

Abstract
The flavin hydroperoxide at the active site of the mixed-function oxidase 2-aminobenzoyl-CoA monooxygenase/reductase (Azoarcus evansii) transfers an oxygen to the 5-position of the 2-aminobenzoyl-CoA substrate to provide the alkoxide intermediate II−. Hydrogen migration from C5 to C6 follows this monooxygenation. The nature of the monooxygenation intermediate and plausible competing reactions leading to hydrogen migration have been considered. Ab initio molecular orbital theory has been used to calculate structures and electron distributions in intermediate and transition state structures. Electrostatic potential surface calculations establish that the transition state and product, associated with the C5 to C6 hydrogen transfer, are stabilized by electron distribution to the benzoyl-CoA thioester carbonyl oxygen. This is not so for the transition state and product associated with hydrogen transfer from C5 to C4. The activation energy for the 5,6-shift is 2.5 kcal/mol lower than that for the 5,4-shift. In addition, the product of the hydrogen 5,6-shift is more stable than is the product of the hydrogen 5,4-shift, by ≈6 kcal/mol. These results explain why only the shift of hydrogen from C5 to C6 is observed experimentally. Oxygen transfer and hydrogen migration almost coincide in the gas phase (activation energy of ≈0.6 kcal/mol, equivalent to a single bond vibration). Enzymatic formation of alkoxide II− requires its stabilization; thus, the rate constant for its breakdown would be slower than in the gas phase.
Oxygenase enzymes are separated into two classes: dioxygenases and mixed-function oxidases. Dioxygenases transfer both oxygens from molecular oxygen to the substrate to provide product. Mixed-function oxidases transfer one oxygen from molecular oxygen to the substrate, and the remaining oxygen is incorporated into a water molecule. For flavin mixed-function oxidases, Hamilton proposed that oxygen reacts with enzyme-bound dihydroflavin to provide a 4a-hydroperoxyflavin (4a-FlH-OOH) (1). Kemal and Bruice synthesized a 4a-hydroperoxide [4a-hydroperoxy-5-ethyl-3-methyllumiflavin (4a-FlEt-OOH)] by reaction of hydrogen peroxide with the flavinium cation, N(5)-ethyl-3-methyl-1,5-dihydrolumiflavin (2). These studies made it possible to explore oxygen transfer from 4a-hydroperoxides to substrates (3). The reaction of 1,5-dihydroflavins to form 4a-hydroperoxideds has been shown to involve O2⨪ and a flavin radical as intermediates (4, 5). It is generally accepted that flavin-4a-hydroperoxides form in the active sites of flavin-dependent mixed-function oxidases when reduced flavin reacts with molecular oxygen (6–12). It was determined that the monooxygen transfer involving 4a-hydroperoxyflavins does not occur from a carbonyl oxide formed on ring opening of the isoalloxazine ring between C4 and C4a (13, 14), as originally proposed (1), but by nucleophilic attack on the terminal oxygen of the flavin 4a-hydroperoxide (15). Dioxygen transfer from the oxygen anion 4a-FlEt-OO− is known in model reactions (3, 16–20), but there is no evidence for the existence of a flavin dioxygenase enzyme.
The flavoenzyme mixed-function oxygenase 2-aminobenzoyl-CoA monooxygenase/reductase (ACMR) (EC 1.13.14.40) from Azoarcus evansii catalyzes both the oxygenation and hydrogenation reactions provided in Eqs. 1 and 2. The biological ![]()
role of the enzyme is tryptophan biosynthesis and degradation as well as the degradation of electron-rich aromatic substrates. ACMR is composed of two identical subunits, each comprised of an 85-kDa chain and containing one flavin and one nicotinamide cofactor. It was found that, if one flavin cofactor was removed from the enzyme, monooxygenation activity was retained but the hydrogenation activity was abolished (21). Thus, it was determined that one flavin cofactor is responsible for monooxygenation whereas the other is involved in the hydrogenation of the substrate. The best substrate for ACMR is 2-aminobenzoyl-CoA (AC), but the enzyme can hydroxylate a number of electron rich aromatic compounds (21, 22). The transfer of an oxygen from 3O2 to substrate can be separated into three distinct steps (Eqs. 3–5).
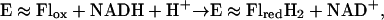 |
3 |
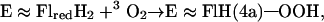 |
4 |
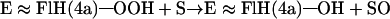 |
5 |
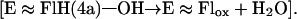 |
In Eq. 5, the terminal oxygen of the hydroperoxide is added to the substrate (Scheme S1) to provide an intermediate that rearranges with hydrogen transfer (Scheme S2). Hartmann and coworkers have examined the reaction using [5-2H]AC (I) with ACMR (23). It was noted that, within the limits of detection, the migration of the hydrogen occurs only from C5 to C6 of AC (Scheme S3). For the identity of SO, one must consider a hydroxylated product (II− in Scheme S1), formed by nucleophilic attack of the 2-aminobenzoyl-CoA on the terminal oxygen of flavin hydroperoxide, or oxygen insertion to provide arene oxide(s) (III and IV of Scheme S2).
Scheme 1.

Scheme 2.

Scheme 3.

In this study, we examine the reactions of Scheme S2 by using ab initio molecular orbital calculations. Quantum mechanical calculations are carried out in the gas phase. Mechanistic conclusions are drawn by the observation of competing pathways and comparing structures with like charge distributions.
Theoretical Procedure
All ab initio molecular orbital calculations were performed by using the gaussian 98 program (24). Geometry optimizations were carried out at the Hartree-Fock (HF)/6 − 31 + G(d, p) level of theory. Inclusion of diffuse functions in the calculations were essential because of the presence of oxygen anions. In addition, polarizable functions were included on the hydrogens as the migration of a hydrogen atom is of interest (25). The large CoA thioester moiety of 2-aminobenzoyl-CoA was modeled by using —C(⩵O)SCH3. Compounds in which this replacement occurs are labeled with a. Transition states were located by using the Synchronous Transit-Guided Quasi-Newton method (26). All stationary points were identified as minima or transition states by using vibrational analysis. Transition states were characterized by the presence of one negative force constant corresponding to the interconversion of the reactant and the respective product. To confirm that the located transition state structure is in fact the one corresponding to the hydrogen migration reactions (5, 6-shift or 5, 4-shift), intrinsic reaction coordinate analyses were performed. The electrostatic potential calculations were performed at the HF/6 − 31 + G(d, p) level.
Results and Discussion
The structure proposed by Langkau et al. (27) as the final product of the reaction of 2-aminobenzoyl-CoA monooxygenase/reductase (ACMR) with 2-aminobenzoyl-CoA (AC) was confirmed by Hartmann et al. (23). They determined that, within detectable limits, migration of 2H on C5 occurs to C6, providing 2-amino-5-oxocyclohex-1-ene-1-carboxyl-CoA; no C5 to C4 migration was observed. Attempts to realize the experimental epoxidation of 2,3-dimethyl-2-butene by 4a-FlEt-OOH were not successful (15). This alkene is particularly subject to epoxidation even in the air. This suggests that formation of arene oxides III and IV by oxygen insertion from ACMR by bound flavin-4a-hydroperoxide should not be favorable. The initial product of monooxygenation is most likely II− (Scheme S1). However, the possibility that arene oxides III and/or IV arise from II− and undergo hydrogen [1,2]-sigmatropic migrations is considered (Scheme S2).
The optimized reactant structure (IIa−) and structure of products corresponding to the 5,6-shift (Va) and 5,4-shift (VIa) at the HF/6 − 31 + G(d, p) level of theory are shown in Fig. 1, with the calculated relative energies presented in Table 1. The HF/6 − 31 + G(d, p) method predicts highly exothermic reactions for formation of products from both the hydrogen 5,6-shift and 5,4-shift. The optimized reactant structure and 5,6-shift product both display the thioester moiety in the plane of the aromatic ring. In the optimized 5,4-shift product, however, the thioester is twisted out of the plane of the aromatic ring by ≈73°. This twisting is likely associated with a lack of resonance interaction with the ring system and lack of enamine stabilization (see below). The calculations reveal that the 5,6-migration product Va is 5.94 kcal/mol more stable than the 5,4-migration product.
Figure 1.

The optimized geometry's for IIa−, Va, and VIa at the HF/6 − 31 + G(d, p) level of theory. Relevant distances for each structure are given in angstroms. The distance corresponding to the hydrogen that migrates is depicted with a curved arrow in each structure.
Table 1.
Calculated relative energies (in kcal/mol) in the gas phase at the HF/6 − 31 + G(d, p) level of theory
| Compound | Relative energy |
|---|---|
| Reactant, IIa | 0 |
| 5,4-shift product, VIa | −59.32 |
| 5,6-shift product, Va | −53.38 |
| 5,6-arene oxide, IIIa | −28.80 |
| 5,6-shift TS, VII | 0.63 |
| 5,4-shift TS, VIII | 3.12 |
The transition state structures corresponding to each product were then calculated. The transition state structures for the 5,6-shift (VII) and 5,4-shift (VIII) reactions are shown in Fig. 2. As expected for highly exothermic reactions, both transition state structures are early and, thus, reactant-like. A schematic drawing of the potential energy surface of the gas phase reaction of both hydrogen migration pathways is shown in Fig. 3. The transition state for the 5,6-shift is 0.63 kcal/mol higher in energy than the reactant II− whereas the transition state for the 5,4-shift is 3.12 kcal/mol higher in energy than II−. This is in accord with the experimental observation that the only product observed is that formed by a hydrogen 5,6-shift (23). The migration of the hydrogen on C5 to C6 is, thus, both kinetically and thermodynamically favored over migration to the C4 position.
Figure 2.

The calculated transition state structures for the 5,6-shift (VI) and the 5,4-shift (VII) pathways.
Figure 3.

The calculated potential energy surface in the gas phase at the HF/6 − 31 + G(d, p) level of theory for the reactions involving migration of the hydrogen on C5 of 2-aminobenzoyl-CoA to either the C4 or C6 position.
Electrostatic potential surfaces may be used to reflect changes that occur in the electron distribution when going from starting state to transition state and then to product. The electrostatic potentials corresponding to the structures in Figs. 1 and 2 were calculated (Fig. 4). In the zwitterion IIa−, the formal negative charge is on the —O− substituent of C5, and the formal positive charge is on the hydrogens of the 2-imino group. As the reaction proceeds to the transition state on the path to the 5,6-shift product, the negative charge is delocalized into the thioester moiety. This can be seen by examination of the electron distribution of the transition state VII (Fig. 4). Although the transition state is early and its overall structure is similar to the reactant, one can clearly see the increase in electron density on the —C(⩵O)SCH3 carbonyl oxygen. This increase in electron density becomes pronounced at both the —C(⩵O)SCH3 carbonyl oxygen and —NH2 moiety in the 5, 6-shift product (Va). Further inspection shows only small changes in electron densities at these positions in the transition state or product ground state in the reaction forming the 5,4-shift product (VIII and VIa). In addition, there is stabilization of the 5,6-shift product attributable to electron delocalization from —NH2 to the —C(⩵O)SCH3 carbonyl oxygen, as represented in Scheme S2, V. Thus, favoring of a formal hydride shift from position C5 to C6 appears to be attributable to stabilization of transition state and product by electron delocalization to the —C(⩵O)SCH3 carbonyl oxygen.
Figure 4.

Structures of the reactant (IIa−), transition states (VII and VIII), and products for the 5,6- (Va) and 5,4-shift (VIa) reactions, respectively, with electrostatic potentials projected onto the molecular surfaces of each. The electrostatic potential spectrum is from −0.05 (blue) to 0.35 (red) with and electron density of 0.002 e/bohr3, with blue representing negative electrostatic potential and red representing positive electrostatic potential.
Can a satisfactory explanation for the product of this oxidation be obtained if an arene oxide is involved? Direct formation of an arene oxide has been judged to be unlikely (loc. cit.) such that the only paths to consider for arene oxide formation are epoxide ring closures of II− to III or IV. If this were the case, an explanation for formation of III being more favorable than IV is required. The transition state for formation of the arene oxide III would likely be resonance stabilized by electron delocalization to both —NH2 and —C(⩵O)SCH3 functionalities. The transition state for formation of IV from II− would not be so stabilized. In the NIH shift (28) of the hydrogen from the 5 to the 6 position from arene oxide III, there would be no resonance stabilization of the transition state, and we have seen that the 5,6 shift from IIa− is favored by resonance stabilization of the product. The relative energies of IIIa and IIa− were compared, and IIIa was found to be ≈29 kcal/mol lower in energy (Table 1). However, the reaction trajectory for conversion of IIa− to product Va is characterized by a very small reaction barrier (≈0.6 kcal mol), and Va is more stable than IIa− by ≈60 kcal/mol. Thus, II− represents an unstable intermediate that proceeds directly to the product V such that arene oxides III and IV cannot be formed from II−. Ignoring entropy, an activation energy of 0.6 kcal/mol is equivalent to a rate constant of ≈2 × 1013 s−1, which is in the range for a single bond vibration in the gas phase.
Conclusions
It is known that 2-aminobenzoyl-CoA (AC) undergoes oxygen addition at C5 by the flavin mixed-function oxidase AC monooxygenase/reductase (ACMR), and as a following step, a hydride is transferred from C5 to C6. It is reasoned that a logical mechanism for the enzymatic oxygen transfer is nucleophilic attack of the 5-position carbon of AC on the terminal oxygen of the enzyme-bound 4a-FlH-OOH to provide II− (Scheme S1). This is an established mechanism in both model (15) and mixed-function oxidase enzymes (9). The results of our ab initio calculations provide a likely explanation of why hydride transfer in II− is from C5 to C6 rather than from C5 to C4. The C5 to C6 rearrangement is favored because of stabilization of both the transition state and product by electron delocalization into the carbonyl carbon of the —C(⩵O)SCH3 substituent. The C5 to C6 migration of hydrogen in IIa− is shown to be more favorable than the ring closure to provide arene oxide (i.e., IIIa), which could then undergo a C5 to C6 NIH shift. Indeed, the exceedingly small barrier for conversion of IIa− to Va (equivalent to a single bond vibration) in the gas phase shows that II− has virtually no lifetime. Because the formation of the anion II− in the gas phase would have a very large activation barrier, we suggest that formation of II− at the active site of ACMR is assisted by positive electrostatic forces. A distinct possibility for this stabilization is an adjacent guanidine function of an arginine residue. Protonation by general acid catalysis would also stabilize the transition state, but the intermediate IIH would be too stable to undergo the 5 to 6 hydrogen transfer. Electrostatic stabilization of II− will increase the kinetic barrier for II− to undergo the hydrogen 5 to 6 migration to give V. This should be of little concern, however, because the rearrangement of unstabilized II− has essentially no activation barrier.
Scheme 4.

Acknowledgments
The authors thank the National Center for Supercomputing Applications (Urbana–Champaign, IL) and the National Science Foundation through the University of California at Santa Barbara Supercomputer Grant (CDA96-01954) and Silicon Graphics, Inc., who made these computations possible. Valuable conversations were carried out with Drs. Ya-Jun Zheng and Kalju Khan. Appreciation is expressed to the National Institutes of Health for a supplement to grant DK09171 for support of R.A.T.
Abbreviation
- ACMR
2-aminobenzoyl-CoA monooxygenase/reductase
References
- 1.Hamilton G A. In: Progress in Bioorganic Chemistry. Kaiser E T, Kezdy F J, editors. Vol. 1. New York: Wiley Interscience; 1971. pp. 83–157. [Google Scholar]
- 2.Kemal C, Bruice T C. Proc Natl Acad Sci USA. 1976;73:995–999. doi: 10.1073/pnas.73.4.995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kemal C, Bruice T C. J Am Chem Soc. 1979;101:1635–1638. [Google Scholar]
- 4.Kemal C, Chan T W, Bruice T C. J Am Chem Soc. 1977;99:7272–7286. doi: 10.1021/ja00464a030. [DOI] [PubMed] [Google Scholar]
- 5.Eberlein G, Bruice T C. J Am Chem Soc. 1983;105:6685–6697. [Google Scholar]
- 6.Entsch B, Ballou D P, Massey V. J Biol Chem. 1976;251:2250–2563. [PubMed] [Google Scholar]
- 7.Hastings J W, Balny C, Le Peuch C, Douzou P. Proc Natl Acad Sci USA. 1973;70:3468–3472. doi: 10.1073/pnas.70.12.3468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Poulsen L L, Ziegler D M. J Biol Chem. 1979;254:6449–6455. [PubMed] [Google Scholar]
- 9.Ortiz-Maldonado M, Ballou D P, Massey V. Biochemistry. 1999;38:8124–8137. doi: 10.1021/bi990560e. [DOI] [PubMed] [Google Scholar]
- 10.Spector T, Massey V. J Biol Chem. 1972;247:5632–5636. [PubMed] [Google Scholar]
- 11.Strickland S, Massey V. J Biol Chem. 1973;248:2953–2962. [PubMed] [Google Scholar]
- 12.Massey V. J Biol Chem. 1994;269:22459–22462. [PubMed] [Google Scholar]
- 13.Wessiak A, Bruice T C. J Am Chem Soc. 1983;105:4809–4825. [Google Scholar]
- 14.Wessiak A, Noar J B, Bruice T C. Proc Natl Acad Sci USA. 1984;81:332–336. doi: 10.1073/pnas.81.2.332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bruice T C, Noar J B, Ball S S, Venkataram U V. J Am Chem Soc. 1983;105:2452–2463. [Google Scholar]
- 16.Keum S K, Gregory D H, Bruice T C. J Am Chem Soc. 1990;112:2711–2715. [Google Scholar]
- 17.Muto S, Bruice T C. J Am Chem Soc. 1980;102:4472–4480. [Google Scholar]
- 18.Muto S, Bruice T C. J Am Chem Soc. 1980;102:7559–7564. [Google Scholar]
- 19.Muto S, Bruice T C. J Am Chem Soc. 1982;104:2284–2290. [Google Scholar]
- 20.Zheng Y-J, Bruice T C. Bioorganic Chem. 1997;25:331–336. [Google Scholar]
- 21.Langkau B, Vock P, Massey V, Fuchs G, Ghisla S. Eur J Biochem. 1995;230:676–685. [PubMed] [Google Scholar]
- 22.Buder R, Fuchs G. Eur J Biochem. 1989;185:629–635. doi: 10.1111/j.1432-1033.1989.tb15159.x. [DOI] [PubMed] [Google Scholar]
- 23.Hartmann S, Hultschig C, Eisenreich W, Fuchs G, Bacher A, Ghisla S. Proc Natl Acad Sci USA. 1999;96:7831–7836. doi: 10.1073/pnas.96.14.7831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Frisch M J, Trucks G W, Schlegel H B, Scuseria G E, Robb M A, Cheeseman J R, Zakrzewski V G, Montgomery J A J, Stratmann R E, Burant J C, et al. gaussian 98. Pittsburgh: Gaussian; 1998. [Google Scholar]
- 25.Foresman J B, Frisch Æ. Exploring Chemistry with Electronic Structure Methods. Pittsburgh: Gaussian; 1995. [Google Scholar]
- 26.Peng C, Schlegel H B. Isr J Chem. 1993;33:449–454. [Google Scholar]
- 27.Langkau B, Ghisla S, Buder R, Ziegler K, Fuchs G. Eur J Biochem. 1990;191:365–371. doi: 10.1111/j.1432-1033.1990.tb19131.x. [DOI] [PubMed] [Google Scholar]
- 28.Gurdoff G, Daly J W, Jerina D M, Renson J, Witkop B, Udenfriend S. Science. 1967;157:1524–1530. doi: 10.1126/science.157.3796.1524. [DOI] [PubMed] [Google Scholar]