GADD)45 [209, 210]. In contrast, it has been reported that H2O2 resulted in acetylation of FOXO4 in association with decreases in transcriptional activity (although the same study also reported that H2O2 stimulates binding of sirt1 to acetylated FOXO4 [211]). Taken together, these data raise the possibility that FOXO4 transcription is regulated in a bi-phasic manner. Notably in this regard, mild transgenic overexpression of sirt1 in mouse hearts was associated with protection against oxidative stress, and ageing, while high overexpressors displayed enhanced cardiomyopathy [212]. In addition to regulation of the sirtuin class of histone deacetylases by oxidative stress, HDACs linked to transcriptional activities of NF-kB, and AP-1 [213–215]) may be inhibited by H2O2 (or environmental stresses), and a role for S-nitrosylation has likewise been identified with HDACs in the context of CREB induced gene expression [216].
While all these observations provide compelling links between H2O2, changes in histone acetylation and, enhanced transcriptional activity, some of these studies examined conditions of overt stress, characterized by decreases in levels of GSH, increases in GSSG, and evidence of tyrosine nitration [214, 215], and highlight the need for additional studies to understand redox regulation of chromatin remodeling factors under physiological settings, which also should entail identification of target amino acids that are oxidized.
Criteria for Signaling: SNO and more
The pharmacological tools in NO biology, including mutant animals (eNOS, nNOS, eNOS, GSNOR) and multiple classes of highly specific NOS inhibitors have established beyond doubt NO’s broad-based signaling function in physiology and disease. Further, compelling data revealing the presence of S-nitrosylated proteins by multiple techniques –chemical assays, SNO antibodies, mercury-coupled photolysis chemiluminescence —have helped to firmly establish S-nitrosylation as an omnipresent signal. Among the various posttranslational modifications, phosphorylation, ubiquitylation and S-nitrosylation, are truly ubiquitous. By contrast, pharmacological tools such as inhibitors or NADPH oxidases, and NOX knock out animals are not completely available and alternative cysteine oxidations have been diffcult to assay. Many of the current research findings, particularly as they relate to glutathionylation and sulfenic acid oxidations are limited to test tube settings. In addition, whereas the assays for SNO are relatively specific, those for alternative oxidations do not readily differentiate SNO, SOH or SS. Consequently the functional importance of individual cysteine oxidations in homeostasis or pathophysiology remains largely unknown; the physiological significance of redox signaling at large is, therefore, still an open question. More specifically, the stringent demonstration of agonist-coupled, reversible modifcations of Cys residues by glutathionylation or hydroxylation that subserves diverse functions or that serve as part of signal transduction cascades involving multiple proteins, which convey a redox-based signal, has not been shown. In this regard, it is important to note that the activation of kinases through oxidative inhibiton of phosphatases indeed constitutes redox-based regulation, but not signal transduction per se: signals are being transduced by phophorylation (kinases). By contrast, the activation of ASK by oxidation of Trx (discussed above) has the incipient fingerprint of oxidative signal transduction. In addition, it is key to understand whether responses are physiological signals or adaptive responses to stress. At present, the signaling function identified with NO biology, particularly S-nitrosylation, which is broad based, has not been replicated by alternative modifications, although recently much exciting headway suggest this is a real possibility.
Methodologies and to detect reversible cysteine oxidations in cells and tissues
Technical hurdles stem from the fact that cysteine oxidations relevant to signal transduction are reversed by commonly used reductants, such as beta-mercaptoethanol or dithiotreitol. Standard assays including, protein purifications, Western blots, (e.g. to assess kinase activity) and gel shifts (transcription factors binding to DNA), which routinely incude these reagents, obscure the effect of oxidations. Mass spectrometric evaluation of complex mixtures also entails the use of reductants and changes in pH that destabilize Cys-based oxidations; further, an SNO does not often survive electrospray or MALDI (electrical current and ionization) treatments.
Thiol-specific alkylating reagents such as iodoacetic acid (IAA) or N-ethyl maleamide (NEM) which irreversibly bind to sulfhydryls, while not affecting oxidized cysteines [29, 217–219] have opened the field to inquiry. The subsequent addition of reductant coupled to the use of a thiol-specific labeling agent such as biotinylated IAA or biotinylated NEM will then identify the oxidized Cys (Figure 3, top panel) [47]. This methodology, also referred to as “the thiol trapping technique” [220] has been successfully applied to cells in culture and demonstrated reversible oxidation and inactivation of tyrosine phosphatases in response to stimulation with growth factor receptors [29, 116, 117, 219]. A similar strategy has been developed to detect S-nitrosylated proteins, widely known as the “biotin switch method” (Figure 3, middle panel) [56, 221]. This approach also relies on thiol specific blocking and labeling reagents, but utilizes ascorbate to specifically denitrosylate S-nitroso bonds (formally a transnitrosation reaction, not a reduction). Indeed, Jaffrey and co-workers originally identified a number of proteins that were biotinylated following ascorbate treatment in wild type but not NOS1 knock-out mice, demonstrating a requirement of neuronal nitric oxide synthase (nNOS) in the ascorbate-dependent labeling of proteins, and establishing protein S-nitrosylation as a physiological event in the brain [56]. Although the specificity of the biotin switch method has been challenged due to concerns about the potential reactivity of ascorbate with disulfides or sulfenic acids [222, 223], those concerns were not well grounded. Reduction of disulfides, sulfenic acids and SNOs by ascorbate is not thermodynamically favorable (the transnitrosation reaction with SNO is not a redox reaction, hence the specificity). A recent comparative analysis of differentially oxidized or S-nitrosylated forms of proteins verified that the assay is highly specific for S-nitrosylated proteins; artifactual ascorbate-dependent signals were formed through exposure to indirect sunlight, likely induced by the semidehydroascorbate radical. One caveat with the biotin switch is the sensitivity: ascorbate does not effectively denitrosylate all S-nitrosothiols [127, 224]. The coupling of the biotin switch coupled to SNO-photolysis (instead of ascorbate) provides an alternative means to detect S-nitrosylated proteins [127]. Overall, the technique has been a major advance, not only opening the field to outside disciplines but also serving to to verify earlier work through the 1990’s using more cumbersome techniques (mercury-coupled photolysis chemiluminescence and various other chemical and spectroscopic methods). Today, anti SNO-antibodies are also commercially available and have been used both in various histological analysis, Westerns, and immunoprecipitations [14, 225, 226].
Figure 3.
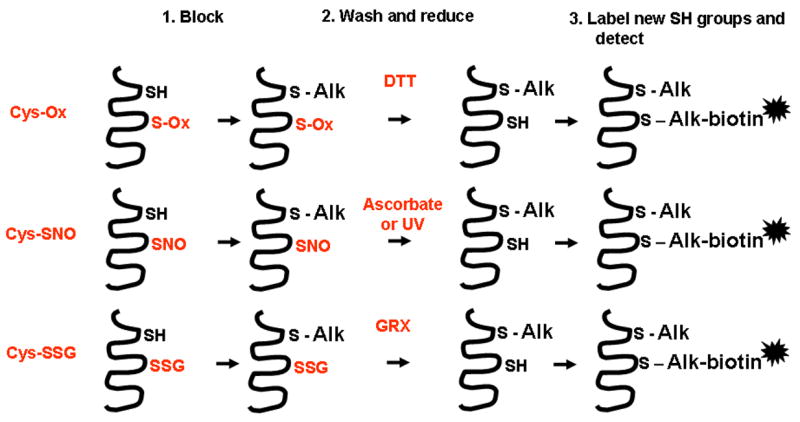
Reduction strategies used to detect various cysteine oxidations. These thiol trapping or biotin switch procedures involve sequential steps of thiol blocking (1), a wash step to remove the chemical blocking agents, followed by a reduction step (2). This reduction step can employ non-specific chemical reagents e.g. dithiotreitol (DTT) to decompose sulfenic acids, disulfides, S-nitrosylated cysteines, or S-glutathionylated cysteines (top). Alternatively, the reduction step utilizes ascorbate or UV to reduce S-nitroso bonds (middle). GRX-catalyzed reduction (bottom) is used to reduce S-glutathionylated proteins. In a labeling step (3), the newly reduced sulfhydryl groups are reacted with a thiol specific labeling agent. Subsequently, by capturing the labeled cysteines, proteins can be identified using proteomic procedures. Labeled proteins can be precipitated, electrophoresed and blotted, and specific antibodies can then be used to detect targets. These methods can also be used to evaluate reversible cysteine oxidations in situ, in cells or tissues, using microscopy approaches (See Figures 4 and 5). Alk denotes a thiol specific alkylating agent, such as N-ethyl maleamide (NEM).
Similar approaches have been taken towards the identification of S-glutathionylated proteins. In this endeavor, catalytically active Grx is used to reduce S-glutathionylated derivatives to the sylfhydryl group, followed by labeling of the newly reduced cysteine with a specific tag (Figure 3, bottom panel). This approach has been used in proteomic studies and identified a number of proteins targeted by S-glutathionylation, in addition to identification of specific sites of S-glutathionylation [154, 227]. Related approaches to detect protein S-glutathionylation include the use of an antibody directed against GSH, GSH affinity purification, glutathione-S-transferase overlay, or the use of cellular labeling strategies with tagged and cell permeable glutathione, such as glutathione ethyl ester which becomes incorporated into proteins following an oxidative event and therefore is useful to detect proteins that are targets for S-glutathionylation [110, 228–230]. It should be highlighted that the latter approach requires the use of an exogenous labeling agent (whose processing and cellular metabolism is not known) and does not allow detection of S-glutathionylated proteins that exist endogenously prior to the presence of GSH-labeling agent. The latter procedure also is problematic when evaluating S-glutathionylated proteins in tissues.
Thiol trapping methodologies have been adapted to visualize reversible cysteine oxidations in intact cells or tissues using microscopy approaches, in order to detect regional or kinetic changes in cysteine oxidations in subcellular compartments or in tissues in vivo [55]. Using these strategies, cells or tissues are first fixed in paraformaldehyde, permeabilized with triton-X100 in presence of the thiol blocking agent, NEM. Subsequently, S-nitrosylated cysteines are reduced with ascorbate followed by incubation with biotinylated NEM. Using streptavidin conjugated to a fluorophore, the location of the S-nitrosylated cysteine is then detected via confocal laser scanning cytometry [55] (Figure 4). Intriguingly, in cultured epithelial cells, substantial reactivity is detected in the nuclear compartment of cells, based upon co-localization with a DNA stain, and indeed the nuclear small GTPase Ran was demonstrated to be a target for S-nitrosylation [55]. Utilization of Grx as the reducing agent to detect S-glutathionylated cysteines yielded different staining patterns, with predominant staining in the cytoplasm or membrane ruffles, and marked increases in reactivity after exposure to H2O2, or expression of NOX1. The specificity of the Grx-catalyzed reduction for S-glutathionylated cysteines was highlighted using S-glutathionylated bovine serum albumin (BSA), a specific competitor in the reaction, vs. fully reduced or cysteinylated BSA, which did not affect the staining [100] (Figure 5).
Figure 4.
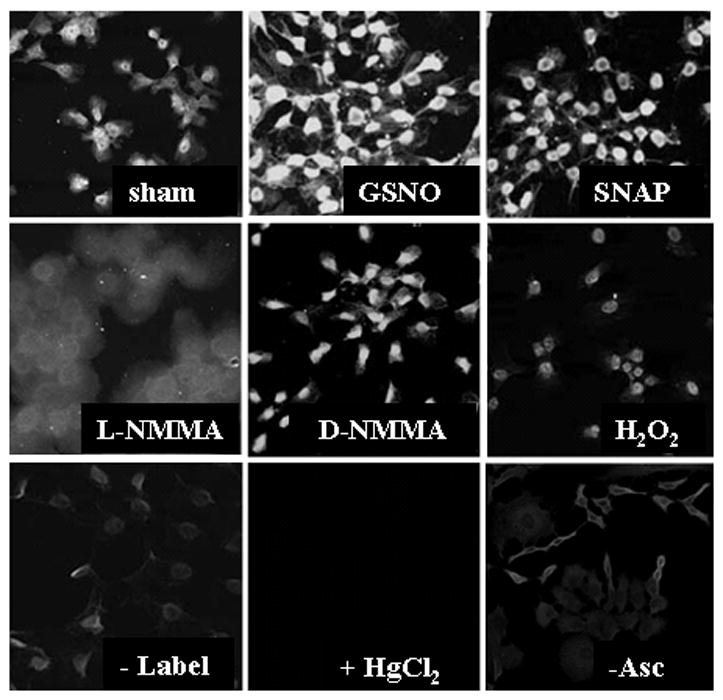
In situ visualization of S-nitrosylated proteins in lung expithelial cells using confocal laser scanning cytometry. Cells were exposed to 1mM SNAP or GSNO for 1 hour, or to the NOS inhibitor, L-NMMA, or reagent control D-NMMA. As additional controls, the biotin-HPDP label (-label), or ascorbate (-Asc) were omitted, or HgCl2 (which depletes SNO), was added before blocking to decompose S-nitrosothiols. As an additional control, cells were treated with H2O2 for 15 minutes (See [55] for experimental details). Reprinted from Nitric Oxide, Volume 11, In situ detection and visualization of S-nitrosylated proteins following chemical derivatization: Identification of Ran GTPase as a target for S-nitrosylation, Ckless K. et al., 216–227, Copyright 2004, with permission from Elsevier.
Figure 5.
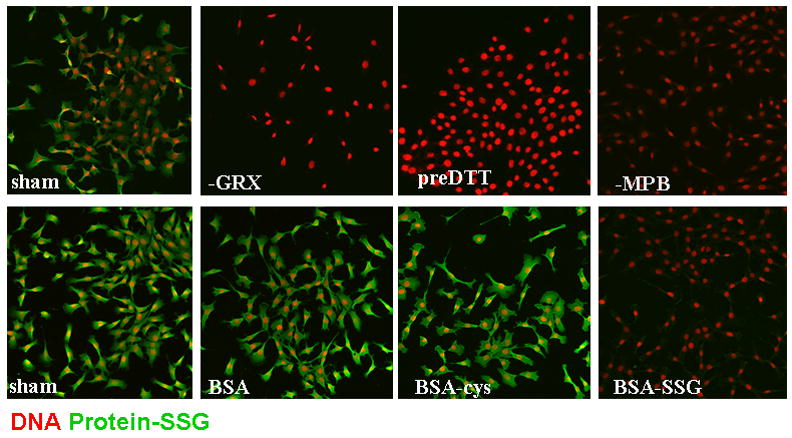
In situ visualization of S-glutathionylated proteins in lung expithelial cells using GRX-catalyzed cysteine derivatization, and visualization via confocal laser scanning cytometry. Top panels: Lung epithelial cells were left untreated and as reagent controls, GRX (-GRX) or MPB (-MBP) were omitted out of the procedure. As an additional control, cells were treated with 1mM DTT prior to blocking with NEM (preDTT). Bottom panels: During the reversal of cysteine oxidations by GRX, fully reduced Bovine Serum Albumin (BSA), Cysteinylated BSA (BSA-cys) or S-glutathionylated BSA (BSA-SSG) were co-incubated in the reaction mixture to evaluate competition with endogenous substrates for GRX1-catalyzed reduction. (See [100] for experimental details). Protein S-glutathionylation is visualized in green, and nuclei are indicated in red. Reprinted from Biochimica et Biophysica Acta, Volume 1760, In situ detection of S-glutathionylated proteins following glutaredoxin-1 catalyzed cysteine derivatization. Reynaert N. et al., 380–387, Copyright 2006, with permission from Elsevier.
Physiological relevance and pitfalls of models used to study redox signaling
Well over 3000 manuscripts have been published in the area of oxidants and “signaling”, but most of these manuscripts rely on the use of externally administered oxidants to cells in culture. Many investigators have raised concerns about the physiological relevance of those model systems. These concerns are warranted when excessive concentrations (>mM) of oxidants are utilized. However, the relevance of a particular experimental design depends on the nature of the hypothesis being tested [121]. If the experimental goal is to elucidate the nature of target cell responses triggered by high levels of reactive oxidants generated by inflammatory cells, exposing target cells to high amounts of oxidants probably represents a reasonable model system. However, if the experimental question is to determine how H2O2 or NO/SNO, generated from NOX or NOS, respectively under physiological settings, evoke physiological signaling, administration of NO or H2O2 externally, may produce spurious results. Exogenous agents are only useful to the extent they recapitulate the known physiology. As was highlighted in the previous section, oxidation events and antioxidant defenses are carefully balanced to allow a transient redox signaling event in precisely targeted locations, coordinated by the presence of adaptor proteins or chaperones that exist in signaling platforms within specific subcellular compartments. Given these considerations, it is unlikely that administration of oxidants extracellularly, can accurately model the signaling function of physiologically generated by H2O2 (irrespective of the concentration of oxidant applied) because different cysteines will be targeted. A hypothetical schematic that visualizes this concern is contained in Figure 6. On the other hand, the application of NO donors has recapitulated physiology more often than would be expected. It may be that NO cannot readily change cellular redox state and instead has been adapted to react with a subset of key thiols across a wide concentration range.
Figure 6.
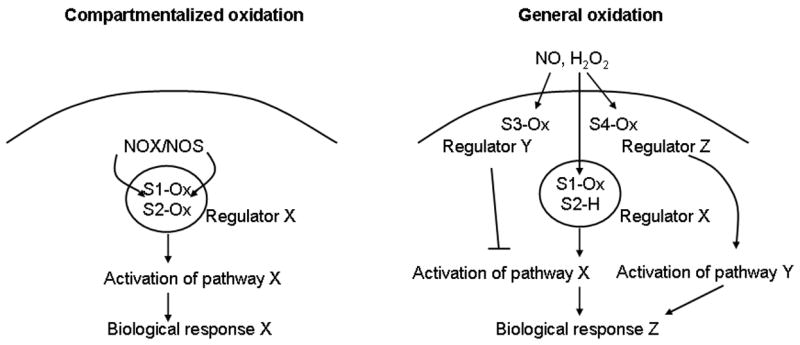
Visualization of the differential impact of compartmentalized cysteine oxidation events, as compared to diffuse oxidation events on signal transduction cascades. The left panel represents a hypothetical schematic illustrating the compartmentalized cysteine oxidation events, S1-Ox, and S2-Ox, following the localized activation of NOX or NOS, which lead to activation of regulator X, which in turn evokes response X. The panel on the right represents diverse intracellular cysteine oxidation events S1-Ox, S3-Ox, and S4-Ox that occur in response to extracellularly encountered H2O2 or NO/SNO. This scenario leads to a different biological response (Z). Activation of Regulator X does not occur, because of lack of oxidation S2, and the oxidation of S3 which activates regulator Y, which in turn inhibits Pathway X. Instead, because of oxidation event S4, activation of regulator Z occurs, leading to activation of pathway Y, and response Z. The scenarios illustrated are strictly hypothetical.
Assessment of the functional importance of cysteine oxidation in vivo
The relevance of cysteine oxidations of target proteins in the regulation of biological responses in vivo remains largely untested to date. Although many claims exist about the in vivo relevance of cysteine oxidation, most of the studies conducted so far have been limited to cells in culture. Many experiments, including work in our own laboratories, have relied on the expression of mutant constructs in which a reactive cysteine is mutated to an alanine or serine residue. In numerous cases, expression of the mutant construct produces a biological response that is distinct from the response triggered by the wild type construct, which have lead to conclusions that oxidation of that particular cysteine is important in vivo. Compelling as this argument may be, conclusions can only be drawn with caution because of the stretched meaning of the “in vivo” scenario. Furthermore, in most studies, transfection of the mutated or wild type proteins occurred in a “transgenic” fashion where expression levels were not regulated or controlled, and the endogenous protein target was also present (experiments that were conducted in mouse embryonic fibroblasts derived from various knock-out mice would be the exception). Given these pitfalls, the question arises about how much we truly know about the functional relevance of cysteine oxidation events in vivo?
Drawing analogy to other areas of signal transduction, one approach used to overcome some of these pitfalls relies on transgenic mouse technologies. For example, in order to understand the impact of phosphorylation of a specific serine residue in a specific protein, the serine residue is mutated to a non-phosphorylatable residue, such as an alanine, and this way a “knock-in” mouse is created where the endogenous protein is replaced by the mutant version of the protein. This amino acid replacement strategy has the advantage that no interference exists with the endogenous protein, and that the mutant protein is regulated, and presumably expressed in the same manner as the wild type protein. An example of this strategy is the JunAA mice in which serines 63 and 73, the targets of phosphorylation of JNK, are mutated to alanines [231]. These mice have been instrumental in understanding the functional significance of phosphorylation of c-Jun in a number of pathophysiological settings [231–233]. Other examples of this strategy are available [234–237]. Similar strategies to evaluate the in vivo importance of specific cysteine oxidation targets are largely lacking, and only a few knock-in mice with mutated cysteine knock-ins have been reported. Rhodopsin is a target for palmitoylation at cysteine residues 322 and 323, and knock-in mice in which these sites were mutated to threonine and serine residues were reported to have enhanced shut-off phototransduction [238]. A mouse containing a single amino acid substitution (Cys249Trp) in the human Crumbs homolog-1 gene displayed in retinal degeneration [239]. Whether these phenotypes are linked to altered redox signaling remains to be determined. One knock-in mouse was generated to specifically address the functional significance of redox regulation in vivo. The protein, Ref-1 participates both in DNA repair pathways as well as in redox regulation. The redox function of Ref-1 involves reduction of oxidized cysteine residues within the DNA binding domains of several transcription factors, including Fos and Jun, and based upon elegant biochemical studies, cysteine 64 of mouse Ref-1 was identified as the redox catalytic site [240]. However, knock-in mice containing a cysteine-to-alanine point mutation into the Ref-1 gene displayed no evidence of aberrant redox regulation of c-Fos and c-Jun in vivo, questioning the in vivo relevance of the role of Ref-1 in redox regulation [241]. This finding was surprising because the biochemical analyses of the redox function of Ref-1 in cell culture models were very elegant. Intriguingly, a recent study suggested that other cysteines in Ref-1 can be S-nitrosylated [242], potentially explaining the lack of a phenotype in the Ref-1 cysteine 62 mutant mouse.
Conclusions and promises for the future
The area of redox-based signaling has arrived at an exciting juncture, bolstered by the concept that oxidative processes can be precisely orchestrated to evoke diverse functional responses. Newly recognized regulatory enzyme systems that operate together with classical antioxidant defenses serve to create independent modes of redox regulation within distinct cellular compartments. Furthermore, new technologies provide investigators with the opportunity, for the first time, to identify sites of oxidative modification within proteins with physiological and pathophysiological effects. While these studies are yielding exciting insights, NO/SNO remain more clearly linked with physiological signaling, and H2O2 with stress responses. These conclusions, however, have been derived largely from cell culture models and bioassays. Exploration of oxidative events in organisms will require implementation of large-scale proteomic studies in vivo, including assessment of the spacial-temporal relationships between oxidative changes and organ (patho)physiology. Analyses of S-nitrosylation in NOS knockout animals with diverse pathophysiology has been an important step forward; by analogy, assessments of alternative S-oxidations in transgenic animals will no doubt be revealing. It will be exciting to watch developments that delineate the extent to which S-NO, S-OH and SSG are involved in health and disease.
Acknowledgments
I dedicate this manuscript to my father, Jo Janssen, who continued to motivate and inspire me until the end of his life, and this chapter – Yvonne Janssen-Heininger.
This work was funded by National Institutes of Health (NIH) grants R01HL60014 (YJH), R01HL079331 (YJH), R01HL074295 (AvdV and YJH), R01HL068865 (AvdV), P01HL67004 (BTM) from NHLBI, and a grant from the Mesothelioma Applied Research Foundation (BTM); R01ES05511 (HJF), and TRDRP14RT-0059 (HJF)
Abbreviations
- ADAM
A Disintegrin and metalloprotease
- AHR
Airway hyperresponsiveness
- Alk
Alkylation
- AMPA
alpha-amino-3-hydroxy-5-methylisoxasole-4-propionic acid
- AP-1
Activator protein-1
- ASK-1
Signal-regulating kinase-1
- ARE
Antioxidant responsive element
- Asc
Ascorbate
- BSA
Bovine serum albumin
- cGMP
Cyclic GMP
- Cys-S-N-R
Sulfenyl amide
- Cys-X-X-Cys
Common dithiol/disulfide active site motif
- DUOX
Dual oxidase
- EGF
Epidermal growth factor
- ER
Endoplasmic reticulum
- GADD
Growth arrest and DNA damage
- GADPH
glyceraldehyde-3-dehydrogenase
- Gpx
Glutathione peroxidase
- Grx
Glutaredoxin
- GST
Glutathione S-transferase
- GSNO
S-Nitrosoglutathione
- GSSG
Glutathione disulfide
- HAT
Histone acyl transferase
- HDAC
Histone deacetylase
- HIF
Hypoxia-inducible factor
- H2O2
Hydrogen peroxide
- HSF
Heat shock factor
- Hsp
Heat shock protein
- IAA
Iodoacetic acid
- IFN-γ
Interferon-gamma
- LPS
Lipopolysaccharide
- MAPK
Mitogen activated protein kinase
- MKP
Mitogen activated protein kinase phosphatase
- MMP
Matrix metalloproteinase
- NAD
Nicotinamide adenine dinucleotide
- NEM
N-ethyl maleamide
- NF-κB
Nuclear factor kappa B
- NH3
Ammonia
- NMDA
N-Methyl-D-Aspartate
- nNOS
Neuronal nitric oxide synthase
- NO
Nitrogen monoxide/nitric oxide
- NOS
Nitric oxide synthase
- NOX
Non-phagocytic oxidase
- Nrf2
NF-E2-related factor 2
- Nrx
Nucleoredoxin
- O2·
Superoxide anion
- PDGF
Platelet derived growth factor
- PDI
Protein disulfide isomerase
- Prx
Peroxiredoxin
- PTP
protein tyrosine phosphatase
- PSSG
Protein-mixed disulfides/glutationylated cysteine Sirt, Sirtuin
- SNO
S-nitrosylation, S-nitrosothiol, S-nitrosylated protein
- SOH
Sulfenic acid
- SO2H
Sulfinic acid
- SO3H
Sulfonic acid
- S-Ox
Oxidized cysteine
- S-S
Disulfide
- TGF-β1
Transforming growth factor-beta 1
- TCF
T cell factor
- TLR
Toll like receptor
- TNF-R1
Tumor necrosis factor receptor-1
- Trx
Thioredoxin
- Srx
Sulfiredoxin
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hess DT, Matsumoto A, Kim SO, Marshall HE, Stamler JS. Protein S-nitrosylation: purview and parameters. Nat Rev Mol Cell Biol. 2005;6:150–166. doi: 10.1038/nrm1569. [DOI] [PubMed] [Google Scholar]
- 2.Finkel T. Oxidant signals and oxidative stress. Curr Opin Cell Biol. 2003;15:247–254. doi: 10.1016/s0955-0674(03)00002-4. [DOI] [PubMed] [Google Scholar]
- 3.Rhee SG. Cell signaling. H2O2, a necessary evil for cell signaling. Science. 2006;312:1882–1883. doi: 10.1126/science.1130481. [DOI] [PubMed] [Google Scholar]
- 4.Rhee SG, Bae YS, Lee SR, Kwon J. Hydrogen peroxide: a key messenger that modulates protein phosphorylation through cysteine oxidation. Sci STKE 2000. 2000:PE1. doi: 10.1126/stke.2000.53.pe1. [DOI] [PubMed] [Google Scholar]
- 5.Bolotina VM, Najibi S, Palacino JJ, Pagano PJ, Cohen RA. Nitric oxide directly activates calcium-dependent potassium channels in vascular smooth muscle. Nature. 1994;368:850–853. doi: 10.1038/368850a0. [DOI] [PubMed] [Google Scholar]
- 6.Moro MA, Russel RJ, Cellek S, Lizasoain I, Su Y, Darley-Usmar VM, Radomski MW, Moncada S. cGMP mediates the vascular and platelet actions of nitric oxide: confirmation using an inhibitor of the soluble guanylyl cyclase. Proc Natl Acad Sci U S A. 1996;93:1480–1485. doi: 10.1073/pnas.93.4.1480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cohen RA, Adachi T. Nitric-oxide-induced vasodilatation: regulation by physiologic s-glutathiolation and pathologic oxidation of the sarcoplasmic endoplasmic reticulum calcium ATPase. Trends in cardiovascular medicine. 2006;16:109–114. doi: 10.1016/j.tcm.2006.02.001. [DOI] [PubMed] [Google Scholar]
- 8.Adachi T, Weisbrod RM, Pimentel DR, Ying J, Sharov VS, Schoneich C, Cohen RA. S-Glutathiolation by peroxynitrite activates SERCA during arterial relaxation by nitric oxide. Nature medicine. 2004;10:1200–1207. doi: 10.1038/nm1119. [DOI] [PubMed] [Google Scholar]
- 9.Liu L, Yan Y, Zeng M, Zhang J, Hanes MA, Ahearn G, McMahon TJ, Dickfeld T, Marshall HE, Que LG, Stamler JS. Essential roles of S-nitrosothiols in vascular homeostasis and endotoxic shock. Cell. 2004;116:617–628. doi: 10.1016/s0092-8674(04)00131-x. [DOI] [PubMed] [Google Scholar]
- 10.Stamler JS, Simon DI, Osborne JA, Mullins ME, Jaraki O, Michel T, Singel DJ, Loscalzo J. S-nitrosylation of proteins with nitric oxide: synthesis and characterization of biologically active compounds. Proc Natl Acad Sci U S A. 1992;89:444–448. doi: 10.1073/pnas.89.1.444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stamler JS. Redox signaling: nitrosylation and related target interactions of nitric oxide. Cell. 1994;78:931–936. doi: 10.1016/0092-8674(94)90269-0. [DOI] [PubMed] [Google Scholar]
- 12.Mayer B, Pfeiffer S, Schrammel A, Koesling D, Schmidt K, Brunner F. A new pathway of nitric oxide/cyclic GMP signaling involving S-nitrosoglutathione. J Biol Chem. 1998;273:3264–3270. doi: 10.1074/jbc.273.6.3264. [DOI] [PubMed] [Google Scholar]
- 13.Sayed N, Baskaran P, Ma X, van den Akker F, Beuve A. Desensitization of soluble guanylyl cyclase, the NO receptor, by S-nitrosylation. Proc Natl Acad Sci U S A. 2007;104:12312–12317. doi: 10.1073/pnas.0703944104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gow AJ, Chen Q, Hess DT, Day BJ, Ischiropoulos H, Stamler JS. Basal and stimulated protein S-nitrosylation in multiple cell types and tissues. J Biol Chem. 2002;277:9637–9640. doi: 10.1074/jbc.C100746200. [DOI] [PubMed] [Google Scholar]
- 15.Huang Y, Man HY, Sekine-Aizawa Y, Han Y, Juluri K, Luo H, Cheah J, Lowenstein C, Huganir RL, Snyder SH. S-nitrosylation of N-ethylmaleimide sensitive factor mediates surface expression of AMPA receptors. Neuron. 2005;46:533–540. doi: 10.1016/j.neuron.2005.03.028. [DOI] [PubMed] [Google Scholar]
- 16.Cheah JH, Kim SF, Hester LD, Clancy KW, Patterson SE, 3rd, Papadopoulos V, Snyder SH. NMDA receptor-nitric oxide transmission mediates neuronal iron homeostasis via the GTPase Dexras1. Neuron. 2006;51:431–440. doi: 10.1016/j.neuron.2006.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xu D, Rovira II, Finkel T. Oxidants painting the cysteine chapel: redox regulation of PTPs. Dev Cell. 2002;2:251–252. doi: 10.1016/s1534-5807(02)00132-6. [DOI] [PubMed] [Google Scholar]
- 18.Iwakiri Y, Satoh A, Chatterjee S, Toomre DK, Chalouni CM, Fulton D, Groszmann RJ, Shah VH, Sessa WC. Nitric oxide synthase generates nitric oxide locally to regulate compartmentalized protein S-nitrosylation and protein trafficking. Proc Natl Acad Sci U S A. 2006;103:19777–19782. doi: 10.1073/pnas.0605907103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gaston B, Singel D, Doctor A, Stamler JS. S-nitrosothiol signaling in respiratory biology. Am J Respir Crit Care Med. 2006;173:1186–1193. doi: 10.1164/rccm.200510-1584PP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Que LG, Liu L, Yan Y, Whitehead GS, Gavett SH, Schwartz DA, Stamler JS. Protection from experimental asthma by an endogenous bronchodilator. Science. 2005;308:1618–1621. doi: 10.1126/science.1108228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sundaresan M, Yu ZX, Ferrans VJ, Irani K, Finkel T. Requirement for generation of H2O2 for platelet-derived growth factor signal transduction. Science. 1995;270:296–299. doi: 10.1126/science.270.5234.296. [DOI] [PubMed] [Google Scholar]
- 22.Bae YS, Kang SW, Seo MS, Baines IC, Tekle E, Chock PB, Rhee SG. Epidermal growth factor (EGF)-induced generation of hydrogen peroxide. Role in EGF receptor-mediated tyrosine phosphorylation. J Biol Chem. 1997;272:217–221. [PubMed] [Google Scholar]
- 23.Ushio-Fukai M, Alexander RW, Akers M, Yin Q, Fujio Y, Walsh K, Griendling KK. Reactive oxygen species mediate the activation of Akt/protein kinase B by angiotensin II in vascular smooth muscle cells. J Biol Chem. 1999;274:22699–22704. doi: 10.1074/jbc.274.32.22699. [DOI] [PubMed] [Google Scholar]
- 24.Ricciardolo FL, Sterk PJ, Gaston B, Folkerts G. Nitric oxide in health and disease of the respiratory system. Physiol Rev. 2004;84:731–765. doi: 10.1152/physrev.00034.2003. [DOI] [PubMed] [Google Scholar]
- 25.Lambeth JD. NOX enzymes and the biology of reactive oxygen. Nat Rev Immunol. 2004;4:181–189. doi: 10.1038/nri1312. [DOI] [PubMed] [Google Scholar]
- 26.Bienert GP, Schjoerring JK, Jahn TP. Membrane transport of hydrogen peroxide. Biochim Biophys Acta. 2006;1758:994–1003. doi: 10.1016/j.bbamem.2006.02.015. [DOI] [PubMed] [Google Scholar]
- 27.Bienert GP, Moller AL, Kristiansen KA, Schulz A, Moller IM, Schjoerring JK, Jahn TP. Specific aquaporins facilitate the diffusion of hydrogen peroxide across membranes. J Biol Chem. 2007;282:1183–1192. doi: 10.1074/jbc.M603761200. [DOI] [PubMed] [Google Scholar]
- 28.Herrera M, Hong NJ, Garvin JL. Aquaporin-1 transports NO across cell membranes. Hypertension. 2006;48:157–164. doi: 10.1161/01.HYP.0000223652.29338.77. [DOI] [PubMed] [Google Scholar]
- 29.Meng TC, Fukada T, Tonks NK. Reversible oxidation and inactivation of protein tyrosine phosphatases in vivo. Mol Cell. 2002;9:387–399. doi: 10.1016/s1097-2765(02)00445-8. [DOI] [PubMed] [Google Scholar]
- 30.Poole LB, Karplus PA, Claiborne A. Protein sulfenic acids in redox signaling. Annu Rev Pharmacol Toxicol. 2004;44:325–347. doi: 10.1146/annurev.pharmtox.44.101802.121735. [DOI] [PubMed] [Google Scholar]
- 31.Forman HJ, Fukuto JM, Torres M. Redox signaling: thiol chemistry defines which reactive oxygen and nitrogen species can act as second messengers. Am J Physiol Cell Physiol. 2004;287:C246–256. doi: 10.1152/ajpcell.00516.2003. [DOI] [PubMed] [Google Scholar]
- 32.Kim SO, Merchant K, Nudelman R, Beyer WF, Jr, Keng T, DeAngelo J, Hausladen A, Stamler JS. OxyR: a molecular code for redox-related signaling. Cell. 2002;109:383–396. doi: 10.1016/s0092-8674(02)00723-7. [DOI] [PubMed] [Google Scholar]
- 33.Jacob C, Giles GI, Giles NM, Sies H. Sulfur and selenium: the role of oxidation state in protein structure and function. Angewandte Chemie (International ed. 2003;42:4742–4758. doi: 10.1002/anie.200300573. [DOI] [PubMed] [Google Scholar]
- 34.Mallis RJ, Thomas JA. Effect of S-nitrosothiols on cellular glutathione and reactive protein sulfhydryls. Arch Biochem Biophys. 2000;383:60–69. doi: 10.1006/abbi.2000.2048. [DOI] [PubMed] [Google Scholar]
- 35.Ji Y, Akerboom TP, Sies H, Thomas JA. S-nitrosylation and S-glutathiolation of protein sulfhydryls by S-nitroso glutathione. Arch Biochem Biophys. 1999;362:67–78. doi: 10.1006/abbi.1998.1013. [DOI] [PubMed] [Google Scholar]
- 36.Sitia R, Molteni SN. Stress, protein (mis)folding, and signaling: the redox connection. Sci STKE 2004. 2004:pe27. doi: 10.1126/stke.2392004pe27. [DOI] [PubMed] [Google Scholar]
- 37.Paget MS, Buttner MJ. Thiol-based regulatory switches. Annu Rev Genet. 2003;37:91–121. doi: 10.1146/annurev.genet.37.110801.142538. [DOI] [PubMed] [Google Scholar]
- 38.Georgiou G. How to flip the (redox) switch. Cell. 2002;111:607–610. doi: 10.1016/s0092-8674(02)01165-0. [DOI] [PubMed] [Google Scholar]
- 39.Rhee SG, Chae HZ, Kim K. Peroxiredoxins: a historical overview and speculative preview of novel mechanisms and emerging concepts in cell signaling. Free Radic Biol Med. 2005;38:1543–1552. doi: 10.1016/j.freeradbiomed.2005.02.026. [DOI] [PubMed] [Google Scholar]
- 40.Haridas V, Kim SO, Nishimura G, Hausladen A, Stamler JS, Gutterman JU. Avicinylation (thioesterification): a protein modification that can regulate the response to oxidative and nitrosative stress. Proc Natl Acad Sci U S A. 2005;102:10088–10093. doi: 10.1073/pnas.0504430102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rossi A, Kapahi P, Natoli G, Takahashi T, Chen Y, Karin M, Santoro MG. Anti-inflammatory cyclopentenone prostaglandins are direct inhibitors of IkappaB kinase. Nature. 2000;403:103–108. doi: 10.1038/47520. [DOI] [PubMed] [Google Scholar]
- 42.Finkelstein EI, Ruben J, Koot CW, Hristova M, van der Vliet A. Regulation of constitutive neutrophil apoptosis by the alpha, beta-unsaturated aldehydes acrolein and 4-hydroxynonenal. Am J Physiol Lung Cell Mol Physiol. 2005;289:L1019–1028. doi: 10.1152/ajplung.00227.2005. [DOI] [PubMed] [Google Scholar]
- 43.Resh MD. Palmitoylation of ligands, receptors, and intracellular signaling molecules. Sci STKE 2006. 2006:re14. doi: 10.1126/stke.3592006re14. [DOI] [PubMed] [Google Scholar]
- 44.Nedospasov A, Rafikov R, Beda N, Nudler E. An autocatalytic mechanism of protein nitrosylation. Proc Natl Acad Sci U S A. 2000;97:13543–13548. doi: 10.1073/pnas.250398197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Foster MW, McMahon TJ, Stamler JS. S-nitrosylation in health and disease. Trends in molecular medicine. 2003;9:160–168. doi: 10.1016/s1471-4914(03)00028-5. [DOI] [PubMed] [Google Scholar]
- 46.Tannenbaum SR, White FM. Regulation and specificity of S-nitrosylation and denitrosylation. ACS chemical biology. 2006;1:615–618. doi: 10.1021/cb600439h. [DOI] [PubMed] [Google Scholar]
- 47.Shelton MD, Chock PB, Mieyal JJ. Glutaredoxin: role in reversible protein s-glutathionylation and regulation of redox signal transduction and protein translocation. Antioxid Redox Signal. 2005;7:348–366. doi: 10.1089/ars.2005.7.348. [DOI] [PubMed] [Google Scholar]
- 48.Watabe S, Kohno H, Kouyama H, Hiroi T, Yago N, Nakazawa T. Purification and characterization of a substrate protein for mitochondrial ATP-dependent protease in bovine adrenal cortex. Journal of biochemistry. 1994;115:648–654. doi: 10.1093/oxfordjournals.jbchem.a124390. [DOI] [PubMed] [Google Scholar]
- 49.Yang KS, Kang SW, Woo HA, Hwang SC, Chae HZ, Kim K, Rhee SG. Inactivation of human peroxiredoxin I during catalysis as the result of the oxidation of the catalytic site cysteine to cysteine-sulfinic acid. J Biol Chem. 2002;277:38029–38036. doi: 10.1074/jbc.M206626200. [DOI] [PubMed] [Google Scholar]
- 50.Rhee SG, Kang SW, Jeong W, Chang TS, Yang KS, Woo HA. Intracellular messenger function of hydrogen peroxide and its regulation by peroxiredoxins. Curr Opin Cell Biol. 2005;17:183–189. doi: 10.1016/j.ceb.2005.02.004. [DOI] [PubMed] [Google Scholar]
- 51.Claiborne A, Yeh JI, Mallett TC, Luba J, Crane EJ, 3rd, Charrier V, Parsonage D. Protein-sulfenic acids: diverse roles for an unlikely player in enzyme catalysis and redox regulation. Biochemistry. 1999;38:15407–15416. doi: 10.1021/bi992025k. [DOI] [PubMed] [Google Scholar]
- 52.Forrester MT, Stamler JS. A classification scheme for redox-based modifications of proteins. Am J Respir Cell Mol Biol. 2007;36:135–137. doi: 10.1165/rcmb.2006-001ED. [DOI] [PubMed] [Google Scholar]
- 53.Mannick JB, Hausladen A, Liu L, Hess DT, Zeng M, Miao QX, Kane LS, Gow AJ, Stamler JS. Fas-induced caspase denitrosylation. Science. 1999;284:651–654. doi: 10.1126/science.284.5414.651. [DOI] [PubMed] [Google Scholar]
- 54.Takahashi H, Shin Y, Cho SJ, Zago WM, Nakamura T, Gu Z, Ma Y, Furukawa H, Liddington R, Zhang D, Tong G, Chen HS, Lipton SA. Hypoxia enhances S-nitrosylation-mediated NMDA receptor inhibition via a thiol oxygen sensor motif. Neuron. 2007;53:53–64. doi: 10.1016/j.neuron.2006.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ckless K, Reynaert NL, Taatjes DJ, Lounsbury KM, van der Vliet A, Janssen-Heininger Y. In situ detection and visualization of S-nitrosylated proteins following chemical derivatization: identification of Ran GTPase as a target for S-nitrosylation. Nitric Oxide. 2004;11:216–227. doi: 10.1016/j.niox.2004.06.002. [DOI] [PubMed] [Google Scholar]
- 56.Jaffrey SR, Erdjument-Bromage H, Ferris CD, Tempst P, Snyder SH. Protein S-nitrosylation: a physiological signal for neuronal nitric oxide. Nat Cell Biol. 2001;3:193–197. doi: 10.1038/35055104. [DOI] [PubMed] [Google Scholar]
- 57.Eu JP, Sun J, Xu L, Stamler JS, Meissner G. The skeletal muscle calcium release channel: coupled O2 sensor and NO signaling functions. Cell. 2000;102:499–509. doi: 10.1016/s0092-8674(00)00054-4. [DOI] [PubMed] [Google Scholar]
- 58.Kim SF, Huri DA, Snyder SH. Inducible nitric oxide synthase binds, S-nitrosylates, and activates cyclooxygenase-2. Science. 2005;310:1966–1970. doi: 10.1126/science.1119407. [DOI] [PubMed] [Google Scholar]
- 59.Patel JM, Zhang J, Block ER. Nitric oxide-induced inhibition of lung endothelial cell nitric oxide synthase via interaction with allosteric thiols: role of thioredoxin in regulation of catalytic activity. Am J Respir Cell Mol Biol. 1996;15:410–419. doi: 10.1165/ajrcmb.15.3.8810647. [DOI] [PubMed] [Google Scholar]
- 60.Ravi K, Brennan LA, Levic S, Ross PA, Black SM. S-nitrosylation of endothelial nitric oxide synthase is associated with monomerization and decreased enzyme activity. Proc Natl Acad Sci U S A. 2004;101:2619–2624. doi: 10.1073/pnas.0300464101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wood ZA, Poole LB, Karplus PA. Peroxiredoxin evolution and the regulation of hydrogen peroxide signaling. Science. 2003;300:650–653. doi: 10.1126/science.1080405. [DOI] [PubMed] [Google Scholar]
- 62.Choi MH, Lee IK, Kim GW, Kim BU, Han YH, Yu DY, Park HS, Kim KY, Lee JS, Choi C, Bae YS, Lee BI, Rhee SG, Kang SW. Regulation of PDGF signalling and vascular remodelling by peroxiredoxin II. Nature. 2005;435:347–353. doi: 10.1038/nature03587. [DOI] [PubMed] [Google Scholar]
- 63.Budanov AV, Sablina AA, Feinstein E, Koonin EV, Chumakov PM. Regeneration of peroxiredoxins by p53-regulated sestrins, homologs of bacterial AhpD. Science. 2004;304:596–600. doi: 10.1126/science.1095569. [DOI] [PubMed] [Google Scholar]
- 64.Chang TS, Jeong W, Woo HA, Lee SM, Park S, Rhee SG. Characterization of mammalian sulfiredoxin and its reactivation of hyperoxidized peroxiredoxin through reduction of cysteine sulfinic acid in the active site to cysteine. J Biol Chem. 2004;279:50994–51001. doi: 10.1074/jbc.M409482200. [DOI] [PubMed] [Google Scholar]
- 65.Jeong W, Park SJ, Chang TS, Lee DY, Rhee SG. Molecular mechanism of the reduction of cysteine sulfinic acid of peroxiredoxin to cysteine by mammalian sulfiredoxin. J Biol Chem. 2006;281:14400–14407. doi: 10.1074/jbc.M511082200. [DOI] [PubMed] [Google Scholar]
- 66.Biteau B, Labarre J, Toledano MB. ATP-dependent reduction of cysteine-sulphinic acid by S. cerevisiae sulphiredoxin. Nature. 2003;425:980–984. doi: 10.1038/nature02075. [DOI] [PubMed] [Google Scholar]
- 67.Phalen TJ, Weirather K, Deming PB, Anathy V, Howe AK, van der Vliet A, Jonsson TJ, Poole LB, Heintz NH. Oxidation state governs structural transitions in peroxiredoxin II that correlate with cell cycle arrest and recovery. The Journal of cell biology. 2006;175:779–789. doi: 10.1083/jcb.200606005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hirotsu S, Abe Y, Okada K, Nagahara N, Hori H, Nishino T, Hakoshima T. Crystal structure of a multifunctional 2-Cys peroxiredoxin heme-binding protein 23 kDa/proliferation-associated gene product. Proc Natl Acad Sci U S A. 1999;96:12333–12338. doi: 10.1073/pnas.96.22.12333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Veal EA, Findlay VJ, Day AM, Bozonet SM, Evans JM, Quinn J, Morgan BA. A 2-Cys peroxiredoxin regulates peroxide-induced oxidation and activation of a stress-activated MAP kinase. Mol Cell. 2004;15:129–139. doi: 10.1016/j.molcel.2004.06.021. [DOI] [PubMed] [Google Scholar]
- 70.Kim YJ, Lee WS, Ip C, Chae HZ, Park EM, Park YM. Prx1 suppresses radiation-induced c-Jun NH2-terminal kinase signaling in lung cancer cells through interaction with the glutathione S-transferase Pi/c-Jun NH2-terminal kinase complex. Cancer Res. 2006;66:7136–7142. doi: 10.1158/0008-5472.CAN-05-4446. [DOI] [PubMed] [Google Scholar]
- 71.Egler RA, Fernandes E, Rothermund K, Sereika S, de Souza-Pinto N, Jaruga P, Dizdaroglu M, Prochownik EV. Regulation of reactive oxygen species, DNA damage, and c-Myc function by peroxiredoxin 1. Oncogene. 2005;24:8038–8050. doi: 10.1038/sj.onc.1208821. [DOI] [PubMed] [Google Scholar]
- 72.Cao Z, Roszak AW, Gourlay LJ, Lindsay JG, Isaacs NW. Bovine mitochondrial peroxiredoxin III forms a two-ring catenane. Structure. 2005;13:1661–1664. doi: 10.1016/j.str.2005.07.021. [DOI] [PubMed] [Google Scholar]
- 73.Jang HH, Kim SY, Park SK, Jeon HS, Lee YM, Jung JH, Lee SY, Chae HB, Jung YJ, Lee KO, Lim CO, Chung WS, Bahk JD, Yun DJ, Cho MJ. Phosphorylation and concomitant structural changes in human 2-Cys peroxiredoxin isotype I differentially regulate its peroxidase and molecular chaperone functions. FEBS Lett. 2006;580:351–355. doi: 10.1016/j.febslet.2005.12.030. [DOI] [PubMed] [Google Scholar]
- 74.Kim HJ, Chae HZ, Kim YJ, Kim YH, Hwangs TS, Park EM, Park YM. Preferential elevation of Prx I and Trx expression in lung cancer cells following hypoxia and in human lung cancer tissues. Cell Biol Toxicol. 2003;19:285–298. doi: 10.1023/b:cbto.0000004952.07979.3d. [DOI] [PubMed] [Google Scholar]
- 75.Kim YJ, Ahn JY, Liang P, Ip C, Zhang Y, Park YM. Human prx1 gene is a target of Nrf2 and is up-regulated by hypoxia/reoxygenation: implication to tumor biology. Cancer Res. 2007;67:546–554. doi: 10.1158/0008-5472.CAN-06-2401. [DOI] [PubMed] [Google Scholar]
- 76.Das KC, Pahl PM, Guo XL, White CW. Induction of peroxiredoxin gene expression by oxygen in lungs of newborn primates. Am J Respir Cell Mol Biol. 2001;25:226–232. doi: 10.1165/ajrcmb.25.2.4314. [DOI] [PubMed] [Google Scholar]
- 77.Phelan SA, Beier DR, Higgins DC, Paigen B. Confirmation and high resolution mapping of an atherosclerosis susceptibility gene in mice on Chromosome 1. Mamm Genome. 2002;13:548–553. doi: 10.1007/s00335-002-2196-1. [DOI] [PubMed] [Google Scholar]
- 78.Liu L, Hausladen A, Zeng M, Que L, Heitman J, Stamler JS. A metabolic enzyme for S-nitrosothiol conserved from bacteria to humans. Nature. 2001;410:490–494. doi: 10.1038/35068596. [DOI] [PubMed] [Google Scholar]
- 79.Haqqani AS, Do SK, Birnboim HC. The role of a formaldehyde dehydrogenase-glutathione pathway in protein S-nitrosation in mammalian cells. Nitric Oxide. 2003;9:172–181. doi: 10.1016/j.niox.2003.11.003. [DOI] [PubMed] [Google Scholar]
- 80.Whalen EJ, Foster MW, Matsumoto A, Ozawa K, Violin JD, Que LG, Nelson CD, Benhar M, Keys JR, Rockman HA, Koch WJ, Daaka Y, Lefkowitz RJ, Stamler JS. Regulation of beta-adrenergic receptor signaling by S-nitrosylation of G-protein-coupled receptor kinase 2. Cell. 2007;129:511–522. doi: 10.1016/j.cell.2007.02.046. [DOI] [PubMed] [Google Scholar]
- 81.Wu H, Romieu I, Sienra-Monge JJ, Estela Del Rio-Navarro B, Anderson DM, Jenchura CA, Li H, Ramirez-Aguilar M, Del Carmen Lara-Sanchez I, London SJ. Genetic variation in S-nitrosoglutathione reductase (GSNOR) and childhood asthma. The Journal of allergy and clinical immunology. 2007;120:322–328. doi: 10.1016/j.jaci.2007.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kaneki M, Shimizu N, Yamada D, Chang K. Nitrosative stress and pathogenesis of insulin resistance. Antioxid Redox Signal. 2007;9:319–329. doi: 10.1089/ars.2006.1464. [DOI] [PubMed] [Google Scholar]
- 83.Berndt C, Lillig CH, Holmgren A. Thiol-based mechanisms of the thioredoxin and glutaredoxin systems: implications for diseases in the cardiovascular system. Am J Physiol Heart Circ Physiol. 2007;292:H1227–1236. doi: 10.1152/ajpheart.01162.2006. [DOI] [PubMed] [Google Scholar]
- 84.Seemann S, Hainaut P. Roles of thioredoxin reductase 1 and APE/Ref-1 in the control of basal p53 stability and activity. Oncogene. 2005;24:3853–3863. doi: 10.1038/sj.onc.1208549. [DOI] [PubMed] [Google Scholar]
- 85.Harper R, Wu K, Chang MM, Yoneda K, Pan R, Reddy SP, Wu R. Activation of nuclear factor-kappa b transcriptional activity in airway epithelial cells by thioredoxin but not by N-acetyl-cysteine and glutathione. Am J Respir Cell Mol Biol. 2001;25:178–185. doi: 10.1165/ajrcmb.25.2.4471. [DOI] [PubMed] [Google Scholar]
- 86.Hayashi T, Ueno Y, Okamoto T. Oxidoreductive regulation of nuclear factor kappa B. Involvement of a cellular reducing catalyst thioredoxin. J Biol Chem. 1993;268:11380–11388. [PubMed] [Google Scholar]
- 87.Hwang CY, Ryu YS, Chung MS, Kim KD, Park SS, Chae SK, Chae HZ, Kwon KS. Thioredoxin modulates activator protein 1 (AP-1) activity and p27Kip1 degradation through direct interaction with Jab1. Oncogene. 2004;23:8868–8875. doi: 10.1038/sj.onc.1208116. [DOI] [PubMed] [Google Scholar]
- 88.Hayashi S, Hajiro-Nakanishi K, Makino Y, Eguchi H, Yodoi J, Tanaka H. Functional modulation of estrogen receptor by redox state with reference to thioredoxin as a mediator. Nucleic Acids Res. 1997;25:4035–4040. doi: 10.1093/nar/25.20.4035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Makino Y, Yoshikawa N, Okamoto K, Hirota K, Yodoi J, Makino I, Tanaka H. Direct association with thioredoxin allows redox regulation of glucocorticoid receptor function. J Biol Chem. 1999;274:3182–3188. doi: 10.1074/jbc.274.5.3182. [DOI] [PubMed] [Google Scholar]
- 90.Masutani H, Hirota K, Sasada T, Ueda-Taniguchi Y, Taniguchi Y, Sono H, Yodoi J. Transactivation of an inducible anti-oxidative stress protein, human thioredoxin by HTLV-I Tax. Immunol Lett. 1996;54:67–71. doi: 10.1016/s0165-2478(96)02651-x. [DOI] [PubMed] [Google Scholar]
- 91.Rubartelli A, Bajetto A, Allavena G, Wollman E, Sitia R. Secretion of thioredoxin by normal and neoplastic cells through a leaderless secretory pathway. J Biol Chem. 1992;267:24161–24164. [PubMed] [Google Scholar]
- 92.Callister ME, Burke-Gaffney A, Quinlan GJ, Nicholson AG, Florio R, Nakamura H, Yodoi J, Evans TW. Extracellular thioredoxin levels are increased in patients with acute lung injury. Thorax. 2006;61:521–527. doi: 10.1136/thx.2005.053041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Wahlgren CM, Pekkari K. Elevated thioredoxin after angioplasty in peripheral arterial disease. Eur J Vasc Endovasc Surg. 2005;29:281–286. doi: 10.1016/j.ejvs.2004.12.020. [DOI] [PubMed] [Google Scholar]
- 94.Jikimoto T, Nishikubo Y, Koshiba M, Kanagawa S, Morinobu S, Morinobu A, Saura R, Mizuno K, Kondo S, Toyokuni S, Nakamura H, Yodoi J, Kumagai S. Thioredoxin as a biomarker for oxidative stress in patients with rheumatoid arthritis. Mol Immunol. 2002;38:765–772. doi: 10.1016/s0161-5890(01)00113-4. [DOI] [PubMed] [Google Scholar]
- 95.Pekkari K, Holmgren A. Truncated thioredoxin: physiological functions and mechanism. Antioxid Redox Signal. 2004;6:53–61. doi: 10.1089/152308604771978345. [DOI] [PubMed] [Google Scholar]
- 96.Bertini R, Howard OM, Dong HF, Oppenheim JJ, Bizzarri C, Sergi R, Caselli G, Pagliei S, Romines B, Wilshire JA, Mengozzi M, Nakamura H, Yodoi J, Pekkari K, Gurunath R, Holmgren A, Herzenberg LA, Ghezzi P. Thioredoxin, a redox enzyme released in infection and inflammation, is a unique chemoattractant for neutrophils, monocytes, and T cells. J Exp Med. 1999;189:1783–1789. doi: 10.1084/jem.189.11.1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Pekkari K, Goodarzi MT, Scheynius A, Holmgren A, Avila-Carino J. Truncated thioredoxin (Trx80) induces differentiation of human CD14+ monocytes into a novel cell type (TAMs) via activation of the MAP kinases p38, ERK, and JNK. Blood. 2005;105:1598–1605. doi: 10.1182/blood-2004-04-1577. [DOI] [PubMed] [Google Scholar]
- 98.Schwertassek U, Balmer Y, Gutscher M, Weingarten L, Preuss M, Engelhard J, Winkler M, Dick TP. Selective redox regulation of cytokine receptor signaling by extracellular thioredoxin-1. Embo J. 2007;26:3086–3097. doi: 10.1038/sj.emboj.7601746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Fernandes AP, Holmgren A. Glutaredoxins: glutathione-dependent redox enzymes with functions far beyond a simple thioredoxin backup system. Antioxid Redox Signal. 2004;6:63–74. doi: 10.1089/152308604771978354. [DOI] [PubMed] [Google Scholar]
- 100.Reynaert NL, Ckless K, Guala AS, Wouters EF, van der Vliet A, Janssen-Heininger YM. In situ detection of S-glutathionylated proteins following glutaredoxin-1 catalyzed cysteine derivatization. Biochim Biophys Acta. 2006;1760:380–387. doi: 10.1016/j.bbagen.2006.01.006. [DOI] [PubMed] [Google Scholar]
- 101.Gravina SA, Mieyal JJ. Thioltransferase is a specific glutathionyl mixed disulfide oxidoreductase. Biochemistry. 1993;32:3368–3376. doi: 10.1021/bi00064a021. [DOI] [PubMed] [Google Scholar]
- 102.Findlay VJ, Townsend DM, Morris TE, Fraser JP, He L, Tew KD. A novel role for human sulfiredoxin in the reversal of glutathionylation. Cancer Res. 2006;66:6800–6806. doi: 10.1158/0008-5472.CAN-06-0484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Okuda M, Inoue N, Azumi H, Seno T, Sumi Y, Hirata K, Kawashima S, Hayashi Y, Itoh H, Yodoi J, Yokoyama M. Expression of glutaredoxin in human coronary arteries: its potential role in antioxidant protection against atherosclerosis. Arterioscler Thromb Vasc Biol. 2001;21:1483–1487. doi: 10.1161/hq0901.095550. [DOI] [PubMed] [Google Scholar]
- 104.Nonaka K, Kume N, Urata Y, Seto S, Kohno T, Honda S, Ikeda S, Muroya T, Ikeda Y, Ihara Y, Kita T, Kondo T. Serum levels of S-glutathionylated proteins as a risk-marker for arteriosclerosis obliterans. Circ J. 2007;71:100–105. doi: 10.1253/circj.71.100. [DOI] [PubMed] [Google Scholar]
- 105.Reynaert NL, Wouters EF, Janssen-Heininger YM. Modulation of glutaredoxin-1 expression in a mouse model of allergic airway disease. Am J Respir Cell Mol Biol. 2007;36:147–151. doi: 10.1165/rcmb.2006-0259RC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Hashemy SI, Johansson C, Berndt C, Lillig CH, Holmgren A. Oxidation and S-Nitrosylation of Cysteines in Human Cytosolic and Mitochondrial Glutaredoxins: EFFECTS ON STRUCTURE AND ACTIVITY. J Biol Chem. 2007;282:14428–14436. doi: 10.1074/jbc.M700927200. [DOI] [PubMed] [Google Scholar]
- 107.Haendeler J, Hoffmann J, Tischler V, Berk BC, Zeiher AM, Dimmeler S. Redox regulatory and anti-apoptotic functions of thioredoxin depend on S-nitrosylation at cysteine 69. Nat Cell Biol. 2002;4:743–749. doi: 10.1038/ncb851. [DOI] [PubMed] [Google Scholar]
- 108.Sumbayev VV. S-nitrosylation of thioredoxin mediates activation of apoptosis signal-regulating kinase 1. Arch Biochem Biophys. 2003;415:133–136. doi: 10.1016/s0003-9861(03)00199-1. [DOI] [PubMed] [Google Scholar]
- 109.Casagrande S, Bonetto V, Fratelli M, Gianazza E, Eberini I, Massignan T, Salmona M, Chang G, Holmgren A, Ghezzi P. Glutathionylation of human thioredoxin: a possible crosstalk between the glutathione and thioredoxin systems. Proc Natl Acad Sci U S A. 2002;99:9745–9749. doi: 10.1073/pnas.152168599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Fratelli M, Demol H, Puype M, Casagrande S, Eberini I, Salmona M, Bonetto V, Mengozzi M, Duffieux F, Miclet E, Bachi A, Vandekerckhove J, Gianazza E, Ghezzi P. Identification by redox proteomics of glutathionylated proteins in oxidatively stressed human T lymphocytes. Proc Natl Acad Sci U S A. 2002;99:3505–3510. doi: 10.1073/pnas.052592699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Mitchell DA, Morton SU, Fernhoff NB, Marletta MA. Thioredoxin is required for S-nitrosation of procaspase-3 and the inhibition of apoptosis in Jurkat cells. Proc Natl Acad Sci U S A. 2007;104:11609–11614. doi: 10.1073/pnas.0704898104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Mitchell DA, Marletta MA. Thioredoxin catalyzes the S-nitrosation of the caspase-3 active site cysteine. Nature chemical biology. 2005;1:154–158. doi: 10.1038/nchembio720. [DOI] [PubMed] [Google Scholar]
- 113.Nikitovic D, Holmgren A. S-nitrosoglutathione is cleaved by the thioredoxin system with liberation of glutathione and redox regulating nitric oxide. J Biol Chem. 1996;271:19180–19185. doi: 10.1074/jbc.271.32.19180. [DOI] [PubMed] [Google Scholar]
- 114.Jones DP, Go YM, Anderson CL, Ziegler TR, Kinkade JM, Jr, Kirlin WG. Cysteine/cystine couple is a newly recognized node in the circuitry for biologic redox signaling and control. Faseb J. 2004;18:1246–1248. doi: 10.1096/fj.03-0971fje. [DOI] [PubMed] [Google Scholar]
- 115.Wang G, Moniri NH, Ozawa K, Stamler JS, Daaka Y. Nitric oxide regulates endocytosis by S-nitrosylation of dynamin. Proc Natl Acad Sci U S A. 2006;103:1295–1300. doi: 10.1073/pnas.0508354103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Lee SR, Kwon KS, Kim SR, Rhee SG. Reversible inactivation of protein-tyrosine phosphatase 1B in A431 cells stimulated with epidermal growth factor. J Biol Chem. 1998;273:15366–15372. doi: 10.1074/jbc.273.25.15366. [DOI] [PubMed] [Google Scholar]
- 117.Kwon J, Lee SR, Yang KS, Ahn Y, Kim YJ, Stadtman ER, Rhee SG. Reversible oxidation and inactivation of the tumor suppressor PTEN in cells stimulated with peptide growth factors. Proc Natl Acad Sci U S A. 2004;101:16419–16424. doi: 10.1073/pnas.0407396101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Sullivan DM, Wehr NB, Fergusson MM, Levine RL, Finkel T. Identification of oxidant-sensitive proteins: TNF-alpha induces protein glutathiolation. Biochemistry. 2000;39:11121–11128. doi: 10.1021/bi0007674. [DOI] [PubMed] [Google Scholar]
- 119.Barrett WC, DeGnore JP, Konig S, Fales HM, Keng YF, Zhang ZY, Yim MB, Chock PB. Regulation of PTP1B via glutathionylation of the active site cysteine 215. Biochemistry. 1999;38:6699–6705. doi: 10.1021/bi990240v. [DOI] [PubMed] [Google Scholar]
- 120.Rinna A, Torres M, Forman HJ. Stimulation of the alveolar macrophage respiratory burst by ADP causes selective glutathionylation of protein tyrosine phosphatase 1B. Free Radic Biol Med. 2006;41:86–91. doi: 10.1016/j.freeradbiomed.2006.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Forman HJ. Use and abuse of exogenous H2O2 in studies of signal transduction. Free Radic Biol Med. 2007;42:926–932. doi: 10.1016/j.freeradbiomed.2007.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Li S, Whorton AR. Regulation of protein tyrosine phosphatase 1B in intact cells by S-nitrosothiols. Arch Biochem Biophys. 2003;410:269–279. doi: 10.1016/s0003-9861(02)00696-3. [DOI] [PubMed] [Google Scholar]
- 123.Xian M, Wang K, Chen X, Hou Y, McGill A, Zhou B, Zhang ZY, Cheng JP, Wang PG. Inhibition of protein tyrosine phosphatases by low-molecular-weight S-nitrosothiols and S-nitrosylated human serum albumin. Biochem Biophys Res Commun. 2000;268:310–314. doi: 10.1006/bbrc.2000.2117. [DOI] [PubMed] [Google Scholar]
- 124.Lim S, Clement MV. Phosphorylation of the survival kinase Akt by superoxide is dependent on an ascorbate-reversible oxidation of PTEN. Free Radic Biol Med. 2007;42:1178–1192. doi: 10.1016/j.freeradbiomed.2007.01.013. [DOI] [PubMed] [Google Scholar]
- 125.Mikkelsen RB, Wardman P. Biological chemistry of reactive oxygen and nitrogen and radiation-induced signal transduction mechanisms. Oncogene. 2003;22:5734–5754. doi: 10.1038/sj.onc.1206663. [DOI] [PubMed] [Google Scholar]
- 126.Barrett DM, Black SM, Todor H, Schmidt-Ullrich RK, Dawson KS, Mikkelsen RB. Inhibition of protein-tyrosine phosphatases by mild oxidative stresses is dependent on S-nitrosylation. J Biol Chem. 2005;280:14453–14461. doi: 10.1074/jbc.M411523200. [DOI] [PubMed] [Google Scholar]
- 127.Forrester MT, Foster MW, Stamler JS. Assessment and application of the biotin switch technique for examining protein S-nitrosylation under conditions of pharmacologically induced oxidative stress. J Biol Chem. 2007 doi: 10.1074/jbc.M609684200. [DOI] [PubMed] [Google Scholar]
- 128.von Montfort C, Sharov VS, Metzger S, Schoneich C, Sies H, Klotz LO. Singlet oxygen inactivates protein tyrosine phosphatase-1B by oxidation of the active site cysteine. Biol Chem. 2006;387:1399–1404. doi: 10.1515/BC.2006.175. [DOI] [PubMed] [Google Scholar]
- 129.Kamata H, Honda S, Maeda S, Chang L, Hirata H, Karin M. Reactive oxygen species promote TNFalpha-induced death and sustained JNK activation by inhibiting MAP kinase phosphatases. Cell. 2005;120:649–661. doi: 10.1016/j.cell.2004.12.041. [DOI] [PubMed] [Google Scholar]
- 130.Seth D, Rudolph J. Redox regulation of MAP kinase phosphatase 3. Biochemistry. 2006;45:8476–8487. doi: 10.1021/bi060157p. [DOI] [PubMed] [Google Scholar]
- 131.Levinthal DJ, Defranco DB. Reversible oxidation of ERK-directed protein phosphatases drives oxidative toxicity in neurons. J Biol Chem. 2005;280:5875–5883. doi: 10.1074/jbc.M410771200. [DOI] [PubMed] [Google Scholar]
- 132.Fox GC, Shafiq M, Briggs DC, Knowles PP, Collister M, Didmon MJ, Makrantoni V, Dickinson RJ, Hanrahan S, Totty N, Stark MJ, Keyse SM, McDonald NQ. Redox-mediated substrate recognition by Sdp1 defines a new group of tyrosine phosphatases. Nature. 2007;447:487–492. doi: 10.1038/nature05804. [DOI] [PubMed] [Google Scholar]
- 133.Mannick JB, Schonhoff C, Papeta N, Ghafourifar P, Szibor M, Fang K, Gaston B. S-Nitrosylation of mitochondrial caspases. The Journal of cell biology. 2001;154:1111–1116. doi: 10.1083/jcb.200104008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Lee YJ, Shacter E. Hydrogen peroxide inhibits activation, not activity, of cellular caspase-3 in vivo. Free Radic Biol Med. 2000;29:684–692. doi: 10.1016/s0891-5849(00)00366-x. [DOI] [PubMed] [Google Scholar]
- 135.Pan S, Berk BC. Glutathiolation regulates tumor necrosis factor-alpha-induced caspase-3 cleavage and apoptosis: key role for glutaredoxin in the death pathway. Circ Res. 2007;100:213–219. doi: 10.1161/01.RES.0000256089.30318.20. [DOI] [PubMed] [Google Scholar]
- 136.Hristova M, Heuvelmans S, van der Vliet A. GSH-dependent regulation of Fas-mediated caspase-8 activation by acrolein. FEBS Lett. 2007;581:361–367. doi: 10.1016/j.febslet.2006.12.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Gu Z, Kaul M, Yan B, Kridel SJ, Cui J, Strongin A, Smith JW, Liddington RC, Lipton SA. S-nitrosylation of matrix metalloproteinases: signaling pathway to neuronal cell death. Science. 2002;297:1186–1190. doi: 10.1126/science.1073634. [DOI] [PubMed] [Google Scholar]
- 138.Zhang Z, Oliver P, Lancaster JJ, Schwarzenberger PO, Joshi MS, Cork J, Kolls JK. Reactive oxygen species mediate tumor necrosis factor alpha-converting, enzyme-dependent ectodomain shedding induced by phorbol myristate acetate. Faseb J. 2001;15:303–305. doi: 10.1096/fj.00-0371fje. [DOI] [PubMed] [Google Scholar]
- 139.Shao MX, Nadel JA. Dual oxidase 1-dependent MUC5AC mucin expression in cultured human airway epithelial cells. Proc Natl Acad Sci U S A. 2005;102:767–772. doi: 10.1073/pnas.0408932102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Okamoto T, Akuta T, Tamura F, van Der Vliet A, Akaike T. Molecular mechanism for activation and regulation of matrix metalloproteinases during bacterial infections and respiratory inflammation. Biol Chem. 2004;385:997–1006. doi: 10.1515/BC.2004.130. [DOI] [PubMed] [Google Scholar]
- 141.Fu X, Kassim SY, Parks WC, Heinecke JW. Hypochlorous acid oxygenates the cysteine switch domain of pro-matrilysin (MMP-7). A mechanism for matrix metalloproteinase activation and atherosclerotic plaque rupture by myeloperoxidase. J Biol Chem. 2001;276:41279–41287. doi: 10.1074/jbc.M106958200. [DOI] [PubMed] [Google Scholar]
- 142.Fu X, Kassim SY, Parks WC, Heinecke JW. Hypochlorous acid generated by myeloperoxidase modifies adjacent tryptophan and glycine residues in the catalytic domain of matrix metalloproteinase-7 (matrilysin): an oxidative mechanism for restraining proteolytic activity during inflammation. J Biol Chem. 2003;278:28403–28409. doi: 10.1074/jbc.M304739200. [DOI] [PubMed] [Google Scholar]
- 143.Michaelis J, Vissers MC, Winterbourn CC. Different effects of hypochlorous acid on human neutrophil metalloproteinases: activation of collagenase and inactivation of collagenase and gelatinase. Arch Biochem Biophys. 1992;292:555–562. doi: 10.1016/0003-9861(92)90030-z. [DOI] [PubMed] [Google Scholar]
- 144.Winter J, Jakob U. Beyond transcription--new mechanisms for the regulation of molecular chaperones. Crit Rev Biochem Mol Biol. 2004;39:297–317. doi: 10.1080/10409230490900658. [DOI] [PubMed] [Google Scholar]
- 145.Bouwmeester T, Bauch A, Ruffner H, Angrand PO, Bergamini G, Croughton K, Cruciat C, Eberhard D, Gagneur J, Ghidelli S, Hopf C, Huhse B, Mangano R, Michon AM, Schirle M, Schlegl J, Schwab M, Stein MA, Bauer A, Casari G, Drewes G, Gavin AC, Jackson DB, Joberty G, Neubauer G, Rick J, Kuster B, Superti-Furga G. A physical and functional map of the human TNF-alpha/NF-kappa B signal transduction pathway. Nat Cell Biol. 2004;6:97–105. doi: 10.1038/ncb1086. [DOI] [PubMed] [Google Scholar]
- 146.Broemer M, Krappmann D, Scheidereit C. Requirement of Hsp90 activity for IkappaB kinase (IKK) biosynthesis and for constitutive and inducible IKK and NF-kappaB activation. Oncogene. 2004;23:5378–5386. doi: 10.1038/sj.onc.1207705. [DOI] [PubMed] [Google Scholar]
- 147.Jakob U, Muse W, Eser M, Bardwell JC. Chaperone activity with a redox switch. Cell. 1999;96:341–352. doi: 10.1016/s0092-8674(00)80547-4. [DOI] [PubMed] [Google Scholar]
- 148.Jakob U, Eser M, Bardwell JC. Redox switch of hsp33 has a novel zinc-binding motif. J Biol Chem. 2000;275:38302–38310. doi: 10.1074/jbc.M005957200. [DOI] [PubMed] [Google Scholar]
- 149.Winter J, Linke K, Jatzek A, Jakob U. Severe oxidative stress causes inactivation of DnaK and activation of the redox-regulated chaperone Hsp33. Mol Cell. 2005;17:381–392. doi: 10.1016/j.molcel.2004.12.027. [DOI] [PubMed] [Google Scholar]
- 150.Cumming RC, Andon NL, Haynes PA, Park M, Fischer WH, Schubert D. Protein disulfide bond formation in the cytoplasm during oxidative stress. J Biol Chem. 2004;279:21749–21758. doi: 10.1074/jbc.M312267200. [DOI] [PubMed] [Google Scholar]
- 151.Diaz-Latoud C, Buache E, Javouhey E, Arrigo AP. Substitution of the unique cysteine residue of murine Hsp25 interferes with the protective activity of this stress protein through inhibition of dimer formation. Antioxid Redox Signal. 2005;7:436–445. doi: 10.1089/ars.2005.7.436. [DOI] [PubMed] [Google Scholar]
- 152.Hoppe G, Chai YC, Crabb JW, Sears J. Protein s-glutathionylation in retinal pigment epithelium converts heat shock protein 70 to an active chaperone. Exp Eye Res. 2004;78:1085–1092. doi: 10.1016/j.exer.2004.02.001. [DOI] [PubMed] [Google Scholar]
- 153.Nardai G, Sass B, Eber J, Orosz G, Csermely P. Reactive cysteines of the 90-kDa heat shock protein, Hsp90. Arch Biochem Biophys. 2000;384:59–67. doi: 10.1006/abbi.2000.2075. [DOI] [PubMed] [Google Scholar]
- 154.Hamnell-Pamment Y, Lind C, Palmberg C, Bergman T, Cotgreave IA. Determination of site-specificity of S-glutathionylated cellular proteins. Biochem Biophys Res Commun. 2005;332:362–369. doi: 10.1016/j.bbrc.2005.04.130. [DOI] [PubMed] [Google Scholar]
- 155.Martinez-Ruiz A, Villanueva L, Gonzalez de Orduna C, Lopez-Ferrer D, Higueras MA, Tarin C, Rodriguez-Crespo I, Vazquez J, Lamas S. S-nitrosylation of Hsp90 promotes the inhibition of its ATPase and endothelial nitric oxide synthase regulatory activities. Proc Natl Acad Sci U S A. 2005;102:8525–8530. doi: 10.1073/pnas.0407294102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Ellgaard L, Ruddock LW. The human protein disulphide isomerase family: substrate interactions and functional properties. EMBO Rep. 2005;6:28–32. doi: 10.1038/sj.embor.7400311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Gruber CW, Cemazar M, Heras B, Martin JL, Craik DJ. Protein disulfide isomerase: the structure of oxidative folding. Trends Biochem Sci. 2006;31:455–464. doi: 10.1016/j.tibs.2006.06.001. [DOI] [PubMed] [Google Scholar]
- 158.Tsai B, Rodighiero C, Lencer WI, Rapoport TA. Protein disulfide isomerase acts as a redox-dependent chaperone to unfold cholera toxin. Cell. 2001;104:937–948. doi: 10.1016/s0092-8674(01)00289-6. [DOI] [PubMed] [Google Scholar]
- 159.Lumb RA, Bulleid NJ. Is protein disulfide isomerase a redox-dependent molecular chaperone? Embo J. 2002;21:6763–6770. doi: 10.1093/emboj/cdf685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Zai A, Rudd MA, Scribner AW, Loscalzo J. Cell-surface protein disulfide isomerase catalyzes transnitrosation and regulates intracellular transfer of nitric oxide. J Clin Invest. 1999;103:393–399. doi: 10.1172/JCI4890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Ramachandran N, Root P, Jiang XM, Hogg PJ, Mutus B. Mechanism of transfer of NO from extracellular S-nitrosothiols into the cytosol by cell-surface protein disulfide isomerase. Proc Natl Acad Sci U S A. 2001;98:9539–9544. doi: 10.1073/pnas.171180998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Uehara T, Nakamura T, Yao D, Shi ZQ, Gu Z, Ma Y, Masliah E, Nomura Y, Lipton SA. S-nitrosylated protein-disulphide isomerase links protein misfolding to neurodegeneration. Nature. 2006;441:513–517. doi: 10.1038/nature04782. [DOI] [PubMed] [Google Scholar]
- 163.Sliskovic I, Raturi A, Mutus B. Characterization of the S-denitrosation activity of protein disulfide isomerase. J Biol Chem. 2005;280:8733–8741. doi: 10.1074/jbc.M408080200. [DOI] [PubMed] [Google Scholar]
- 164.Morrison DK, Davis RJ. Regulation of MAP kinase signaling modules by scaffold proteins in mammals. Annu Rev Cell Dev Biol. 2003;19:91–118. doi: 10.1146/annurev.cellbio.19.111401.091942. [DOI] [PubMed] [Google Scholar]
- 165.Whitmarsh AJ, Davis RJ. Structural organization of MAP-kinase signaling modules by scaffold proteins in yeast and mammals. Trends Biochem Sci. 1998;23:481–485. doi: 10.1016/s0968-0004(98)01309-7. [DOI] [PubMed] [Google Scholar]
- 166.Liu H, Nishitoh H, Ichijo H, Kyriakis JM. Activation of apoptosis signal-regulating kinase 1 (ASK1) by tumor necrosis factor receptor-associated factor 2 requires prior dissociation of the ASK1 inhibitor thioredoxin. Mol Cell Biol. 2000;20:2198–2208. doi: 10.1128/mcb.20.6.2198-2208.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Saitoh M, Nishitoh H, Fujii M, Takeda K, Tobiume K, Sawada Y, Kawabata M, Miyazono K, Ichijo H. Mammalian thioredoxin is a direct inhibitor of apoptosis signal-regulating kinase (ASK) 1. Embo J. 1998;17:2596–2606. doi: 10.1093/emboj/17.9.2596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Liu H, Zhang H, Iles KE, Rinna A, Merrill G, Yodoi J, Torres M, Forman HJ. The ADP-stimulated NADPH oxidase activates the ASK-1/MKK4/JNK pathway in alveolar macrophages. Free radical research. 2006;40:865–874. doi: 10.1080/10715760600758514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Liu Y, Min W. Thioredoxin promotes ASK1 ubiquitination and degradation to inhibit ASK1-mediated apoptosis in a redox activity-independent manner. Circ Res. 2002;90:1259–1266. doi: 10.1161/01.res.0000022160.64355.62. [DOI] [PubMed] [Google Scholar]
- 170.Pantano C, Shrivastava P, McElhinney B, Janssen-Heininger Y. Hydrogen peroxide signaling through tumor necrosis factor receptor 1 leads to selective activation of c-Jun N-terminal kinase. J Biol Chem. 2003;278:44091–44096. doi: 10.1074/jbc.M308487200. [DOI] [PubMed] [Google Scholar]
- 171.Pantano C, Anathy V, Ranjan P, Heintz NH, Janssen-Heininger YM. Nonphagocytic oxidase 1 causes death in lung epithelial cells via a TNF-RI-JNK signaling axis. Am J Respir Cell Mol Biol. 2007;36:473–479. doi: 10.1165/rcmb.2006-0109OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Zhang DD. Mechanistic studies of the Nrf2-Keap1 signaling pathway. Drug Metab Rev. 2006;38:769–789. doi: 10.1080/03602530600971974. [DOI] [PubMed] [Google Scholar]
- 173.Itoh K, Tong KI, Yamamoto M. Molecular mechanism activating Nrf2-Keap1 pathway in regulation of adaptive response to electrophiles. Free Radic Biol Med. 2004;36:1208–1213. doi: 10.1016/j.freeradbiomed.2004.02.075. [DOI] [PubMed] [Google Scholar]
- 174.Dinkova-Kostova AT, Holtzclaw WD, Cole RN, Itoh K, Wakabayashi N, Katoh Y, Yamamoto M, Talalay P. Direct evidence that sulfhydryl groups of Keap1 are the sensors regulating induction of phase 2 enzymes that protect against carcinogens and oxidants. Proc Natl Acad Sci U S A. 2002;99:11908–11913. doi: 10.1073/pnas.172398899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Wakabayashi N, Dinkova-Kostova AT, Holtzclaw WD, Kang MI, Kobayashi A, Yamamoto M, Kensler TW, Talalay P. Protection against electrophile and oxidant stress by induction of the phase 2 response: fate of cysteines of the Keap1 sensor modified by inducers. Proc Natl Acad Sci U S A. 2004;101:2040–2045. doi: 10.1073/pnas.0307301101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Kobayashi A, Kang MI, Okawa H, Ohtsuji M, Zenke Y, Chiba T, Igarashi K, Yamamoto M. Oxidative stress sensor Keap1 functions as an adaptor for Cul3-based E3 ligase to regulate proteasomal degradation of Nrf2. Mol Cell Biol. 2004;24:7130–7139. doi: 10.1128/MCB.24.16.7130-7139.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Tong KI, Kobayashi A, Katsuoka F, Yamamoto M. Two-site substrate recognition model for the Keap1-Nrf2 system: a hinge and latch mechanism. Biol Chem. 2006;387:1311–1320. doi: 10.1515/BC.2006.164. [DOI] [PubMed] [Google Scholar]
- 178.Padmanabhan B, Tong KI, Ohta T, Nakamura Y, Scharlock M, Ohtsuji M, Kang MI, Kobayashi A, Yokoyama S, Yamamoto M. Structural basis for defects of Keap1 activity provoked by its point mutations in lung cancer. Mol Cell. 2006;21:689–700. doi: 10.1016/j.molcel.2006.01.013. [DOI] [PubMed] [Google Scholar]
- 179.Singh A, Misra V, Thimmulappa RK, Lee H, Ames S, Hoque MO, Herman JG, Baylin SB, Sidransky D, Gabrielson E, Brock MV, Biswal S. Dysfunctional KEAP1-NRF2 interaction in non-small-cell lung cancer. PLoS Med. 2006;3:e420. doi: 10.1371/journal.pmed.0030420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Matsumoto A, Comatas KE, Liu L, Stamler JS. Screening for nitric oxide-dependent protein-protein interactions. Science. 2003;301:657–661. doi: 10.1126/science.1079319. [DOI] [PubMed] [Google Scholar]
- 181.Ischiropoulos H, Zhu L, Beckman JS. Peroxynitrite formation from macrophage-derived nitric oxide. Arch Biochem Biophys. 1992;298:446–451. doi: 10.1016/0003-9861(92)90433-w. [DOI] [PubMed] [Google Scholar]
- 182.Pacher P, Beckman JS, Liaudet L. Nitric oxide and peroxynitrite in health and disease. Physiol Rev. 2007;87:315–424. doi: 10.1152/physrev.00029.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Benhar M, Stamler JS. A central role for S-nitrosylation in apoptosis. Nat Cell Biol. 2005;7:645–646. doi: 10.1038/ncb0705-645. [DOI] [PubMed] [Google Scholar]
- 184.Hara MR, Agrawal N, Kim SF, Cascio MB, Fujimuro M, Ozeki Y, Takahashi M, Cheah JH, Tankou SK, Hester LD, Ferris CD, Hayward SD, Snyder SH, Sawa A. S-nitrosylated GAPDH initiates apoptotic cell death by nuclear translocation following Siah1 binding. Nat Cell Biol. 2005;7:665–674. doi: 10.1038/ncb1268. [DOI] [PubMed] [Google Scholar]
- 185.Hara MR, Snyder SH. Nitric oxide-GAPDH-Siah: a novel cell death cascade. Cellular and molecular neurobiology. 2006;26:527–538. doi: 10.1007/s10571-006-9011-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Funato Y, Michiue T, Asashima M, Miki H. The thioredoxin-related redox-regulating protein nucleoredoxin inhibits Wnt-beta-catenin signalling through dishevelled. Nat Cell Biol. 2006;8:501–508. doi: 10.1038/ncb1405. [DOI] [PubMed] [Google Scholar]
- 187.Delaunay A, Pflieger D, Barrault MB, Vinh J, Toledano MB. A thiol peroxidase is an H2O2 receptor and redox-transducer in gene activation. Cell. 2002;111:471–481. doi: 10.1016/s0092-8674(02)01048-6. [DOI] [PubMed] [Google Scholar]
- 188.Wang T, Arifoglu P, Ronai Z, Tew KD. Glutathione S-transferase P1-1 (GSTP1-1) inhibits c-Jun N-terminal kinase (JNK1) signaling through interaction with the C terminus. J Biol Chem. 2001;276:20999–21003. doi: 10.1074/jbc.M101355200. [DOI] [PubMed] [Google Scholar]
- 189.Adler V, Yin Z, Fuchs SY, Benezra M, Rosario L, Tew KD, Pincus MR, Sardana M, Henderson CJ, Wolf CR, Davis RJ, Ronai Z. Regulation of JNK signaling by GSTp. Embo J. 1999;18:1321–1334. doi: 10.1093/emboj/18.5.1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Gate L, Majumdar RS, Lunk A, Tew KD. Increased myeloproliferation in glutathione S-transferase pi-deficient mice is associated with a deregulation of JNK and Janus kinase/STAT pathways. J Biol Chem. 2004;279:8608–8616. doi: 10.1074/jbc.M308613200. [DOI] [PubMed] [Google Scholar]
- 191.Manevich Y, Feinstein SI, Fisher AB. Activation of the antioxidant enzyme 1-CYS peroxiredoxin requires glutathionylation mediated by heterodimerization with pi GST. Proc Natl Acad Sci U S A. 2004;101:3780–3785. doi: 10.1073/pnas.0400181101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Na HK, Surh YJ. Transcriptional regulation via cysteine thiol modification: a novel molecular strategy for chemoprevention and cytoprotection. Mol Carcinog. 2006;45:368–380. doi: 10.1002/mc.20225. [DOI] [PubMed] [Google Scholar]
- 193.Christman MF, Morgan RW, Jacobson FS, Ames BN. Positive control of a regulon for defenses against oxidative stress and some heat-shock proteins in Salmonella typhimurium. Cell. 1985;41:753–762. doi: 10.1016/s0092-8674(85)80056-8. [DOI] [PubMed] [Google Scholar]
- 194.Choi H, Kim S, Mukhopadhyay P, Cho S, Woo J, Storz G, Ryu S. Structural basis of the redox switch in the OxyR transcription factor. Cell. 2001;105:103–113. doi: 10.1016/s0092-8674(01)00300-2. [DOI] [PubMed] [Google Scholar]
- 195.Lee C, Lee SM, Mukhopadhyay P, Kim SJ, Lee SC, Ahn WS, Yu MH, Storz G, Ryu SE. Redox regulation of OxyR requires specific disulfide bond formation involving a rapid kinetic reaction path. Nat Struct Mol Biol. 2004;11:1179–1185. doi: 10.1038/nsmb856. [DOI] [PubMed] [Google Scholar]
- 196.Hausladen A, Privalle CT, Keng T, DeAngelo J, Stamler JS. Nitrosative stress: activation of the transcription factor OxyR. Cell. 1996;86:719–729. doi: 10.1016/s0092-8674(00)80147-6. [DOI] [PubMed] [Google Scholar]
- 197.Klatt P, Molina EP, De Lacoba MG, Padilla CA, Martinez-Galesteo E, Barcena JA, Lamas S. Redox regulation of c-Jun DNA binding by reversible S-glutathiolation. Faseb J. 1999;13:1481–1490. doi: 10.1096/fasebj.13.12.1481. [DOI] [PubMed] [Google Scholar]
- 198.Pineda-Molina E, Klatt P, Vazquez J, Marina A, Garcia de Lacoba M, Perez-Sala D, Lamas S. Glutathionylation of the p50 subunit of NF-kappaB: a mechanism for redox-induced inhibition of DNA binding. Biochemistry. 2001;40:14134–14142. doi: 10.1021/bi011459o. [DOI] [PubMed] [Google Scholar]
- 199.Marshall HE, Stamler JS. Inhibition of NF-kappa B by S-nitrosylation. Biochemistry. 2001;40:1688–1693. doi: 10.1021/bi002239y. [DOI] [PubMed] [Google Scholar]
- 200.Matthews JR, Botting CH, Panico M, Morris HR, Hay RT. Inhibition of NF-kappaB DNA binding by nitric oxide. Nucleic Acids Res. 1996;24:2236–2242. doi: 10.1093/nar/24.12.2236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Kelleher ZT, Matsumoto A, Stamler JS, Marshall HE. NOS2 regulation of NF-kappa B by S-nitrosylation of p65*. J Biol Chem. 2007 doi: 10.1074/jbc.M705929200. [DOI] [PubMed] [Google Scholar]
- 202.Rainwater R, Parks D, Anderson ME, Tegtmeyer P, Mann K. Role of cysteine residues in regulation of p53 function. Mol Cell Biol. 1995;15:3892–3903. doi: 10.1128/mcb.15.7.3892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Sun XZ, Vinci C, Makmura L, Han S, Tran D, Nguyen J, Hamann M, Grazziani S, Sheppard S, Gutova M, Zhou F, Thomas J, Momand J. Formation of disulfide bond in p53 correlates with inhibition of DNA binding and tetramerization. Antioxid Redox Signal. 2003;5:655–665. doi: 10.1089/152308603770310338. [DOI] [PubMed] [Google Scholar]
- 204.Buzek J, Latonen L, Kurki S, Peltonen K, Laiho M. Redox state of tumor suppressor p53 regulates its sequence-specific DNA binding in DNA-damaged cells by cysteine 277. Nucleic Acids Res. 2002;30:2340–2348. doi: 10.1093/nar/30.11.2340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Sumbayev VV, Budde A, Zhou J, Brune B. HIF-1 alpha protein as a target for S-nitrosation. FEBS Lett. 2003;535:106–112. doi: 10.1016/s0014-5793(02)03887-5. [DOI] [PubMed] [Google Scholar]
- 206.Yasinska IM, Sumbayev VV. S-nitrosation of Cys-800 of HIF-1alpha protein activates its interaction with p300 and stimulates its transcriptional activity. FEBS Lett. 2003;549:105–109. doi: 10.1016/s0014-5793(03)00807-x. [DOI] [PubMed] [Google Scholar]
- 207.Ahn SG, Thiele DJ. Redox regulation of mammalian heat shock factor 1 is essential for Hsp gene activation and protection from stress. Genes Dev. 2003;17:516–528. doi: 10.1101/gad.1044503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Brunet A, Sweeney LB, Sturgill JF, Chua KF, Greer PL, Lin Y, Tran H, Ross SE, Mostoslavsky R, Cohen HY, Hu LS, Cheng HL, Jedrychowski MP, Gygi SP, Sinclair DA, Alt FW, Greenberg ME. Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science. 2004;303:2011–2015. doi: 10.1126/science.1094637. [DOI] [PubMed] [Google Scholar]
- 209.Wang F, Nguyen M, Qin FX, Tong Q. SIRT2 deacetylates FOXO3a in response to oxidative stress and caloric restriction. Aging Cell. 2007 doi: 10.1111/j.1474-9726.2007.00304.x. [DOI] [PubMed] [Google Scholar]
- 210.Kobayashi Y, Furukawa-Hibi Y, Chen C, Horio Y, Isobe K, Ikeda K, Motoyama N. SIRT1 is critical regulator of FOXO-mediated transcription in response to oxidative stress. Int J Mol Med. 2005;16:237–243. [PubMed] [Google Scholar]
- 211.van der Horst A, Tertoolen LG, de Vries-Smits LM, Frye RA, Medema RH, Burgering BM. FOXO4 is acetylated upon peroxide stress and deacetylated by the longevity protein hSir2(SIRT1) J Biol Chem. 2004;279:28873–28879. doi: 10.1074/jbc.M401138200. [DOI] [PubMed] [Google Scholar]
- 212.Alcendor RR, Gao S, Zhai P, Zablocki D, Holle E, Yu X, Tian B, Wagner T, Vatner SF, Sadoshima J. Sirt1 regulates aging and resistance to oxidative stress in the heart. Circ Res. 2007;100:1512–1521. doi: 10.1161/01.RES.0000267723.65696.4a. [DOI] [PubMed] [Google Scholar]
- 213.Rahman I, Marwick J, Kirkham P. Redox modulation of chromatin remodeling: impact on histone acetylation and deacetylation, NF-kappaB and pro-inflammatory gene expression. Biochem Pharmacol. 2004;68:1255–1267. doi: 10.1016/j.bcp.2004.05.042. [DOI] [PubMed] [Google Scholar]
- 214.Rahman I, Gilmour PS, Jimenez LA, MacNee W. Oxidative stress and TNF-alpha induce histone acetylation and NF-kappaB/AP-1 activation in alveolar epithelial cells: potential mechanism in gene transcription in lung inflammation. Molecular and cellular biochemistry. 2002;234–235:239–248. [PubMed] [Google Scholar]
- 215.Ito K, Hanazawa T, Tomita K, Barnes PJ, Adcock IM. Oxidative stress reduces histone deacetylase 2 activity and enhances IL-8 gene expression: role of tyrosine nitration. Biochem Biophys Res Commun. 2004;315:240–245. doi: 10.1016/j.bbrc.2004.01.046. [DOI] [PubMed] [Google Scholar]
- 216.Riccio A, Alvania RS, Lonze BE, Ramanan N, Kim T, Huang Y, Dawson TM, Snyder SH, Ginty DD. A nitric oxide signaling pathway controls CREB-mediated gene expression in neurons. Mol Cell. 2006;21:283–294. doi: 10.1016/j.molcel.2005.12.006. [DOI] [PubMed] [Google Scholar]
- 217.Wu Y, Kwon KS, Rhee SG. Probing cellular protein targets of H2O2 with fluorescein-conjugated iodoacetamide and antibodies to fluorescein. FEBS Lett. 1998;440:111–115. doi: 10.1016/s0014-5793(98)01415-x. [DOI] [PubMed] [Google Scholar]
- 218.Kim JR, Yoon HW, Kwon KS, Lee SR, Rhee SG. Identification of proteins containing cysteine residues that are sensitive to oxidation by hydrogen peroxide at neutral pH. Analytical biochemistry. 2000;283:214–221. doi: 10.1006/abio.2000.4623. [DOI] [PubMed] [Google Scholar]
- 219.Hess DT, Lin LH, Freeman JA, Norden JJ. Modification of cysteine residues within G(o) and other neuronal proteins by exposure to nitric oxide. Neuropharmacology. 1994;33:1283–1292. doi: 10.1016/0028-3908(94)90028-0. [DOI] [PubMed] [Google Scholar]
- 220.Leichert LI, Jakob U. Protein thiol modifications visualized in vivo. PLoS Biol. 2004;2:e333. doi: 10.1371/journal.pbio.0020333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Jaffrey SR, Snyder SH. The biotin switch method for the detection of S-nitrosylated proteins. Sci STKE 2001. 2001:PL1. doi: 10.1126/stke.2001.86.pl1. [DOI] [PubMed] [Google Scholar]
- 222.Landino LM, Koumas MT, Mason CE, Alston JA. Ascorbic acid reduction of microtubule protein disulfides and its relevance to protein S-nitrosylation assays. Biochem Biophys Res Commun. 2006;340:347–352. doi: 10.1016/j.bbrc.2005.12.013. [DOI] [PubMed] [Google Scholar]
- 223.Huang B, Chen C. An ascorbate-dependent artifact that interferes with the interpretation of the biotin switch assay. Free Radic Biol Med. 2006;41:562–567. doi: 10.1016/j.freeradbiomed.2006.03.006. [DOI] [PubMed] [Google Scholar]
- 224.Zhang Y, Keszler A, Broniowska KA, Hogg N. Characterization and application of the biotin-switch assay for the identification of S-nitrosated proteins. Free Radic Biol Med. 2005;38:874–881. doi: 10.1016/j.freeradbiomed.2004.12.012. [DOI] [PubMed] [Google Scholar]
- 225.Sun J, Xin C, Eu JP, Stamler JS, Meissner G. Cysteine-3635 is responsible for skeletal muscle ryanodine receptor modulation by NO. Proc Natl Acad Sci U S A. 2001;98:11158–11162. doi: 10.1073/pnas.201289098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Matsushita K, Morrell CN, Cambien B, Yang SX, Yamakuchi M, Bao C, Hara MR, Quick RA, Cao W, O’Rourke B, Lowenstein JM, Pevsner J, Wagner DD, Lowenstein CJ. Nitric oxide regulates exocytosis by S-nitrosylation of N-ethylmaleimide-sensitive factor. Cell. 2003;115:139–150. doi: 10.1016/s0092-8674(03)00803-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Lind C, Gerdes R, Hamnell Y, Schuppe-Koistinen I, von Lowenhielm HB, Holmgren A, Cotgreave IA. Identification of S-glutathionylated cellular proteins during oxidative stress and constitutive metabolism by affinity purification and proteomic analysis. Arch Biochem Biophys. 2002;406:229–240. doi: 10.1016/s0003-9861(02)00468-x. [DOI] [PubMed] [Google Scholar]
- 228.Niture SK, Velu CS, Bailey NI, Srivenugopal KS. S-thiolation mimicry: quantitative and kinetic analysis of redox status of protein cysteines by glutathione-affinity chromatography. Arch Biochem Biophys. 2005;444:174–184. doi: 10.1016/j.abb.2005.10.013. [DOI] [PubMed] [Google Scholar]
- 229.Cheng G, Ikeda Y, Iuchi Y, Fujii J. Detection of S-glutathionylated proteins by glutathione S-transferase overlay. Arch Biochem Biophys. 2005;435:42–49. doi: 10.1016/j.abb.2004.12.016. [DOI] [PubMed] [Google Scholar]
- 230.Fratelli M, Gianazza E, Ghezzi P. Redox proteomics: identification and functional role of glutathionylated proteins. Expert Rev Proteomics. 2004;1:365–376. doi: 10.1586/14789450.1.3.365. [DOI] [PubMed] [Google Scholar]
- 231.Behrens A, Sibilia M, Wagner EF. Amino-terminal phosphorylation of c-Jun regulates stress-induced apoptosis and cellular proliferation. Nat Genet. 1999;21:326–329. doi: 10.1038/6854. [DOI] [PubMed] [Google Scholar]
- 232.Behrens A, Jochum W, Sibilia M, Wagner EF. Oncogenic transformation by ras and fos is mediated by c-Jun N-terminal phosphorylation. Oncogene. 2000;19:2657–2663. doi: 10.1038/sj.onc.1203603. [DOI] [PubMed] [Google Scholar]
- 233.Brecht S, Kirchhof R, Chromik A, Willesen M, Nicolaus T, Raivich G, Wessig J, Waetzig V, Goetz M, Claussen M, Pearse D, Kuan CY, Vaudano E, Behrens A, Wagner E, Flavell RA, Davis RJ, Herdegen T. Specific pathophysiological functions of JNK isoforms in the brain. Eur J Neurosci. 2005;21:363–377. doi: 10.1111/j.1460-9568.2005.03857.x. [DOI] [PubMed] [Google Scholar]
- 234.Koziczak-Holbro M, Joyce C, Gluck A, Kinzel B, Muller M, Tschopp C, Mathison JC, Davis CN, Gram H. IRAK-4 kinase activity is required for IL-1 receptor- and Toll-like receptor 7-mediated signaling and gene expression. J Biol Chem. 2007 doi: 10.1074/jbc.M700548200. [DOI] [PubMed] [Google Scholar]
- 235.Sano Y, Nakaya T, Pedrini S, Takeda S, Iijima-Ando K, Iijima K, Mathews PM, Itohara S, Gandy S, Suzuki T. Physiological Mouse Brain Abeta Levels Are Not Related to the Phosphorylation State of Threonine-668 of Alzheimer’s APP. PLoS ONE. 2006;1:e51. doi: 10.1371/journal.pone.0000051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Feng L, Hollstein M, Xu Y. Ser46 phosphorylation regulates p53-dependent apoptosis and replicative senescence. Cell Cycle. 2006;5:2812–2819. doi: 10.4161/cc.5.23.3526. [DOI] [PubMed] [Google Scholar]
- 237.Tanemura K, Chui DH, Fukuda T, Murayama M, Park JM, Akagi T, Tatebayashi Y, Miyasaka T, Kimura T, Hashikawa T, Nakano Y, Kudo T, Takeda M, Takashima A. Formation of tau inclusions in knock-in mice with familial Alzheimer disease (FAD) mutation of presenilin 1 (PS1) J Biol Chem. 2006;281:5037–5041. doi: 10.1074/jbc.M509145200. [DOI] [PubMed] [Google Scholar]
- 238.Wang Z, Wen XH, Ablonczy Z, Crouch RK, Makino CL, Lem J. Enhanced shutoff of phototransduction in transgenic mice expressing palmitoylation-deficient rhodopsin. J Biol Chem. 2005;280:24293–24300. doi: 10.1074/jbc.M502588200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.van de Pavert SA, Meuleman J, Malysheva A, Aartsen WM, Versteeg I, Tonagel F, Kamphuis W, McCabe CJ, Seeliger MW, Wijnholds J. A single amino acid substitution (Cys249Trp) in Crb1 causes retinal degeneration and deregulates expression of pituitary tumor transforming gene Pttg1. J Neurosci. 2007;27:564–573. doi: 10.1523/JNEUROSCI.3496-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Xanthoudakis S, Miao G, Wang F, Pan YC, Curran T. Redox activation of Fos-Jun DNA binding activity is mediated by a DNA repair enzyme. Embo J. 1992;11:3323–3335. doi: 10.1002/j.1460-2075.1992.tb05411.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Ordway JM, Eberhart D, Curran T. Cysteine 64 of Ref-1 is not essential for redox regulation of AP-1 DNA binding. Mol Cell Biol. 2003;23:4257–4266. doi: 10.1128/MCB.23.12.4257-4266.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Qu J, Liu GH, Huang B, Chen C. Nitric oxide controls nuclear export of APE1/Ref-1 through S-nitrosation of cysteines 93 and 310. Nucleic Acids Res. 2007;35:2522–2532. doi: 10.1093/nar/gkl1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Rizzo MA, Piston DW. Regulation of beta cell glucokinase by S-nitrosylation and association with nitric oxide synthase. The Journal of cell biology. 2003;161:243–248. doi: 10.1083/jcb.200301063. [DOI] [PMC free article] [PubMed] [Google Scholar]