

Abstract
There has been much debate on the contribution of processes such as the persistence of antigens, cross-reactive stimulation, homeostasis, competition between different lineages of lymphocytes, and the rate of cell turnover on the duration of immune memory and the maintenance of the immune repertoire. We use simple mathematical models to investigate the contributions of these various processes to the longevity of immune memory (defined as the rate of decline of the population of antigen-specific memory cells). The models we develop incorporate a large repertoire of immune cells, each lineage having distinct antigenic specificities, and describe the dynamics of the individual lineages and total population of cells. Our results suggest that, if homeostatic control regulates the total population of memory cells, then, for a wide range of parameters, immune memory will be long-lived in the absence of persistent antigen (T1/2 > 1 year). We also show that the longevity of memory in this situation will be insensitive to the relative rates of cross-reactive stimulation, the rate of turnover of immune cells, and the functional form of the term for the maintenance of homeostasis.
Although the ability to maintain memory after an encounter with an antigen is one of the central features of the immune system, the mechanism(s) by which immune memory is maintained are not yet fully understood. The initial view (1) suggesting that “memory” lymphocytes might be very long-lived cells could be rejected because lymphocytes have turnover rates much shorter than the lifespan of the host (2, 3). The relatively high rates observed for the turnover particularly of antigen-specific cells after stimulation led to the hypothesis that maintenance of an elevated population of antigen-specific immune cells might require restimulation, either by persistent antigen or by repeated exposure to antigen. Several observations were marshaled in support of this hypothesis. First, antigen–antibody complexes were found to remain on follicular dendritic cells long after initial exposure to the antigen (4). Second, the transfer of lymphocytes (either B cells or CD4+ T-helper or CD8+ cytotoxic T lymphocyte) from an antigen-stimulated to a naive host (in the absence of transferred antigen) was followed by a rapid decline in the population of these cells (5–7). Although persistent antigen or repeated stimulation is likely to result in long-lasting immune memory, the question of the duration of immune memory in the absence of such restimulation remained. Several lines of evidence in support of the hypothesis that immune memory may be long-lived in the absence of persisting antigen include the long-standing “natural history” studies, which showed long-lived immunity to viruses such as the measles virus and yellow fever virus persisted for decades after infection under conditions in which repeated exposure was highly unlikely (8–10), and recent experimental studies that have followed populations of antigen-specific immune cells in the absence of their specific antigen (11–20).
It has been suggested that the interplay between processes, including the stimulation of antigen-specific lineages and their interaction with other populations of cells, both via cross-reactive stimulation and homeostatic competition for space, will be important in determining the longevity of immune memory and the repertoire (2, 9, 21–23). However, there is no consensus regarding the interplay between these various processes for the maintenance of memory. We use simple models to allow us to rigorously analyze the outcomes arising from a set of well defined assumptions for the underlying biology of immune cells on the dynamics of memory and the repertoire. In particular, we examine how the population dynamics of antigen-specific cells will depend on the following: the rate of input from progenitor cells (via the thymus for T cells and bone marrow for B cells), the death rate of cells, the mechanism and rate of homeostasis, and the level of cross-reactive stimulation of cells. For simplicity, we thus have defined immune memory as the maintenance of an elevated population of antigen-specific cells, noting that this is a major but not the sole requirement for protective immunity (24, 25).
In this paper, we begin by melding a simple “antigen-specific” immune response into a model of the immune repertoire. The dynamics of a simple antigen-specific immune response during an acute infection (see Fig. 1 and ref. 21 for details) has an “effector phase” and “memory phase.” The effector phase begins with rapid clonal expansion (≈4–5 logs in magnitude) of antigen-specific cells, and the generation of effector and possibly memory cells, and ends with the rapid contraction (≈2 logs in magnitude) of the population of antigen-specific lymphocytes, presumably by apoptosis of the effector cells and possibly via differentiation of some antigen-specific cells into memory cells. The second, or memory, phase of the immune response is characterized by much slower changes in the densities of antigen-specific cells in comparison to the first phase of the immune response. We begin by developing a “single compartment” model in which cells have the same characteristics except their antigen specificity. This model allows us to study how the different factors, such as homeostatic regulation and cell turnover, will affect the rate of loss of a single antigen-specific lineage. We then extend the model to examine the consequences of including naive and memory compartments.
Figure 1.

A schematic view of the dynamics of an antigen-specific immune response after stimulation.
Our models differs from earlier mathematical models that have focused on the population dynamics of antigen-specific immune cells (26–29) and as a first approximation have assumed that the dynamics of these cells is independent of the rest of the immune system. Other models that have considered the interaction between different antigen-specific cells for the most part have focused on idiotype–antiidiotype networks (30–32). In this paper, we develop models that combine the dynamics of antigen-specific stimulation and interaction between different lineages (see refs. 33–35) and use these models to examine the duration of immune memory and the maintenance of the repertoire.
Simple Single Compartment Model
We begin with a simple model in which immune cells have distinct antigen specificities but otherwise have the same properties (identical intrinsic growth rates, background death rates, and so forth). We let n equal the total number of possible lineages of immune cells (the expressed repertoire), xi (i = 1, 2… . n) equal the number of immune cells in the ith lineage, and X = Σxi equal the total population of immune cells.
The following processes determine the dynamics of a single lineage: (i) Input (from the thymus for T cells or bone marrow for B cells) is set at a low rate a per lineage per unit time. Because input is small (i.e., a ≪ 1 per day), we introduce such input by a stochastic term. For a given lineage i, the input a*i is modeled by the addition of one cell at random times with an exponential distribution of waiting times between successive inputs and a mean waiting time of 1/a. The average total number of cells input from the thymus or bone marrow equals na per day. (ii) Next is proliferation in response to stimulation, either by pathogen or antigen. A given lineage only infrequently encounters the specific antigen, so we let the probability of such encounter for specific antigen equal q per unit time. Because q ≪ 1, we introduce this event by a stochastic term. For a given lineage i, the waiting time between two antigen-specific stimulations q*i has an exponential distribution with a mean of 1/q. On stimulation with antigen, we increase the specific lineage by an amount m (because the timescale for this effector phase is short compared with that of memory, we allow this increase to be of magnitude m and occur instantly). On average, nq clones are stimulated per day. (iii) Cross-reactive stimulation is incorporated in the model by assuming that, simultaneously with the high degree of stimulation of cells with very high affinity for that antigen (mentioned in ii), other lineages are stimulated to a lesser extent. Clearly for cross-reactive stimulation to be effective in maintaining memory, it must be relatively frequent, and we introduce it by a deterministic term. If every time an antigen is introduced, the cross-reactive stimulation results in proliferation of a fraction f of all lineages by a factor m′ (m′ smaller than m), then the average rate of cross-reactive stimulation per lineage per unit time will be given by m′fq. We rewrite this amount as cq with c = m′f. (iv) The death rate of immune cells is set to d per unit time. (v) Homeostatic regulation is incorporated by introduction of a density-dependent term S(X), which maintains the total number of immune cells near the “carrying capacity.” It can operate by changing either the growth or death rates of immune cells.
With these variables and parameters, we have the following set of equations for the dynamics of each lineage:
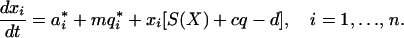 |
1 |
We obtain the rate of change of X by summing over all lineages. Because the number of lineages, n, is very large, we can replace the stochastic terms a*i and q*i by their averages na and mnq:
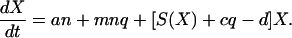 |
2 |
The dynamics of X under Eq. 2 is simple. Any positive solution of Eq. 2 exponentially fast reaches a steady-state level, X̂, which can be found by solving
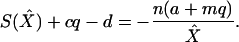 |
3 |
Because the dynamics of Eq. 2 are fast, and we consider a much longer timescale, we may assume that X is held at its steady-state level, that is, X = X̂. Using Eq. 3 and setting q*i ≡ 0 and X = X̂ in Eq. 1, we obtain:
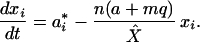 |
4 |
To study the long-term average behavior of xi under Eq. 4, we can replace the stochastic term a*i by its expectation per unit time a. We find that xi(t) will approach the value x̂i at an exponential rate, R, independent of the antigenic specificity, i:
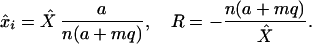 |
5 |
This tells us that the rate of loss of memory, R, is directly proportional to the rate of generation of new immune cells either by input from the thymus or by antigenic stimulation and is inversely proportional to the total population of immune cells. Because the variations in c and d contribute exclusively to the value of X̂, which persists near the carrying capacity of the immune system, we find that the rate of loss of immune memory is insensitive to the death rate of immune cells and the rate of cross-reactive stimulation. Of interest, although homeostasis is required for immune memory, the duration of immune memory is independent of a particular form of the function for homeostasis S(X) as far as the total population X is maintained near a given density. We can estimate the rate of decline of immune (memory) cells for various rates of stimulation with antigen (q) and the magnitude of the antigen-specific expansion in the population of immune cells (m). As seen in Fig. 2, for a wide range of parameters, the population of antigen-specific immune cells declines with a half-life in excess of 2 years. The model also suggests that turnover rates only indirectly influence immune memory caused by homeostasis and will do so through the drift in the population. Although the population is relatively high (as it must be when immune memory is present), drift will not affect the longevity of immune memory.
Figure 2.

The duration of immune memory in the single compartment model. We show how the duration of immune memory, plotted as the half-life of the population of pathogen-specific cells after stimulation, T1/2, will depend on the average frequency of infection with different pathogens, average clonal expansion per pathogen, and input from the thymus. Parameters: Total number of cells X = 108, total input from thymus 105 cells per day, and rates of stimulation with pathogens and extent of expansion of pathogen specific memory cells as indicated.
In Fig. 3, we plot the dynamics of the immune repertoire. We find that the rate of loss of the repertoire depends on the input from the thymus. If the level of input from the thymus is sufficiently high, so that x̂i > 1, then unstimulated lineages will persist near the level x̂i, with small stochastic fluctuations, and conversely, if the level of input from the thymus is sufficiently low (x̂i < 1), then we expect lineages that have not been stimulated to be driven to extinction over time, and there will be a dramatic reduction in the repertoire over time.
Figure 3.

The dynamics of the immune repertoire for a single compartment model. We plot the repertoire of immune cells (scaled so that the maximum repertoire equals unity) as a function of time. We find that the maintenance of the repertoire over a long duration depends on the input from the thymus. Parameters: In the simulation, we examined the dynamics of 105 lineages of cells with a total of ≈106 cells (this amounts to ≈1/100th of the immune system of a mouse). Probability of stimulation per lineage per day, q = 10−6; input from thymus stochastic with probabilities, a = 0.0, 0.001, and 0.01 per lineage per day; background death rate, d = 0.003 per day; cross-reactivity, c = 0.001 per day; homeostasis being incorporated by a logistic function, S(X) = s(1 − X/k), with s = 1.0 per day and k = 106 lymphocytes; magnitude of the specific immune response (burst size), m = 104 cells per lineage.
Model with Naive and Memory Compartments
Naive cells and memory cells have different characteristics. We incorporate the differences between naive and immune cells by introducing two subpopulations for each lineage of antigen-specific cells. We denote the lineage of ith specificity in the naive and memory compartments by xi and yi, respectively, and introduce the subscripts x and y to represent parameters for the these compartments, respectively. On stimulation, naive cells in the specific lineage expand to form m cells in the memory compartment independently of the initial size of the corresponding naive compartment. As the sizes of naive and memory populations remain relatively stable, we allow for independent homeostasis in naive and memory compartments (see ref. 36 and Discussion for a more detailed consideration of this assumption):
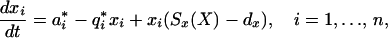 |
6 |
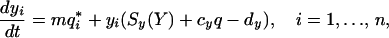 |
7 |
Consequently, the equations for X = ∑xi and Y = ∑yi will be:
 |
8 |
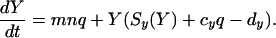 |
9 |
If homeostatic regulation operating on the memory compartment maintains its size at a certain level Ŷ, then, in the absence of subsequent stimulation to the ith lineage, the rate of change in its population yi can be estimated as
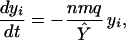 |
10 |
This is very similar to Eq. 5 for the single compartment model: the rate of loss of immune memory is expected to be directly proportional to the number of new cells generated by exposure to specific antigen and inversely proportional to the total size of the memory compartment (Fig. 4). The difference with the single compartment model is that the rate of input of naive cells from the thymus does not directly affect the duration of immune memory, but the size of the memory compartment is smaller than the total population of naive and memory cells.
Figure 4.

The duration of immune memory for a two-compartment model with independent homeostatic regulation in naive and memory compartments. In this model, the naive and memory cells differ in their properties, but the homeostasis acts independently in both compartments. We plot how the duration of immune memory (represented by the half-life of the population of pathogen-specific cells after stimulation, T1/2) will depend on the frequency of infection with different pathogens and the extent of clonal expansion to memory cells per pathogen. Parameters as in Fig. 2, with the carrying capacity of 108 cells equally divided in naive and memory compartments and input from thymus set to zero.
The dynamics of the naive and memory repertoire is shown in Fig. 5. Changes in the naive repertoire depend on the rate of input of new cells from the thymus and the rate of loss of lineages caused either by conversion of naive cells to memory cells on stimulation or by drift. If antigenic stimulation does not result in the conversion of all naive cells to memory cells, or if there is a very low rate of reversion from memory to naive phenotype (37, 38), then the loss of the naive repertoire will, in this model, result only because of the drift in the populations of cells in different lineages. The low rate of turnover observed in the naive cells will minimize the rate of loss of the naive repertoire by drift. The repertoire of the memory compartment initially will increase at the rate of stimulation with new antigens and saturates at time t̃ and repertoire rm given by:
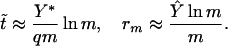 |
11 |
The maximal repertoire rm in the memory compartment increases (linearly) with the size of the memory compartment, decreases with an increase in the extent of expansion of a given lineage on antigenic stimulation, and does not depend on the rate of stimulation with antigens. The distribution of clone size in the memory compartment after this time will be an exponential decay with those clones that most recently have been stimulated having the highest populations and the populations of the other clones decaying exponentially with time since their last stimulation.
Figure 5.

Dynamics of naive and memory repertoires in the two-compartment model. We plot the repertoire in the naive and memory compartments and the total number of lineages stimulated as a function of time. We find that independent homeostasis in naive and memory compartments allows the repertoire to be maintained even in the absence of input from the thymus, which is set to zero in the simulations. In a, the naive and memory compartments are assumed to have a fixed size, and in b, the size of the naive compartment decreases and that of the memory compartment increases linearly with time. Parameters as in Fig. 3 except input from thymus, a = 0; levels of cross-reactivity, cx = 0.001 (naive) and cy = 0.05 (memory); background death rates, dx = 0.003 and dy = 0.05; the total population, kx + ky = 106 cells, is assumed to be divided equally between naive and memory compartments (kx = ky = 5 × 105 cells) in a whereas in b there is a slow shift from naive to memory populations, with 80% of the cells having a naive phenotype initially and then declining to 20% by the end of the simulations.
Provided that naive and memory compartments are of similar sizes, the longevities of immune memory will be comparable for the single compartment model and one with independent regulation of homeostasis in naive and memory compartments. However, the latter model can allow for the maintenance of the naive repertoire to be much larger than the in single compartment model. Consequently, a model with independent homeostasis in naive and memory compartments will have a better compromise between the maintenance of memory and the repertoire.
Discussion
In this section, we discuss the implications of this model on the current debate on the mechanism for immune memory and the trade-off between the longevity of immune memory and the maintenance of the immune repertoire. We then consider how our model fits into the current theoretical and experimental literature, critically discussing some of the limitations on the model and outlining how it may be extended in the future. Finally, we describe potential ways in which the model may be experimentally tested.
Our models indicate that, if homeostasis maintains the size of the memory compartment, then the rate of loss of memory will be directly proportional to the average rate of generation of memory cells to other antigens and inversely proportional to the average size of the memory compartment. We have used the model to estimate the duration of memory in the absence of persistent antigen–and have obtained a half-life for antigen-specific immune cells to be in excess of 1 year (and upward to 100 years) (Figs. 2 and 4). This agrees with observations that show the persistence of immune memory in the absence of antigen in many experimental model systems (11–18, 20). Our models thus predict that the maintenance of a constant average size of the memory compartment (homeostasis) is central for the maintenance of memory and the duration of immune memory.
The maintenance of homeostasis is not yet fully understood and may be influenced by many factors, including input from the thymus (39), competition of cells for major histocompatibility complex (17), possibly in conjunction with self-antigen (35), resource (40), and cross-reactive (bystander) stimulation (22). The factors responsible for maintenance of homeostasis mentioned above may work by regulating (in a density-dependent fashion) the proliferation or death rates (or both) of immune cells. Our models formally show that the rate of decline of memory will be independent of the precise mechanisms involved. In particular, we note that cross-reactive or bystander stimulation may be required for memory only indirectly through maintaining homeostasis. Our model assumes that the parameters for homeostasis of different lineages within the naive (or memory) compartments are approximately equal and independent of the repertoire. Differences in the parameters governing homeostasis for different lineages will result in competition between lineages in a manner dependent on the mechanism of homeostasis. We currently are modifying our model to include competition between lineages for self-antigen (35) and partial Lotka–Volterra competition between naive and memory compartments (34, 40). Our preliminary observations suggest that, provided that the diversity of the repertoire (and the number of self-antigens) is sufficiently high, the basic results of this model for the longevity of immune memory are relatively robust with respect to incorporation of such forms of competition. One basic assumption of our model, namely that the population of cells in both naive and memory compartments is relatively stable, is consistent with numerous experimental studies (36, 41). This assumption does not, however, take into account the very gradual decline in size of the naive compartment or the increase in size of the memory compartment with age. If we assume that the size of the memory compartment increases linearly with time, that is, Ŷ(t) = Ŷ(0) + βt >0, then the decline of a memory lineage, yi, after specific stimulation at time t0 will be slightly slower than previously determined (and subexponential, given by
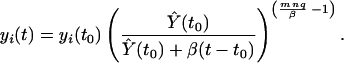 |
12 |
Because biologically we expect β to be small (i.e., the memory compartment to only slowly increase in size), we expect only a small increase in the duration of immune memory, accompanied by a decline of the naive repertoire at higher ages (Fig. 5b).
In the current paper, we have focused on the duration of immune memory and have touched only briefly on the consequences for the immune repertoire. In particular, we have not incorporated changes in the immune response as the repertoire changes. In particular, we have assumed that each lineage is stimulated with a very low probability per unit time, and that this is independent of the “repertoire,” and have not considered explicitly at what point the repertoire is not sufficient to control new invading pathogens. Nor have we considered the consequences of stochastic drift in the immune lineages into our model. Although drift does not result in a change in the duration of memory (because these lineages have a relatively large size), it may, however, have a more important effect on the naive repertoire, which is composed of lineages that may have a relatively small size.
The model thus lends strong support to the hypothesis first proposed by Freitas, Rocha, and colleagues (2, 36) that the duration of immune memory is governed principally by the homeostasis and that independent (or almost independent) regulation of homeostasis in the naive and memory compartments could optimize the tradeoff between the duration of immune memory and the repertoire. We also note that the model rules out cross-reactive stimulation from regulating immune memory except indirectly insofar as it is necessary for the maintenance of homeostasis.
The model makes testable predictions. We briefly list these and examine how they fit in with existing experimental observations. First, our model predicts that immune memory may last several years in the absence of either persistent antigen or subsequent antigenic stimulations. This is consistent with the experiments described in the introduction of the paper (11–18, 20). The estimates for the half-life of immune memory suggest that persistent antigen or repeated exposure to antigen may not be required for the maintenance of immune memory in short lived vertebrates such as mice; however, depending on the precise region of parameter space (i.e., the rate and extent of stimulation with new pathogens), repeated exposure may play an additional role in the maintenance of memory of long-lived vertebrates.
Second, if homeostasis is responsible for the maintenance of immune memory, the model suggests that the rate of loss of immune memory to a given antigen-specific lineage will be directly proportional to the product of the rate of encounter with other antigens and the magnitude of stimulation per exposure. This would allow discrimination from the situation in which cross-reactive stimulation was responsible for maintaining immune memory. In the latter case, we would expect the average (i.e., in the long term) duration of immune memory to increase with increases in the rate of stimulation with other, potentially cross-reactive antigens. Selin et al. (42) followed virus-specific CD8+ cells after LCMV infection and found that their frequency declined after infection of the host with unrelated viruses, suggesting that homeostasis rather than cross-reactivity is responsible for the maintenance of memory. It remains to be determined whether the rate of loss of memory depends on the number of memory cells produced on stimulation with the unrelated viruses, as predicted by our model.
Third, our model assumes that the extent of cross-reactivity to self-antigens is similar for different memory lineages. The alternative possibility is that different degrees of self-reactivity maintain memory-cell lineages at levels dependent on the degree of cross-reactivity with self (see ref. 35). These alternatives can be tested by determining whether the relative magnitudes of immune memory to different epitopes on the same proteins (or pathogens) decline at the same rate or at different rates after immunization. Recent results for CD8+ T-cell memory after LCMV infection (43, 44) suggest that the rate of loss of CD8+ memory cells to different epitopes does not vary significantly, suggesting that our assumption is valid for CD8+ cells.
Finally, our models suggest that, when the frequency of antigenic stimulation from other infectious agents is very high, the duration of immune memory is likely to be relatively low. This may be tested in several ways. At the experimental level we expect that sufficiently frequent exposure to new pathogens will result in a relatively high rate of decline of memory to a given pathogen. Heterogeneity in the immune system itself may result in some locations, such as the gut-associated lymphoid tissue, receiving a disproportionately higher frequency of antigenic stimulation in comparison to other lymphoid tissue. Consequently, we expect the duration of memory to gut infections to be relatively short-lived, unless the pathogens or antigens of the pathogen migrate to the systemic immune system, where the frequency of antigenic insult is relatively low and, in accord with our models, the duration of immune memory is relatively high.
Table 1.
Variables and parameters in the model
| Symbol and parameter | Range |
|---|---|
| xi, number of cells in ith lineage | at t = 0 random with average k/n |
| X = Σ xi number of cells | X large, X ≈ k |
| k, carrying capacity | k large, ≈ 108 in mice |
| n, maximal repertoire | (105 − 109) 107 in simulations |
| a, thymic input per lineage per day | a ≪ 1 |
| p, frequency of parasite stimulation per day | p < 1 (stochastic) (10−2 − 1) |
| g, lineages per parasite | g > 1, (2 − 20) |
| q, frequency of stimulation per lineage per day | q = gp/n, stochastic (10−8 − 10−6) |
| mp, clonal expansion after stimulation | k/10 > mp ≫ c/n, (104 − 106) |
| m, clonal expansion per lineage | m = mp/g, (103 − 105) |
| c, cross-reactivity | cq < 1 |
| s, maximum turnover rate for homeostasis per day | 1.0 |
| d, death rate for immune cells, dx, dy, for naive and memory cells, per day | dx < dy < 1 (0.1–0.001) |
Estimates of immune system parameters for mice. To reduce simulation time we reduced the repertoire to 105 and rescaled all of the related quantities correspondingly. The parameters we used are: k = 106; n = 105; q = 10−6; and m = 104.
Acknowledgments
This project was supported by National Institutes of Health research grants R29-GM-54268 (to R. Antia) and AI-30048 (to R. Ahmed).
Footnotes
This paper was submitted directly (Track II) to the Proceedings Office.
References
- 1.Hood L, Weissman I L, Wood W B, Wilson J H. Immunology. Menlo Park, California: Benjamin/Cummings; 1984. [Google Scholar]
- 2.Freitas A A, Rocha B B. Immunol Today. 1993;14:25–29. doi: 10.1016/0167-5699(93)90320-K. [DOI] [PubMed] [Google Scholar]
- 3.Tough D F, Sprent J. Immunol Res. 1995;14:1–12. doi: 10.1007/BF02918494. [DOI] [PubMed] [Google Scholar]
- 4.Tew J G, Phipps R P, Mandel T E. Immunol Rev. 1980;53:173–201. doi: 10.1111/j.1600-065x.1980.tb01044.x. [DOI] [PubMed] [Google Scholar]
- 5.Gray D, Skarvall H. Nature (London) 1988;336:70–73. doi: 10.1038/336070a0. [DOI] [PubMed] [Google Scholar]
- 6.Gray D, Matzinger P. J Exp Med. 1991;174:969–974. doi: 10.1084/jem.174.5.969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Oehen S, Waldner H, Kundig T M, Hengartner H, Zinkernagel R M. J Exp Med. 1992;176:1273–1281. doi: 10.1084/jem.176.5.1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sawyer W A. J Prev Med. 1931;5:413–428. [Google Scholar]
- 9.Matzinger P. Science. 1994;369:605–606. doi: 10.1038/369605a0. [DOI] [PubMed] [Google Scholar]
- 10.Panum P L. Arch Pathol Anat Physiol Klin Med. 1847;1:492–512. [Google Scholar]
- 11.Hou S, Hyland L, Ryan K W, Portner A, Doherty P C. Nature (London) 1994;369:652–654. doi: 10.1038/369652a0. [DOI] [PubMed] [Google Scholar]
- 12.Lau L L, Jamieson B D, Somasundaram T, Ahmed R. Nature (London) 1994;369:648–652. [Google Scholar]
- 13.Mullbacher A. J Exp Med. 1994;179:317–321. doi: 10.1084/jem.179.1.317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bruno L, Kirberg J, von Boehmer H. Immunity. 1995;2:37–43. doi: 10.1016/1074-7613(95)90077-2. [DOI] [PubMed] [Google Scholar]
- 15.Asano M S, Ahmed R. J Exp Med. 1996;183:2165–2174. doi: 10.1084/jem.183.5.2165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Markiewicz M A, Girao C, Opferman J T, Sun J, Hu Q, Agulnik A A, Bishop C E, Thompson C B, Ashton-Rickardt P G. Proc Natl Acad Sci USA. 1998;95:306–3070. doi: 10.1073/pnas.95.6.3065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tanchot C, Lemonnier F A, Perarnau B, Freitas A A, Rocha B. Science. 1997;276:2057–2062. doi: 10.1126/science.276.5321.2057. [DOI] [PubMed] [Google Scholar]
- 18.Di Rosa F, Matzinger P. J Exp Med. 1996;183:2153–2163. doi: 10.1084/jem.183.5.2153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dutton R W, Bradley L M, Swain S L. Annu Rev Immunol. 1998;16:201–233. doi: 10.1146/annurev.immunol.16.1.201. [DOI] [PubMed] [Google Scholar]
- 20.Ke Y, Ma H, Kapp J A. J Exp Med. 1998;187:49–57. doi: 10.1084/jem.187.1.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ahmed R, Gray D. Science. 1996;272:54–60. doi: 10.1126/science.272.5258.54. [DOI] [PubMed] [Google Scholar]
- 22.Tough D F, Borrow P, Sprent J. Science. 1996;272:1947–1950. doi: 10.1126/science.272.5270.1947. [DOI] [PubMed] [Google Scholar]
- 23.Beverly P C L. Immunol Today. 1990;11:203–205. doi: 10.1016/0167-5699(90)90083-l. [DOI] [PubMed] [Google Scholar]
- 24.Kundig T M, Bachmann M F, Oehen S, Hoffmann U W, Simard J J L, Kalberer C P, Pircher H, Ohashi P S, Hengartner H, Zinkernagel R M. Proc Natl Acad Sci USA. 1996;93:9716–9723. doi: 10.1073/pnas.93.18.9716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bachmann M F, Kundig T M, Hengartner H, Zinkernagel R M. Proc Natl Acad Sci USA. 1997;94:640–645. doi: 10.1073/pnas.94.2.640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bell G I. Math Biosci. 1973;16:291–314. [Google Scholar]
- 27.McLean A R. J Theor Biol. 1994;170:63–74. doi: 10.1006/jtbi.1994.1168. [DOI] [PubMed] [Google Scholar]
- 28.Schweitzer A N, Anderson R M. Proc R Soc Lond Ser B. 1992;247:107–112. doi: 10.1098/rspb.1992.0015. [DOI] [PubMed] [Google Scholar]
- 29.De Boer R J, Perelson A S. J Theor Biol. 1995;175:567–576. doi: 10.1006/jtbi.1995.0165. [DOI] [PubMed] [Google Scholar]
- 30.De Boer R J, Hogeweg P. J Theor Biol. 1989;139:17–38. doi: 10.1016/s0022-5193(89)80055-4. [DOI] [PubMed] [Google Scholar]
- 31.Perelson A S. Immunol Rev. 1989;110:5–36. doi: 10.1111/j.1600-065x.1989.tb00025.x. [DOI] [PubMed] [Google Scholar]
- 32.Weisbuch G, De Boer R J, Perelson A S. J Theor Biol. 1990;146:483–499. doi: 10.1016/s0022-5193(05)80374-1. [DOI] [PubMed] [Google Scholar]
- 33.Merrill S J, De Boer R J, Perelson A S. Rocky Mountain J Math. 1994;24:213–231. [Google Scholar]
- 34.Mittler J, Levin B, Antia R. J Acquired Immune Defic Syndr. 1996;12:233–248. doi: 10.1097/00042560-199607000-00003. [DOI] [PubMed] [Google Scholar]
- 35.De Boer R J, Perelson A S. Int Immunol. 1997;9:779–790. doi: 10.1093/intimm/9.5.779. [DOI] [PubMed] [Google Scholar]
- 36.Tanchot C, Rocha B. Eur J Immunol. 1995;25:2127–2136. doi: 10.1002/eji.1830250802. [DOI] [PubMed] [Google Scholar]
- 37.Bell E B, Sparshott S M. Nature (London) 1990;348:163–166. doi: 10.1038/348163a0. [DOI] [PubMed] [Google Scholar]
- 38.Michie C A, McLean A, Alcock C, Beverley P C L. Nature (London) 1992;360:264–265. doi: 10.1038/360264a0. [DOI] [PubMed] [Google Scholar]
- 39.Tanchot C, Rocha B. J Exp Med. 1997;186:1099–1106. doi: 10.1084/jem.186.7.1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.McLean A R, Rosado M M, Agenes F, Vasconcello R, Freitas A A. Proc Natl Acad Sci USA. 1997;94:5792–5797. doi: 10.1073/pnas.94.11.5792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mackall C L, Hakim F T, Gress R E. Immunol Today. 1997;18:245–251. doi: 10.1016/s0167-5699(97)81664-7. [DOI] [PubMed] [Google Scholar]
- 42.Selin L K, Vergilis K, Welsh R M, Nahill S R. J Exp Med. 1996;183:2489–2499. doi: 10.1084/jem.183.6.2489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Murali-Krishna K, Altman J D, Suresh M, Sourdive D J D, Zajac A J, Miller J D, Slansky J, Ahmed R. Immunity. 1998;8:177–187. doi: 10.1016/s1074-7613(00)80470-7. [DOI] [PubMed] [Google Scholar]
- 44.Busch D H, Pilip I M, Vijh S, Pamer E G. Immunity. 1998;8:353–62. doi: 10.1016/s1074-7613(00)80540-3. [DOI] [PubMed] [Google Scholar]