

Abstract
A system of cluster analysis for genome-wide expression data from DNA microarray hybridization is described that uses standard statistical algorithms to arrange genes according to similarity in pattern of gene expression. The output is displayed graphically, conveying the clustering and the underlying expression data simultaneously in a form intuitive for biologists. We have found in the budding yeast Saccharomyces cerevisiae that clustering gene expression data groups together efficiently genes of known similar function, and we find a similar tendency in human data. Thus patterns seen in genome-wide expression experiments can be interpreted as indications of the status of cellular processes. Also, coexpression of genes of known function with poorly characterized or novel genes may provide a simple means of gaining leads to the functions of many genes for which information is not available currently.
The rapid advance of genome-scale sequencing has driven the development of methods to exploit this information by characterizing biological processes in new ways. The knowledge of the coding sequences of virtually every gene in an organism, for instance, invites development of technology to study the expression of all of them at once, because the study of gene expression of genes one by one has already provided a wealth of biological insight. To this end, a variety of techniques has evolved to monitor, rapidly and efficiently, transcript abundance for all of an organism’s genes (1–3). Within the mass of numbers produced by these techniques, which amount to hundreds of data points for thousands or tens of thousands of genes, is an immense amount of biological information. In this paper we address the problem of analyzing and presenting information on this genomic scale.
A natural first step in extracting this information is to examine the extremes, e.g., genes with significant differential expression in two individual samples or in a time series after a given treatment. This simple technique can be extremely efficient, for example, in screens for potential tumor markers or drug targets. However, such analyses do not address the full potential of genome-scale experiments to alter our understanding of cellular biology by providing, through an inclusive analysis of the entire repertoire of transcripts, a continuing comprehensive window into the state of a cell as it goes through a biological process. What is needed instead is a holistic approach to analysis of genomic data that focuses on illuminating order in the entire set of observations, allowing biologists to develop an integrated understanding of the process being studied.
A natural basis for organizing gene expression data is to group together genes with similar patterns of expression. The first step to this end is to adopt a mathematical description of similarity. For any series of measurements, a number of sensible measures of similarity in the behavior of two genes can be used, such as the Euclidean distance, angle, or dot products of the two n-dimensional vectors representing a series of n measurements. We have found that the standard correlation coefficient (i.e., the dot product of two normalized vectors) conforms well to the intuitive biological notion of what it means for two genes to be “coexpressed;” this may be because this statistic captures similarity in “shape” but places no emphasis on the magnitude of the two series of measurements.
It is not the purpose of this paper to survey the various methods available to cluster genes on the basis of their expression patterns, but rather to illustrate how such methods can be useful to biologists in the analysis of gene expression data. We aim to use these methods to organize, but not to alter, tables containing primary data; we have thus used methods that can be reduced, in the end, to a reordering of lists of genes. Clustering methods can be divided into two general classes, designated supervised and unsupervised clustering (4). In supervised clustering, vectors are classified with respect to known reference vectors. In unsupervised clustering, no predefined reference vectors are used. As we have little a priori knowledge of the complete repertoire of expected gene expression patterns for any condition, we have favored unsupervised methods or hybrid (unsupervised followed by supervised) approaches.
Although various clustering methods can usefully organize tables of gene expression measurements, the resulting ordered but still massive collection of numbers remains difficult to assimilate. Therefore, we always combine clustering methods with a graphical representation of the primary data by representing each data point with a color that quantitatively and qualitatively reflects the original experimental observations. The end product is a representation of complex gene expression data that, through statistical organization and graphical display, allows biologists to assimilate and explore the data in a natural intuitive manner.
To illustrate this approach, we have applied pairwise average-linkage cluster analysis (5) to gene expression data collected in our laboratories. This method is a form of hierarchical clustering, familiar to most biologists through its application in sequence and phylogenetic analysis. Relationships among objects (genes) are represented by a tree whose branch lengths reflect the degree of similarity between the objects, as assessed by a pairwise similarity function such as that described above. In sequence comparison, these methods are used to infer the evolutionary history of sequences being compared. Whereas no such underlying tree exists for expression patterns of genes, such methods are useful in their ability to represent varying degrees of similarity and more distant relationships among groups of closely related genes, as well as in requiring few assumptions about the nature of the data. The computed trees can be used to order genes in the original data table, so that genes or groups of genes with similar expression patterns are adjacent. The ordered table can then be displayed graphically, as above, with a representation of the tree to indicate the relationships among genes.
MATERIALS AND METHODS
Sources of Experimental Data.
Data analyzed here were collected on spotted DNA microarrays (6, 7). Gene expression in the budding yeast Saccharomyces cerevisiae was studied during the diauxic shift (8), the mitotic cell division cycle (9), sporulation (10), and temperature and reducing shocks (P.T.S., P.O.B., and D.B., unpublished results) by using microarrays containing essentially every ORF from this fully sequenced organism (8). Gene expression of primary human fibroblasts stimulated with serum following serum starvation was studied by using a microarray with 9,800 cDNAs representing approximately 8,600 distinct human transcripts (11). In all experiments, RNA from experimental samples (taken at selected times during the process) was labeled during reverse transcription with the red-fluorescent dye Cy5 (Amersham) and was mixed with a reference sample labeled in parallel with the green-fluorescent dye Cy3 (Amersham) (the reference sample was time 0 for all experiments, except for the yeast cell cycle where asynchronous cells were used). After hybridization and appropriate washing steps, separate images were acquired for each fluor, and fluorescence intensity ratios were obtained for all target elements.
We maintain and update master data tables for all experiments conducted in our labs; in these tables, rows represent all genes for which data has been collected, columns represent individual array experiments (e.g., single timepoints or conditions), and each cell represents the measured Cy5/Cy3 fluorescence ratio at the corresponding target element on the appropriate array. All ratio values are log transformed (base 2 for simplicity) to treat inductions or repressions of identical magnitude as numerically equal but with opposite sign. Individual observations are rejected in some cases, on the basis of measurement quality parameters produced by the image analysis software. For the analyses presented here, smaller tables containing selected genes and experiments were produced; full descriptions are given in the appropriate figure legends. These tables are available on the PNAS web site at (www.pnas.org) or at http://rana.stanford.edu/clustering.
Metrics.
The gene similarity metric we use is a form of correlation coefficient. Let Gi equal the (log-transformed) primary data for gene G in condition i. For any two genes X and Y observed over a series of N conditions, a similarity score can be computed as follows:
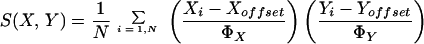 |
where
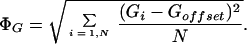 |
When Goffset is set to the mean of observations on G, then ΦG becomes the standard deviation of G, and S(X, Y) is exactly equal to the Pearson correlation coefficient of the observations of X and Y. Values of Goffset which are not the average over observations on G are used when there is an assumed unchanged or reference state represented by the value of Goffset, against which changes are to be analyzed; in all of the examples presented here, Goffset is set to 0, corresponding to a fluorescence ratio of 1.0.
Hierarchical Clustering.
The hierarchical clustering algorithm used is based closely on the average-linkage method of Sokal and Michener (5), which was developed for clustering correlation matrixes such as those used here. The object of this algorithm is to compute a dendrogram that assembles all elements into a single tree. For any set of n genes, an upper-diagonal similarity matrix is computed by using the metric described above, which contains similarity scores for all pairs of genes. The matrix is scanned to identify the highest value (representing the most similar pair of genes). A node is created joining these two genes, and a gene expression profile is computed for the node by averaging observation for the joined elements (missing values are omitted and the two joined elements are weighted by the number of genes they contain). The similarity matrix is updated with this new node replacing the two joined elements, and the process is repeated n-1 times until only a single element remains. Software implementation of this algorithm can be obtained from the authors at http://rana.stanford.edu/clustering.
Ordering of Data Tables.
For any dendrogram of n elements, there are 2n−1 linear orderings consistent with the structure of the tree (at each node, either of the two elements joined by the node can be ordered ahead of the other). An optimal linear ordering, one that maximizes the similarity of adjacent elements in the ordering, is impractical to compute. However, to consistently arrange nodes between analyses, we use simple methods of weighting genes, such as average expression level, time of maximal induction, or chromosomal position, and we place the element with the lower average weight earlier in the final ordering.
Display.
Using any ordering, the primary data table is represented graphically by coloring each cell on the basis of the measured fluorescence ratio. Cells with log ratios of 0 (ratios of 1.0 − genes unchanged) are colored black, increasingly positive log ratios with reds of increasing intensity, and increasingly negative log ratios with greens of increasing intensity. A representation of the dendrogram is appended to the colored table to indicate the nature of the computed relationship among genes in the table.
RESULTS
We applied this method to two sets of data, a single time course (Fig. 1) of a canonical model of the growth response in human cells (11) and an aggregation of data from experiments on the budding yeast S. cerevisiae (Fig. 2), including time courses of the mitotic cell division cycle (9), sporulation (10), the diauxic shift (8), and shock responses (P.T.S., P.O.B., and D.B., unpublished results). A striking property of the clustered images in Figs. 1 and 2 is the presence of large contiguous patches of color representing groups of genes that share similar expression patterns over multiple conditions. To verify that this structure is of biological origin and is not an artifact of the clustering procedure, the initial data from the human growth response experiment were randomized in three different ways and were clustered by using the same procedure (Fig. 3). No similar structure resulted from any of these randomized data sets, indicating that the patterns seen in Figs. 1 and 2 depict biological order in the gene expression response of the organism during the studied processes.
Figure 1.

Clustered display of data from time course of serum stimulation of primary human fibroblasts. Experimental details are described elsewhere (11). Briefly, foreskin fibroblasts were grown in culture and were deprived of serum for 48 hr. Serum was added back and samples taken at time 0, 15 min, 30 min, 1 hr, 2 hr, 3 hr, 4 hr, 8 hr, 12 hr, 16 hr, 20 hr, 24 hr. The final datapoint was from a separate unsynchronized sample. Data were measured by using a cDNA microarray with elements representing approximately 8,600 distinct human genes. All measurements are relative to time 0. Genes were selected for this analysis if their expression level deviated from time 0 by at least a factor of 3.0 in at least 2 time points. The dendrogram and colored image were produced as described in the text; the color scale ranges from saturated green for log ratios −3.0 and below to saturated red for log ratios 3.0 and above. Each gene is represented by a single row of colored boxes; each time point is represented by a single column. Five separate clusters are indicated by colored bars and by identical coloring of the corresponding region of the dendrogram. As described in detail in ref. 11, the sequence-verified named genes in these clusters contain multiple genes involved in (A) cholesterol biosynthesis, (B) the cell cycle, (C) the immediate–early response, (D) signaling and angiogenesis, and (E) wound healing and tissue remodeling. These clusters also contain named genes not involved in these processes and numerous uncharacterized genes. A larger version of this image, with gene names, is available at http://rana.stanford.edu/clustering/serum.html.
Figure 2.

Cluster analysis of combined yeast data sets. Data from separate time courses of gene expression in the yeast S. cerevisiae were combined and clustered. Data were drawn from time courses during the following processes: the cell division cycle (9) after synchronization by alpha factor arrest (ALPH; 18 time points); centrifugal elutriation (ELU; 14 time points), and with a temperature-sensitive cdc15 mutant (CDC15; 15 time points); sporulation (10) (SPO, 7 time points plus four additional samples); shock by high temperature (HT, 6 time points); reducing agents (D, 4 time points) and low temperature (C; 4 time points) (P. T. S., J. Cuoczo, C. Kaiser, P.O. B., and D. B., unpublished work); and the diauxic shift (8) (DX, 7 time points). All data were collected by using DNA microarrays with elements representing nearly all of the ORFs from the fully sequenced S. cerevisiae genome (8); all measurements were made against a time 0 reference sample except for the cell-cycle experiments, where an unsynchronized sample was used. All genes (2,467) for which functional annotation was available in the Saccharomyces Genome Database were included (12). The contribution to the gene similarity score of each sample from a given process was weighted by the inverse of the square root of the number of samples analyzed from that process. The entire clustered image is shown in A; a larger version of this image, along with dendrogram and gene names, is available at http://rana.stanford.edu/clustering/yeastall.html. Full gene names are shown for representative clusters containing functionally related genes involved in (B) spindle pole body assembly and function, (C) the proteasome, (D) mRNA splicing, (E) glycolysis, (F) the mitochondrial ribosome, (G) ATP synthesis, (H) chromatin structure, (I) the ribosome and translation, (J) DNA replication, and (K) the tricarboxylic acid cycle and respiration. The full-color range represents log ratios of −1.2 to 1.2 for the cell-cycle experiments, −1.5 to 1.5 for the shock experiments, −2.0 to 2.0 for the diauxic shift, and −3.0 to 3.0 for sporulation. Gene name, functional category, and specific function are from the Saccharomyces Genome Database (13). Cluster I contains 112 ribosomal protein genes, seven translation initiation or elongation factors, three tRNA synthetases, and three genes of apparently unrelated function.
Figure 3.

To demonstrate the biological origins of patterns seen in Figs. 1 and 2, data from Fig. 1 were clustered by using methods described here before and after random permutation within rows (random 1), within columns (random 2), and both (random 3).
A central feature of Figs. 1 and 2 is that one can look at such images, identify patterns of interest, and readily zoom in on the detailed expression patterns and identities of the genes contributing to these patterns. An important test of the value of this approach comes when we examine the identity of the clustered genes at varying levels of identity.
Redundant Representations of Genes Cluster Together.
At the finest level, we have found repeatedly that genes represented by more than one array element or genes with high degrees of sequence identity are clustered next to, or in the immediate vicinity of, each other. Thus the exact representation of a gene on the array (alternate cDNA clones of differing length in the case of human arrays or highly homologous genes in S. cerevisiae) makes little difference in the observed pattern of gene expression. Moreover, even though groups of genes may show very similar patterns of expression, as is seen in Figs. 1 and 2, in general individual genes can be distinguished from all other genes on the basis of subtle differences in their regulation. Finally, this result also indicates that noise present in single observations does not contribute significantly when genes are compared across even a relatively small number of nonidentical conditions. Therefore, when designing experiments, it may be more valuable to sample a wide variety of conditions than to make repeat observations on identical conditions.
Genes of Similar Function Cluster Together.
A far more striking result is found when larger groups of clustered genes are examined, where we observe a strong tendency for these genes to share common roles in cellular processes. This relationship is clearest in data from experiments on the budding yeast S. cerevisiae, where arrays representing essentially all of the genes from this organism are available (8) and for which a large fraction of the identified genes (more than 35%) have been studied in some detail. Fig. 2A represents a clustering analysis of 2,467 genes, all the genes that currently have a functional annotation in the Saccharomyces Genome Database (12). As can be seen in Fig. 2 B–K, numerous groups of coexpressed genes representing diverse expression patterns across the sampled conditions are involved in common cellular processes. Although one might be concerned about the possibility of crosshybridization, it is clear in the examples below that genes of unrelated sequence but similar function cluster tightly together.
A particularly dramatic example is the extensive cluster (shown in Fig. 2I) of 126 genes strongly down-regulated in response to stress (after each of the shocks, at the latter stages of the diauxic shift where glucose levels are diminished, and after transfer to nutrient-limited sporulation media), and which covary throughout the cell cycle. This cluster is dominated by genes encoding ribosomal proteins (112 genes) and other proteins involved in translation (initiation and elongation factors and tRNA synthetases). It has been reported that yeast responds to favorable growth conditions by increasing the production of ribosomes (13) through transcriptional regulation of genes encoding ribosomal proteins (14).
Mitochondrial protein synthesis genes were also expressed concordantly along with a number of genes involved in respiration (Fig. 2F) in a pattern roughly similar to a large cluster of coexpressed genes involved in ATP synthesis (predominantly members of the F1F0 ATPase complex) and oxidative phosphorylation (Fig. 2G). Oxygen-related transcriptional regulation of genes involved in oxygen utilization has been characterized extensively (15).
The genes encoding the bulk of the components of the proteasome (Fig. 2C) and the mini-chromosome maintenance DNA replication complex (Fig. 2J) are also coexpressed. In addition, there are many examples of coexpressed genes that share a common or related function but are not members of large protein complexes, such as genes encoding numerous glycolytic enzymes (Fig. 2E), genes involved in the tricarboxylic acid cycle and oxidative phosphorylation (Fig. 2K), and genes involved in mating (not shown). These examples emphasize that the observed coregulation occurs primarily at the level of cellular function and not only with the exact protein function (e.g., enzymic reaction catalyzed) of the gene product.
Finally, there is an extremely tight cluster of eight histone genes (duplicates of each of histones H2A, H2B, H3, and H4). It is well known that these genes are coregulated and are transcribed at a particular point in the cell cycle (16).
In human data sets, relationships among the functions of genes in clusters are obscured somewhat by the less complete functional annotation of human gene sequences. Nonetheless, when the composition of the clusters is examined, they are often found to contain genes known to share a common role in the cell. This observation is well illustrated in the data from the response of human tissue culture cells to serum after serum starvation (Fig. 1). When available functional information on the genes studied in this experiment was examined, keeping in mind the often poor state of annotation of the human genome, the clusters of genes indicated by colored branches were found to generally contain genes involved in cholesterol biosynthesis (cluster A), the cell cycle (cluster B), the immediate–early response (cluster C), signaling and angiogenesis (cluster D), and tissue remodeling and wound healing (cluster E); a detailed description of these observations is contained in ref. 11.
DISCUSSION
Microarray-based genomic surveys and other high-throughput approaches (ranging from genomics to combinatorial chemistry) are becoming increasingly important in biology and chemistry. As a result, we need to develop our ability to “see” the information in the massive tables of quantitative measurements that these approaches produce. Our approach to this problem can be generalized as follows. First, we use a common-sense approach to organize the data, based on order inherent in the data. Next, recognizing that the rate-limiting step in exploring and searching large tables of numerical data is a trivial one: reading the numbers (human brains are not well adapted to assimilating quantitative data by reading digits), we represent the quantitative values in the table by using a naturalistic color scale rather than numbers. This alternative encoding preserves all the quantitative information, but transmits it to our brains by way of a much higher-bandwidth channel than the “number-reading” channel.
A natural way of viewing complex data sets is first to scan and survey the large-scale features and then to focus in on the interesting details. What we have found to be the most valuable feature of the approach described here is that it allows this natural and intuitive process to be applied to genomic data sets. The approach is a general one, with no inherent specificity to the particular method used to acquire data or even to gene-expression data. It is therefore likely that very similar approaches may be applied to many other kinds of very large data sets. In each case, it may be necessary to find alternative algorithms and computation methods to bring out inherent structures in the data, and, equally important, to find dense naturalistic visual representations that convey the quantitative information effectively. We recognize that the particular clustering algorithm we used is not the only, or even the best, method available. We have used and are actively exploring alternatives such as parametric ordering of genes (9) and supervised clustering methods based on representative hand-picked or computer-generated expression profiles (10). The success of these very simple approaches has given us confidence to face the coming flood of functional genomic data.
The examples presented here demonstrate a feature of gene expression that makes these methods particularly useful, namely the tendency of expression data to organize genes into functional categories. It is, of course, not very surprising that genes that are expressed together share common functions. Nonetheless, the extent to which gene expression patterns suffice to separate genes into functional categories across a relatively small and redundant collection of conditions is surprising. It seems likely that the addition of more and diverse conditions can only enhance these observations. When the clustering analysis described here is applied to all of the approximately 6,200 genes of S. cerevisiae, the clusters of functionally related genes are maintained, but are usually expanded with the addition of uncharacterized genes (the results of this analysis will be the subject of a subsequent report). On the basis of our observations here, it is probable that many of these genes will also share common functions. While not based on biological necessity, similarity of pattern of expression may be the easiest available means of making at least provisional attribution of function on a genomic scale.
Finally, the functional concordance of coexpressed genes imparts biological significance to the broad patterns seen in images like those of Figs. 1 and 2. For example, the representation of the transcriptional response of human fibroblasts to serum shown in Fig. 1 is not simply a list of genes and their associated expression patterns, nor is it an arbitrary structure that is being seen, but rather it is a comprehensive representation of the state of the cell throughout its response to serum. Likewise, for yeast experiments, information on the state of many cellular processes can be inferred quickly by combining and comparing new experiments with the data presented here.
Supplementary Material
Acknowledgments
We thank J. De Risi for excellent technical assistance and many useful suggestions and the staff of the Saccharomyces Genome Database. We also thank J. Cuoczo and C. Kaiser for the use of unpublished results. This work was supported by a grants from the National Institutes of Health (GM 46406, HG 00983, and CA77097). P.O.B. is an associate investigator with the Howard Hughes Medical Institute. P.T.S. was supported by a training grant from the National Eye Institute (Bethesda, MD). M.B.E. was supported by a postdoctoral fellowship from the Alfred E. Sloan Foundation (New York, NY).
References
- 1.Schena M, Shalon D, Davis R W, Brown P O. Science. 1995;270:467–470. doi: 10.1126/science.270.5235.467. [DOI] [PubMed] [Google Scholar]
- 2.Velculescu V E, Zhang L, Vogelstein B, Kinzler K W. Science. 1995;270:484–487. doi: 10.1126/science.270.5235.484. [DOI] [PubMed] [Google Scholar]
- 3.Lockhart D J, Dong H, Byrne M C, Follettie M T, Gallo M V, Chee M S, Mittmann M, Wang C, Kobayashi M, Horton H, et al. Nat Biotechnol. 1996;14:1675–1680. doi: 10.1038/nbt1296-1675. [DOI] [PubMed] [Google Scholar]
- 4.Kohonen T. Self-Organizing Maps. New York: Springer; 1997. [Google Scholar]
- 5.Sokal R R, Michener C D. Univ Kans Sci Bull. 1958;38:1409–1438. [Google Scholar]
- 6.Schena M, Shalon D, Heller R, Chai A, Brown P O, Davis R W. Proc Natl Acad Sci USA. 1996;93:10614–10619. doi: 10.1073/pnas.93.20.10614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shalon D, Smith S J, Brown P O. Genome Res. 1996;6:639–645. doi: 10.1101/gr.6.7.639. [DOI] [PubMed] [Google Scholar]
- 8.DeRisi J L, Iyer V R, Brown P O. Science. 1997;278:680–686. doi: 10.1126/science.278.5338.680. [DOI] [PubMed] [Google Scholar]
- 9.Spellman, P. T., Sherlock, G., Iyer, V. R., Zhang, M., Anders, K., Eisen, M. B., Brown, P. O., Botstein, D. & Futcher, B. (1998). Mol. Biol. Cell, in press. [DOI] [PMC free article] [PubMed]
- 10.Chu S, DeRisi J, Eisen M, Mulholland J, Botstein D, Brown P O, Herskowitz I. Science. 1998;282:699–705. doi: 10.1126/science.282.5389.699. [DOI] [PubMed] [Google Scholar]
- 11.Iyer, V. R., Eisen, M. B., Ross, D. R., Schuler, G., Moore, T., Lee, J. C. F., Trent, J. M., Hudson, J., Boguski, M., Lashkari, D., et al. (1998) Science, in press. [DOI] [PubMed]
- 12.Cherry J M, Ball C, Weng S, Juvik G, Schmidt R, Adler C, Dunn B, Dwight S, Riles L, Mortimer R K, et al. Nature (London) 1997;387:67–73. [PMC free article] [PubMed] [Google Scholar]
- 13.Kief D R, Warner J R. Mol Cell Biol. 1981;1:1007–1015. doi: 10.1128/mcb.1.11.1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kraakman L S, Griffioen G, Zerp S, Groeneveld P, Thevelein J M, Mager W H, Planta R J. Mol Gen Genet. 1993;239:196–204. doi: 10.1007/BF00281618. [DOI] [PubMed] [Google Scholar]
- 15.Kwast K E, Burke P V, Poyton R O. J Exp Biol. 1998;201:1177–1195. doi: 10.1242/jeb.201.8.1177. [DOI] [PubMed] [Google Scholar]
- 16.Hereford L M, Osley M A, Ludwig T R, 2nd, McLaughlin C S. Cell. 1981;24:367–375. doi: 10.1016/0092-8674(81)90326-3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.