

Abstract
Pyrrolizidine alkaloids are preformed plant defense compounds with sporadic phylogenetic distribution. They are thought to have evolved in response to the selective pressure of herbivory. The first pathway-specific intermediate of these alkaloids is the rare polyamine homospermidine, which is synthesized by homospermidine synthase (HSS). The HSS gene from Senecio vernalis was cloned and shown to be derived from the deoxyhypusine synthase (DHS) gene, which is highly conserved among all eukaryotes and archaebacteria. DHS catalyzes the first step in the activation of translation initiation factor 5A (eIF5A), which is essential for eukaryotic cell proliferation and which acts as a cofactor of the HIV-1 Rev regulatory protein. Sequence comparison provides direct evidence for the evolutionary recruitment of an essential gene of primary metabolism (DHS) for the origin of the committing step (HSS) in the biosynthesis of pyrrolizidine alkaloids.
Keywords: secondary metabolism, gene recruitment, initiation factor 5A, alkaloid phylogeny, plant chemical defense
Pyrrolizidine alkaloids (PAs) are typical secondary compounds that are constitutively expressed in species of highly restricted phylogenetic distribution within the angiosperms. They are preferably found in the plant families Asteraceae (tribes Eupatorieae and Senecioneae), Boraginaceae (many genera), Fabaceae (mainly the genus Crotalaria), and Orchidaceae (nine genera). More than 95% of the PA-containing species investigated thus far belong to these four families. In addition, PAs are also scattered in several other families, where they sometimes occur in a single species (1) (Fig. 1). Thus, from the evolutionary standpoint, PAs appear to be a relatively young group of secondary compounds. Their skew occurrence among angiosperms has been particularly puzzling, i.e., did the common ancestor of all PA-producing plants possess the pathway, followed by many independent losses, or did the PA-biosynthetic pathway evolve several times independently in evolution in distinctly separate lineages?
Figure 1.
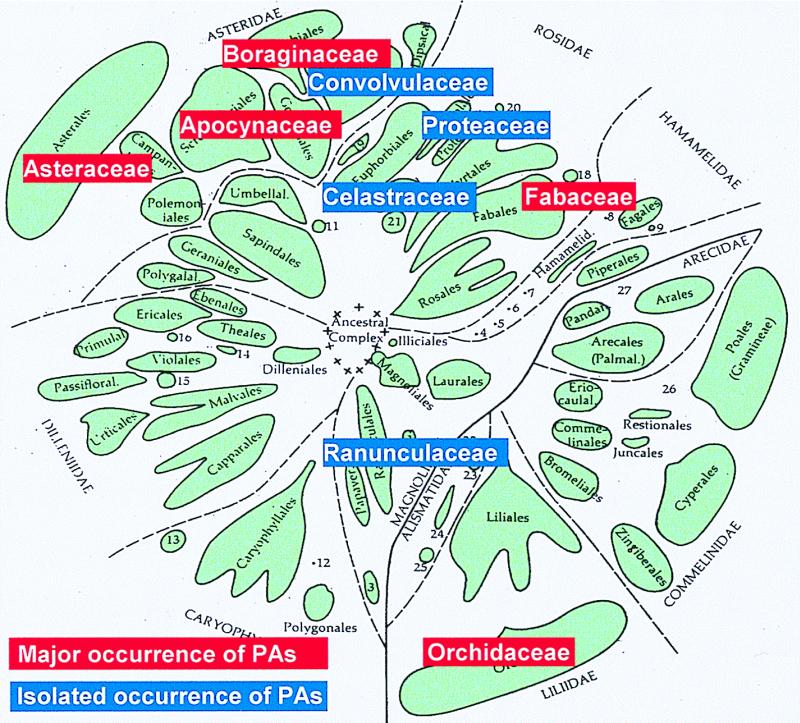
Families with pyrrolizidine alkaloid-containing species within the angiosperms (modified from ref. 34).
PAs produce dynamic interactions between PA-containing plants and their herbivores. Though direct evidence of a protective role of PAs in plant defense is sparse, convincing evidence comes from a number of insect herbivores that have developed impressive adaptations. These not only overcome the PA-mediated defense barrier, but sequester and utilize the alkaloids for their own defense against predators. All PA-adapted insects advertise their unpalatability to potential predators through their conspicuous warning coloration (2).
PAs are found in all plant organs, but in species of the Asteraceae, such as Senecio and Eupatorium, they are synthesized exclusively in the roots (3). From the roots, PAs are translocated into the shoot by means of the phloem-path and channeled to the preferential sites of accumulation, i.e., reproductive organs and peripheral tissues (2, 3). Biochemical studies revealed that the biosynthesis of the necine base, the basic moiety of the PAs, is linked to primary metabolism by putrescine and the aminobutyl moiety of spermidine, both of which are derived from arginine (4). Homospermidine, the first pathway-specific intermediate in PA biosynthesis (5), is formed by homospermidine synthase (HSS) (EC 2.5.1.44) (Fig. 2A). HSS catalyzes the NAD+-dependent transfer of an aminobutyl group of spermidine to putrescine. In contrast to previous views (5, 6) and in contrast to HSS from a bacterial source (7, 8), putrescine cannot replace spermidine as an aminobutyl donor (R. Harms, D.O., T.H., unpublished results).
Figure 2.

Reactions catalyzed by HSS (A) and DHS (B) in their metabolic background. Both enzymes transfer an aminobutyl moiety of spermidine to their individual substrates in an NAD+-dependent reaction. (A) HSS catalyzes the first step in PA biosynthesis, generating homospermidine as the precursor of the necine base, the typical backbone structure of PAs. (B) DHS commences the posttranslational activation of eIF5A by modifying a protein-bound lysine residue to deoxyhypusine, which is hydroxylated in a second step to hypusine by deoxyhypusine monooxygenase.
The biosynthesis of PAs in Asteraceae is a strictly controlled and coordinated process that is tightly linked to root growth, as it is reduced or ceases when root growth stops (4, 9). PAs do not undergo turnover except for diversification of their basic chemical structures (10). Thus, HSS occupies a key regulatory position in PA biosynthesis by controlling the formation of the total amount of alkaloid in the plant.
In this article, we describe the purification of the HSS from Senecio vernalis (Asteraceae), a typical PA-producing plant, to apparent homogeneity, the microsequencing of the protein resulting in four peptides and the cloning of HSS cDNA. The sequence of HSS reveals a surprising evolutionary origin of the biochemical pathway leading to PAs and may lead to a satisfactory explanation of the scattered distribution of these compounds across taxa.
Materials and Methods
Purification of HSS.
Two-week-old root cultures of S. vernalis (5) were used as starting material, and buffer A [50 mM KH2PO4/2 mM dithioerythritol/0.5 mM NAD+/0.5 mM spermidine/0.1 mM EDTA (pH 8.7)] was used as standard buffer in all purification steps. Roots (approximately 500 g fresh weight) were pulverized in a mortar in the presence of liquid nitrogen, and the powder was extracted in buffer A supplemented with 20 mM ascorbate and 25% (wt/wt of root mass) Polyclar AT (Serva) by stirring at 4°C for 60 min. The extract was centrifuged at 20,000 × g for 30 min, frozen at −80°C, and concentrated by lyophilization. The lyophilisate was dissolved in buffer A (1/25th of the initial volume of the extract) and desalted with Sephadex G-50 (Amersham Pharmacia). The desalted solution was applied to a DEAE-Fractogel column (Merck) and eluted with a gradient from 0 M to 0.2 M NaCl. Fractions containing HSS activity were pooled, adjusted to a NaCl concentration of 1.5 M, and loaded to a Phenyl Sepharose CL-4B column (Amersham Pharmacia). After elution with modified buffer A (5 mM instead of 50 mM KH2PO4), the eluate was applied again to a DEAE-Fractogel column, but was now eluted with a pH gradient from pH 8.5 to pH 4.5 by using McIlvaine buffer (25 mM citric acid and 50 mM Na2HPO4) supplemented with 2 mM dithioerythritol, 0.5 mM NAD+, 0.5 mM spermidine, and 0.1 mM EDTA. Fractions containing HSS activity were pooled, rebuffered to buffer A, and subjected to spermidine Sepharose affinity chromatography applying CH-Sepharose 4B (Amersham Pharmacia) coupled with spermidine. HSS was eluted in the presence of 100 mM spermidine and was subsequently applied to a Superdex 200 HR 10/30 column (Amersham Pharmacia). Peak fractions were loaded to a Mono Q column (Amersham Pharmacia) and eluted with a gradient from 0 M to 1.0 M NaCl. Purified HSS was separated by SDS/PAGE and transferred to poly(vinylidene difluoride) membrane (Millipore). The band corresponding to the HSS was subjected to microsequencing by using a method described earlier (7).
RNA Isolation and cDNA Synthesis.
Four-day-old root cultures and young shoot tips of S. vernalis were frozen in liquid nitrogen and stored at −80°C until use. Total RNA was extracted with the RNeasy Plant Mini Kit (Qiagen, Chatsworth, CA), and 2 μg of total RNA was used as template for reverse transcription as described (11).
PCR with Degenerate Primers.
According to an alignment of amino acid sequences of deoxyhypusine synthase (DHS) from various organisms (12), degenerated primers were synthesized (Table 1). In a reaction volume of 25 μl, 1 μl of cDNA was amplified by using all possible combinations of the forward primers P1–P3 and the reverse primers P4–P6. As temperature program, a modified “touch-down” protocol (13) with decreasing annealing temperatures from 55°C to 40°C (0.5°C per cycle, 40°C constant for 10 further cycles) was used. With the primer pair P3–P6, 597-bp and 591-bp fragments were amplified from the cDNA derived from root tissue and shoot tips, respectively. After electrophoretic purification, the PCR products were subcloned with the TA Cloning Kit (Invitrogen), according to the manufacturer's protocol, and sequenced.
Table 1.
Nucleotide sequence, restriction sites, and purpose of all synthesized primers
| Primer | 5′-nucleic acid sequence-3′ |
|---|---|
| P1 for 20 mer, 1536× degeneracy | ACN ATH TTY YTN GGN TAY AC |
| P2 for 19 mer, 1024× degeneracy | GN TAY YTN GTN CAR CAY AA |
| P3 for 18 mer, 1024× degeneracy | GGN GGN MTN GAR GAR GAY |
| P4 rev 18 mer, 384× degeneracy | CAY CAY ATH GCN AAY GCN |
| P5 rev 19 mer, 1024× degeneracy | TA NTC NGC NCC RTT NCK CA |
| P6 rev 18 mer, 1024× degeneracy | CCA NSW NAC NGC YTC RTC |
| P7 for 3′RACE (HSS) | CGA GAC TTC ATA TCT GTA TTG GGC ATA TAA |
| P8 for 3′RACE (HSS) (nested) | AAC AGA TGG CTC TAT CGG AGA CAT GTT |
| P9 rev 5′RACE (HSS) | CCC TTT CGA TCT CAA AT |
| P10 rev 5′RACE (HSS) (nested 1) | TCT CAA ATC AGC TCC GGG TAA AGA A |
| P11 for 5′RACE (HSS) (nested 2) | GTG CTT GCT AGG CAT TTT ATG A |
| P12 for 3′RACE (DHS) | GAA TTG CGT TCG AAA GGA CTA AAT CGT ATT |
| P13 rev 5′RACE (DHS) | CGC ATT ATT AGT TTT GAT |
| P14 rev 5′RACE (DHS) (nested 1) | TTT GTT CTT CCA ACA CTT GGT CAA ATA |
| P15 rev 5′RACE (DHS) (nested 2) | GTT GTC ATT CGG CAC CAA CAA |
| P16 for full-length (HSS), NdeI | TA TAT CAT ATG GCC GAG TCA AAC AAA GAA G |
| P17 rev full-length (HSS) | TT ACC TGA TCA AAA ACC ATT GAC TTT AGA TGC |
| P18 for full-length (DHS) | TA TCT AGA ATG GAA GAA TCT ATG AAA CAA G |
| P19 rev full-length (DHS), BamHI | TAA GGA TCC TCA GGA ACT CGG CTC AGC |
| P20 for 20 mer, 64× degeneracy | ATG TCN GAY GAR GAR CAY CA |
| P21 for full-length (eifsv), NdeI | TA TAT CAT ATG TCG GAT GAG GAG CAC CA |
| P22 rev full-length (eifsv), XhoI | TAA CTC GAG TTT TGG GCC AAT ATC CTT AAG AG |
For the degenerated primers P1–P6 and P20 the following code was used: K = G + T, H = A + T + C, M = A + C, N = A + T + C + G, R = A + G, S = C + G, W = A + T, Y = T + C. Restriction sites are underlined and the start-ATG is shown in bold; for, forward primer; rev, reverse primer.
Generation of Full-Length cDNA Clones.
To identify the 3′ and 5′ ends of the cDNA fragments, 3′-rapid amplification of cDNA ends (RACE) and 5′-RACE techniques were used as described (11). For this purpose, the gene-specific primers P7–P11 were designed for the HSS-coding cDNA, and primers P12–P15 were designed for the DHS-coding cDNA (Table 1). The resulting 685-bp and 900-bp fragments from 3′ RACE contained the 3′ end, including the poly(A) tail of HSS and DHS, respectively. 5′ RACE yielded 535-bp and 740-bp fragments containing the ATG start codon of HSS and DHS, respectively. To amplify the full-length cDNA encoding HSS and DHS, two gene-specific primer pairs were synthesized (P16–P17 and P18–P19, respectively; Table 1). Using these primer pairs (0.4 μM each), the HSS- and DHS-coding cDNAs were amplified with Native Pfu DNA Polymerase (Stratagene). The reaction mixture of 100 μl contained 4 μl of the same cDNA sample, which was successfully used as template to amplify the corresponding cDNA fragment with the degenerate primers. In the case of the HSS-coding cDNA, the resulting 1,129-bp fragment was digested with NdeI. For ligation, the expression vector pET3a (Novagen) was linearized with BamHI, filled in with Klenow fragment of DNA polymerase I, purified, and digested with NdeI. The ligation constructs were transformed into Escherichia coli NM522 cells (Stratagene), screened with primers P16 and P17 for the correct insert, and purified for sequencing and for transformation of E. coli BL21(DE3) (Stratagene) for overexpression. The 1,133-bp cDNA fragment coding the DHS was digested with BamHI and ligated in pET3a, as described above, with the exception that the vector was linearized with NdeI and digested with BamHI after treatment with Klenow fragment and that positive transformants were screened with the primers P18 and P19.
Purification of Recombinant HSS and DHS.
The overexpressed proteins were purified to apparent homogeneity by a simplified purification procedure, as described for the purification of recombinant tobacco DHS (11).
Cloning and Expression of cDNA Encoding eifsv1.
Using an alignment of amino acid sequences of all plant eukaryotic initiation factor 5A (eIF5A) precursor proteins from the database (blast search), a degenerated primer was synthesized containing the start ATG (P20). The amplification of 1 μl of S. vernalis root cDNA was performed in a reaction volume of 25 μl with primer P20 and an oligo(dT)17 primer (5′-dGTCGACTCGAGAATTC(T)17-3′) with an annealing temperature of 60°C. The resulting fragment of 720 bp was subcloned after electrophoretic purification with the TOPO TA Cloning Kit (Invitrogen). After sequence analysis, two gene-specific primers were synthesized that contained a NdeI restriction site together with the start ATG (P21) and a XhoI restriction site together with the stop codon (P22). The full-length eIF5A precursor protein cDNA was amplified by using the root cDNA as template. The resulting 491-bp fragment was cloned into the pET-23b expression vector (Novagen) and expressed with a C-terminal His-Tag motive, as described for the cloning of the tobacco eIF5A (11). The overexpressed protein was purified with Ni-NTA-agarose (Qiagen), according to the manufacturer's instructions.
Enzyme Assays.
HSS and DHS activity was measured as described (11) with spermidine, eifsv1, and putrescine concentrations of 40 μM each (DHS assay; substrate saturation) and putrescine and spermidine concentrations of 400 μM each (HSS assay; substrate saturation).
Results and Discussion
Purification of HSS and Microsequencing.
To purify the HSS of root cultures of S. vernalis, a seven-step purification procedure was established. Problems encountered in purifying HSS from other Asteraceae that resulted in low specific activity and poor stability (5) were overcome by freeze-drying the crude extract. The lyophilisate could be stored at −20°C without significant loss of enzyme activity over 3 mo and thus allowed us to accumulate enough plant material. Moreover, this procedure allowed a gentle and effective concentration necessary for purifying detectable amounts of the enzyme. Fig. 3 shows an SDS/PAGE gel of the different steps of the purification procedure, which resulted in a single prominent band of an apparent molecular mass of 44.5 kDa. Using isoelectric focusing and native PAGE procedures followed by testing the sliced gel for HSS-activity, it was shown that this protein is homogenous HSS (data not shown). The purified enzyme exhibited a pH optimum between 9.0 and 9.5. By size exclusion chromatography on a Superdex 200 column, the molecular mass of the native HSS was determined to be 174 kDa, indicating that Senecio HSS is probably a homotetramer. Because microsequencing of the N terminus of the purified HSS was not possible, the protein was digested with endopeptidase LysC prior to sequencing. Four peptides were obtained: 1, EAIDSARSNVFK; 2, XXLPGADLRSK; 3, (E)INNETSYL(Q)GAYK; and 4, TEIIILEEELPK (amino acids marked by an X could not be identified; those in parentheses were uncertain).
Figure 3.

Purification of HSS from roots of S. vernalis. Indian ink stain of an SDS/PAGE on a 12% gel blotted onto a poly(vinylidene difluoride)-membrane. A 10-kDa protein ladder (Life Technologies, Grand Island, NY) (lane 1, the 50-kDa band is indicated by an arrow), crude extract (76.5 μg, lane 2), and the pooled fractions containing HSS activity of DEAE Fractogel (NaCl-elution, 9.5 μg, lane 3), Phenyl Sepharose (1.6 μg, lane 4), DEAE Fractogel (pH-elution, 1.5 μg, lane 5), spermidine-affinity chromatography and gel filtration (0.6 μg, lane 6), and Mono Q (0.6 μg, lane 7).
Homology Analysis of the Protein Peptides.
Of the resulting four peptides, two (peptides 2 and 3) showed homology in database searches to DHS, an enzyme involved in the posttranslational activation of eIF5A (see below). DHS has been intensively studied from human (14–16), different animal (17) and fungal species (18–21), and very recently from tobacco (11). Comparison of these two enzymes revealed striking similarities concerning their reaction mechanisms and some further biochemical properties. Both enzymes transfer, in NAD+-dependent reactions, the aminobutyl moiety of spermidine to a primary amino group of their respective substrates, either the diamine putrescine (HSS) or the ɛ-amino group of a specific lysine residue in the eIF5A-precursor protein (DHS) (Fig. 2). Furthermore, the in vivo activities of the two enzymes seem to be correlated with cell growth (9, 22). Both enzymes consist of four subunits with a native molecular mass of approximately 180 kDa (14, 20) and show a pH optimum between 9.0 and 9.5 (23).
Cloning, Expression, and Purification of Recombinant HSS and DHS.
Because of the striking similarities between HSS and DHS and because of the homology of two of the peptides of HSS to DHS, we generated an alignment of the highly conserved DHS amino acid sequences from human, yeast, Neurospora crassa, and the archaebacterium Methanococcus jannaschii (12) to design degenerate primers for cloning and comparative biochemistry of HSS and DHS. These were used with total RNA from different tissues of S. vernalis for reverse transcription-PCR. From root tissue and shoot tips, two different fragments (597 bp and 591 bp, respectively) were amplified containing ORFs, both of which showed high homology to the DHS amino acid sequences. Using this sequence information, gene-specific primers were synthesized for 3′ and 5′ RACE techniques, allowing the elucidation of full-length cDNAs specific to both amplification products, which later turned out to code for HSS and DHS, respectively. For both cDNA sequences, a forward primer containing the ATG start codon (primers P16 and P18) and reverse primers containing the stop codon (primers P17 and P19) were constructed to amplify the full-length sequences with Pfu DNA polymerase for overexpression and sequence confirmation. The overexpressed proteins were purified with a simplified purification procedure to apparent homogeneity. The biochemical characterization of the purified proteins and the analysis of the cDNAs for the peptides sequenced from purified HSS revealed that the cDNA cloned from roots of S. vernalis encodes HSS and the cDNA cloned from the shoot tips encodes for DHS.
Sequence Analysis of HSS and DHS cDNA.
The cDNA clone encoding HSS contains an ORF for a protein of 370 amino acids with a molecular mass of 40.7 kDa. Of the four peptides sequenced from the purified HSS, peptides 1, 2, and 3 were found in the ORF, revealing that the ambiguous first two amino acids of peptide 2 are phenylalanine (F) and serine (S) and that, in peptide 3, the amino acids 10 and 11, of which one was uncertain, could be identified as tyrosine (Y) and tryptophan (W) instead of glutamine (Q) and glycine (G) (Fig. 4). Peptide 4 also contains ambiguities in that the ORF contains glycine (G) instead of glutamate (E) residues. That these differences are peptide sequencing uncertainties and that the ORF encodes authentic HSS was shown by expression of the ORF and biochemical characterization of the overexpressed enzyme (see below). The 5′- and 3′-untranslated regions of the HSS cDNA are of 27 bp and 106 bp in length, respectively, as determined by sequencing the cDNA fragments resulting from 3′- and 5′-RACE methods. Sequence analysis of the full-length cDNA revealed different ORFs for each cDNA clone, which mainly differed in the third position of the codons, indicating that more than one gene for HSS is expressed in Senecio roots. This is consistent with Southern blots, which show several hybridizing bands for HSS (in contrast to the DHS) (S. Moll, D.O., T.H., unpublished results). The cDNA clone encoding DHS contains an ORF for a protein of 371 amino acids with molecular mass of 41.4 kDa and 5′- and 3′-untranslated regions of 174 bp and 222 bp, respectively.
Figure 4.

Alignment of the amino acid sequences of HSS and DHS from S. vernalis (Sv-HSS and Sv-DHS, respectively), DHS from tobacco (Nt-DHS), and DHS from human (Hs-DHS). Identical amino acids in all sequences are shown in black boxes, conservative replacements according to the criteria of BLOSUM matrix (35) are framed. The four peptides sequenced from the purified HSS of S. vernalis are indicated by black bars below the alignment. The amino acid sequence homology of Sv-DHS to Nt-DHS and Hs-DHS is 80% and 61% identity, respectively. The amino acid sequence homology of Sv-HSS to Sv-DHS, Nt-DHS, and Hs-DHS is 79%, 74%, and 61% identity, respectively.
The amino acid sequence of DHS from S. vernalis shows high homology to the amino acid sequences of DHS from tobacco (80% identity) and human (61%) (Fig. 4), as well as DHS from yeast (58%), Neurospora (60%), and Methanococcus (47%). The amino acid sequence of HSS from S. vernalis fits precisely into the DHS alignment (Fig. 4). It shows homology to DHS from various sources: S. vernalis (79% identity), tobacco (74%), human (61%), yeast (53%), Neurospora (56%), and Methanococcus (47%). This indicates that HSS (involved in alkaloid biosynthesis) and DHS (involved in posttranslational modification of a highly conserved translation initiation factor) are related by means of gene duplication.
Cloning and Expression of S. vernalis eIF5A-Precursor Protein.
It is known from the literature that the eIF5A-precursor proteins from different eukaryotic sources are not only similar at the sequence level, but can substitute for each other functionally as well (24). To compare the kinetic properties of HSS and DHS from S. vernalis, we decided to use the authentic eIF5A-precursor protein from the same organism as substrate for DHS, to avoid difficulties with eIF5A-precursor proteins from unrelated eukaryotes. Thus, we created an amino acid alignment of all eIF5A-cDNAs of a plant source available in the database to design a degenerate primer (P20) that contained the start-ATG because of high homology of the sequences at the N terminus. Together with the oligo(dT)17-primer, a 720-bp fragment was amplified, containing an ORF and a 3′-untranslated region of 220 bp. The ORF encodes a protein of 159 amino acids with a molecular mass of 17.3 kDa, which shows 91% amino acid identity to eIF5A-precursor protein from tobacco. The cDNA coding this protein was cloned into pET-22b and expressed in E. coli with a 6xHis-tag motive at the C-terminus, allowing the purification of the expressed protein (subsequently called eifsv1) by metal chelate-affinity chromatography for use as substrate in DHS-activity assays.
Biochemical Characterization of Recombinant HSS and DHS.
Overexpressed and purified HSS and DHS from S. vernalis were analyzed in their efficiencies to transfer an aminobutyl moiety of spermidine to eifsv1 and putrescine. Table 2 shows the specific enzyme activities, illustrating that DHS catalyzes the formation of both homospermidine and deoxyhypusine with almost the same activity, whereas HSS is inactive with eifsv1 but catalyzes the formation of homospermidine with almost the same specific activity as DHS. The observation that DHS from S. vernalis efficiently produces homospermidine from putrescine is consistent with recent results obtained for DHS from tobacco (11), a plant devoid of PAs.
Table 2.
Specific enzyme activities of homogeneously purified recombinant DHS and HSS from S. vernalis
| Substrate
|
Specific activity (pkat/mg)
|
||
|---|---|---|---|
| Aminobutyl donor (μM) | Aminobutyl acceptor (μM) | DHS | HSS |
| Spermidine (40) | eifsv1 (40) | 318 | ND* |
| Spermidine (40) | Putrescine (40) | 138 | 590 |
| Spermidine (400) | Putrescine (400) | 737 | 3206 |
Enzyme assays were performed under identical conditions in the presence of 1 mM NAD+ and two different substrate concentrations: (i) 40 μM, which is saturating and optimal in the DHS assay with eifsv 1 as substrate (eifsv 1 concentrations >40 μM cause substrate inhibition), and (ii) 400 μM, which is saturating in the HSS assay with putrescine (11).
*ND, not detectable.
Conclusions
This study describes the purification, microsequencing, cloning, overexpression, and biochemical characterization of HSS and DHS from the PA-producing plant S. vernalis, two enzymes involved in totally different segments of plant metabolism. HSS is the key enzyme for the biosynthesis of PAs, a group of secondary compounds that accumulate constitutively as defensive compounds against herbivory. DHS is involved in the activation of a translation initiation factor and is ubiquitous among eukaryotes and archaebacteria (12). This factor, eIF5A, is activated by a two-step posttranslational modification of a specific lysine residue [Lys-50 in the human eIF5A (25)] to the unusual amino acid hypusine (Fig. 2B), which is one of the two most specific posttranslational modifications known to date (26). The first modification step is catalyzed by DHS, and the resulting deoxyhypusine is hydroxylated by deoxyhypusine monooxygenase in the second step. The factor and its activation mechanism are conserved among all eukaryotes as well as in archaebacteria (12). Active eIF5A is essential for cell viability in the yeast Saccharomyces cerevisiae (27) and is a cellular cofactor required for the function of the HIV type-1 (HIV-1) (28). Though the function of eIF5A is not understood in detail (29), it was shown that depletion of eIF5A from yeast cells causes an immediate inhibition of cell growth but only a moderate inhibition of protein biosynthesis, leading to the conclusion that this factor is not required for global protein synthesis (30). It appears that eIF5A may function as a specific factor promoting the expression of a subset of genes (30) possibly involved in cell-cycle progression (31).
The sequences of the cDNAs coding for HSS and DHS from S. vernalis show amino acid and nucleic acid identities of 79% and 83%, respectively, demonstrating that the two enzymes are closely related. Furthermore, the kinetic results show that HSS and DHS are both able to synthesize homospermidine. These data suggest that in PA-producing plants like S. vernalis, the HSS gene evolved from the conserved DHS gene by gene duplication. It may have survived under the selection pressure of herbivory, to initiate the first step in PA biosynthesis. Because plant DHS seems to possess HSS activity as an inherent property, as shown for a non-PA-producing plant (11), there was no evolutionary need to shape and optimize the preexisting DHS for its function as HSS. The selection pressure for retaining the gene clearly rests on its HSS activity, which, in turn, is the prerequisite for synthesizing PAs in plant chemical defense. The HSS gene lost its ability to aminobutylate the eIF5A precursor protein of about 17 kDa in size, probably because of some amino acid exchanges that influence protein–protein interactions with eIF5A. The only other known example of recruitment of a ubiquitous primary enzyme for a secondary function is putrescine N-methyltransferase, an enzyme committed to nicotine biosynthesis in tobacco (32). This enzyme shows higher sequence homology to spermidine synthases from different plant sources as to spermidine synthases from mammals and E. coli. Thus, it is suggested that putrescine N-methyltransferase has evolved from plant spermidine synthase (33).
These findings provide an unexpected link between conserved essential cell functions (DHS) and secondary metabolism (HSS) and allow a glimpse into the hitherto untouched area of evolution of alkaloid biosynthetic pathways. The independent recruitment of HSS from the essential gene for DHS in various angiosperm lineages would provide a straightforward explanation for the sparse occurrence of these defense compounds within the angiosperms. To test this hypothesis of multiple independent origins of PAs, work is in progress to identify cDNA sequences encoding HSS and DHS from different PA-containing species and plant families to allow a sequence-based evolutionary analysis.
Acknowledgments
We thank Dr. J. Kellermann for microsequencing the protein peptides, A. Backenköhler for her excellent technical assistance, and Dr. W. Martin for critical discussions and for reading of the manuscript. This work was supported by grants from the Deutsche Forschungsgemeinschaft and Fonds der Chemischen Industrie.
Abbreviations
- PA
pyrrolizidine alkaloid
- HSS
homospermidine synthase
- DHS
deoxyhypusine synthase
- eIF5A
eukaryotic initiation factor 5A
- eifsv1
eIF5A precursor protein of Senecio vernalis with unmodified lysine residue
- RACE
rapid amplification of cDNA ends
Footnotes
Data deposition: The sequences reported in this paper have been submitted to the EMBL Nucleotide Sequence Database [accession nos. AJ238622 (S. vernalis mRNA for deoxyhypusine synthase), AJ238623 (S. vernalis mRNA for homospermidine synthase), and AJ238624 (S. vernalis mRNA for translation initiation factor 5A (eIF5A) precursor protein)].
References
- 1.Hartmann T, Witte L. In: Alkaloids: Chemical and Biological Perspectives. Pelletier S W, editor. Vol. 9. Oxford: Pergamon; 1995. pp. 155–233. [Google Scholar]
- 2.Hartmann T. Planta. 1999;207:483–495. [Google Scholar]
- 3.Hartmann T, Ehmke A, Eilert U, von Borstel K, Theuring C. Planta. 1989;177:98–107. doi: 10.1007/BF00392159. [DOI] [PubMed] [Google Scholar]
- 4.Hartmann T, Sander H, Adolph R-D, Toppel G. Planta. 1988;175:82–90. doi: 10.1007/BF00402884. [DOI] [PubMed] [Google Scholar]
- 5.Böttcher F, Adolph R-D, Hartmann T. Phytochemistry. 1993;32:679–689. [Google Scholar]
- 6.Böttcher F, Ober D, Hartmann T. Can J Chem. 1994;72:80–85. [Google Scholar]
- 7.Tholl D, Ober D, Martin W, Kellermann J, Hartmann T. Eur J Biochem. 1996;240:373–379. doi: 10.1111/j.1432-1033.1996.0373h.x. [DOI] [PubMed] [Google Scholar]
- 8.Ober D, Tholl D, Martin W, Hartmann T. J Gen Appl Microbiol. 1996;42:411–419. [Google Scholar]
- 9.Sander H, Hartmann T. Plant Cell Tissue Organ Cult. 1989;18:19–31. [Google Scholar]
- 10.Hartmann T, Dierich B. Planta. 1998;206:443–451. [Google Scholar]
- 11.Ober D, Hartmann T. J Biol Chem. 1999;274:32040–32047. doi: 10.1074/jbc.274.45.32040. [DOI] [PubMed] [Google Scholar]
- 12.Chen K Y, Liu A Y C. Biol Signals. 1997;6:105–109. doi: 10.1159/000109115. [DOI] [PubMed] [Google Scholar]
- 13.Don R H, Cox P T, Wainwright B J, Baker K, Mattick J S. Nucleic Acids Res. 1991;19:4008. doi: 10.1093/nar/19.14.4008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Klier H, Csonga R, Steinkasserer A, Wöhl T, Lottspeich F, Eder J. FEBS Lett. 1995;364:207–210. doi: 10.1016/0014-5793(95)00394-o. [DOI] [PubMed] [Google Scholar]
- 15.Joe Y A, Wolff E C, Park M H. J Biol Chem. 1995;270:22386–22392. doi: 10.1074/jbc.270.38.22386. [DOI] [PubMed] [Google Scholar]
- 16.Yan Y P, Tao Y, Chen K Y. Biochem J. 1996;315:429–434. doi: 10.1042/bj3150429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wolff E C, Lee Y B, Chung S I, Folk J E, Park M H. J Biol Chem. 1995;270:8660–8666. doi: 10.1074/jbc.270.15.8660. [DOI] [PubMed] [Google Scholar]
- 18.Sasaki K, Abid M R, Miyazaki M. FEBS Lett. 1996;384:151–154. doi: 10.1016/0014-5793(96)00310-9. [DOI] [PubMed] [Google Scholar]
- 19.Abid M R, Sasaki K, Titani K, Miyazaki M. J Biochem. 1997;121:769–778. doi: 10.1093/oxfordjournals.jbchem.a021652. [DOI] [PubMed] [Google Scholar]
- 20.Tao Y, Chen K Y. J Biol Chem. 1995;270:383–386. doi: 10.1074/jbc.270.1.383. [DOI] [PubMed] [Google Scholar]
- 21.Tao Y, Chen K Y. J Biol Chem. 1995;270:23984–23987. doi: 10.1074/jbc.270.41.23984. [DOI] [PubMed] [Google Scholar]
- 22.Park M H, Joe Y A, Kang K R, Lee Y B, Wolff E C. Amino Acids. 1996;10:109–121. doi: 10.1007/BF00806584. [DOI] [PubMed] [Google Scholar]
- 23.Murphey R J, Gerner E W. J Biol Chem. 1987;262:15033–15036. [PubMed] [Google Scholar]
- 24.Magdolen V, Klier H, Wöhl T, Klink F, Hirt H, Hauber J, Lottspeich F. Mol Gen Genet. 1994;244:646–652. doi: 10.1007/BF00282755. [DOI] [PubMed] [Google Scholar]
- 25.Smit-McBride Z, Dever T E, Hershey J W B, Merrick W C. J Biol Chem. 1989;264:1578–1583. [PubMed] [Google Scholar]
- 26.Krishna R G, Wold F. Adv Enzymol. 1993;67:265–298. doi: 10.1002/9780470123133.ch3. [DOI] [PubMed] [Google Scholar]
- 27.Schnier J, Schwelberger H G, Smit-McBride Z, Kang H A, Hershey J W B. Mol Cell Biol. 1991;11:3105–3114. doi: 10.1128/mcb.11.6.3105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ruhl M, Himmelspach M, Bahr G M, Hammerschmid F, Jaksche H, Wolff B, Aschauer H, Farrington G K, Probst H, Bevec D, Hauber J. J Cell Biol. 1993;123:1309–1320. doi: 10.1083/jcb.123.6.1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Park M H, Lee Y B, Joe A. Biol Signals. 1997;6:115–123. doi: 10.1159/000109117. [DOI] [PubMed] [Google Scholar]
- 30.Kang H A, Hershey W B. J Biol Chem. 1994;269:3934–3940. [PubMed] [Google Scholar]
- 31.Park M H, Wolff E C, Folk J K. Biofactors. 1993;4:95–104. [PubMed] [Google Scholar]
- 32.Hibi N, Higashiguchi S, Hashimoto T, Yamada Y. Plant Cell. 1994;6:723–735. doi: 10.1105/tpc.6.5.723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hashimoto T, Tamaki K, Suzuki K, Yamada Y. Plant Cell Physiol. 1998;39:73–79. doi: 10.1093/oxfordjournals.pcp.a029291. [DOI] [PubMed] [Google Scholar]
- 34.Stebbins G L. Flowering Plants: Evolution Above the Species Level. MA: Cambridge; 1974. [Google Scholar]
- 35.Henikoff J G, Henikoff S. Methods Enzymol. 1996;266:88–105. doi: 10.1016/s0076-6879(96)66008-x. [DOI] [PubMed] [Google Scholar]