


Abstract
The roles and target genes of many transcription factors (TFs) are still unknown. To predict the roles of TFs, we present a computational method for associating Gene Ontology (GO) terms with TF-binding motifs. The method works by ranking all genes as potential targets of the TF, and reporting GO terms that are significantly associated with highly ranked genes. We also present an approach, whereby these predicted GO terms can be used to improve predictions of TF target genes. This uses a novel gene-scoring function that reflects the insight that genes annotated with GO terms predicted to be associated with the TF are more likely to be its targets. We construct validation sets of GO terms highly associated with known targets of various yeast and human TF. On the yeast reference sets, our prediction method identifies at least one correct GO term for 73% of the TF, 49% of the correct GO terms are predicted and almost one-third of the predicted GO terms are correct. Results on human reference sets are similarly encouraging. Validation of our target gene prediction method shows that its accuracy exceeds that of simple motif scanning.
INTRODUCTION
Interactions between trans- and cis-acting elements bestow the eukaryotic cell with a flexible mechanism to control gene expression. Experimental data elucidating the relationship between transcription factors (TFs) and their target genes is sparse, but paints a non-trivial picture. It often involves multiple factors and epigenetic modifications, imparting both positive and negative influences on expression levels (1,2).
Extensive research has explored the computational discovery and prediction of sites at which TFs bind. A TF-binding site (TFBS) is usually found upstream of the coding region. Sites are typically short and their motifs are consequently non-specific. When used for screening large genomic regions, TFBS motifs often require further qualification to be useful. Complementary approaches leveraging co-expression data and conservation patterns offer modest improvements for binding site discovery and prediction (3–7). However, drawing on recent experimental technologies, such as chromatin immunoprecipitation-chip (ChIP-chip) experiments, predictive tools enable the tentative construction of gene-regulatory networks (1,8).
This study aims to improve the problematic signal-to-noise ratio of TFBS prediction, not by building a more accurate model of transcription, but by leveraging a functional profile drawn from secondary information resources such as the Gene Ontology (GO). We first predict the targets of a TF in silico using a position weight matrix [PWM; (9)] description of its binding site preferences and the upstream regions of an entire genome. Then, by assuming that factors tend to bind genes of related biological purpose, we validate such relationships by inspecting the functional profile of the set of predicted targets. More specifically, we determine the statistical significance of a correlation between a set of putative transcription binding events and the functional terms transitively associated with the transcribed genes, i.e. the probability of such a correlation occurring at random.
Our proposed method will allow us to assign putative roles to a TF given its binding motif. This will provide insights into the functions of binding motifs discovered ab initio using either whole genome approaches (3,5), or discovered in sequence sets prepared based on SELEX [systematic evolution of ligands by exponential amplification; cf. (10)] experiments, expression data (e.g. upstream regions of genes with similar expression profiles), or ChIP-chip experiments. It may be argued that experimental data like ChIP-chip and gene expression can be directly correlated with annotations. However, operating at a finer-grained resolution (i.e. motifs as opposed to genes), the proposed method may assist in resolving ambiguities among candidate binding sites identified by motif discovery tools in nominated genes.
In addition to suggesting possible and specific roles of a transcription factor, the method delivers complementary evidence to validate binding of the usually large number of predicted binding sites. Intuitively, matches in the upstream regions of (putative) target genes with identical or semantically similar annotations (fetched from our secondary information resource) attain more support than those that do not.
To evaluate alternative implementations of the principal idea, we first identify a reliable and authoritative set of TF-gene annotations of the yeast genome (Saccharomyces cerevisiae). Next, predictor configurations are quantitatively assessed, and their performance and limitations are investigated. The configuration exhibiting the most reliable predictions of target function is then applied to the human genome (Homo sapiens), revealing putative roles of several TFs some of which are verified from recent experimental work. Finally, we establish the method's; ability to assist in identifying target genes in the recently proposed yeast gene-regulatory network (8). We make several observations that help illustrate the capacity of the method to infer true roles of a factor given its binding motif, and its potential to improve the specificity of in silico target gene prediction.
MATERIALS AND METHODS
Overview of method
The GO consists of three separate dictionaries of terms (collectively referred to as GO terms) that describe cellular component, biological process and molecular function (11).
We develop a method for predicting GO terms associated with (the targets of) a TF. The method takes a TFBS motif in the form of a PWM, and the upstream regions of all genes in a genome, and produces a (possibly empty) set of GO terms. Each such predicted GO term is significantly associated (over- or under-represented) with putative target genes. We then use the method to improve in silico prediction of the targets of a TF by increasing the scores of putative target genes that are annotated with the significant GO terms associated with the TF, resulting in improved target prediction specificity.
The steps of our GO term prediction method are (i) construct a ranked list of (putative) target genes by in silico prediction and (ii) find significant GO terms using the target gene ranking:
To evaluate our method, we require a ‘correct’ list of GO terms associated with a given TF. To create such a list, we use a variant of the above approach that uses a set of target genes, rather than a ranked list:
Here, the set of target genes is an input to the method, rather than being predicted by it.
In the following sections, we first describe how we perform in silico prediction of TFBSs. This is the essential first step in our method for associating TFs with GO terms. Next, we describe our statistical approach to predicting over- and under-represented GO terms linked (transitively) to a TF. Then, we describe our method for improving target gene prediction using predicted GO term associations. Finally, we describe the sources of PWMs and sequence data used in evaluating our methods.
Predicting gene targets of TFs in silico
We consider several approaches for predicting the gene targets of a TF. All of the methods are based on predicting the propensity of the upstream region of a given gene to bind to the TF. Each of the methods uses a TFBS motif described in the form of a PWM, which encodes the propensity of different, fixed-length DNA sequences to bind to the TF (9). Each gene receives a score based on the match between the motif and the upstream sequence of the gene. The motif is defined in terms of a probability matrix, q, where w is the width in base pairs, and q(k,X) is the probability of observing the nucleotide X∈{A,C,G,T } at position k∈{1,...,w}.
We explore three gene-scoring methods and variations of them. In each scoring method and for each position in the upstream region relative to the gene's transcription start site (including both strands), we compute the odds of the position being a binding site versus random (as specified by a zero-order background model and described below). Two of our three scoring methods compute the maximum and average odds, respectively, for all positions in a given gene's upstream region. We refer to these methods as ‘Max-Odds’ and ‘Avg-Odds’, respectively. The third method computes the number of positions with odds score above a given threshold (‘hits’), and we refer to this scoring method as ‘Hit-Count’. This score accommodates multiple binding sites while requiring that their strength exceeds a threshold. In order to make the thresholds for different PWMs more consistent, the Hit-Count threshold is expressed as a P-value—the probability of a single position being a random match. Max-Odds and Avg-Odds share the benefit of not requiring a threshold. Max-Odds considers only one putative binding site, whereas Avg-Odds accounts for multiple sites within the same regulatory region. The disadvantage of averaging over all positions is that sensitivity drops when sequences are long. The disadvantage of the Hit-Count method is that it requires selecting a suitable P-value threshold.
To maximize transparency, we have chosen to include only the aforementioned baseline approaches, popular in several motif-scanning software, e.g. MAST (12) (Max-Odds and Hit-count) and Clover (13) (Avg-Odds). Alternative scoring functions are also applicable and may even incorporate additional resources such as phylogenetic and expression profiles.
Each of the three gene-scoring methods requires the specification of a TFBS motif model (the PWM) and a background model. In all instances, we use a zero-order Markov background model that reflects the prior probability of each possible nucleotide. We consider two variants of each scoring method, one using ‘Global’ background, which reflects the global occurrence of each nucleotide in the upstream regions of genes, and the other using a ‘Local’ background that is re-computed for each sequence. The Local background model-based scores attempt to compensate for regional biases in nucleotide composition along the chromosome. For example, genes whose upstream regions have high GC-content will tend to receive lower scores when scored with GC-rich motifs. Each background model is constructed by estimating the probabilities of nucleotides A, C, G and T as the average number of each letter in the relevant sequence(s).
As another way of reducing potential problems caused by sequence composition biases, we also investigate two additional variants of the Avg-Odds score. Each of these is based on the standard Z-score of the Avg-Odds score. The Z-score is computed using the mean and SD estimated using either (i) 30 Avg-Odds scores generated on zero-order shuffled sequence data (Avg-Odds-ZS) or (ii) 30 Avg-Odds scores generated when the columns of the PWM are randomly reordered (Avg-Odds-ZM).
In addition to the three motif-based scores (Max-Odds, Avg-Odds and Hit-Count), we also consider a non-informative score as a negative control. We do this because there is reason to be cautious with GO term analysis. It is well-known that the base-composition of the DNA sequence is far from uniform. Some genomic regions have high GC-content and others are rich in AT. Likewise, motifs have varying preference for sequence regions with high (or low) GC-content. Significant correlations between some TF motifs and certain GO terms are possibly due to base composition alone. As a negative control score, we simply use the GC-content (expressed as a fraction) of the upstream region.
The mathematical details of our various gene-scoring functions are as follows. The binding motif (PWM) for a TF is specified by q(), where q( j,a) is the probability of base a at position j of the width-w motif. Similarly, p(a) represents the background probability of base a. For all three scores, each position i∈{1,...,L(g)} on both strands of the L(g)-base pair upstream sequence of gene g∈G are screened, where G is the set of genes for a species. All three scoring functions use the same fundamental odds score,
where N(g,k) returns the kth nucleotide in the upstream sequence of gene g∈G. The odds score for the reverse strand, S′O, is defined analogously using q′ and p′ counterparts of the above that are flipped to reflect the complementarity of nucleotide base pairing. The Avg-Odds score is the average odds of each position being a binding site versus random sequence,
Max-Odds scores each position on both strands and selects the highest score,
The Hit-Count score takes an upstream sequence and counts the number of times a motif scores above a specified threshold on either strand. It is defined as
where the threshold X(p) is chosen such that the probability (P-value) of obtaining an SO score at least as extreme as X(p) by chance is p (14).
Sometimes referred to as standardizing, a Z-score is derived by subtracting the population mean from an individual raw score and then dividing the difference by the population SD. A Z-score can be determined from any of scoring methods by using as the individual raw score the method's; score on the original data and then determining a ‘random score’ population mean and deviation by repeatedly applying the method on either (i) randomly shuffled sequences, retaining composition of the original sequence or (ii) randomly reordered columns of the original motif, retaining composition and information content. The Z-score indicates how many SD an actual observation is above or below the mean. The expectation is that a true motif match receives a higher Z-score than a random match (represented by the populations generated in either of the two ways above). The score is defined as
where μ is the population mean and σ is its SD.
Predicting GO terms associated with a TF
The GO database contains a finite set of GO terms, T. A specific gene, g∈G, is associated with zero, one or more GO terms, i.e. forms a set Tg = {t:annot(g,t)∧t∈T}, where the predicate annot() pairs genes with their terms. Using one of the scoring functions described in the previous section, we can rank all genes according to their (putative) tendency to be a target of a given TF by scoring the gene's; upstream region using the PWM for the TF. To determine if a particular GO term is significant, we then label each gene ‘+’ if it is annotated with that term, and ‘−’ otherwise. Our null hypothesis is that the order of the ‘+’ and ‘−’ genes in the list is random. If the ‘+’ genes tend to have high scores (low ranks in the list), this would indicate that the (predicted) targets of the TF are frequently annotated with the given GO term. We refer to this as the GO term being over-represented. We apply the Mann–Whitney U-test (also known as the Wilcoxon Ranksum-test) to determine whether we can reject the null hypothesis and state that the GO term is significantly associated with either high- or low-scoring genes. It should be noted that it is not completely clear that low scoring genes have any biological meaning, but they are included here for completeness.
We repeat the above process for each term in the Gene Ontology. To each term we assign as its score the P-value of the U-test. To account for multiple testing, we use a Bonferroni correction to compute the E-value of the GO term—the number of terms that would score as well by chance in the entire GO database. For a given E-value, E, and TF, f, we denote the set of predicted terms as 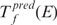 .
.
Constructing a gold standard of TF–GO term associations
In order to construct a reference set of associations between a TF and GO terms, we require a list of the TF's; target genes. To determine if a given GO term is significantly over- or under-represented in such a list, we construct a two-by-two contingency table and apply Fisher's; Exact test. The rows of the table are counts of ‘target’ and ‘non-target’ genes. The columns are ‘has GO term t’ and ‘does not have GO term t’. The Fisher's; Exact Test determines whether the two groups differ in the proportion with which they fall into the categories with or without GO term t. Rejecting the null hypothesis (that they do not differ) is acceptable only if the P-value is small. As before, we compute E-values to correct for multiple tests. We designate all GO terms enriched with an E-value of 0.05 or below as belonging to target level 1. All terms with E-values of 0.01 or below, belong to target level 2. Finally, all terms with E-values of 0.001 or below, belong to the strictest level, target level 3.
Improving target gene prediction using GO term associations
Suppose we knew what GO terms a TF was associated with. We would then expect genes with these terms to be a priori more probable targets. We can leverage this intuition to create an improved target gene predictor. First, we predict TF–GO term associations to TF f using one of the scoring methods described above. We then use the following score to re-score all genes, g, as potential targets of TF f:
where E is the GO term E-value.
GO terms, genomic sequences and PWMs
We use the color GO (24 August, 2007) OBO version 1.2. In total, there are 13 911 terms for biological processes, 7892 terms for molecular functions and 2007 terms for cellular components. color The terms are linked with ‘is-a’ and ‘part-of’ relations. When appropriate, the closure of such transitive relations is determined to ascribe terms to genes, e.g. if a gene is associated with the term nuclear exosome (GO:0000176), which ‘is-a’ nuclear part (GO:0044428), it is (per transitive inference) also associated with this more generic term. As a consequence of collapsing terms this way, the method will use a more generic description of each GO term, no longer specific to the level in the GO hierarchy at which it appears. Furthermore, we do not distinguish between evidence classes in the GO, but simply include them all. Including less corroborated annotations (e.g. those inferred by homology) could compromise the quality of predictions. However, we argue that since inference requires broad statistical support, the use of less reliable data may instead alleviate the problem of sparsely annotated genomes.
To ensure generality of our approach, we test it on both yeast and human regulatory regions. We chose yeast and human as examples because their genomes are well-studied and data-accessible (see below), and because they are relatively extreme representatives of eukaryotic regulatory complexity. We use the 1000-bp upstream regions all genes in S. cerevisiae (www.yeastgenome.org; 6 September 2007; 5883 sequences) and H. sapiens (genome.ucsc.edu; version hg18; 23 570 sequences). According to the GO, there are 4490 unique terms associated with the S. cerevisiae sequences, and 8387 with H. sapiens. There are terms for 6475 S. cerevisiae genes and 16 251 H. sapiens genes.
Using ChIP data of Harbison et al. (15) and the merger of two highly sensitive binding site discovery algorithms incorporating evolutionary conservation, a high-confidence yeast regulatory network was recently suggested (8). Statistical scores of motif matches were stringently based on empirical P-values and limited to regions known to be bound by the corresponding factor. The resulting network expands that of Harbison et al. (15) without compromising specificity. Of the 172 transcription factors with at least four bound probes in the Harbison study, MacIsaac et al. (8) found 98 significant motifs (of which 64 were validated from the literature). Augmented by another 26 TF specificities from the literature, the network consists of a total 124 factors with binding sites. We use the complete regulatory map [http://fraenkel.mit.edu/improved_map (8)] as a gold standard.
The regulatory map thus specifies a substantial number of TFs and their binding sites, here converted into position-specific scoring matrices. When assembling the target gene set for a given TF, we distinguish between MacIsaac and colleagues' high confidence gene set (HC-set), filtered via their strictest controls (motif match P ≤ 0.001 and their highest sequence conservation level ‘2’), and their low confidence gene set (LC-set; motif match P ≤ 0.05). The statistical significance of each GO term associated with these target genes is determined in the context of the full set of yeast genes.
For the human gold standard, we use a total of 51 TF motifs for H. sapiens taken from JASPAR (16). We also use published target gene data sets for NFKB1 (17), SRY (18), CREB1 (19), and TP53 (20).
To further validate predictions in human genes, JASPAR identifies applicable TFs linked to UniProt. We use the Function statement in the Comment field of the entry (when available) to subjectively match predicted terms with experimental knowledge.
Classification performance
We present methods for predicting (i) GO terms and (ii) target genes from TF-binding motifs. The discrete nature of such predictions allows us to view them as classifications. In our case, the set of predicted terms or genes are all ‘positives’. All terms or genes that are not part of the predicted set can be viewed as ‘negatives’. For the binding motifs in our gold standard sets, we have access to the terms and genes that should ideally be in the predicted set. We thus distinguish between ‘true positives’ (correct predictions), ‘false positives’ (incorrect predictions), ‘true negatives’ (terms and genes that are correctly not included as predictions) and ‘false negatives’ (terms or genes that should have been but are not in the predicted set). When viewed over all motifs, the number of each is determined, here referred to as tp, fp, tn and fn, respectively.
The true positive rate is then defined as
The false positive rate is defined as
Receiver operating characteristic (ROC) curves illustrate how the true positive rate changes with the false positive rate for all possible thresholds (separating positives from negatives). The ROC50 (21) is simply the same curve but only shows the true positive rate up to a maximum of 50 false positives. The area under the ROC (ROC50) curve is referred to as AUC (AUC50), and gives a single measure of classification accuracy that considers all possible tradeoffs of sensitivity and specificity. AUC50 illustrates the ability of the methods to identify positives before falsely identifying many negatives. It is more appropriate than conventional ROC when what is important is predicting a few correct relationships, rather than predicting all relationships.
For GO term prediction, we report on recall and precision. Recall (also known as sensitivity) is equivalent to the true positive rate above. Precision is defined as
and, similar to the false positive rate, illustrates the integrity of the predictions. However, the GO term prediction problem has a large number of ‘easy’ negatives which motivates the stricter precision-measure.
RESULTS
Gold standard of GO terms associated with TFs
In order to evaluate our method for in silico prediction of the GO terms associated with a TF, we require reliable annotation of this type for a set of TFs. To our knowledge, there is currently no resource that contains annotation of TF with the GO terms associated with the genes they regulate. Thus, we created such a resource for a large number of yeast TFs and for four human TFs for which sufficient knowledge of their target genes exists. To create the gold standard for a given TF, we use the set of ‘known’ targets of the TF, and apply the statistical method described in Materials and methods section, in order to determine the set of GO terms significantly associated with the target set. Details of all target GO terms are provided in the Supplementary Material.
In yeast, we use the target genes for each of the 124 TF in the gene-regulatory network of MacIsaac et al. (8). For each TF, we determine the statistical enrichment of GO terms amongst the target genes in their most conservative mapping (the HC-set) only. Table 1 panel a summarizes how many significant TF–Go term relationships for yeast TFs our gold standard contains.
Table 1.
Gold standards of TF–GO term associations used in this study
| Panel (a) | |||
|---|---|---|---|
| Yeast gold standard | |||
| Target level | E-value | TFs | GO terms per TF |
| 1 | ≤0.05 | 70 | 13.1 |
| 2 | ≤0.01 | 57 | 12.4 |
| 3 | ≤0.001 | 43 | 12.0 |
| Panel (b) | |||
|---|---|---|---|
| Human gold standard | |||
| TF | Target genes | GO terms | |
| NFKB1 | 99 | 214 | |
| SRY | 906 | 82 | |
| CREB1 | 2197 | 168 | |
| TP53 | 62 | 40 | |
Panel (a) shows for each target level in the yeast transcription network the E-value range defining the level, the number of TFs with one or more signficant GO terms and the average number of GO terms per TF. Panel (b) shows the human TF gene name, the number of known target genes and the number of significantly over-represented GO terms (E ≤ 0.05) associated with that TF.
For creating the human gold standard, we use recent experimentally determined target gene lists for NFKB1 (17), SRY (18), CREB1 (19) and TP53 (20). NFKB1 is a member of the family of NF-kappa-B TFs, which are involved in inflammatory and immune system mechanisms, cellular stress reactions, as well as apoptosis (22). SRY is a genetic switch in male development (23). CREB1 is a TF involved in many viral and cellular promoters. TP53 is a tumour suppressor involved in several cancer types. Table 1 panel b summarizes the number of significant TF–GO term relationships in our gold standard for the four human TFs.
Negative control: GO terms associated with biased base composition
We designed the Z-score variants of our Avg-Odds gene-scoring method to try to eliminate spurious correlations between sequences and motifs with biased base compositions. In particular, it is well known that CpG islands (large clusters of the dinucleotide CpG) are common in the promoter regions of genes in many eukaryotes (24).
To give an upper bound on TF–GO term associations that could be caused by associations between GO terms and genes with biased base composition in their upstream regions, we apply our GC-content score to both our S. cerevisiae and H. sapiens upstream regions. We rank genes using this simple composition score, and compute significant GO terms as described in Materials and methods section. Using this GC-score, five GO terms are found as over- or under-represented (in this context ‘under-represented’ means ‘over-represented’ using an AT-context score.) in S. cerevisiae and 138 terms were similarly singled out in H. sapiens (each statistically significant E < 0.05). A truly random score should result in no significant GO terms in either genome. Hence, matches for short and unspecific motifs may similarly result in spurious GO terms with deceptively small E-values.
Based on these results, we expect that the Z-score variants of the Avg-Odds method may be useful with the human genome, but probably will have little effect with the yeast genome, since so few terms are predicted in yeast using the GC-score. All terms identified in this negative control are provided in the Supplementary Material.
Identifying GO terms associated with TF targets
The gene-scoring methods described in Materials and methods section provide each gene with a numerical score. Since we look at binding events only for a fixed-length upstream region, all genes are a priori equally probable. Our test for over- and under-represented GO terms involves ranking the genes according to their scores and observing the prevalence of each GO term. More specifically, for each motif, the U-test assigns an E-value to each GO term, indicating if the genes with that GO term are dispersed randomly (the null hypothesis) or if they appear non-randomly in the ranked list, either at the top (over-represented) or the bottom (under-represented).
Table 2 shows the accuracy of the different variations of our method for predicting associations between TFs and GO terms in yeast. To instill maximum confidence in our GO term prediction evaluation, we focus solely on the 43 TFs that have GO terms associated with them at all levels, and illustrate results using our ‘Target Level 3’ gold standard. (The observations below are essentially unchanged when using the less stringent target levels. The Supplementary Material contains extended tables of results at all levels, Tables S1 and S2.) Table 2 shows results when we only consider predictions with an E-value smaller than 10, as well as when we consider all possible score cutoffs up to the fiftieth false positive (AUC50).
Table 2.
Accuracy of predicted TF–GO term associations in yeast
| TF–GO term predictions in yeast | |||||
|---|---|---|---|---|---|
| Scoring method |
E = 10 cutoff |
AUC50 | |||
| Pred. | 1 TP | Rec. | Prec. | ||
| Hit-Count | |||||
| 10−3 Local | 38.4 | 0.64 | 0.33 | 0.12 | 0.31 |
| 10−3 Global | 34.2 | 0.76 | 0.44 | 0.16 | 0.39 |
| 10−4 Local | 16.3 | 0.73 | 0.49 | *0.37 | 0.57 |
| 10−4 Global | 13.0 | 0.64 | 0.40 | *0.37 | 0.54 |
| 10−5 Local | 3.1 | 0.29 | 0.11 | *0.67 | 0.24 |
| 10−5 Global | 2.3 | 0.22 | 0.09 | *0.63 | 0.21 |
| Avg-Odds | |||||
| Local | 47.8 | 0.87 | 0.63 | 0.17 | 0.56 |
| Global | 54.0 | 0.87 | 0.66 | 0.15 | 0.56 |
| ZS Local | 39.1 | 0.84 | 0.63 | 0.20 | 0.56 |
| ZS Global | 39.9 | 0.84 | 0.62 | 0.20 | 0.57 |
| ZM Local | 36.7 | 0.87 | 0.60 | 0.21 | 0.56 |
| ZM Global | 37.1 | 0.84 | 0.59 | 0.20 | 0.55 |
| Max-Odds | |||||
| Local | 39.9 | 0.82 | 0.60 | 0.19 | 0.54 |
| Global | 44.2 | 0.84 | 0.63 | 0.17 | 0.54 |
The average results for yeast TFs using different methods of scoring target genes. Each row shows results at a significance level of E = 10, as well as overall results measured using the area under the ROC50 curve. The columns indicate the number of predictions returned (Pred.), the probability of predicting at least one true positive (1 TP), the recall (Rec.), the precision (Prec.) and AUC50. The best value for each metric is shown in bold-face. When one or more TFs render no predictions they are excluded from the precision average (marked with ‘*’; based on a different data source, such values are not included when determining the best for each metric).
In terms of their ability to predict at least one correct GO term (‘1 TP’) when the E-value cutoff is set at ten, the methods differ considerably. The Avg-Odds methods perform best according to this metric, achieving rates up to 87%. The best Max-Odds method is slightly less accurate at this task (84%). The Hit-Count method performance depends strongly on the choice of the motif-match threshold, but the ‘1 TP’ rate is only 76% at best. When we compare the methods using the AUC50 accuracy metric, the Avg-Odds, Max-Odds and Hit-Count (motif-match threshold 10−4) methods are all statistically indistinguishable according to a paired t-test comparing the AUC50 for each of the 43 yeast binding motifs.
In terms of the precision of the predictions, the Z-score variants of Avg-Odds work best. The precision of the Avg-Odds ZM Local method is 0.21 at this E-value cutoff, compared with 0.15 for the Avg-Odds Global method. However, this increased precision comes at the expense of lower sensitivity (recall is only 0.60, compared with 0.66 for Avg-Odds Global). On average, all Avg-Odds-based methods predict roughly the same number of GO terms. In general, the Max-Odds score variants do not perform as well as the Avg-Odds methods in the experiment with yeast TFs. The differences in accuracy are not significant in practice.
In terms of overall accuracy (as measured by AUC50), there is very little difference among the scoring methods when applied to the yeast genome. In particular, the performance of the Z-score variants of the Avg-Odds scoring methods are essentially indistinguishable from that of the simpler methods on which they are based. This is not too surprising given the fact that our negative control (using the GC-content score) gave only five spurious GO term predictions in yeast.
As noted earlier, the Hit-Count methods are extremely sensitive to the choice of the motif-match threshold, and perform poorly except when the threshold is set to 10− 4. Tuning this threshold may improve accuracies slightly but in turn result in a parameter selection bias (tied to the target data). When the threshold is strict enough, positive predictions are rare. If no positives are predicted for one or more TFs, average precision excludes such TFs (marked with ‘*’ in Table 2). This however may lead to optimistic estimates. We also tested Z-score variants of the Hit-Count method, but AUC50 scores were inferior to those of the simpler variant described here so they are not shown.
Many TF-binding motifs without target GO terms still predict convincing lists of GO terms. The following examples are predicted by Avg-Odds Global. Forkhead homologues 1 and 2 (FKH1 and FKH2) are known to modulate the yeast cell cycle and for controlling cell morphology (25). Rightly, predictions include seven GO terms (FKH1; 11 for FKH2), all related to these target roles, but in some cases more specific than what is apparent from the literature (e.g. ‘M phase of mitotic cell cycle’ and ‘microtubule cytoskeleton’). Similarly, Sucrose transporter SUT1 is not only predicted by our method to trigger a range of sugar-related processes, but its targets' cellular compartment is also confirmed to relate to the plasma membrane (26). Finally, GATA-1 is known to regulate nitrogen metabolism, and our predictor (correctly) lists ‘Allantoin metabolic process’ and ‘Heterocyclic catabolic process’ (27) as significant GO terms associated with it.
Our gold standard for TF–GO term associations for human TFs comprises the four well-studied TFs NFKB1, SRY, CREB1 and TP53. Table 3 summarizes the performance of the Avg-Odds Global prediction method using the corresponding JASPAR PWMs for these factors at prediction E-value cutoffs of 10 and 0.05 (the target GO term E-value cutoff is 0.05 in all human tests).
Table 3.
Accuracy of predicted TF–GO term associations in human using the avg-odds global method
| TF-GO Term Predictions in Human | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Gene name |
E = 10 cutoff |
E = 0.05 cutoff |
AUC50 | ||||||
| Pred. | 1 TP | Rec. | Prec. | Pred. | 1 TP | Rec. | Prec. | ||
| NFKB1 | 120 | 1 | 0.19 | 0.32 | 18 | 1 | 0.07 | 0.78 | 0.10 |
| SRY | 147 | 1 | 0.28 | 0.15 | 28 | 1 | 0.08 | 0.21 | 0.07 |
| CREB1 | 212 | 1 | 0.69 | 0.52 | 78 | 1 | 0.37 | 0.76 | 0.40 |
| TP53 | 50 | 1 | 0.03 | 0.02 | 2 | 0 | 0.00 | 0.00 | 0.02 |
| Mean | 132.2 | 1 | 0.30 | 0.25 | 31.5 | 0.75 | 0.13 | 0.44 | 0.15 |
Each row shows results at significance level of E = 10 and E = 0.05, as well as overall results measured using the area under the ROC50 curve. The columns indicate the number of predictions returned (Pred.), the probability of predicting at least one true positive (1 TP), the recall (Rec.), the precision (Prec.) and AUC50, for four human TFs.
On this small set of human TFs, with E ≤ 10 the average recall is lower than with the yeast TFs (0.30 versus 0.66) but the average precision is higher (0.25 versus 0.15; refer to Table 2 for yeast results and Table 3 for human results). On a cautionary note and in line with the negative control, not only are more terms predicted than for yeast, but the number of targets is also higher (refer to Table 1). At E ≤ 0.05, the number of human GO term predictions is comparable to that of yeast when E ≤ 10. At this level, for all but one factor (TP53), the Avg-Odds Global prediction method correctly identifies at least one TF–GO term association. For the three factors where a correct association is predicted, the result is judged highly significant by a one-tailed Fisher's; Exact test (P < 10− 7) (data not shown). However, these P-values should be interpreted with caution. GO terms are organized into rich networks manifesting their relations, reporting dependencies that are not considered when P-values are determined.
Since the Avg-Odds Global scoring method performs at least as well (except for precision) as the other methods, we further investigate its behavior at different precision levels. Table 4 shows the GO term prediction performance of this method when the prediction E-value cutoff is varied. As expected, higher precision is observed at more stringent (lower) E-value cutoffs, at the expense of lower recall and lower probability that the predictions include at least one true positive. In fact, at E ≤ 0.1, there are TFs for which there are no predicted GO terms. At E ≤ 1, i.e. with one-expected chance positive, all of our four human TFBS motif searches result in at least one correct GO term (73% in yeast). Moreover, of the predicted terms, one-third represent a true role of the TF (27% in yeast). At the more conservative cutoff E = 0.01, we do not always get a prediction, but when we do, about 60% are correct in human (61% in yeast).
Table 4.
Accuracy of predicted TF–GO terms associations at different E-value cutoffs using the Avg-Odds global method
| E-value cutoff | Yeast |
Human |
||||||
|---|---|---|---|---|---|---|---|---|
| Pred. | 1 TP | Rec. | Prec. | Pred. | 1 TP | Rec. | Prec. | |
| 0.01 | 5.4 | 0.56 | 0.27 | *0.61 | 22.8 | 0.75 | 0.10 | *0.60 |
| 0.05 | 8.2 | 0.58 | 0.32 | *0.49 | 31.5 | 0.75 | 0.13 | 0.44 |
| 0.1 | 10.0 | 0.60 | 0.36 | *0.44 | 36.8 | 0.75 | 0.14 | 0.40 |
| 1 | 20.2 | 0.73 | 0.49 | 0.27 | 72.3 | 1 | 0.22 | 0.33 |
| 10 | 54.0 | 0.87 | 0.66 | 0.15 | 132.3 | 1 | 0.30 | 0.25 |
| 50 | 144.6 | 0.93 | 0.78 | 0.07 | 250.3 | 1 | 0.37 | 0.17 |
The columns indicate the E-value cutoff, number of TF–GO term association predictions returned by the method at that cutoff, the probability of predicting at least one true positive (1 TP; higher is better), the recall (higher is better), and the precision (higher is better). The values for yeast are averages calculated from the 43 TFs for which target GO terms exist. The values for human are averages calculated from NFKB1, SRY, CREB1 and TP53. When one or more TFs render no predictions they are excluded from the precision average (marked with ‘*’).
Although we do not have a TF–GO term gold standard for all 51 human TFs in JASPAR, we also report here on using our prediction method with each of them. With a stringent cutoff of E = 0.05, 43 of the human PWMs in JASPAR result in one or more predicted GO terms (the details are provided in the Supplementary Material). On average, 32 GO terms are identified per motif. Hence, the average number of predicted terms is almost four times higher than in yeast with the same E-value (Table 4). We evaluate these predictions subjectively based on (i) a generic summary over about half the motifs, filtered to include those with short and specific lists of GO terms and (ii) a detailed assessment of a few motifs with experimentally determined target genes. For each of the motifs for H. sapiens, we extract the entry for the TF (as specified in JASPAR) from UniProt. We manually examine the Function statement of the UniProt entry to identify keywords relating to the target function of the factor. In a few cases, we were unable to find any relevant target role.
All motifs where our prediction method predicts more than 25 GO terms are likely to involve terms included by chance alone. To increase the confidence in our subjective evaluation, such motifs were removed entirely from consideration. This leaves 21 motifs, of which 15 have predicted GO terms that (subjectively) match the UniProt documentation for the TF. The 21 JASPAR motifs, the corresponding TF and the documented and predicted target roles are listed in Supplementary Material Table S3 and S4.
Our negative control (using the GC-content score) indicates that the compositional bias of many human promoters may lead to large numbers of spurious GO term predictions for motifs that also have biased basecomposition. Due to the small number of human TFs in our study, it is impossible to draw strong conclusions, but the results in Table 5 suggest that using the Z-score variant of the Avg-Odds score may be beneficial with compositionally-biased motifs. The accuracy (AUC50) of the predicted GO terms improves substantially using the Z-score variant for the two motifs with very high compositional bias (NFKB1 76% GC and SRY 76% AT). The improvement is especially notable for the AT-rich motif SRY, where the AUC50 score increases from 0.07 to 0.34. On the other hand, using the Z-score variant of the scoring method causes a large decrease in accuracy for the GC-rich motif CREB1. This effect may be due to the fact that the promoters of CREB1 target genes are very rich in CpG islands—44% of the target upstream regions are CpG islands. Because the CREB1 is GC-rich (59% GC), we expect the Z-score variant to decrease the scores of these true targets relative to the scores of promoters that do not contain CpG islands. This would tend to decrease the accuracy of GO term predictions. This suggests that it may be dangerous to use the Z-score variants with GC-rich motifs in genomes that contain many promoter-associated CpG islands.
Table 5.
Effect on accuracy of using the Z-score variant of Avg-Odds global scores in human
| Gene name | AUC50 |
Motif content | Target CpG islands (%) | |
|---|---|---|---|---|
| Avg-Odds | Avg-Odds ZS | |||
| NFKB1 | 0.10 | 0.16 | 76% GC | 27 |
| SRY | 0.07 | 0.34 | 76% AT | 40 |
| CREB1 | 0.40 | 0.05 | 59% GC | 44 |
| TP53 | 0.02 | 0.00 | 60% GC | 48 |
The table compares the AUC50 accuracy score of predicted TF–GO term associations in Human using the Avg-Odds Global method and its shuffled-sequence Z-score variant (ZS) for four TFs. For each TF, the average GC- (or AT-) content of the motif (the average total probability of the two bases in the PWM) and the percentage of the known targets of the TF that are CpG islands [according to the definition of Takai and Jones (24)] are also shown.
Improving target gene prediction using GO term associations
In this section, we ask if predicted TF–GO associations can be used to improve TF target gene prediction. We use the augmented scoring function S_GO(g,f) given in the Materials and methods section, where the underlying score is Avg-Odds Global. We refer to the overall gene-scoring method as ‘Avg-Odds-GO’. To test the approach, we use the 63 TFs in the regulatory map of MacIsaac et al. (8) for which our method predicts one or more GO terms and that have at least one gene in MacIsaac's; high confidence target set (HC-set), with which we measure prediction accuracy.
Figure 1 shows the average ROC50 curves for predicting targets of these 63 yeast TFs using the Avg-Odds-GO, Avg-Odds and Max-Odds scoring functions. The AUC are 0.172, 0.162 and 0.120 for Avg-Odds-GO, Avg-Odds and Max-Odds, respectively (higher is better, maximum is 1.0). This shows that the inclusion of GO terms is cleary beneficial, although the advantage of the Avg-Odds-GO scoring function compared to the Avg-Odds function is rather small, especially at low false positive rates. The individual AUC50 scores of each method on each of the 63 motifs are provided in Supplementary Material Tables S5 and S6 together with two examples Tables S7 and S8.
Figure 1.

Accuracy (ROC50) predicting yeast TF target genes. Curves show the ROC50 plots for three methods of predicting target genes of 63 Yeast TFs. Error bars show the standard error.
DISCUSSION
Hvidsten et al. (28) observed that genes bound to by the same TFs are more strongly associated with the same GO terms than with common binding sites or expression patterns. Corá and colleagues (5,29) investigated using GO annotation as well as expression data for de novo discovery of TFBS binding sites by first identifying over-represented short DNA sequences in upstream regions of known genes. Genes were subsequently grouped by shared putative binding motifs and validated by requiring that microarray expression data support co-regulation and that functional annotations be in agreement (per the GO). Gene sets showing a statistically significant functional characterization are then viewed as exhibiting a valid binding site for a particular TF. Several of the identified over-represented patterns were known TFBS or variations thereof. Hu et al. (30) and Long et al. (31) similarly used GO terms as a means of validating their binding site predictions. Hvidsten et al. (28) used GO terms to evaluate combinations of transcription events described by rules. The GO thus appears to offer a rich secondary information resource to validate primary data analysis.
This article presents a method by which GO terms associated with a TF's; target genes are predicted using a TF-binding motif. TF–GO term associations are predicted using a two-stage process. The first stage consists of ranking potential target genes by considering matches to the transcription factor binding motif. The second stage consists of finding GO terms significantly associated with genes appearing at the top (or bottom) of the ranked list.
In tests using S. cerevisiae TFBS motifs, our predictor predicts about 20 GO terms per TF at an E-value cutoff of 1. At this cutoff, there is a 73% chance of predicting at least one correct GO term. In fact, at this E-value, the predictor picks up 49% of known TF–GO term associations. Conversely, 27% of the predicted terms are true positives, attesting to the method's; usefulness with yeast.
We illustrate the broader applicability of the method by predicting GO terms for human TF-binding motifs. The method produces more predictions than on the yeast genome, but highly useful rates of precision and recall are achieved, and the probability of predicting at least one correct association is satisfyingly close to 1. colorWe note that compositional biases in sequence data pose an obstacle and that improved accuracy may result from compensating for a motif's; GC-content and its association with CpG islands.
On a cautionary note, the regulatory map for yeast is partially refined through sequence analysis, biasing the choice of target genes and, indirectly, target GO terms. Since the proposed method is based on motif searching, reported accuracies of predicting GO terms may be slightly inflated. However, the human target gene sets we use (other than for SRY) do not appear to have been identified in a sequence-biased fashion, so the afore mentioned bias should not be present in our results for human TFs.
Finally, we incorporate our TF–GO term prediction method into a novel scoring method for predicting the gene targets of a TF. This scoring method gives priority to genes that are annotated with the GO terms predicted to be associated with the binding motif. We show that the augmented score increases the accuracy of TF target gene prediction in the yeast genome. It should be noted that we did not attempt to tune the E-value used by our augmented scoring function, in order to achieve maximum target gene prediction accuracy. A refined scoring function could consider the number and quality of GO term predictions in combination with the preliminary score to provide a possibly improved ability to identify actual target genes. One possible approach would be filtering predicted GO terms to maximize their coherence, e.g. using semantic distance measures between terms (32,33). We further anticipate that an augmented score that takes GO terms associated with a query TF-binding motif into account could be used in an iterated manner.
Using aligned genomic sequences from related species can benefit TFBS prediction (34). Our GO term predictor could similarly benefit from comparative genomics by (i) altering the motif model in the scoring function to use phylogenetic footprinting or (ii) running the native scoring function independently on each genomic sequence and then combining the species-specific GO term predictions. The latter strategy would also alleviate concerns with sparsely annotated genomes if more well-studied and evolutionary related sequences are indeed available.
The GO is a secondary information resource that has so far been under-utilized for improving our ability to discover true TFBS. Representing a clear-cut complement to the incorporation of phylogenetic profiles and epigenetic modifications, this article starts to leverage the ever-expanding GO annotations to devise a novel and powerful scoring method for TF target genes and, eventually, binding sites. We anticipate that putative TF binding sites from SELEX and ChIP experiments, and gene-expression analyses can be directly corroborated by the proposed method.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
ACKNOWLEDGEMENTS
The authors acknowledge Zuzana Cienikova who conducted a pilot investigation into using PWMs to identify GO terms associated with TFs, and two anonymous reviewers for their constructive advice. Australian Research Council Centre of Excellence in Bioinformatics; National Institutes of Health (RO-1 RR021692-01 to T.L.B). Funding to pay the Open Access publication charges for this article was provided by National Institutes of Health (RO-1 RR021692-01).
Conflict of interest statement. None declared.
REFERENCES
- 1.Tsankov AM, Brown CR, Yu MC, Win MZ, Silver PA, Casolari JM. Communication between levels of transcriptional control improves robustness and adaptivity. Mol. Syst. Biol. 2006;2:65. doi: 10.1038/msb4100106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Misteli T. Beyond the sequence: cellular organization of genome function. Cell. 2007;128:787–800. doi: 10.1016/j.cell.2007.01.028. [DOI] [PubMed] [Google Scholar]
- 3.Kellis M, Patterson N, Endrizzi M, Birren B, Lander ES. Sequencing and comparison of yeast species to identify genes and regulatory elements. Nature. 2003;423:241–254. doi: 10.1038/nature01644. [DOI] [PubMed] [Google Scholar]
- 4.Wasserman WW, Sandelin A. Applied bioinformatics for the identification of regulatory elements. Nat. Rev. Genet. 2004;5:276–287. doi: 10.1038/nrg1315. [DOI] [PubMed] [Google Scholar]
- 5.Corà D, Cunto FD, Provero P, Silengo L, Caselle M. Computational identification of transcription factor binding sites by functional analysis of sets of genes sharing overrepresented upstream motifs. BMC Bioinform. 2004;5:57. doi: 10.1186/1471-2105-5-57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Xie X, Lu J, Kulbokas EJ, Golub TR, Mootha V, Lindblad-Toh K, Lander ES, Kellis M. Systematic discovery of regulatory motifs in human promoters and 3′ UTRs by comparison of several mammals. Nature. 2005;434:338–345. doi: 10.1038/nature03441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Blanco E, Messeguer X, Smith TF, Guigó R. Transcription factor map alignment of promoter regions. PLoS Comput. Biol. 2006;2:e49. doi: 10.1371/journal.pcbi.0020049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.MacIsaac KD, Wang T, Gordon DB, Gifford DK, Stormo GD, Fraenkel E. An improved map of conserved regulatory sites for Saccharomyces cerevisiae. BMC Bioinform. 2006;7:113. doi: 10.1186/1471-2105-7-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stormo GD. DNA binding sites: representation and discovery. Bioinformatics. 2000;16:16–23. doi: 10.1093/bioinformatics/16.1.16. [DOI] [PubMed] [Google Scholar]
- 10.Lazebnik MB, Tussie-Luna M-I, Roy AL. Determination and functional analysis of the consensus binding site for TFII-I family member BEN, implicated in Williams-Beuren syndrome. J. Biol. Chem. 2008;283:11078–11082. doi: 10.1074/jbc.C800049200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, et al. Gene Ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bailey TL, Gribskov M. Methods and statistics for combining motif match scores. J. Comput. Biol. 1998;5:211–221. doi: 10.1089/cmb.1998.5.211. [DOI] [PubMed] [Google Scholar]
- 13.Frith MC, Fu Y, Yu L, Chen J-F, Hansen U, Weng Z. Detection of functional DNA motifs via statistical over-representation. Nucleic Acids Res. 2004;32:1372–1381. doi: 10.1093/nar/gkh299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bailey TL, Gribskov M. Combining evidence using p-values: application to sequence homology searches. Bioinformatics. 1998;14:48–54. doi: 10.1093/bioinformatics/14.1.48. [DOI] [PubMed] [Google Scholar]
- 15.Harbison CT, Gordon DB, Lee TI, Rinaldi NJ, Macisaac KD, Danford TW, Hannett NM, Tagne J-B, Reynolds DB, ,,,,,,,,, Yoo J, et al. Transcriptional regulatory code of a eukaryotic genome. Nature. 2004;431:99–104. doi: 10.1038/nature02800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sandelin A, Alkema W, Engström P, Wasserman WW, Lenhard B. JASPAR: an open-access database for eukaryotic transcription factor binding profiles. Nucleic Acids Res. 2004;32 (Database issue):D91–D94. doi: 10.1093/nar/gkh012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Defrance M, Touzet H. Predicting transcription factor binding sites using local over-representation and comparative genomics. BMC Bioinform. 2006;7:396. doi: 10.1186/1471-2105-7-396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jin VX, O'Geen H, Iyengar S, Green R, Farnham PJ. Identification of an OCT4 and SRY regulatory module using integrated computational and experimental genomics approaches. Genome Res. 2007;17:807–817. doi: 10.1101/gr.6006107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Odom DT, Dowell RD, Jacobsen ES, Nekludova L, Rolfe PA, Danford TW, Gifford DK, Fraenkel E, Bell GI, Young RA. Core transcriptional regulatory circuitry in human hepatocytes. Mol. Syst. Biol. 2006;2:2006–0017. doi: 10.1038/msb4100059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wei C-L, Wu Q, Vega VB, Chiu KP, Ng P, Zhang T, Shahab A, Yong HC, Fu Y, Weng Z, et al. A global map of p53 transcription-factor binding sites in the human genome. Cell. 2006;124:207–219. doi: 10.1016/j.cell.2005.10.043. [DOI] [PubMed] [Google Scholar]
- 21.Gribskov M, Veretnik S. Identification of sequence pattern with profile analysis. Methods Enzymol. 1996;266:198–212. doi: 10.1016/s0076-6879(96)66015-7. [DOI] [PubMed] [Google Scholar]
- 22.Pahl HL. Activators and target genes of Rel/NF-kappaB transcription factors. Oncogene. 1999;18:6853–6866. doi: 10.1038/sj.onc.1203239. [DOI] [PubMed] [Google Scholar]
- 23.Wilhelm D, Palmer S, Koopman P. Sex determination and gonadal development in mammals. Physiol. Rev. 2007;87:1–28. doi: 10.1152/physrev.00009.2006. [DOI] [PubMed] [Google Scholar]
- 24.Takai D, Jones PA. Comprehensive analysis of cpg islands in human chromosomes 21 and 22. Proc. Natl Acad. Sci. USA. 2002;99:3740–3745. doi: 10.1073/pnas.052410099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hollenhorst PC, Bose ME, Mielke MR, Müller U, Fox CA. Forkhead genes in transcriptional silencing, cell morphology and the cell cycle. Overlapping and distinct functions for fkh1 and fkh2 in Saccharomyces cerevisiae. Genetics. 2000;154:1533–1548. doi: 10.1093/genetics/154.4.1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Reinders A, Schulze W, Thaminy S, Stagljar I, Frommer WB, Ward JM. Intra- and intermolecular interactions in sucrose transporters at the plasma membrane detected by the split-ubiquitin system and functional assays. Structure. 2002;10:763–772. doi: 10.1016/s0969-2126(02)00773-6. [DOI] [PubMed] [Google Scholar]
- 27.Marzluf GA. Genetic regulation of nitrogen metabolism in the fungi. Microbiol. Mol. Biol. Rev. 1997;61:17–32. doi: 10.1128/mmbr.61.1.17-32.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hvidsten TR, Wilczynski B, Kryshtafovych A, Tiuryn J, Komorowski J, Fidelis K. Discovering regulatory binding-site modules using rule-based learning. Genome Res. 2005;15:856–866. doi: 10.1101/gr.3760605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Corà D, Herrmann C, Dieterich C, Cunto FD, Provero P, Caselle M. Ab initio identification of putative human transcription factor binding sites by comparative genomics. BMC Bioinformatics. 2005;6:110. doi: 10.1186/1471-2105-6-110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hu Z, Hu B, Collins JF. Prediction of synergistic transcription factors by function conservation. Genome Biol. 2007;8:R257. doi: 10.1186/gb-2007-8-12-r257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Long F, Liu H, Hahn C, Sumazin P, Zhang MQ, Zilberstein A. Genome-wide prediction and analysis of function-specific transcription factor binding sites. In Silico Biol. 2004;4:395–410. [PubMed] [Google Scholar]
- 32.Wang JZ, Du Z, Payattakool R, Yu PS, Chen C-F. A new method to measure the semantic similarity of go terms. Bioinform. 2007;23:1274–1281. doi: 10.1093/bioinformatics/btm087. [DOI] [PubMed] [Google Scholar]
- 33.Guo X, Liu R, Shriver CD, Hu H, Liebman MN. Assessing semantic similarity measures for the characterization of human regulatory pathways. Bioinform. 2006;22:967–973. doi: 10.1093/bioinformatics/btl042. [DOI] [PubMed] [Google Scholar]
- 34.Lenhard B, Sandelin A, Mendoza L, Engström P, Jareborg N, Wasserman WW. Identification of conserved regulatory elements by comparative genome analysis. J. Biol. 2003;2:13. doi: 10.1186/1475-4924-2-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.