

Abstract
Staphylococcus aureus is a versatile pathogen capable of causing a wide range of human diseases. However, the role of different virulence factors in the development of staphylococcal infections remains incompletely understood. Some clonal types are well equipped to cause disease across the globe, whereas others are facile at causing disease among community members. In this review, general aspects of staphylococcal pathogenesis are addressed, with emphasis on methicillin-resistant strains. Although methicillin-resistant S. aureus (MRSA) strains are not necessarily more virulent than methicillin-sensitive S. aureus strains, some MRSA strains contain factors or genetic backgrounds that may enhance their virulence or may enable them to cause particular clinical syndromes. We examine these pathogenic factors.
OVERVIEW OF THE PATHOGENESIS OF STAPHYLOCOCCUS AUREUS
This article summarizes the pathogenesis of S. aureus disease and specifically addresses the pathogenesis of infections caused by methicillin-resistant S. aureus (MRSA) strains originating in health care settings (hospital-acquired MRSA [HA-MRSA]) and in the community (community-acquired MRSA [CA-MRSA]). S. aureus pathogenesis is reviewed before the discussion of the pathogenesis of MRSA, because MRSA virulence factors are generally not unique to MRSA. Nonetheless, certain MRSA strains appear to contain particular factors or genetic backgrounds that enhance their virulence or enable them to cause particular clinical syndromes.
Colonization and disease
S. aureus is both a commensal organism and a pathogen. The anterior nares are the main ecological niche for S. aureus. Approximately 20% of individuals are persistently nasally colonized with S. aureus, and 30% are intermittently colonized. However, numerous other sites may be colonized, including the axillae, groin, and gastrointestinal tract. Colonization provides a reservoir from which bacteria can be introduced when host defenses are breached, whether by shaving, aspiration, insertion of an indwelling catheter, or surgery. Colonization clearly increases the risk for subsequent infection [1, 2]. Those with S. aureus infections are generally infected with their colonizing strain [3]. In a study of bacteremia, blood isolates were identical to nasal isolates in 82% of patients [4]. Colonization also allows S. aureus to be transmitted among individuals in both health care and community settings. The basis for S. aureus colonization is complex and incompletely understood but appears to involve the host's contact with S. aureus (e.g., other carriers) and the ability of S. aureus to adhere to host cells and to evade the immune response (reviewed by Wertheim et al. [1]).
Virulence factors and disease
The armamentarium of virulence factors of S. aureus is extensive, with both structural and secreted products playing a role in the pathogenesis of infection (figure 1). Selected examples of these factors are described in table 1. Two noteworthy features of staphylococci are that a virulence factor may have several functions in pathogenesis and that multiple virulence factors may perform the same function. In establishing an infection, S. aureus has numerous surface proteins, called “microbial surface components recognizing adhesive matrix molecules” (MSCRAMMs), that mediate adherence to host tissues. MSCRAMMs bind molecules such as collagen, fibronectin, and fibrinogen, and different MSCRAMMs may adhere to the same host-tissue component. MSCRAMMs appear to play a key role in initiation of endovascular infections, bone and joint infections, and prosthetic-device infections. Different S. aureus strains may have different constellations of MSCRAMMs and so may be predisposed to causing certain kinds of infections [5–8].
Figure 1.
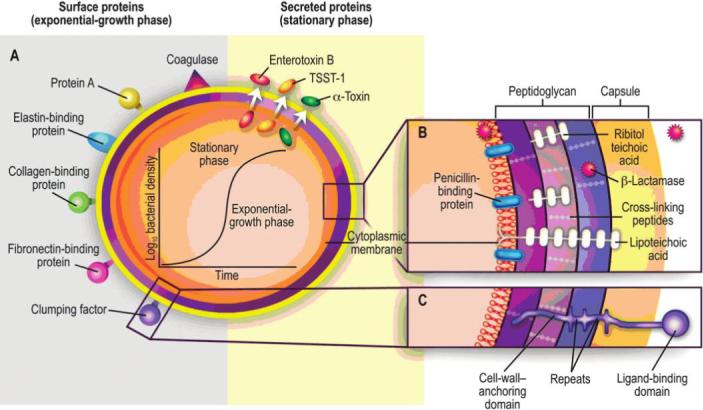
Pathogenic factors of Staphylococcus aureus, with structural and secreted products both playing roles as virulence factors. A, Surface and secreted proteins. B and C, Cross-sections of the cell envelope. TSST-1, toxic shock syndrome toxin 1. Reprinted from [32], with permission from the Massachusetts Medical Society. Copyright 1998 Massachusetts Medical Society. All rights reserved.
Table 1.
Selected Staphylococcus aureus virulence factors.
| Type of virulence factors | Selected factorsa | Genes | Associated clinical syndromes | Reference(s) |
|---|---|---|---|---|
| Involved in attachment | MSCRAMMs (e.g., clumping factors, fibronectin-binding proteins, collagen, and bone sialoprotein-binding proteins) | clfA, clfB, fnbA, fnbB, cna, sdr, bbp | Endocarditis, osteomyelitis, septic arthritis, and prosthetic-device and catheter infections | [5-8] |
| Involved in persistence | Biofilm accumulation (e.g., polysaccharide intercellular adhesion), small-colony variants, and intracellular persistence | ica locus, hemB mutation | Relapsing infections, cystic fibrosis, and syndromes as described above for attachment | [9-15] |
| Involved in evading/destroying host defenses | Leukocidins (e.g., PVL and γ-toxin), capsular polysaccharides (e.g., 5 and 8), protein A, CHIPS, Eap, and phenol-soluble modulins | lukS-PV, lukF-PV, hlg, cap5 and 8 gene clusters, spa, chp, eap, psm-α gene cluster | Invasive skin infections and necrotizing pneumonia (CA-MRSA strains that cause these are often associated with PVL) abscesses (associated with capsular polysaccharides) | [16-20] |
| Involved in tissue invasion/penetration | Proteases, lipases, nucleases, hyaluronate lyase, phospholipase C, and metalloproteases (elastase) | V8, hysA, hla, plc, sepA | Tissue destruction and metastatic infections | [21] |
| Involved in toxin-mediated disease and/or sepsis | Enterotoxins, toxic shock syndrome toxin-1, exfoliative toxins A and B, α-toxin, peptidoglycan, and lipoteichoic acid | sea-q (no sef), tstH, eta, etb, hla | Food poisoning, toxic shock syndrome, scalded skin syndrome, bullous impetigo, and sepsis syndrome | [22-26] |
| With poorly defined role in virulence | Coagulase, ACME, and bacteriocin | arc cluster, opp-3 cluster, bsa | [27, 28] |
NOTE. ACME, arginine catabolic mobile element; CA-MRSA, community-acquired methicillin-resistant S. aureus; CHIPS, chemotaxis inhibitory protein of staphylococci; Eap, extracellular adherence protein; MSCRAMMs, microbial surface components recognizing adhesive matrix molecules; PVL, Panton-Valentine leukocidin. Adapted from Projan and Novick [21] and Archer [29].
Several factors may have >1 role in S. aureus pathogenesis.
Once S. aureus adheres to host tissues or prosthetic materials, it is able to grow and persist in various ways. S. aureus can form biofilms (slime) on host and prosthetic surfaces, enabling it to persist by evading host defenses and antimicrobials [9]. The ability to form and reside in biofilms is one reason why prosthetic-device infections, for example, can be so difficult to eradicate without removal of the device. In vitro, S. aureus can also invade and survive inside epithelial cells, including endothelial cells, which theoretically may also allow it to escape host defenses, particularly in endocarditis [10–12, 30]. S. aureus is also able to form small-colony variants (SCVs), which may contribute to persistent and recurrent infection. In vitro, SCVs are able to “hide” in host cells without causing significant host-cell damage and are relatively protected from antibiotics and host defenses. They can later revert to the more virulent wild-type phenotype, possibly resulting in recurrent infection [13–15].
S. aureus has many other characteristics that help it evade the host immune system during an infection (reviewed by Foster [16]). Its main defense is production of an antiphagocytic microcapsule (most clinical isolates produce type 5 or 8). The zwitterionic capsule (both positively and negatively charged) can also induce abscess formation [17, 18]. The MSCRAMM protein A binds the Fc portion of immunoglobulin [31] and, as a result, may prevent opsonization. S. aureus may also secrete chemotaxis inhibitory protein of staphylococci or the extracellular adherence protein, which interfere with neutrophil extravasation and chemotaxis to the site of infection (reviewed by Foster [16]). In addition, S. aureus produces leukocidins that cause leukocyte destruction by the formation of pores in the cell membrane [19].
During infection, S. aureus produces numerous enzymes, such as proteases, lipases, and elastases, that enable it to invade and destroy host tissues and metastasize to other sites. S. aureus is also capable of producing septic shock. It does this by interacting with and activating the host immune system and coagulation pathways. Peptidoglycan, lipoteichoic acid, and α-toxin may all play a role [22–24] (reviewed by Lowy [32]). In addition to causing septic shock, some S. aureus strains produce superantigens, resulting in various toxinoses, such as food poisoning and toxic shock syndrome [25, 33]. Unlike the structural components noted earlier, these superantigens can produce a sepsis-like syndrome by initiating a “cytokine storm.” Some strains also produce epidermolysins or exfoliative toxins capable of causing scalded skin syndrome or bullous impetigo [26].
Regulation of expression of staphylococcal virulence factors plays a central role in pathogenesis. To reduce undue metabolic demands, expression occurs in a coordinated fashion—only when required by the bacterium. Expression of MSCRAMMs generally occurs during logarithmic growth (replication), whereas secreted proteins, such as toxins, are produced during the stationary phase. During infection, the early expression of the MSCRAMM proteins facilitates initial colonization of tissue sites, whereas the later elaboration of toxins facilitates spread. The accessory gene regulator (agr) is a quorum-sensing system that plays a critical role in the regulation of staphylococcal virulence. It has been studied extensively and has been reviewed by Yarwood and Schlievert [34] and Novick [35], among others. The agr mutants appear to have diminished virulence, and certain agr types are associated with particular clinical syndromes [36]. Other important regulators include the staphylococcal accessory regulator [37], ArlR and ArlS [38], SaeRS [39, 40], Rot [41], and mgr [42].
Host factors may also affect susceptibility to staphylococcal disease but, in general, are poorly characterized. In one large study, S. aureus nasal carriage and subsequent development of S. aureus bacteremia and mortality were assessed in nonsurgical, hospitalized patients. Among those who developed S. aureus bacteremia, noncarriers had mortality higher than that among carriers. Because most infections among carriers occurred with their colonizing strains, colonization may confer some protective immunity if staphylococcal infection develops [43]. Antibodies also appear to protect against the development of toxic shock syndrome, which occurs almost exclusively in those who lack antibodies to the implicated toxin at the time of acute illness [33].
As described, S. aureus has numerous mechanisms to produce disease and to evade host defenses. However, it is important to note that not all S. aureus strains are created equal. Different strains may contain different adhesins or toxins or may differ in their ability to produce biofilms and resist phagocytosis. The distribution of some virulence factors is related to clonal type, whereas the presence of others is unrelated to genetic background [44]. In this regard, it is important to note that there is limited information on the expression of these genes during infection.
PATHOGENESIS OF HA-MRSA
History of MRSA
Methicillin was first introduced in 1959−1960, and, within a year, methicillin-resistant isolates were reported [45]. Methicillin resistance is conferred by the mecA gene, which encodes a penicillin-binding protein (PBP2A) with decreased affinity for β-lactam antibiotics. mecA is part of a mobile genetic element called the “staphylococcal cassette chromosome (SCC) mec.” SCCmec is flanked by cassette chromosome recombinase genes (ccrA/ccrB or ccrC) that permit intra- and interspecies horizontal transmission of SCCmec. The initial reservoir of SCCmec is unclear but may have been a coagulase-negative staphylococcal species [46–48].
A limited number of MRSA lineages has emerged from the transfer of SCCmec into successful methicillin-susceptible S. aureus (MSSA) clones. Using multilocus sequence typing (comparing the internal sequences of 7 housekeeping genes), Enright et al. [49] demonstrated that MRSA clones evolved from 5 different groups of related genotypes or clonal complexes, each arising from a distinct ancestral genotype. The earliest MRSA isolates evolved from sequence type (ST) 8-MSSA, which, after a point mutation, evolved into ST250-MSSA. This MSSA was likely the first recipient of SCCmec (specifically, type I) to yield the first MRSA, labeled ST250-MRSA-I [49]. As in the work of Enright et al. [49], Crisóstomo et al. [50] identified probable recipient MSSA strains for early MRSA strains in another collection of isolates. Select MRSA clones are described in table 2.
Table 2.
Details of select important methicillin-resistant Staphylococcus aureus (MRSA) clones and their clonal complexes.
| Clone namea | Clonal complex | Other names of cloneb |
|---|---|---|
| ST1-MRSA-IV | 1 | USA400, MW2 |
| ST5-MRSA-I | 5 | UK EMRSA-3 |
| ST5-MRSA-II | 5 | New York/Japanese, GISA, and USA100 |
| ST5-MRSA-IV | 5 | USA800 and Pediatric |
| ST228-MRSA-I | 5 | Southern Germany |
| ST8-MRSA-II | 8 | Irish-1 |
| ST8-MRSA-IV | 8 | UK EMRSA-2, -6, USA300, and USA500 |
| ST239-MRSA-III | 8 | UK EMRSA-1, -4, -11, Portuguese, Brazilian, and Viennese |
| ST247-MRSA-I | 8 | UK EMRSA-5, -17, and Iberian |
| ST250-MRSA-I | 8 | First MRSA and Archaic |
| ST22-MRSA-IV | 22 | UK EMRSA-15 and Barnim |
| ST36-MRSA-II | 30 | UK EMRSA-16 and USA200 |
| ST30-MRSA-IV | 30 | Southwest Pacific |
| ST45-MRSA-IV | 45 | Berlin and USA600 |
| ST72-MRSA-IV | ... | USA700 |
NOTE. EMRSA, epidemic MRSA; GISA, glycopeptide-intermediate S. aureus. Adapted from [51], with permission from Elsevier.
The clone name is comprised of the sequence type (ST), which is the multilocus sequence type based on the sequences of 7 housekeeping genes, and the MRSA staphylococcal cassette chromosome (SCC) mec type.
HA-MRSA infections historically have been caused by internationally disseminated clones, including 5 major clones (the Iberian, Brazilian, Hungarian, New York/Japan, and Pediatric clones) that have been described in several ways (e.g., by multilocus sequence typing and PFGE) with the use of different nomenclature. Subsequently, these multidrug-resistant clones were disseminated globally and accounted for the majority of HA-MRSA infections in several regions. For example, the Brazilian clone spread to Portugal, Argentina, Uruguay, Chile, and the Czech Republic [55]. It remains unclear why particular clones are so transmissible and are able to become the “established” HA-MRSA strains in certain regions. Certainly, resistance to multiple antibiotics plays a role in establishing dominance in hospital settings. However, investigators have also postulated that these clones have enhanced virulence, as denoted by their increased transmissibility or ability to colonize hosts.
One example of a successful clonal type is phage type 80/81, which was responsible for pandemic S. aureus nosocomial and community-acquired infections throughout the 1950s. Its prevalence began to fade in the 1960s after methicillin became available. Phage type 80/81 is ST30 and contains the Panton-Valentine leukocidin (PVL) gene. This highly successful clone is related to the southwest Pacific (SWP) clone, a CA-MRSA clone that is also ST30 and contains SCCmec IV as well as PVL. Given the similar genetic backgrounds of these strains and the previous epidemicity of phage type 80/81, one would expect the SWP clone to have great potential to cause widespread disease. Of note, this clone has already appeared in the United Kingdom. Phage type 80/81 also is a likely close relative of the hospital-acquired, epidemic MRSA-16 strain (ST36-MRSA-II) [56].
HA-MRSA virulence: the Brazilian clone
The Brazilian clone (also known as Brazilian epidemic clonal complex [BECC]), PFGE type A1, became the major cause of invasive staphylococcal infections at João Barros Barreto University Hospital (Belém, Brazil) in the 1990s. In 1995, it accounted for 38% of S. aureus isolates and, by 1998, 79% of isolates. Investigators compared BECC A1 strains to MSSA and sporadic MRSA strains (rarely detected in hospitals) in several in vitro experiments. BECC A1 strains produced significantly more biofilm than did the other strains. They also had higher adhesion to polystyrene, as well as to bronchial epithelial cells, and were more likely to invade these cells. The presence of accessible fibronectin-binding domains appeared to be necessary for a high level of invasion. These in vitro studies suggest that this particular clone may be successful because it has an enhanced ability to bind, persist, and invade [57]. Whether these attributes are present in other HA-MRSA epidemic clones is unknown.
PATHOGENESIS OF CA-MRSA INFECTION
Until the 1990s, MRSA rarely caused infections among community members without exposure to the health care setting (one exception is injection drug users). An outbreak of CA-MRSA infections occurred between 1989 and 1991 among indigenous Australians in western Australia without health care contact [58]. CA-MRSA infections were also reported in people from neighboring regions [59]. In the late 1990s, several cases of aggressive MRSA infection also occurred among individuals in the United States without established risk factors for MRSA. Four children died of CA-MRSA infections in Minnesota and North Dakota from 1997 to 1999. All the cases were rapidly fatal and were associated with necrotizing pneumonia or pulmonary abscesses and sepsis [60]. The strain responsible for these infections was ST1 and PFGE type USA400 (also known as the MW2 strain) [52]. Subsequently, clonal outbreaks of skin and soft-tissue infection caused by CA-MRSA were also reported among prison inmates, men who have sex with men, soldiers, and athletes, particularly football players [61–64]. The strain responsible for these infections was ST8 and PFGE type USA300 [53]. Cases of CA-MRSA skin infection and necrotizing pneumonia were reported internationally as well [65, 66].
In addition to causing necrotizing pneumonia, CA-MRSA has recently been reported to cause infections or infectious complications in situations in which S. aureus or MRSA is an unusual pathogen. These have included cases of necrotizing fasciitis caused by PFGE type USA300 [67], as well as cases of pyomyositis [68, 69], purpura fulminans with toxic shock syndrome [70], and Waterhouse-Friderichsen syndrome [71].
The number of CA-MRSA infections appears to be increasing, and the strains responsible for these infections have now entered the health care setting, blurring the line between “community” and “hospital” strains [72, 73]. The strains that cause these virulent infections carry SCCmecIV (sometimes SCCmecV), the smallest of the SCCs that confer methicillin resistance, and are generally susceptible to several non–β-lactam antibiotics. This is in contrast to the multidrug-resistant nosocomial MRSA strains that carry larger SCCmec types [74, 75]. CA-MRSA strains may also have a growth advantage over HA-MRSA strains [27, 76].
Although SCCmecIV has appeared in several different genetic backgrounds [55], PFGE types USA300 (ST8) and USA400 (ST1)—both agr type III—accounted for the vast majority of CA-MRSA infections in individuals without the usual MRSA risk factors or health care contact in the United States [52, 77]. USA300 is now the predominant strain. Of interest, some of these USA300 isolates that cause infections are PVL positive but methicillin susceptible [78].
Worldwide, there are other prevalent CA-MRSA strains, such as ST80 (France-Switzerland), ST30 (SWP clone), and ST93 (Australia Queensland clone) [65]. Said-Salim et al. [77] identified additional “community-acquired strains” (CA-MRSA strains defined as containing SCCmecIV); however, these were in individuals with MRSA risk factors or health care contact.
The basis for the apparent increased virulence of CA-MRSA strains is incompletely understood. Numerous factors have been proposed, such as increased fitness, improved evasion of the host immune system, and unique toxin production. The genes and mechanisms by which CA-MRSA strains may cause aggressive disease are discussed in the sections that follow. Because these strains usually contain PVL, which is usually absent in HA-MRSA strains, some researchers postulate that this protein, with leukocytolytic and dermonecrotic activity, is responsible.
The role of PVL versus other virulence determinants
There is a strong epidemiological association between PVL and the emergence of CA-MRSA infections. PVL is uncommonly found in MSSA and HA-MRSA isolates [79–83]. In a study of 593 S. aureus isolates in France, PVL was absent in HA-MRSA isolates but was associated with all CA-MRSA strains [83]. In another study, PVL was ubiquitous in a large sample of CA-MRSA isolates collected from across the globe [65]. It is usually present in USA300 and USA400 [27, 53, 77] and is often harbored by other SCCmecIV-containing strains [77]. The outbreaks of skin and soft-tissue infections and necrotizing pneumonia mentioned above were caused by PVL-positive strains.
Lina et al. [66] determined the presence of lukS-PV and lukF-PV (the cotranscribed genes for PVL) in 172 S. aureus strains collected from patients with a variety of clinical syndromes. PVL was significantly associated with community-acquired pneumonia (85% of strains), compared with hospital-acquired pneumonia (0%). PVL was also significantly associated with strains causing invasive skin infections such as furunculosis (93%) and cutaneous abscess (50%), compared with superficial folliculitis (0%). PVL was not observed in strains associated with infective endocarditis, urinary tract infections, toxic shock syndrome, or mediastinitis, although few strains were tested [66]. Diep et al. [80] reported a similar association of PVL and skin and soft-tissue infections caused by MRSA isolated from inpatients and outpatients from San Francisco General Hospital and inmates in county jails.
In addition to the epidemiological evidence suggesting that PVL may be a virulence factor in CA-MRSA, there is a scientific rationale for this association. Staphylococcal leukotoxins, including PVL, are secreted as bicomponent toxins consisting of S and F proteins [16, 84]. Depending on the combination of particular S and F proteins, a toxin is formed with varying leukocytolytic, erythrocytolytic, and dermonecrotic properties [84, 85]. PVL consists of LukS-PV and LukF-PV and 4 units of each form of octameric β-barrel pores in leukocyte membranes in vitro, resulting in cell lysis [19, 86–88]. This may cause cells such as neutrophils to release inflammatory enzymes and cytokines (sublytic concentrations of PVL also appear to induce the release of these substances) [88–90]. PVL also appears to induce apoptosis of neutrophils via a mitrochondrial pathway at lower concentrations, whereas, at higher concentrations, PVL induces necrosis [91]. In vivo, PVL causes dermonecrosis when injected intradermally in rabbits [92].
Given this evidence and the strong epidemiological association between PVL-containing CA-MRSA strains and necrotizing pneumonia and skin and soft-tissue infections, it is plausible that PVL is partly responsible for the enhanced virulence of CA-MRSA (other leukocidins may also play a role). However, recent studies comparing the virulence of PVL-positive and PVL-negative strains have had conflicting results.
Saïd-Salim et al. [77] compared human polymorphonuclear cell lysis among PVL-positive and PVL-negative CA-MRSA strains with similar genetic backgrounds and found no difference in polymorphonuclear lysis. Voyich et al. [93] compared PVL-positive strains and PVL-negative strains with similar genetic backgrounds in mouse sepsis and abscess models, as well as PVL knockouts created for the USA300 and USA400 strains. There was no difference in survival in the mouse sepsis model. In the abscess model, PVL-negative strains unexpectedly caused slightly larger abscesses than did the PVL-positive strains. Isogenic pvl strains of USA300 and USA400 showed no difference in the ability to cause polymorphonuclear lysis in vitro. The authors concluded that the PVL “...toxin is not the major determinant of disease caused by these prominent CA-MRSA strains” [93, p. 1769]. It is possible that the mouse models used in this study were not optimal to assess the in vivo effects of PVL, or, as the authors suggested, that PVL either is a marker for other virulence factors present in these strains or is one of many factors causing the enhanced virulence of particular CA-MRSA strains.
PVL was investigated in a mouse pneumonia model by Labandeira-Rey et al. [94]. Mice were infected with isogenic PVL-positive and PVL-negative (non–CA-MRSA) strains. PVL-positive strains caused necrotizing pneumonia similar to that seen in humans, whereas PVL-negative strains showed only some leukocytic invasion. When PVL-negative mutants were complemented with plasmids containing the PVL operon, massive tissue damage and mortality resulted. In mice, exposure to LukS-PV and LukF-PV toxin was sufficient to cause lung damage, weight loss, and increased mortality in a concentration-dependent fashion [94]. In these studies, however, a single non–CA-MRSA strain was used.
In contrast, Bubeck Wardenburg et al. [95] recently reported conflicting results. They demonstrated that α-hemolysin and not PVL was responsible for mortality in a mouse pneumonia model, using USA300 and USA400 CA-MRSA strains.
These studies suggest that the association of PVL with enhanced S. aureus virulence is complex and controversial and warrants further investigation. Furthermore, Wang et al. [20] recently discovered that phenol-soluble modulins, a previously unrecognized class of secreted S. aureus peptides, are up-regulated in CA-MRSA strains, compared with the level in HAMRSA strains; cause inflammation; destroy neutrophils; and are responsible for virulence in mouse abscess and bacteremia models. Other toxins, such as the enterotoxins, may also play an important role in these infections.
Virulence of USA400
USA400 (or MW2) is a highly virulent CA-MRSA strain. This is apparent not only in human disease but also in animal models [27, 93]. Initially, its only resistance genes were mec and blaZ, which encodes penicillinase. Researchers sequenced USA400 and compared its sequence with the sequences of 5 other strains (N315, a Japanese MRSA; Mu50, a vancomycin-resistant MRSA; E-MRSA-16, an epidemic MRSA in the United Kingdom; COL, a MRSA strain; and NCTC8325, a widely used reference strain) to identify potential virulence factors associated with this strain. USA400 was the only strain to contain the PVL operon. In addition, it contained 16 unique superantigen genes, including 11 exotoxin genes and 5 enterotoxin genes. These genes had at least a 2% difference in their amino acids, compared with their homologues. One exception was staphylococcal enterotoxin H (seh), which was unique to USA400 [27] and can cause a toxic-shock–like syndrome [96]. USA400 also contained a novel gene cluster dubbed “bacteriocin of S. aureus” (bsa). bsa encodes a potential bacteriocin, or antibacterial agent. This bacteriocin could help USA400 compete with other colonizing flora and increase the chance of infection with this strain [27]. These data suggest that there are several factors that may contribute to the virulence of USA400 and that these factors are ripe for future investigation.
Virulence of USA300
Like USA400, USA300 is associated with virulent disease [93]; however, USA300 causes far more incident cases of CA-MRSA infection and is becoming resistant to several non–β-lactam antibiotics [28]. The genome of USA300 was sequenced by Diep et al. [28] and compared with 10 previously sequenced S. aureus strains as well as 4 coagulase-negative strains to identify factors potentially associated with its high virulence. Of interest, there were minimal differences between the core sequences of USA300 and COL, an early MRSA. In addition to harboring SCCmecIV and the PVL operon, USA300 contained homologues closely related to staphylococcal enterotoxins Q and K, designated SEQ2 and SEK2. Like COL and USA400, USA300 also has a genome that includes a bacteriocin gene cluster. Most notably, USA300 contains a genomic island, termed “arginine catabolic mobile element” (ACME), which encodes an arginine deaminase pathway that converts l-arginine to carbon dioxide, adenosine triphosphate, and ammonia. Arginine deaminase, a known virulence factor in other pathogens, may enhance the virulence of USA300 by enabling it to (1) survive more easily on acidic, human skin; (2) proliferate more easily in conditions low in oxygen, such as abscesses; and (3) evade host defenses by inhibiting production of nitric oxide and mononuclear cell proliferation as in Streptococcus pyogenes [28, 97]. Further investigation of ACME may help elucidate the remarkable success and virulence of the USA300 strain.
Colonization and CA-MRSA
As discussed above, the anterior nares are the classic reservoir for nosocomial S. aureus infections, including HA-MRSA. However, data suggest that other sites of colonization or modes of transmission play an important and underappreciated role in the development of CA-MRSA infection. Heterosexual contact was recently identified as a mode of transmission of CA-MRSA. Most cases had genital CA-MRSA colonization without nasal colonization [98]. In an outbreak investigation of CA-MRSA abscesses among St. Louis Rams football players, no MRSA was isolated from nasal or environmental samples. Perhaps other sites of colonization, shared items, or an unsampled environmental site played a role in transmission [64]. Future epidemiological investigations of CA-MRSA should include sampling of several environmental and body sites in addition to the anterior nares.
IS MRSA MORE VIRULENT THAN MSSA?
There is an active debate as to whether MRSA is more virulent than MSSA. Some epidemiologic studies, including a meta-analysis, found increased morbidity and/or mortality from nosocomial MRSA (e.g., bloodstream infections, surgical-site infections, and pneumonia), compared with those from MSSA [99–102]; however, these studies may be confounded because not all accounted for important factors such as time to initiation of appropriate therapy or patient comorbidities. A recent retrospective review found increased mortality for MRSA bacteremia but not MRSA pneumonia [103]. Other studies did not demonstrate increased mortality associated with nosocomial MRSA bacteremia [104] or ventilator-associated pneumonia [105], compared with MSSA infections. An investigation that compared CA-MRSA skin infections and CA-MSSA skin infections did not find more serious outcomes for the CA-MRSA infections [106]. To date, there is no compelling evidence that MRSA, in general, is more virulent than MSSA. Although this issue remains unresolved, invasive MRSA infection is associated with greater costs [101, 102, 104] and limited treatment options.
UNANSWERED QUESTIONS
Although considerable progress has been made in understanding the pathogenesis of S. aureus infection, numerous questions remain unanswered. The role of many virulence factors in the pathogenesis of staphylococcal disease is unclear. This is a result, in part, of the redundancy of function and/or the ubiquitous nature of many virulence factors in addition to the complex nature of virulence factor regulation. In particular, the role of PVL in staphylococcal virulence remains uncertain. Also, as discussed above, particular clonal strains have the ability to persist for years and to establish themselves globally. Why certain clonal types have this ability remains unknown. Other clonal types have become established among otherwise healthy community members. Understanding what enables these strains to do this, what their reservoirs are, and what their means of transmission are requires further investigation. We hope that, in the future, a better understanding of the pathogenesis of staphylococcal disease will lead to improved prevention and treatment strategies.
Acknowledgments
We thank Linda K. McDougal for her assistance with table 2. Dr. David DeVellis and Hilary Selby Polk provided assistance in editing the manuscript.
Financial support. R.J.G. was supported by National Institutes of Health (NIH) grant 1K08AI072043-01A1. F.D.L. was supported by Centers for Disease Control and Prevention grant CCR223380; NIH grants DA-15018 and HL077096-01 from the National Heart, Lung, and Blood Institute, NIH-Specialized Center for Clinically Oriented Research; and grant P20 RR020616 from the National Center for Research Resources, NIH, which supports the Center for Interdisciplinary Research on Antimicrobial Resistance.
Supplement sponsorship. This article was published as part of a supplement entitled “Methicillin-Resistant Staphylococcus aureus: An Evolving Clinical Challenge,” sponsored by the Boston University School of Medicine and supported by an unrestricted educational grant from Cubist Pharmaceuticals, Inc.
Footnotes
Potential conflicts of interests. F.D.L. has received research support from Cubist Pharmaceuticals, GlaxoSmithKline, and Pfizer and has been on advisory panels for Pfizer, Nabi Biopharmaceuticals, and Wyeth. R.J.G.: no conflicts.
References
- 1.Wertheim HF, Melles DC, Vos MC, et al. The role of nasal carriage in Staphylococcus aureus infections. Lancet Infect Dis. 2005;5:751–62. doi: 10.1016/S1473-3099(05)70295-4. [DOI] [PubMed] [Google Scholar]
- 2.Kluytmans J, van Belkum A, Verbrugh H. Nasal carriage of Staphylococcus aureus: epidemiology, underlying mechanisms, and associated risks. Clin Microbiol Rev. 1997;10:505–20. doi: 10.1128/cmr.10.3.505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Williams REO, Jevons MP, Shooter RA, et al. Nasal staphylococci and sepsis in hospital patients. Br Med J. 1959;2:658–62. doi: 10.1136/bmj.2.5153.658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Von Eiff C, Becker K, Machka K, Stammer H, Peters G. Nasal carriage as a source of Staphylococcus aureus bacteremia. N Engl J Med. 2001;344:11–6. doi: 10.1056/NEJM200101043440102. [DOI] [PubMed] [Google Scholar]
- 5.Patti JM, Allen BL, McGavin MJ, Hook M. MSCRAMM-mediated adherence of microorganisms to host tissues. Annu Rev Microbiol. 1994;48:585–617. doi: 10.1146/annurev.mi.48.100194.003101. [DOI] [PubMed] [Google Scholar]
- 6.Foster TJ, Hook M. Surface protein adhesins of Staphylococcus aureus. Trends Microbiol. 1998;6:484–8. doi: 10.1016/s0966-842x(98)01400-0. [DOI] [PubMed] [Google Scholar]
- 7.Menzies BE. The role of fibronectin binding proteins in the pathogenesis of Staphylococcus aureus infections. Curr Opin Infect Dis. 2003;16:225–9. doi: 10.1097/00001432-200306000-00007. [DOI] [PubMed] [Google Scholar]
- 8.Tung H, Guss B, Hellman U, Persson L, Rubin K, Ryden C. A bone sialoprotein-binding protein from Staphylococcus aureus: a member of the staphylococcal Sdr family. Biochem J. 2000;345:611–9. [PMC free article] [PubMed] [Google Scholar]
- 9.Donlan RM, Costerton JW. Biofilms: survival mechanisms of clinically relevant microorganisms. Clin Microbiol Rev. 2002;15:167–93. doi: 10.1128/CMR.15.2.167-193.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ogawa SK, Yurberg ER, Hatcher VB, Levitt MA, Lowy FD. Bacterial adherence to human endothelial cells in vitro. Infect Immun. 1985;50:218–24. doi: 10.1128/iai.50.1.218-224.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hamill RJ, Vann JM, Proctor RA. Phagocytosis of Staphylococcus aureus by cultured bovine aortic endothelial cells: model for postadherence events in endovascular infections. Infect Immun. 1986;54:833–6. doi: 10.1128/iai.54.3.833-836.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Arrecubieta C, Lowy FD. Staphylococcus aureus–eukaryotic cell interactions. In: Fischetti VA, Novick RP, Ferretti JJ, Portnoy DA, Rood JI, editors. Gram-positive pathogens. 2nd ed. ASM Press; Washington, DC: 2006. pp. 517–25. [Google Scholar]
- 13.Kahl B, Herrmann M, Everding AS, et al. Persistent infection with small colony variant strains of Staphylococcus aureus in patients with cystic fibrosis. J Infect Dis. 1998;177:1023–9. doi: 10.1086/515238. [DOI] [PubMed] [Google Scholar]
- 14.Proctor RA, Peters G. Small colony variants in staphylococcal infections: diagnostic and therapeutic implications. Clin Infect Dis. 1998;27:419–22. doi: 10.1086/514706. [DOI] [PubMed] [Google Scholar]
- 15.Proctor RA, van Langevelde P, Kristjansson M, Maslow JN, Arbeit RD. Persistent and relapsing infections associated with small-colony variants of Staphylococcus aureus. Clin Infect Dis. 1995;20:95–102. doi: 10.1093/clinids/20.1.95. [DOI] [PubMed] [Google Scholar]
- 16.Foster TJ. Immune evasion by staphylococci. Nat Rev Microbiol. 2005;3:948–58. doi: 10.1038/nrmicro1289. [DOI] [PubMed] [Google Scholar]
- 17.O'Riordan K, Lee JC. Staphylococcus aureus capsular polysaccharides. Clin Microbiol Rev. 2004;17:218–34. doi: 10.1128/CMR.17.1.218-234.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tzianabos AO, Wang JY, Lee JC. Structural rationale for the modulation of abscess formation by Staphylococcus aureus capsular polysaccharides. Proc Natl Acad Sci USA. 2001;98:9365–70. doi: 10.1073/pnas.161175598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gladstone GP, VanHeyningen WE. Staphylococcal leucocidins. BrJExp Pathol. 1957;38:123–37. [PMC free article] [PubMed] [Google Scholar]
- 20.Wang R, Braughton KR, Kretschmer D, et al. Identification of novel cytolytic peptides as key virulence determinants for community-associated MRSA. Nat Med. 2007;13:1510–4. doi: 10.1038/nm1656. [DOI] [PubMed] [Google Scholar]
- 21.Projan SJ, Novick RP. The molecular basis of pathogenicity. In: Crossley KB, Archer GL, editors. The staphylococci in human disease. Churchill Livingstone; New York: 1997. pp. 55–81. [Google Scholar]
- 22.Timmerman CP, Mattsson E, Martinez-Martinez L, et al. Induction of release of tumor necrosis factor from human monocytes by staphylococci and staphylococcal peptidoglycans. Infect Immun. 1993;61:4167–72. doi: 10.1128/iai.61.10.4167-4172.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Heumann D, Barras C, Severin A, Glauser MP, Tomasz A. Gram-positive cell walls stimulate synthesis of tumor necrosis factor alpha and interleukin-6 by human monocytes. Infect Immun. 1994;62:2715–21. doi: 10.1128/iai.62.7.2715-2721.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bhakdi S, Tranum-Jensen J. Alpha-toxin of Staphylococcus aureus. Microbiol Rev. 1991;55:733–51. doi: 10.1128/mr.55.4.733-751.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dinges MM, Orwin PM, Schlievert PM. Exotoxins of Staphylococcus aureus. Clin Microbiol Rev. 2000;13:16–34. doi: 10.1128/cmr.13.1.16-34.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Prevost G, Couppie P, Monteil H. Staphylococcal epidermolysins. Curr Opin Infect Dis. 2003;16:71–6. doi: 10.1097/00001432-200304000-00002. [DOI] [PubMed] [Google Scholar]
- 27.Baba T, Takeuchi F, Kuroda M, et al. Genome and virulence determinants of high virulence community-acquired MRSA. Lancet. 2002;359:1819–27. doi: 10.1016/s0140-6736(02)08713-5. [DOI] [PubMed] [Google Scholar]
- 28.Diep BA, Gill SR, Chang RF, et al. Complete genome sequence of USA300, an epidemic clone of community-acquired methicillin-resistant Staphylococcus aureus. Lancet. 2006;367:731–9. doi: 10.1016/S0140-6736(06)68231-7. [DOI] [PubMed] [Google Scholar]
- 29.Archer GL. Staphylococcus aureus: a well-armed pathogen. Clin Infect Dis. 1998;26:1179–81. doi: 10.1086/520289. [DOI] [PubMed] [Google Scholar]
- 30.Moreillon P, Que YA, Bayer AS. Pathogenesis of streptococcal and staphylococcal endocarditis. Infect Dis Clin North Am. 2002;16:297–318. doi: 10.1016/s0891-5520(01)00009-5. [DOI] [PubMed] [Google Scholar]
- 31.Deisenhofer J. Crystallographic refinement and atomic models of a human Fc fragment and its complex with fragment B of protein A from Staphylococcus aureus at 2.9- and 2.8-A resolution. Biochemistry. 1981;20:2361–70. [PubMed] [Google Scholar]
- 32.Lowy FD. Staphylococcus aureus infections. N Engl J Med. 1998;339:520–32. doi: 10.1056/NEJM199808203390806. [DOI] [PubMed] [Google Scholar]
- 33.McCormick JK, Yarwood JM, Schlievert PM. Toxic shock syndrome and bacterial superantigens: an update. Annu Rev Microbiol. 2001;55:77–104. doi: 10.1146/annurev.micro.55.1.77. [DOI] [PubMed] [Google Scholar]
- 34.Yarwood JM, Schlievert PM. Quorum sensing in Staphylococcus infections. J Clin Invest. 2003;112:1620–5. doi: 10.1172/JCI20442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Novick RP. Autoinduction and signal transduction in the regulation of staphylococcal virulence. Mol Microbiol. 2003;48:1429–49. doi: 10.1046/j.1365-2958.2003.03526.x. [DOI] [PubMed] [Google Scholar]
- 36.Cheung AL, Eberhardt KJ, Chung E, et al. Diminished virulence of a sar−/agr− mutant of Staphylococcus aureus in the rabbit model of endocarditis. J Clin Invest. 1994;94:1815–22. doi: 10.1172/JCI117530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cheung AL, Koomey JM, Butler CA, Projan SJ, Fischetti VA. Regulation of exoprotein expression in Staphylococcus aureus by a locus (sar) distinct from agr. Proc Natl Acad Sci USA. 1992;89:6462–6. doi: 10.1073/pnas.89.14.6462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fournier B, Hooper DC. A new two-component regulatory system involved in adhesion, autolysis, and extracellular proteolytic activity of Staphylococcus aureus. J Bacteriol. 2000;182:3955–64. doi: 10.1128/jb.182.14.3955-3964.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liang X, Yu C, Sun J, et al. Inactivation of a two-component signal transduction system, SaeRS, eliminates adherence and attenuates virulence of Staphylococcus aureus. Infect Immun. 2006;74:4655–65. doi: 10.1128/IAI.00322-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Giraudo AT, Cheung AL, Nagel R. The sae locus of Staphylococcus aureus controls exoprotein synthesis at the transcriptional level. Arch Microbiol. 1997;168:53–8. doi: 10.1007/s002030050469. [DOI] [PubMed] [Google Scholar]
- 41.Said-Salim B, Dunman PM, McAleese FM, et al. Global regulation of Staphylococcus aureus genes by Rot. J Bacteriol. 2003;185:610–9. doi: 10.1128/JB.185.2.610-619.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Luong TT, Newell SW, Lee CY. Mgr, a novel global regulator in Staphylococcus aureus. J Bacteriol. 2003;185:3703–10. doi: 10.1128/JB.185.13.3703-3710.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wertheim HF, Vos MC, Ott A, et al. Risk and outcome of nosocomial Staphylococcus aureus bacteraemia in nasal carriers versus non-carriers. Lancet. 2004;364:703–5. doi: 10.1016/S0140-6736(04)16897-9. [DOI] [PubMed] [Google Scholar]
- 44.Peacock SJ, Moore CE, Justice A, et al. Virulent combinations of adhesin and toxin genes in natural populations of Staphylococcus aureus. Infect Immun. 2002;70:4987–96. doi: 10.1128/IAI.70.9.4987-4996.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jevons MP. “Celbenin”-resistant staphylococci. Br Med J. 1961;1:124–5. [Google Scholar]
- 46.Wu S, Piscitelli C, de LH, Tomasz A. Tracking the evolutionary origin of the methicillin resistance gene: cloning and sequencing of a homologue of mecA from a methicillin susceptible strain of Staphylococcus sciuri. Microb Drug Resist. 1996;2:435–41. doi: 10.1089/mdr.1996.2.435. [DOI] [PubMed] [Google Scholar]
- 47.Hiramatsu K, Cui L, Kuroda M, Ito T. The emergence and evolution of methicillin-resistant Staphylococcus aureus. Trends Microbiol. 2001;9:486–93. doi: 10.1016/s0966-842x(01)02175-8. [DOI] [PubMed] [Google Scholar]
- 48.Wisplinghoff H, Rosato AE, Enright MC, Noto M, Craig W, Archer GL. Related clones containing SCCmec type IV predominate among clinically significant Staphylococcus epidermidis isolates. Antimicrob Agents Chemother. 2003;47:3574–9. doi: 10.1128/AAC.47.11.3574-3579.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Enright MC, Robinson DA, Randle G, Feil EJ, Grundmann H, Spratt BG. The evolutionary history of methicillin-resistant Staphylococcus aureus (MRSA). Proc Natl Acad Sci USA. 2002;99:7687–92. doi: 10.1073/pnas.122108599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Crisostomo MI, Westh H, Tomasz A, Chung M, Oliveira DC, de Lencastre H. The evolution of methicillin resistance in Staphylococcus aureus: similarity of genetic backgrounds in historically early methicillin-susceptible and -resistant isolates and contemporary epidemic clones. Proc Natl Acad Sci USA. 2001;98:9865–70. doi: 10.1073/pnas.161272898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Enright MC. The evolution of a resistant pathogen—the case of MRSA. Curr Opin Pharmacol. 2003;3:474–9. doi: 10.1016/s1471-4892(03)00109-7. [DOI] [PubMed] [Google Scholar]
- 52.McDougal LK, Steward CD, Killgore GE, Chaitram JM, McAllister SK, Tenover FC. Pulsed-field gel electrophoresis typing of oxacillin-resistant Staphylococcus aureus isolates from the United States: establishing a national database. J Clin Microbiol. 2003;41:5113–20. doi: 10.1128/JCM.41.11.5113-5120.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tenover FC, McDougal LK, Goering RV, et al. Characterization of a strain of community-associated methicillin-resistant Staphylococcus aureus widely disseminated in the United States. J Clin Microbiol. 2006;44:108–18. doi: 10.1128/JCM.44.1.108-118.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Melles DC, Gorkink RF, Boelens HA, et al. Natural population dynamics and expansion of pathogenic clones of Staphylococcus aureus. J Clin Invest. 2004;114:1732–40. doi: 10.1172/JCI23083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Oliveira DC, Tomasz A, de Lencastre H. Secrets of success of a human pathogen: molecular evolution of pandemic clones of methicillin-resistant Staphylococcus aureus. Lancet Infect Dis. 2002;2:180–9. doi: 10.1016/s1473-3099(02)00227-x. [DOI] [PubMed] [Google Scholar]
- 56.Robinson DA, Kearns AM, Holmes A, et al. Re-emergence of early pandemic Staphylococcus aureus as a community-acquired methicillin-resistant clone. Lancet. 2005;365:1256–8. doi: 10.1016/S0140-6736(05)74814-5. [DOI] [PubMed] [Google Scholar]
- 57.Amaral MM, Coelho LR, Flores RP, et al. The predominant variant of the Brazilian epidemic clonal complex of methicillin-resistant Staphylococcus aureus has an enhanced ability to produce biofilm and to adhere to and invade airway epithelial cells. J Infect Dis. 2005;192:801–10. doi: 10.1086/432515. [DOI] [PubMed] [Google Scholar]
- 58.Udo EE, Pearman JW, Grubb WB. Genetic analysis of community isolates of methicillin-resistant Staphylococcus aureus in western Australia. J Hosp Infect. 1993;25:97–108. doi: 10.1016/0195-6701(93)90100-e. [DOI] [PubMed] [Google Scholar]
- 59.Gosbell IB, Mercer JL, Neville SA, et al. Non-multiresistant and multiresistant methicillin-resistant Staphylococcus aureus in community-acquired infections. Med J Aust. 2001;174:627–30. doi: 10.5694/j.1326-5377.2001.tb143470.x. [DOI] [PubMed] [Google Scholar]
- 60.Four pediatric deaths from community-acquired methicillin-resistant Staphylococcus aureus—Minnesota and North Dakota, 1997–1999. MMWR Morb Mortal Wkly Rep. 1999;48:707–10. [PubMed] [Google Scholar]
- 61.Centers for Disease Control and Prevention Outbreaks of community-associated methicillin-resistant Staphylococcus aureus skin infections—Los Angeles County, California, 2002–2003. MMWR Morb Mortal Wkly Rep. 2003;52:88. [PubMed] [Google Scholar]
- 62.Centers for Disease Control and Prevention Methicillin-resistant Staphylococcus aureus infections among competitive sports participants—Colorado, Indiana, Pennsylvania, and Los Angeles County, 2000–2003. MMWR Morb Mortal Wkly Rep. 2003;52:793–5. [PubMed] [Google Scholar]
- 63.Centers for Disease Control and Prevention Methicillin-resistant Staphylococcus aureus skin or soft tissue infections in a state prison—Mississippi, 2000. MMWR Morb Mortal Wkly Rep. 2001;50:919–22. [PubMed] [Google Scholar]
- 64.Kazakova SV, Hageman JC, Matava M, et al. A clone of methicillin-resistant Staphylococcus aureus among professional football players. N Engl J Med. 2005;352:468–75. doi: 10.1056/NEJMoa042859. [DOI] [PubMed] [Google Scholar]
- 65.Vandenesch F, Naimi T, Enright MC, et al. Community-acquired methicillin-resistant Staphylococcus aureus carrying Panton-Valentine leukocidin genes: worldwide emergence. Emerg Infect Dis. 2003;9:978–84. doi: 10.3201/eid0908.030089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lina G, Piemont Y, Godail-Gamot F, et al. Involvement of Panton-Valentine leukocidin–producing Staphylococcus aureus in primary skin infections and pneumonia. Clin Infect Dis. 1999;29:1128–32. doi: 10.1086/313461. [DOI] [PubMed] [Google Scholar]
- 67.Miller LG, Perdreau-Remington F, Rieg G, et al. Necrotizing fasciitis caused by community-associated methicillin-resistant Staphylococcus aureus in Los Angeles. N Engl J Med. 2005;352:1445–53. doi: 10.1056/NEJMoa042683. [DOI] [PubMed] [Google Scholar]
- 68.Ruiz ME, Yohannes S, Wladyka CG. Pyomyositis caused by methicillin-resistant Staphylococcus aureus. N Engl J Med. 2005;352:1488–9. doi: 10.1056/NEJM200504073521417. [DOI] [PubMed] [Google Scholar]
- 69.Fowler A, Mackay A. Community-acquired methicillin-resistant Staphylococcus aureus pyomyositis in an intravenous drug user. J Med Microbiol. 2006;55:123–5. doi: 10.1099/jmm.0.46271-0. [DOI] [PubMed] [Google Scholar]
- 70.Kravitz GR, Dries DJ, Peterson ML, Schlievert PM. Purpura fulminans due to Staphylococcus aureus. Clin Infect Dis. 2005;40:941–7. doi: 10.1086/428573. [DOI] [PubMed] [Google Scholar]
- 71.Adem PV, Montgomery CP, Husain AN, et al. Staphylococcus aureus sepsis and the Waterhouse-Friderichsen syndrome in children. N Engl J Med. 2005;353:1245–51. doi: 10.1056/NEJMoa044194. [DOI] [PubMed] [Google Scholar]
- 72.Seybold U, Kourbatova EV, Johnson JG, et al. Emergence of community-associated methicillin-resistant Staphylococcus aureus USA300 genotype as a major cause of health care–associated blood stream infections. Clin Infect Dis. 2006;42:647–56. doi: 10.1086/499815. [DOI] [PubMed] [Google Scholar]
- 73.Gonzalez BE, Rueda AM, Shelburne SA, III, Musher DM, Hamill RJ, Hulten KG. Community-associated strains of methicillin-resistant Staphylococccus aureus as the cause of healthcare-associated infection. Infect Control Hosp Epidemiol. 2006;27:1051–6. doi: 10.1086/507923. [DOI] [PubMed] [Google Scholar]
- 74.Zetola N, Francis JS, Nuermberger EL, Bishai WR. Community-acquired methicillin-resistant Staphylococcus aureus: an emerging threat. Lancet Infect Dis. 2005;5:275–86. doi: 10.1016/S1473-3099(05)70112-2. [DOI] [PubMed] [Google Scholar]
- 75.Ito T, Ma XX, Takeuchi F, Okuma K, Yuzawa H, Hiramatsu K. Novel type V staphylococcal cassette chromosome mec driven by a novel cassette chromosome recombinase, ccrC. Antimicrob Agents Che-mother. 2004;48:2637–51. doi: 10.1128/AAC.48.7.2637-2651.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Okuma K, Iwakawa K, Turnidge JD, et al. Dissemination of new methicillin-resistant Staphylococcus aureus clones in the community. J Clin Microbiol. 2002;40:4289–94. doi: 10.1128/JCM.40.11.4289-4294.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Said-Salim B, Mathema B, Braughton K, et al. Differential distribution and expression of Panton-Valentine leucocidin among community-acquired methicillin-resistant Staphylococcus aureus strains. J Clin Microbiol. 2005;43:3373–9. doi: 10.1128/JCM.43.7.3373-3379.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Moran GJ, Krishnadasan A, Gorwitz RJ, et al. Methicillin-resistant S. aureus infections among patients in the emergency department. N Engl J Med. 2006;355:666–74. doi: 10.1056/NEJMoa055356. [DOI] [PubMed] [Google Scholar]
- 79.Prevost G, Couppie P, Prevost P, et al. Epidemiological data on Staphylococcus aureus strains producing synergohymenotropic toxins. J Med Microbiol. 1995;42:237–45. doi: 10.1099/00222615-42-4-237. [DOI] [PubMed] [Google Scholar]
- 80.Diep BA, Sensabaugh GF, Somboona NS, Carleton HA, Perdreau-Remington F. Widespread skin and soft-tissue infections due to two methicillin-resistant Staphylococcus aureus strains harboring the genes for Panton-Valentine leucocidin. J Clin Microbiol. 2004;42:2080–4. doi: 10.1128/JCM.42.5.2080-2084.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Johnsson D, Molling P, Stralin K, Soderquist B. Detection of Panton-Valentine leukocidin gene in Staphylococcus aureus by LightCycler PCR: clinical and epidemiological aspects. Clin Microbiol Infect. 2004;10:884–9. doi: 10.1111/j.1469-0691.2004.00976.x. [DOI] [PubMed] [Google Scholar]
- 82.Chini V, Petinaki E, Foka A, Paratiras S, Dimitracopoulos G, Spiliopoulou I. Spread of Staphylococcus aureus clinical isolates carrying Panton-Valentine leukocidin genes during a 3-year period in Greece. Clin Microbiol Infect. 2006;12:29–34. doi: 10.1111/j.1469-0691.2005.01295.x. [DOI] [PubMed] [Google Scholar]
- 83.Dufour P, Gillet Y, Bes M, et al. Community-acquired methicillin-resistant Staphylococcus aureus infections in France: emergence of a single clone that produces Panton-Valentine leukocidin. Clin Infect Dis. 2002;35:819–24. doi: 10.1086/342576. [DOI] [PubMed] [Google Scholar]
- 84.Menestrina G, Dalla SM, Comai M, et al. Ion channels and bacterial infection: the case of b-barrel pore-forming protein toxins of Staphylococcus aureus. FEBS Lett. 2003;552:54–60. doi: 10.1016/s0014-5793(03)00850-0. [DOI] [PubMed] [Google Scholar]
- 85.Prevost G, Cribier B, Couppie P, et al. Panton-Valentine leucocidin and gamma-hemolysin from Staphylococcus aureus ATCC 49775 are encoded by distinct genetic loci and have different biological activities. Infect Immun. 1995;63:4121–9. doi: 10.1128/iai.63.10.4121-4129.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Szmigielski S, Prevost G, Monteil H, Colin DA, Jeljaszewicz J. Leukocidal toxins of staphylococci. Zentralbl Bakteriol. 1999;289:185–201. doi: 10.1016/s0934-8840(99)80105-4. [DOI] [PubMed] [Google Scholar]
- 87.Miles G, Movileanu L, Bayley H. Subunit composition of a bicomponent toxin: staphylococcal leukocidin forms an octameric trans-membrane pore. Protein Sci. 2002;11:894–902. doi: 10.1110/ps.4360102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Colin DA, Mazurier I, Sire S, Finck-Barbancon V. Interaction of the two components of leukocidin from Staphylococcus aureus with human polymorphonuclear leukocyte membranes: sequential binding and subsequent activation. Infect Immun. 1994;62:3184–8. doi: 10.1128/iai.62.8.3184-3188.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Hensler T, Konig B, Prevost G, Piemont Y, Koller M, Konig W. Leukotriene B4 generation and DNA fragmentation induced by leukocidin from Staphylococcus aureus: protective role of granulocyte-macrophage colony-stimulating factor (GM-CSF) and G-CSF for human neutrophils. Infect Immun. 1994;62:2529–35. doi: 10.1128/iai.62.6.2529-2535.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Konig B, Koller M, Prevost G, et al. Activation of human effector cells by different bacterial toxins (leukocidin, alveolysin, and erythrogenic toxin A): generation of interleukin-8. Infect Immun. 1994;62:4831–7. doi: 10.1128/iai.62.11.4831-4837.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Genestier AL, Michallet MC, Prevost G, et al. Staphylococcus aureus Panton-Valentine leukocidin directly targets mitochondria and induces Bax-independent apoptosis of human neutrophils. J Clin Invest. 2005;115:3117–27. doi: 10.1172/JCI22684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Ward PD, Turner WH. Identification of staphylococcal Panton-Valentine leukocidin as a potent dermonecrotic toxin. Infect Immun. 1980;28:393–7. doi: 10.1128/iai.28.2.393-397.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Voyich JM, Otto M, Mathema B, et al. Is Panton-Valentine leukocidin the major virulence determinant in community-associated methicillin-resistant Staphylococcus aureus disease? J Infect Dis. 2006;194:1761–70. doi: 10.1086/509506. [DOI] [PubMed] [Google Scholar]
- 94.Labandeira-Rey M, Couzon F, Boisset S, et al. Staphylococcus aureus Panton-Valentine leukocidin causes necrotizing pneumonia. Science. 2007;315:1130–3. doi: 10.1126/science.1137165. [DOI] [PubMed] [Google Scholar]
- 95.Bubeck Wardenburg J, Bae T, Otto M, DeLeo FR, Schneewind O. Poring over pores: a-hemolysin and Panton-Valentine leukocidin in Staphylococcus aureus pneumonia. Nat Med. 2007;13:1405–6. doi: 10.1038/nm1207-1405. [DOI] [PubMed] [Google Scholar]
- 96.Ren K, Bannan JD, Pancholi V, et al. Characterization and biological properties of a new staphylococcal exotoxin. J Exp Med. 1994;180:1675–83. doi: 10.1084/jem.180.5.1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Degnan BA, Palmer JM, Robson T, et al. Inhibition of human peripheral blood mononuclear cell proliferation by Streptococcus pyogenes cell extract is associated with arginine deiminase activity. Infect Immun. 1998;66:3050–8. doi: 10.1128/iai.66.7.3050-3058.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Cook HA, Furuya EY, Larson E, Vasquez G, Lowy FD. Heterosexual transmission of community-associated methicillin-resistant Staphylococcus aureus. Clin Infect Dis. 2007;44:410–3. doi: 10.1086/510681. [DOI] [PubMed] [Google Scholar]
- 99.Cosgrove SE, Sakoulas G, Perencevich EN, Schwaber MJ, Karchmer AW, Carmeli Y. Comparison of mortality associated with methicillin-resistant and methicillin-susceptible Staphylococcus aureus bacteremia: a meta-analysis. Clin Infect Dis. 2003;36:53–9. doi: 10.1086/345476. [DOI] [PubMed] [Google Scholar]
- 100.Gastmeier P, Sohr D, Geffers C, Behnke M, Daschner F, Ruden H. Mortality risk factors with nosocomial Staphylococcus aureus infections in intensive care units: results from the German Nosocomial Infection Surveillance System (KISS). Infection. 2005;33:50–5. doi: 10.1007/s15010-005-3186-5. [DOI] [PubMed] [Google Scholar]
- 101.Engemann JJ, Carmeli Y, Cosgrove SE, et al. Adverse clinical and economic outcomes attributable to methicillin resistance among patients with Staphylococcus aureus surgical site infection. Clin Infect Dis. 2003;36:592–8. doi: 10.1086/367653. [DOI] [PubMed] [Google Scholar]
- 102.Reed SD, Friedman JY, Engemann JJ, et al. Costs and outcomes among hemodialysis-dependent patients with methicillin-resistant or methicillin-susceptible Staphylococcus aureus bacteremia. Infect Control Hosp Epidemiol. 2005;26:175–83. doi: 10.1086/502523. [DOI] [PubMed] [Google Scholar]
- 103.Shurland S, Zhan M, Bradham DD, Roghmann MC. Comparison of mortality risk associated with bacteremia due to methicillin-resistant and methicillin-susceptible Staphylococcus aureus. Infect Control Hosp Epidemiol. 2007;28:273–9. doi: 10.1086/512627. [DOI] [PubMed] [Google Scholar]
- 104.Cosgrove SE, Qi Y, Kaye KS, Harbarth S, Karchmer AW, Carmeli Y. The impact of methicillin resistance in Staphylococcus aureus bacteremia on patient outcomes: mortality, length of stay, and hospital charges. Infect Control Hosp Epidemiol. 2005;26:166–74. doi: 10.1086/502522. [DOI] [PubMed] [Google Scholar]
- 105.Zahar JR, Clec'h C, Tafflet M, et al. Is methicillin resistance associated with a worse prognosis in Staphylococcus aureus ventilator-associated pneumonia? Clin Infect Dis. 2005;41:1224–31. doi: 10.1086/496923. [DOI] [PubMed] [Google Scholar]
- 106.Miller LG, Quan C, Shay A, et al. A prospective investigation of outcomes after hospital discharge for endemic, community-acquired methicillin-resistant and -susceptible Staphylococcus aureus skin infection. Clin Infect Dis. 2007;44:483–92. doi: 10.1086/511041. [DOI] [PubMed] [Google Scholar]