

Abstract
There is growing concern that bovine spongiform encephalopathy (BSE) may have passed from cattle to humans. We report here that transgenic (Tg) mice expressing bovine (Bo) prion protein (PrP) serially propagate BSE prions and that there is no species barrier for transmission from cattle to Tg(BoPrP) mice. These same mice were also highly susceptible to a new variant of Creutzfeldt–Jakob disease (nvCJD) and natural sheep scrapie. The incubation times (≈250 days), neuropathology, and disease-causing PrP isoforms in Tg(BoPrP)Prnp0/0 mice inoculated with nvCJD and BSE brain extracts were indistinguishable and differed dramatically from those seen in these mice injected with natural scrapie prions. Our findings provide the most compelling evidence to date that prions from cattle with BSE have infected humans and caused fatal neurodegeneration.
There is growing concern that bovine spongiform encephalopathy (BSE) may have passed from cattle to humans, resulting in ≈50 cases of an atypical, new variant of Creutzfeldt–Jakob disease (nvCJD) in teenagers and young adults (1–4). Epidemiological findings (2, 4), gel electrophoresis of the prion protein (PrP) (5), and transmissions to inbred mice (6, 7) and primates (8) have each raised the possibility of a link between BSE and nvCJD.
More than 175,000 cattle, primarily dairy cows, have died of BSE over the past decade (9). How many more cattle were exposed to BSE prions but slaughtered before developing clinical signs is uncertain (10). Given the enormity of the affected cattle population in the United Kingdom, a means of assessing risk to the human population is paramount (9), and more sensitive methods for detection of prions are urgently needed (11). The magnitude of the potential risk to the human population is still speculative, but the death rate from nvCJD per year had remained approximately constant until recently, when a disquietingly high number of deaths from the disease, a total of nine new cases, was reported in the last quarter of 1998 (4). Although it is not yet known whether this trend will continue, the possibility that a large section of the population is at high risk must be seriously entertained.
In assessing this potential risk, it is generally assumed that humans would benefit from some degree of protection from exposure to BSE prions because of the “species barrier.” This term refers to the relative lack of susceptibility of one species to prions derived from another species (12). Transgenic mice expressing PrP transgenes have conclusively shown that the PrP gene is the primary determinant controlling susceptibility to foreign prions (13–18). Knockout of the endogenous mouse PrP gene (19) has facilitated these studies in some instances (20). Transgenic mice expressing human (Hu) PrP with the V129 polymorphism designated Tg(HuPrP)152/Prnp0/0 mice (21) have been reported to be susceptible to human nvCJD prions, with incubation periods of ≈220 days and 25/56 (n/n0) animals developing disease (7). The Tg(HuPrP)152/Prnp0/0 mice were not susceptible to BSE: 10/26 mice reportedly showed signs of neurologic disease ≈600 days after inoculation but no disease-causing isoform of PrP (PrPSc) was found. In contrast, Prnp0/0 mice expressing bovine PrP transgenes (BoPrP) were highly susceptible to BSE prions; 100% of the mice became ill at ≈250 days after inoculation with BSE prions (13). The Tg(BoPrP)Prnp0/0 mice faithfully reproduced the hallmarks of BSE in cattle: neuropathology and accumulation of PrPSc were confined largely to the brainstem (13, 22, 23). Furthermore, PrP fragments identified after limited proteolytic digestion of PrPSc in the brains of Tg(BoPrP) mice were indistinguishable from those found in brains of cattle afflicted with BSE (13).
Materials and Methods
Sources of BSE Inocula and Laboratory Animals.
Mice used in this study and primary transmissions of the PG31/90 and GJ248/85 BSE isolates to Tg(BoPrP)4125/Prnp0/0 and FVB mice were previously described (13). Other BSE isolates were obtained from John Wilesmith at Central Veterinary Laboratory Weybridge. Samples from nvCJD patients were obtained through the U.K. National CJD Surveillance Unit. Sheep scrapie isolates (24) have been described elsewhere except the Oreo and T97–1437 isolates, which were a generous gift from Pat Blanchard (Univ. of California, Davis).
Preparation of Brain Homogenates and Determination of BSE, nvCJD, and Scrapie Incubation Periods.
Brain homogenates [10% (wt/vol) in sterile PBS without Ca or Mg] were prepared by using a sterile disposable tissue grinder (Sage Products, Crystal Lake, IL). Inoculation of 30 μl of a 10% homogenate was carried out with a 27-gauge disposable hypodermic needle inserted into the right parietal lobe. After inoculation, the status of the mice was monitored daily, while the neurologic status was assessed semiweekly as previously described (16, 25). Mice were scored positive for prion disease when two or three signs of neurologic dysfunction were present and progressive deterioration of the animals was apparent by using 16 diagnostic criteria previously described (14, 16, 25).
Western Blotting.
Undigested control samples contained approximately 5 μg of total protein. Samples that were digested with proteinase K contained the equivalent of 25 μg of undigested total protein and were diluted in PBS containing 2% sodium sarcosinate immediately before digestion. Proteinase K (50 μg/ml; Boehringer Mannheim) was added and samples were incubated at 37°C for 1 h. The reaction was terminated by addition of Pefabloc (Boehringer Mannheim) to 1 mM. Undigested control aliquots were added immediately to sample buffer. An equal volume of 2× SDS sample buffer containing 125 mM Tris⋅HCl at pH 7.4, 4% SDS, and 20% (vol/vol) glycerol was added to all samples and each was boiled for 5 min before loading on an SDS/11% polyacrylamide gel. The anti-PrP monoclonal antiserum 6H4 (Prionics, Zurich, Switzerland) was used at a 1:2000 dilution for Western immunoblotting. Procedures for immunoblotting and development using the Amersham enhanced chemiluminescence (ECL) system have been described (16).
Neuropathology, PrP Immunohistochemistry, and Histoblot Analysis.
Brain tissue was immersion fixed in 10% buffered formalin solution after the animals had been sacrificed. The brains were embedded in paraffin, and histological sections were prepared and stained with hematoxylin and eosin (H&E) for evaluation of spongiform degeneration. Vacuolation scores were determined by a single rater (S.J.D.). The vacuolation score is a semiquantitative estimate of the area of a brain region occupied by vacuoles (26). PrP immunohistochemistry of formalin-fixed, paraffin-embedded tissue sections was performed by using the hydrolytic autoclaving technique (27). Histoblot analysis was performed as previously described (28). Immunostaining was performed with the PrP-specific rabbit 9095 antiserum (13).
Results
BSE Prions Are Not Altered by Serial Transmission in Tg(BoPrP) Mice.
Although nontransgenic mice seem moderately susceptible to BSE prions, a considerable change in incubation period from first to second passage is always observed (6, 8). This change is indicative of a species barrier (12) and is clear evidence that the properties, and especially the pathogenicity of the prion, have changed during passage. To demonstrate that expression of BoPrP abolishes the species barrier for passage of BSE from cattle to mice, we have inoculated Tg(BoPrP)4125/Prnp0/0 mice with brain homogenate from Tg(BoPrP)4125/Prnp0/0 animals that became sick after inoculation with BSE prions. As anticipated, virtually identical incubation periods of ≈250 days in Tg(BoPrP) mice were obtained upon serial transmission of BSE (Table 1). A control brain, containing BSE prions that had been transmitted directly to normal Prnpa (FVB) mice, failed to produce disease after >540 days (Table 1). Together, these results show that the properties of BSE prions passaged in Prnpa mice, the type most frequently used (6, 8), can vary widely, depending on the passage history.
Table 1.
Susceptibility of transgenic mice to BSE, nvCJD, and sheep scrapie prions
| Original source | Inoculum | Incubation time, days (n/n0) |
|---|---|---|
| BSE | PG31/90 | 234 ± 8.3 (10/10) |
| BSE | PG31/90 → Tg(BoPrP)E4125 | 217 ± 6.1 (15/15) |
| BSE | GJ248/85 | 265 ± 22.6 (8/8) |
| BSE | GJ248/85 → Tg(BoPrP)E4125 | 257 ± 6.6 (15/15) |
| BSE | SE1809/11 | 254 ± 6.7 (9/9) |
| BSE | 97/1612 | 220 ± 7.8 (10/10) |
| BSE | 97/1997 | 227 ± 5.4 (5/6) |
| nvCJD | RU96/02 | 271 ± 6.1 (5/6) |
| nvCJD | RU96/07 | 274 ± 5.8 (10/10) |
| nvCJD | RU96/110 | 247 ± 4.3 (8/8) |
| Suffolk sheep scrapie | Ewe 15 | 234 ± 55.4 (3/7) |
| Suffolk sheep scrapie | Ram 139 | 215 ± 7.3 (8/8) |
| Suffolk sheep scrapie | Ewe 340 | 206 ± 5.5 (8/8) |
| Suffolk sheep scrapie | Oreo | 203 ± 5.4 (6/10) |
| Suffolk sheep scrapie | T97-1437 | 245 ± 15.9 (4/5) |
| BSE | PG31/90 → FVB | >540 (0/10) |
Incubation times are presented as mean ± SEM for Tg(BoPrP)4125/Prnp0/0 mice. The numbers in parentheses are the ratio of animals scored sick expressed as a fraction of the total animals.
The fact that the incubation periods of two separate BSE isolates were unchanged by serial passage in Tg(BoPrP) mice stands in marked contrast to corresponding studies using nontransgenic mice (6, 8, 29), where a change in incubation period from >300 days to ≈150 days was typically observed from the first to second passage. It is clear from a comparison of the data that expression of BoPrP in mice eliminates the species barrier for transmission of BSE.
Efficient Transmission of Variant CJD from Humans to Tg(BoPrP) Mice.
Prion strain “typing” experiments have been presented as evidence in favor of the hypothesis that nvCJD is caused by the transmission of BSE to humans (5–7). To test this hypothesis, we inoculated Tg(BoPrP) mice with nvCJD prions. We reasoned that the reintroduction of BSE from humans, i.e., nvCJD, into transgenic mice expressing BoPrP might restore the original strain properties of the BSE prions. If this transmission were negotiated successfully, the newly formed nvCJD prions, which would be composed of BoPrPSc, would be indistinguishable from native BSE prions.
Unexpectedly, we found that nvCJD prions readily transmitted to Tg(BoPrP) mice. Each of three separate cases of nvCJD produced an incubation period of 250–270 days on first passage (Table 1), which was comparable to that obtained with both BSE prions from cattle tissue and serially passaged BSE prions obtained from Tg(BoPrP) mice (Table 1). We were initially surprised that the incubation period obtained was so short because the transmission was between species encoding heterologous PrP. It is important to note that almost 100% of animals inoculated developed disease (Table 1) and that the inoculated animals exhibited similar incubation periods with a SEM similar to that observed with BSE or serially passaged BSE prions (Table 1). Furthermore, clinical signs observed in affected animals were indistinguishable from those in Tg(BoPrP) mice inoculated with serially passaged BSE. These data demonstrate that Tg(BoPrP) mice are susceptible to human nvCJD prions.
Susceptibility of Tg(BoPrP) Mice to Natural Scrapie of Suffolk Sheep.
It is widely believed that a change in the processing of offal implemented in the late 1970s may have allowed scrapie prions from sheep to survive rendering and to be passed into cattle (30, 31). Because studies of natural scrapie in the United States have shown that ≈85% of the afflicted sheep are of the Suffolk breed (32, 33), we tested Tg(BoPrP)Prnp0/0 mice for susceptibility to natural scrapie arising in the United States in Suffolk sheep. We found that 5/5 cases of natural Suffolk sheep scrapie, 3 from a herd in Illinois and 2 identified from California, transmitted disease to Tg(BoPrP) mice (Table 1). Each of the five isolates transmitted with a relatively short incubation period of ≈210 days, and in two cases 100% of the inoculated animals developed disease. Transmission efficiency was high (>60%) in two of the other cases, and only one case (ewe no. 15) transmitted relatively inefficiently (Table 1). Susceptibility in Suffolk sheep is primarily governed by the PrP codon 171 polymorphism; only sheep homozygous for Gln (Q) at codon 171 are susceptible to scrapie (24, 34, 35). BoPrP contains Q at the position equivalent to residue 171 in sheep PrP. Because all of the scrapie sheep inocula were also homozygous Q/Q at codon 171, the efficient transmission of sheep scrapie to Tg(BoPrP) mice is consistent with the disposition of this polymorphic residue implicated in controlling susceptibility to sheep prions.
Neuropathology of nvCJD in Tg(BoPrP) Mice Is Indistinguishable from That of Serially Passaged BSE but Distinct from That of Scrapie.
We previously reported that the neuropathological changes of Tg(BoPrP) mice infected with BSE (13) were strikingly similar to those previously reported in cattle affected with BSE (36). After a second transmission to Tg(BoPrP) mice in the present study, the neuroanatomic distribution of vacuolation remained essentially the same; that is, it was confined largely to the brainstem tegmentum and periaqueductal gray (Fig. 1). Although description of the habenula was omitted from the earlier study (13), significant vacuolation occurred in that structure in both the present and previous studies. One neuropathological difference was the presence of PrP-immunopositive amyloid plaques, primarily in the subcallosal region, in the present study and their absence in the first passage (Fig. 2C). The plaques fulfilled the criteria for amyloid, including binding to Congo red dye, which displayed apple-green birefringence in polarized light (data not shown). The first passage of nvCJD in Tg(BoPrP)Prnp0/0 mice resulted in a neuropathological phenotype that was essentially identical to that caused by BSE prions. The most intense vacuolation with both prion inocula occurred in the tegmentum of the pons and midbrain (Fig. 1, BS bar), the periaqueductal gray of the midbrain (Fig. 1, PG), and the habenula (Fig. 1, Hb). Congophilic PrP-immunopositive amyloid plaques were also found in the subcallosal region (Fig. 2E). Very little or no vacuolation was identified in other brain regions, including the neocortex, hippocampus, hypothalamus, and caudate.
Figure 1.

Vacuolation histograms in Tg(BoPrP)Prnp0/0 mice inoculated with BSE, nvCJD, and scrapie prions. The BSE and nvCJD histograms are very similar and both are different from scrapie (Sc). Mean and standard deviations are shown. NC, neocortex; Hp, hippocampus; Th, thalamus; Hb, habenula; Hy, hypothalamus; Cd, caudate nucleus; BF, basal forebrain; BS, pontine and midbrain tegmentum; PG, periaqueductal gray. N = the number of animals analyzed.
Figure 2.
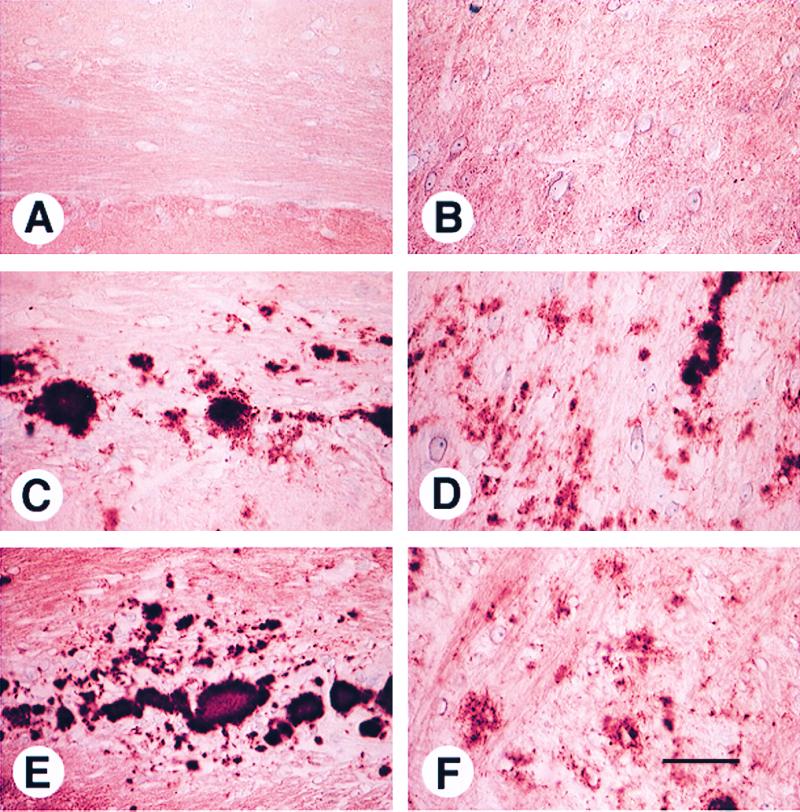
Amyloid plaques in Tg(BoPrP)Prnp0/0 mice inoculated with BSE and nvCJD prions. Subcallosal PrP-immunopositive amyloid plaques (C and E) and coarse nonamyloid PrP deposits (D and F) are characteristic of BSE (C and D) and nvCJD (E and F) and not of scrapie (A and B). Staining was performed as described in Materials and Methods. (Bar in F = 25 μm.)
We were concerned that the highly characteristic and essentially identical pattern of neuropathology found in Tg(BoPrP) mice infected with BSE and nvCJD could be attributable to an unusual property of the host Tg(BoPrP) mice. To test this remote possibility, we also evaluated the neuropathological lesions of Tg(BoPrP) mice that developed neurologic dysfunction after inoculation with sheep scrapie. In these mice, the pattern of neuropathology differed markedly; inocula derived from sheep scrapie resulted in a mild degree of vacuolation in virtually all brain regions and no amyloid plaques (Fig. 2A). The characteristics of nonamyloid PrP deposits in Tg(BoPrP) mice inoculated with sheep scrapie also differed from those of mice inoculated with BSE and nvCJD. Specifically, the nonamyloid PrP deposits with the latter were of the coarse type (Fig. 2 D and F), whereas they were of the finely granular, “synaptic” type with scrapie (Fig. 2B).
Previous histoblotting studies revealed that Tg(BoPrP) mice infected with BSE accumulate PrPSc predominantly in the brainstem (13). When the neuroanatomic distribution of PrPSc deposition was determined by histoblot analysis (Fig. 3), the distribution of PrPSc was virtually identical in Tg(BoPrP) mice inoculated with either serially passaged BSE or nvCJD prions and was dramatically different after inoculation with scrapie prions. The vacuolation profiles with each prion inoculum correlated well with the PrPSc distribution profiles. Common to all three inocula was a uniformly moderate degree of vacuolation in the brainstem tegmentum and periaqueductal gray that corresponded to intense immunostaining for PrPSc in the same regions (compare Figs. 3 B, D, and F). A moderate degree of vacuolation found in the habenula in Tg(BoPrP) mice inoculated with BSE and nvCJD also correlated with intense PrPSc immunostaining (Fig. 3 C and E), whereas there was very modest vacuolation in the habenula of mice inoculated with scrapie prions, a result consistent with the lower intensity of PrPSc staining observed (Fig. 3A). There was a generalized mild vacuolation of many brain regions in mice inoculated with scrapie that correlated with weak, diffusely homogenous PrPSc immunostaining. In contrast, PrPSc immunostaining in mice inoculated with BSE or nvCJD tended to be punctate in many regions, in agreement with the markedly variable and inconsistent pattern of vacuolation that we found by histopathological analysis. Together, the foregoing results establish that nvCJD and BSE produce an identical disease in Tg(BoPrP) mice, and that nvCJD prions appear to have retained the potential to regenerate authentic BSE prions upon retransmission to animals expressing BoPrP.
Figure 3.

Regional distribution of PrPSc in the brains of Tg(BoPrP) mice inoculated with BSE prions. The neuroanatomic distribution of protease-resistant PrPSc was virtually identical in Tg(BoPrP)Prnp0/0 mice inoculated with BSE (C and D) and nvCJD (E and F) prions but different from that found in mice inoculated with sheep scrapie (Sc) prions (A and B). The distribution of PrPSc correlates well with the vacuolation scores in Fig. 1. A, C, and E are coronal sections of the cerebral hemispheres at the level of the thalamus and hippocampus. B, D, and F are coronal sections through the pons and cerebellum. Am, amygdala; GC, granule cell layer of cerebellar cortex; Hb, habenula; Hp, hippocampus; Hy, hypothalamus; IC, internal capsule; NC, neocortex; N7, facial nerve; Py, pyramidal tract; S5, spinal tract of trigeminal nerve; Th, thalamus; ZI, zona incerta.
Convergent Molecular Properties of nvCJD and BSE Prions in Tg(BoPrP) Mice.
Assessment of the size of the deglycosylated, protease-resistant fragment has been used to argue that strain-specific properties of prions are enciphered in the conformation of PrPSc (37, 38). The relative amounts of di- and monoglycosylated PrPSc have been used to characterize prion strains (5, 39) but the efficacy of this approach is questionable (40, 41). Both the size of the unglycosylated PrPSc after limited proteolysis and the relative amount of diglycosylated PrPSc were similar in nvCJD and BSE samples before and after passage to Tg(BoPrP)Prnp0/0 mice. Predominance of diglycosylated PrPSc and the apparent size of the PrP fragments were indistinguishable when bovine PrPSc present in bovine brain (Fig. 4, lane 2) was compared with first- (Fig. 4, lanes 3 and 4) or second-passage BSE (Fig. 4, lanes 5 and 6) or first-passage nvCJD (Fig. 4, lanes 7 and 8) in Tg(BoPrP)Prnp0/0 mice. The amount of diglycosylated PrPSc and size of the PrP fragments were also identical to those found in human nvCJD (Fig. 4, lane 10). In all cases, the size of the unglycosylated PrPSc was ≈19 kDa, strikingly smaller than the ≈21-kDa fragment found in human sCJD (Fig. 4, lane 9). The amount of diglycosylated PrPSc was substantially less in sCJD compared with BSE and nvCJD, as previously reported (5). From these data, it seems clear that a single passage of nvCJD prions from humans to Tg(BoPrP) mice was sufficient to produce a disease that is in all respects indistinguishable from that produced by BSE prions, and the conservation of the 19-kDa protease-resistant fragment size upon passage of nvCJD to Tg(BoPrP) mice argues that the conformation of the prion may be maintained during propagation from the HuPrP to BoPrP background (37, 38).
Figure 4.

Characterization of PrPSc fragments by immunoblot. Western blots of PrP in brains from Tg mice inoculated with BSE, nvCJD, and sporadic CJD (sCJD) prions. Lane 1, undigested PG31/90 BSE brain. Lane 2, PG31/90 BSE brain control. Lanes 3 and 4, Tg(BoPrP)4125/Prnp0/0 mice clinically ill after inoculation with BSE inocula PG31/90 and GJ248/85, respectively. Lanes 5 and 6, Tg(BoPrP)4125/Prnp0/0 mice clinically ill after inoculation with PG31/90 and GJ248/85, respectively, after a single serial passage in Tg(BoPrP)4125/Prnp0/0 mice. Lanes 7 and 8, Tg(BoPrP)4125/Prnp0/0 mice clinically ill after inoculation with prions from nvCJD patient RU96/02. Lane 9, sCJD, patient RG. Lane 10, nvCJD patient RU96/02. A is an exposure selected to clearly show differences in the PrP glycoform ratios. B is a longer exposure of lanes 7–10, selected to make the lower molecular weight unglycosylated fragment more evident.
It is notable that our findings presented here contrast with the results of many studies demonstrating that prion strain properties are frequently altered during passage between hosts encoding PrP of different sequence (42, 43). It is not yet clear whether the apparent stability of the nvCJD prion strain upon transmission from humans to Tg(BoPrP) mice reflects an intrinsic susceptibility of animals expressing BoPrP to foreign prions or whether this is a property of this particular prion strain.
Discussion
BSE and nvCJD Prions in Tg(BoPrP)Prnp0/0 Mice.
The demonstration that there is no significant difference in incubation period when Tg(BoPrP) mice are inoculated with either bovine brain or Tg(BoPrP) mouse-passaged BSE prions confirms that these animals seamlessly and precisely transmit BSE prions, with no detectable alteration of strain or species-specific properties attributable to the host animal. This result supports our assertion that Tg(BoPrP) mice represent the best rodent model available for detecting BSE prions (13). In contrast, studies with nontransgenic mice demonstrate differences in incubation period between first and second passage and between strains of inbred mice (6, 29, 44). Whereas it is apparent from the extreme change in incubation period between first and second passage that mouse-passaged BSE is significantly more pathogenic in inbred mice than BSE prions of bovine origin, the same critique cannot be made of BSE prions passaged in Tg(BoPrP); these retain their original pathogenicity. By obviating the species barrier, expression of BoPrP allows detection of BSE prions from cattle with maximal possible efficiency. In the future, it may prove possible to create transgenic mice that express modified BoPrP, eliminate the species barrier, and provide even shorter incubation periods. However, until such mice are available, Tg(BoPrP) mice represent the best rodent model for BSE.
The extreme susceptibility of Tg(BoPrP)Prnp0/0 mice to BSE is not unexpected, but their susceptibility to both sheep scrapie and human nvCJD prions was surprising (Table 1). Paradoxically, nvCJD prions have been found to transmit relatively inefficiently to Tg(HuPrP) mice (7) and also display protracted incubation periods (≈500 days) in Tg(MHu2M)Prnp0/0 mice (J. A. Mastrianni, G. Telling, P. Parchi, M. Torchia, P.T., P. Bosque, D. Groth, J.I., R. Gabizon, P. Gambetti, et al., unpublished results). While the incubation period for sCJD prions in Tg(MHu2M)Prnp0/0 mice remained ≈200 days on serial passage (17), the incubation time for nvCJD prions decreased from more than 560 days to ≈240 days on second passage (J. A. Mastrianni, et al., unpublished results). In contrast to BoPrP, which can readily adopt the appropriate PrPSc conformation representative of the BSE/nvCJD strain and thereby allow efficient transmission, neither chimeric MHu2M PrP nor HuPrP is so readily transformed into PrPSc. Determining the molecular basis underlying these findings may prove important in advancing our understanding of prion replication and development of improved Tg mice for bioassay of human prions (17).
Exploring the Origins of BSE and nvCJD.
We have demonstrated that nvCJD prions assume an identity indistinguishable from that of BSE prions after a single passage in Tg(BoPrP) mice. Incubation periods obtained after inoculation of human nvCJD brain tissues into Tg(BoPrP) mice were remarkably similar to those obtained after inoculation with BSE prions obtained from either bovine brain or Tg(BoPrP) mice, and clinical signs of disease were indistinguishable. Neuropathological analysis revealed the same distribution of spongiform degeneration, and both the appearance and localization of amyloid plaques and the regional distribution of PrPSc were identical when Tg(BoPrP) mice were inoculated with nvCJD and serially passaged BSE. In addition, unglycosylated, protease-resistant PrPSc remained 19 kDa through multiple passages of BSE prions. Moreover, the predominance of the diglycosylated fragments also remained unchanged on passage of either BSE or nvCJD prions. Thus, by all available criteria, nvCJD and BSE are the same when passaged in Tg(BoPrP) mice.
That human nvCJD prions so precisely duplicate the properties of native bovine BSE prions in their behavior on transmission to Tg(BoPrP)Prnp0/0 mice creates a compelling argument for an etiologic link between BSE and nvCJD. Although earlier proposals of an etiological link between BSE and nvCJD were disquieting (2, 4–8), the investigations reported here raise greater concern that a large section of the United Kingdom population may be at considerable risk.
Whereas it now seems clear that nvCJD arose through exposure of humans to BSE, the origins of BSE remain obscure. One possibility is that scrapie prions from sheep may have survived rendering and passed to cattle via a dietary supplement (30, 31). An experimental isolate of scrapie of sheep and goats was fed to cattle but produced illness substantially different from BSE (45). More recently, when scrapie from Suffolk sheep was passaged to cattle, it was found to produce a disease that was distinct from BSE and whose characteristics remained essentially constant after a serial transmission of the scrapie-infected bovine brain (46). Despite the foregoing, there is still reason to suspect that BSE may have originated in sheep. Western blots of PrPSc reveal a striking similarity between the glycoforms in Cheviot sheep infected with BSE and an experimental scrapie isolate (CH1641) (47). The high degree of susceptibility to sheep scrapie exhibited by Tg(BoPrP)Prnp0/0 mice provides an efficient animal model for screening of natural scrapie isolates for the potential to produce BSE in cattle and, in this regard, it will be of considerable interest to determine whether CH1641 as well as other Cheviot sheep scrapie isolates produce a disease similar to that caused by BSE and nvCJD in Tg(BoPrP) mice. Alternatively, selection of particular PrPSc conformers during rendering may have been responsible for the emergence of the BSE strain (48), a hypothesis that can now be tested by controlled exposure of sheep scrapie prions to denaturants and subsequent transmission to Tg(BoPrP)Prnp0/0 mice.
Acknowledgments
We thank Drs. Gerald Wells, John Wilesmith, and Ray Bradley for the BSE brain tissue; Vincenza Itri and Dr. Jiri Safar for BSE brain extracts; Dr. David Westaway and Dr. Pat Blanchard for natural sheep scrapie samples; and Juliana Cayetano, Ignatius Arellano, and Thomas Lisse for excellent technical assistance. This work was supported by grants from the National Institutes of Health (NS14069, AG08967, AG02132, and NS22786) and the American Health Assistance Foundation, as well as by gifts from the G. Harold and Leila Y. Mathers Foundation, the Sherman Fairchild Foundation, the Bernard Osher Foundation, and Centeon, Inc.
Abbreviations
- BSE
bovine spongiform encephalopathy
- nvCJD
new variant Creutzfeldt–Jacob disease
- sCJD
sporadic CJD
- PrP
prion protein
- Tg
transgenic
- BoPrP
bovine prion protein
- PrPSc
disease-causing isoform of PrP
- HuPrP
human PrP
References
- 1.Chazot G, Broussolle E, Lapras C I, Blättler T, Aguzzi A, Kopp N. Lancet. 1996;347:1181. doi: 10.1016/s0140-6736(96)90638-8. [DOI] [PubMed] [Google Scholar]
- 2.Will R G, Ironside J W, Zeidler M, Cousens S N, Estibeiro K, Alperovitch A, Poser S, Pocchiari M, Hofman A, Smith P G. Lancet. 1996;347:921–925. doi: 10.1016/s0140-6736(96)91412-9. [DOI] [PubMed] [Google Scholar]
- 3.Cousens S N, Vynnycky E, Zeidler M, Will R G, Smith P G. Nature (London) 1997;385:197–198. doi: 10.1038/385197a0. [DOI] [PubMed] [Google Scholar]
- 4.Will R G, Cousens S N, Farrington C P, Smith P G, Knight R S G, Ironside J W. Lancet. 1999;353:979. doi: 10.1016/s0140-6736(99)01160-5. [DOI] [PubMed] [Google Scholar]
- 5.Collinge J, Sidle K C L, Meads J, Ironside J, Hill A F. Nature (London) 1996;383:685–690. doi: 10.1038/383685a0. [DOI] [PubMed] [Google Scholar]
- 6.Bruce M E, Will R G, Ironside J W, McConnell I, Drummond D, Suttie A, McCardle L, Chree A, Hope J, Birkett C, et al. Nature (London) 1997;389:498–501. doi: 10.1038/39057. [DOI] [PubMed] [Google Scholar]
- 7.Hill A F, Desbruslais M, Joiner S, Sidle K C L, Gowland I, Collinge J, Doey L J, Lantos P. Nature (London) 1997;389:448–450. doi: 10.1038/38925. [DOI] [PubMed] [Google Scholar]
- 8.Lasmézas C I, Deslys J-P, Demaimay R, Adjou K T, Lamoury F, Dormont D, Robain O, Ironside J, Hauw J-J. Nature (London) 1996;381:743–744. doi: 10.1038/381743a0. [DOI] [PubMed] [Google Scholar]
- 9.Anderson R M, Donnelly C A, Ferguson N M, Woolhouse M E J, Watt C J, Udy H J, MaWhinney S, Dunstan S P, Southwood T R E, Wilesmith J W, et al. Nature (London) 1996;382:779–788. doi: 10.1038/382779a0. [DOI] [PubMed] [Google Scholar]
- 10.Stekel D J, Nowak M A, Southwood T R E. Nature (London) 1996;381:119. doi: 10.1038/381119a0. [DOI] [PubMed] [Google Scholar]
- 11.Safar J, Wille H, Itri V, Groth D, Serban H, Torchia M, Cohen F E, Prusiner S B. Nat Med. 1998;4:1157–1165. doi: 10.1038/2654. [DOI] [PubMed] [Google Scholar]
- 12.Pattison I H. In: Slow, Latent and Temperate Virus Infections, NINDB Monograph 2. Gajdusek D C, Gibbs C J Jr, Alpers M P, editors. Washington, D.C.: U.S. Government Printing Office; 1965. pp. 249–257. [Google Scholar]
- 13.Scott M R, Safar J, Telling G, Nguyen O, Groth D, Torchia M, Koehler R, Tremblay P, Walther D, Cohen F E, et al. Proc Natl Acad Sci USA. 1997;94:14279–14284. doi: 10.1073/pnas.94.26.14279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Scott M, Foster D, Mirenda C, Serban D, Coufal F, Wälchli M, Torchia M, Groth D, Carlson G, DeArmond S J, et al. Cell. 1989;59:847–857. doi: 10.1016/0092-8674(89)90608-9. [DOI] [PubMed] [Google Scholar]
- 15.Prusiner S B, Scott M, Foster D, Pan K-M, Groth D, Mirenda C, Torchia M, Yang S-L, Serban D, Carlson G A, et al. Cell. 1990;63:673–686. doi: 10.1016/0092-8674(90)90134-z. [DOI] [PubMed] [Google Scholar]
- 16.Scott M, Groth D, Foster D, Torchia M, Yang S-L, DeArmond S J, Prusiner S B. Cell. 1993;73:979–988. doi: 10.1016/0092-8674(93)90275-u. [DOI] [PubMed] [Google Scholar]
- 17.Telling G C, Scott M, Mastrianni J, Gabizon R, Torchia M, Cohen F E, DeArmond S J, Prusiner S B. Cell. 1995;83:79–90. doi: 10.1016/0092-8674(95)90236-8. [DOI] [PubMed] [Google Scholar]
- 18.Race R E, Priola S A, Bessen R A, Ernst D, Dockter J, Rall G F, Mucke L, Chesebro B, Oldstone M B A. Neuron. 1995;15:1183–1191. doi: 10.1016/0896-6273(95)90105-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Büeler H, Fischer M, Lang Y, Bluethmann H, Lipp H-P, DeArmond S J, Prusiner S B, Aguet M, Weissmann C. Nature (London) 1992;356:577–582. doi: 10.1038/356577a0. [DOI] [PubMed] [Google Scholar]
- 20.Prusiner S B, Groth D, Serban A, Koehler R, Foster D, Torchia M, Burton D, Yang S-L, DeArmond S J. Proc Natl Acad Sci USA. 1993;90:10608–10612. doi: 10.1073/pnas.90.22.10608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Telling G C, Scott M, Hsiao K K, Foster D, Yang S-L, Torchia M, Sidle K C L, Collinge J, DeArmond S J, Prusiner S B. Proc Natl Acad Sci USA. 1994;91:9936–9940. doi: 10.1073/pnas.91.21.9936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wells G A H, Scott A C, Johnson C T, Gunning R F, Hancock R D, Jeffrey M, Dawson M, Bradley R. Vet Rec. 1987;121:419–420. doi: 10.1136/vr.121.18.419. [DOI] [PubMed] [Google Scholar]
- 23.Prusiner S B, Fuzi M, Scott M, Serban D, Serban H, Taraboulos A, Gabriel J-M, Wells G, Wilesmith J, Bradley R, et al. J Infect Dis. 1993;167:602–613. doi: 10.1093/infdis/167.3.602. [DOI] [PubMed] [Google Scholar]
- 24.Westaway D, Zuliani V, Cooper C M, Da Costa M, Neuman S, Jenny A L, Detwiler L, Prusiner S B. Genes Dev. 1994;8:959–969. doi: 10.1101/gad.8.8.959. [DOI] [PubMed] [Google Scholar]
- 25.Carlson G A, Goodman P A, Lovett M, Taylor B A, Marshall S T, Peterson-Torchia M, Westaway D, Prusiner S B. Mol Cell Biol. 1988;8:5528–5540. doi: 10.1128/mcb.8.12.5528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Carlson G A, Ebeling C, Yang S-L, Telling G, Torchia M, Groth D, Westaway D, DeArmond S J, Prusiner S B. Proc Natl Acad Sci USA. 1994;91:5690–5694. doi: 10.1073/pnas.91.12.5690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Muramoto T, Kitamoto T, Tateishi J, Goto I. Am J Pathol. 1992;140:1411–1420. [PMC free article] [PubMed] [Google Scholar]
- 28.Taraboulos A, Jendroska K, Serban D, Yang S-L, DeArmond S J, Prusiner S B. Proc Natl Acad Sci USA. 1992;89:7620–7624. doi: 10.1073/pnas.89.16.7620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lasmézas C I, Deslys J-P, Robain O, Jaegly A, Beringue V, Peyrin J-M, Fournier J-G, Hauw J-J, Rossier J, Dormont D. Science. 1997;275:402–405. doi: 10.1126/science.275.5298.402. [DOI] [PubMed] [Google Scholar]
- 30.Wilesmith J W. Semin Virol. 1991;2:239–245. [Google Scholar]
- 31.Kimberlin R H. In: Bovine Spongiform Encephalopathy: The BSE Dilemma. Gibbs C J Jr, editor. New York: Springer; 1996. pp. 155–175. [Google Scholar]
- 32.Hourrigan J, Klingsporn A, Clark W W, de Camp M. In: Slow Transmissible Diseases of the Nervous System. Prusiner S B, Hadlow W J, editors. Vol. 1. New York: Academic; 1979. pp. 331–356. [Google Scholar]
- 33.Westaway D, Neuman S, Zuliani V, Mirenda C, Foster D, Detwiler L, Carlson G, Prusiner S B. In: Prion Diseases of Humans and Animals. Prusiner S B, Collinge J, Powell J, Anderton B, editors. London: Ellis Horwood; 1992. pp. 474–482. [Google Scholar]
- 34.Hunter N, Moore L, Hosie B D, Dingwall W S, Greig A. Vet Rec. 1997;140:59–63. doi: 10.1136/vr.140.3.59. [DOI] [PubMed] [Google Scholar]
- 35.Hunter N, Cairns D, Foster J D, Smith G, Goldmann W, Donnelly K. Nature (London) 1997;386:137. doi: 10.1038/386137a0. [DOI] [PubMed] [Google Scholar]
- 36.Wells G A H, Wilesmith J W. Brain Pathol. 1995;5:91–103. doi: 10.1111/j.1750-3639.1995.tb00580.x. [DOI] [PubMed] [Google Scholar]
- 37.Telling G C, Parchi P, DeArmond S J, Cortelli P, Montagna P, Gabizon R, Mastrianni J, Lugaresi E, Gambetti P, Prusiner S B. Science. 1996;274:2079–2082. doi: 10.1126/science.274.5295.2079. [DOI] [PubMed] [Google Scholar]
- 38.Bessen R A, Marsh R F. J Virol. 1994;68:7859–7868. doi: 10.1128/jvi.68.12.7859-7868.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Somerville R A, Chong A, Mulqueen O U, Birkett C R, Wood S C E R, Hope J. Nature (London) 1997;386:564. doi: 10.1038/386564a0. [DOI] [PubMed] [Google Scholar]
- 40.Mastrianni J A, Nixon R, Layzer R, Telling G C, Han D, DeArmond S J, Prusiner S B. N Engl J Med. 1999;340:1630–1638. doi: 10.1056/NEJM199905273402104. [DOI] [PubMed] [Google Scholar]
- 41.Parchi P, Capellari S, Chin S, Schwarz H B, Schecter N P, Butts J D, Hudkins P, Burns D K, Powers J M, Gambetti P. Neurology. 1999;52:1757–1763. doi: 10.1212/wnl.52.9.1757. [DOI] [PubMed] [Google Scholar]
- 42.Scott M R, Groth D, Tatzelt J, Torchia M, Tremblay P, DeArmond S J, Prusiner S B. J Virol. 1997;71:9032–9044. doi: 10.1128/jvi.71.12.9032-9044.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kimberlin R H, Walker C A, Fraser H. J Gen Virol. 1989;70:2017–2025. doi: 10.1099/0022-1317-70-8-2017. [DOI] [PubMed] [Google Scholar]
- 44.Bruce M, Chree A, McConnell I, Foster J, Pearson G, Fraser H. Phil Trans R Soc London B. 1994;343:405–411. doi: 10.1098/rstb.1994.0036. [DOI] [PubMed] [Google Scholar]
- 45.Robinson M M, Hadlow W J, Knowles D P, Huff T P, Lacy P A, Marsh R F, Gorham J R. J Comp Path. 1995;113:241–251. doi: 10.1016/s0021-9975(05)80039-8. [DOI] [PubMed] [Google Scholar]
- 46.Cutlip R C, Miller J M, Lehmkuhl H D. J Comp Pathol. 1997;117:271–275. doi: 10.1016/s0021-9975(97)80022-9. [DOI] [PubMed] [Google Scholar]
- 47.Hope J, Wood S C, Birkett C R, Chong A, Bruce M E, Cairns D, Goldmann W, Hunter N, Bostock C J. J Gen Virol. 1999;80:1–4. doi: 10.1099/0022-1317-80-1-1. [DOI] [PubMed] [Google Scholar]
- 48.Prusiner S B. Science. 1997;278:245–251. doi: 10.1126/science.278.5336.245. [DOI] [PubMed] [Google Scholar]