

Abstract
Lipopolysaccharide (LPS), which constitutes the outermost layer of Gram-negative bacterial cells as a typical component essential for their life, induces the first line defense system of innate immunity of higher animals. To understand the basic mode of interaction between bacterial LPS and phospholipid cell membranes, distribution patterns were studied by various physical methods of deep rough mutant LPS (ReLPS) of Escherichia coli incorporated in phospholipid bilayers as simple models of cell membranes. Solid-state 31P-NMR spectroscopic analysis suggested that a substantial part of ReLPS is incorporated into 1,2-dimyristoyl-sn-glycero-3-phosphocholine lipid bilayers when multilamellar vesicles were prepared from mixtures of these. In egg L-α-phosphatidylcholine (egg-PC)-rich membranes, ReLPS undergoes micellization. In phosphatidylethanolamine-rich membranes, however, micellization was not observed. We studied by microscopic techniques the location of ReLPS in membranes of ReLPS/egg-PC (1:10 M/M) and ReLPS/egg-PC/1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoglycerol (POPG) (1:9:1 M/M/M). The influence of ReLPS on the physicochemical properties of the membranes was studied as well. Microscopic images of both giant unilamellar vesicles and supported planar lipid bilayers showed that LPS was uniformly incorporated in the egg-PC lipid bilayers. In the egg-PC/POPG (9:1 M/M) lipid bilayers, however, ReLPS is only partially incorporated and becomes a part of the membrane in a form of aggregates (or as mixed aggregates with the lipids) on the bilayer surface. The lipid lateral diffusion coefficient measurements at various molar ratios of ReLPS/egg-PC/POPG indicated that the incorporated ReLPS reduces the diffusion coefficients of the phospholipids in the membrane. The retardation of diffusion became more significant with increasing POPG concentrations in the membrane at high ReLPS/phospholipid ratios. This work demonstrated that the phospholipid composition has critical influence on the distribution of added ReLPS in the respective lipid membranes and also on the morphology and physicochemical property of the resulting membranes. A putative major factor causing these phenomena is reasoned to be the miscibility between ReLPS and individual phospholipid compositions.
INTRODUCTION
Lipopolysaccharide (LPS) is an amphiphilic molecule composed of covalently-linked lipid A (an N,O-polyacylated disaccharide of glucosamine carrying two negatively charged phosphates), core oligosaccharide, and O-specific antigen polysaccharide (1). LPS is a major constituent of the outer leaflet of the outer membrane of Gram-negative bacteria and is known to be a key molecule in the innate immune recognition of bacteria (2); the lipid A moiety of LPS has proved to be responsible for this biological activity. When immunocompetent cells such as macrophages and dendritic cells are exposed to LPS, the cells are stimulated and release inflammatory cytokines. Cellular activation mechanisms by LPS have been investigated and the current understanding is as follows: LPS initially binds to the LPS binding protein (LBP) (3) and then LPS/LBP associates to glycosylphosphatidylinositol-anchored protein CD-14 (4). After this, LPS is transported to the MD2/Toll-like receptor-4 (TLR-4) complex (5–7), where TLR4 recognizes the LPS/MD2 complex at the leucine-rich repeat region of the extracellular domain and then activates a signal transduction cascade consisting of MyD88, IRAK, TRAF6, and NF-κB inside the cells. Finally, the cells synthesize and release cytokines (such as tumor necrosis factor-α and those of the interleukin family). In addition to this, recent studies have shown the importance of lipid domain formation for the innate immune recognition of LPS (8,9). Even though the innate immune response mechanism is now better understood, the relationship between LPS and the target cell membrane is not as clear. Since phospholipids laterally diffuse in membranes rapidly (10), incorporation of LPS into the target cell membrane may proceed upon its encounter with membrane-bound receptor proteins such as CD14 and MD2/TLR-4.
The role of the target cell membrane during LPS action has already been studied by Gutsman et al. (11–13) and Schromm et al. (14) using several techniques. They have characterized the intercalation of LPS into the phospholipid membrane using electrical measurements (11), surface plasmon resonance (12), and fluorescence resonance energy transfer spectroscopy (13), and also characterized the relationship between biological activity and molecular conformation for several types of LPS by applying small-angle x-ray diffraction (14). Roes et al. also characterized LPS domain formation in LBP-containing liposomes using atomic force microscopy (15). Each of these studies emphasized the importance of LBP on the interaction between LPS and membrane. In this study, we attempted to directly observe the presence of LPS in lipid bilayers by means of solid-state nuclear magnetic resonance (NMR) and microscopic techniques and elucidate the location and mode of distribution of LPS in phospholipid membranes. This simple system allows us to clarify the direct interaction between LPS and membrane. For such analyses, the use of a homogeneous substrate is essential. We used LPS of Escherichia coli deep rough (Re) mutant, ReLPS, which is now commercially available in a sufficiently pure form for this purpose (16,17). As shown in Fig. 1, ReLPS consists of lipid A and a 3-deoxy-D-manno-octulosonic acid (Kdo) disaccharide attached to the 6′-position of the former. Although the tendency of ReLPS molecules to form multimeric aggregates is supposed to be stronger than that of common natural LPS, due to the lack of long sugar chains linked to the lipid A, we used ReLPS in this study because no other natural LPS with longer oligosaccharide chains is available in a sufficient homogeneity to enable precise analyses.
FIGURE 1.

Chemical structures of ReLPS, POPC, and POPG.
In this work, the behavior of ReLPS in various lipid membranes prepared in its presence was first examined by 31P NMR spectra with reference to the relative amounts of ReLPS. The distribution of ReLPS in lipid bilayers of giant unilamellar vesicles (GUVs) was next examined by dark-field and fluorescence microscopy; the former is a powerful tool to obtain high-contrast images of GUV in aqueous solution (18–20), whereas the distribution of ReLPS in the lipid bilayer of supported planar lipid bilayers (SPBs) was visualized in more detail by using the latter microscope. We also checked the kinetics of SPBs formation of small unilamellar vesicles (SUVs) on a SiO2 surface by using a quartz crystal microbalance with dissipation monitoring (QCM-D). The influence of ReLPS incorporated in the membrane on the lateral diffusion of phospholipids in SPBs was observed by measuring the lateral diffusion coefficients by the boundary profile evolution (BPE) method.
ReLPS is a negatively charged molecule having two phosphate groups linked to the disaccharide backbone of lipid A and two carboxyl groups of the Kdo disaccharide (Fig. 1). Though one of us (U.S.) has reported on the importance of the negative charges in LPS for its interaction with its receptor proteins (12,21), there has been no study examining the effect of membrane phospholipids charges on the interaction between LPS and the membrane. Therefore, in this study we have focused on the influence of the charge of phospholipid components in the membrane.
MATERIALS AND METHODS
Materials
The solutions 1,2-dimyristoyl-sn-glycero-3-phosphocholine (DMPC); 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoglycerol (POPG); egg L-α-phosphatidylcholine (egg-PC); 1,2-dielaidoyl-sn-glycero-3-phosphoethanolamine (DEPE); E. coli total lipids extract; bovine brain L-α-phosphatidyl-L-serine (Brain-PS); Bovine-sphingomyelin (Bovine-SM); E. coli L-α-phosphatidylethanolamine (E. coli-PE); and deep rough mutant ReLPS from E. coli strain WBB06 were purchased from Avanti Polar Lipids (Alabaster, AL) and used without further purification. The mouse anti-ReLPS antibody, A20 (immunoglobin M, IgM) (22), was a kind gift from H. Brade (Borstel, Germany). The solutions 3,3′-dihexyloxacarbocyanine iodide (DiO) (excitation/emission: 484 nm/519 nm); Texas Red 1,2-hexadecanoyl-sn-glycero-3-phosphoethanolamine (TR-DHPE) (excitation/emission: 583 nm/601 nm); and the Alexa Fluor-555 labeled goat anti-mouse IgM antibody (excitation/emission: 555 nm/565 nm) were purchased from Molecular Probes (Eugene, OR). Hellmanex solution was purchased from Hellma (Müllheim, Germany). Tris buffer (20 mM Tris-HCl, 100 mM NaCl, pH 7.6) was used for all experiments.
Preparation of phospholipid vesicles and fluorescence labeling
Small unilamellar vesicles (SUVs) used both for QCM-D measurements and for SPB observations were prepared as follows. Appropriate amounts of phospholipids or mixtures of phospholipids and ReLPS were dissolved in chloroform/methanol (8:2 v/v) and the mixture dried first with nitrogen gas stream and then under high vacuum overnight. The lipid film obtained was hydrated with Tris buffer to a final lipid concentration of 1 mM, and the lipid suspension was extensively vortexed and freeze-thawed for 10 cycles. Finally, the lipid dispersions were sonicated in ice water for 3–5 min using a Branson Sonifier 150 (Branson Ultrasonics, Danbury, CT) at 5 W. For QCM-D experiments, SUV suspensions were diluted with Tris buffer to a lipid concentration of 140 μM. For observation of SPBs, DiO in dimethyl sulfoxide was added to 0.01 mM (consequently, the final dimethyl-sulfoxide concentrations are 0.05% (v/v)). For lateral diffusion coefficient determination by the BPE method, TR-DHPE was added to 0.01 mM instead of DiO as a component of lipid mixtures.
Multilamellar vesicles (MLVs) for NMR measurements were prepared as follows. Phospholipids and ReLPS were cosolubilized at a molar ratio of 10:1 in chloroform/methanol (8:2 v/v) and the mixture dried first under a nitrogen gas stream and then under high vacuum overnight. After evaporation of the solvent, the lipid film was hydrated with Tris buffer to a final lipid concentration of 50 mM at 40°C, and extensively vortexed. The suspension was freeze-thawed for 10 cycles and transferred to NMR tubes.
Fluorescence-labeled GUVs for dark-field and fluorescence microscopic analyses were prepared as follows (20,23). A total concentration of 10 mM of lipid mixtures in chloroform/methanol (8:2 v/v) were cosolubilized with 1 mM ReLPS in chloroform/methanol (8:2 v/v) at a molar ratio of 10:1. After solvent evaporation under a flow of nitrogen gas onto the walls of a continuously rolled test tube and further vacuum for 3 h, the lipid film was hydrated with Tris buffer to a final lipids concentration of 1 mM and incubated for 3 h at 37°C.
For immunofluorescence labeling of ReLPS, GUV dispersions were incubated for 1 h with the mouse anti-ReLPS antibody A20, which recognizes a terminal Kdo of ReLPS, at a concentration of 1 μg/ml in Tris buffer at room temperature. Samples were sequentially incubated for 1 h with the Alexa Fluor-555-labeled goat anti-mouse IgM antibody (second antibody) at a concentration of 1 μg/ml in Tris buffer at room temperature. GUVs without ReLPS were used as negative controls.
Solid-state NMR spectroscopy
All solid-state NMR spectra were acquired on a CMX Infinity 300 spectrometer (Chemagnetics, Varian, Palo Alto, CA) operating at a proton resonance frequency of 300 MHz. 31P spectra were acquired using a 5 μs single excitation pulse with 30 kHz continuous wave 1H decoupling during acquisition. The dwell time was 50 μs, and 1024–2048 transients were accumulated for each free induction decay with a 3 s delay. The 31P chemical shifts were referenced externally to 85% H3PO4 (0 ppm). The 31P spectra were processed using 50 Hz line broadening.
QCM measurements
The QCM-D measurements were performed with a model No. D300 system with a QAFC 302 axial flow chamber (Q-Sence, Goteborg, Sweden). Before use, the SiO2 QCM sensor was cleaned in 100 mM SDS solution (immersed for 30 min at 30°C) and rinsed with deionized water. Then, the sensor was dried under a nitrogen stream and further cleaned by exposure to UV-produced ozone in air at atmospheric pressure for 20 min (model No. PL16-110, Sen Light, Toyonaka, Japan). After each set of data runs, the fluid cell was cleaned by filling it with a 0.5% Hellmanex solution and then repeatedly rinsing with deionized water. The QCM sensor crystal was oscillated at its resonance frequencies of 15, 25, and 35 MHz, respectively, and the frequency shifts (Δf) and dissipation shifts (ΔD) were monitored. The interval for data acquisition was 0.4 s. The mounted QCM sensor crystal was equilibrated with a degassed Tris buffer at 21.8°C. The buffer solution was subsequently replaced with the vesicle suspension (lipid concentration 140 μM). All QCM data were acquired at 21.8°C.
Preparation of SPBs on glass slides and fluorescence labeling
Glass slides were cleaned by sonicating in a 0.5% Hellmanex solution for 20 min and rinsing with deionized water. They were further treated in a cleaning solution of 0.05:1:5 NH4OH (28%)/H2O2 (30%)/H2O for 10 min at 65°C and rinsed again extensively with deionized water. They were dried in a vacuum oven for 30 min at 80°C. After cleaning by exposure to UV-produced ozone for 30 min, an SUV suspension was applied onto a glass slide. The vesicles were allowed to adsorb and to form SPB on the surface at room temperature for 15 min. SPBs thus formed were subsequently rinsed with the Tris buffer. For immunofluorescence observation of ReLPS, they were further incubated for 1 h at room temperature with the mouse anti-ReLPS antibody A20 at a concentration of 1 μg/ml in Tris buffer. Then, the mixtures were sequentially incubated for 1 h with the Alexa Fluor-555 goat anti-mouse IgM antibody at a concentration of 1 μg/ml in Tris buffer. SUVs without ReLPS were used as negative controls.
Dark-field and fluorescence imaging
Dark-field and fluorescence images of GUVs were acquired as previously described (20,23) with slight modification. A microscope (model No. BX51WI, Olympus, Tokyo, Japan) was equipped with an oil immersion objective lens (Plan Fluor, 100×, NA = 0.6–1.3, Olympus). An oil immersion dark-field condenser (U-D continuous wave, NA = 1.4–1.2, Olympus) was used as the dark-field illumination with a 100 W high-pressure mercury lamp. For fluorescence observation, Alexa Fluor-555 fluorescence was illuminated with a NIGA filter (Olympus). All images were recorded by using an SIT video camera (model No. C-2400, Hamamatsu Photonics, Hamamatsu, Japan) and DVD recorder (model No. DV-HR300, SHARP, Osaka, Japan), further processed with WinDVD (InterVideo Japan, Yokohama, Japan) and Photoshop (Adobe Systems, Mountain View, CA) software.
SPB observation
Fluorescence images of SPBs were acquired with a BX51WI microscope (Olympus) equipped with a water immersion objective lens (60×, NA = 0.9, Olympus) and a 75 W Xenon lamp (LH75XEAPO). NIBA, NIGA, and WIY filters (Olympus) were used to detect DiO, Alexa Fluor-555, and Texas-Red fluorescence, respectively. Photographs of SPB fluorescence were taken with a charge-coupled device camera (DP30BW, Olympus) mounted on the microscope and further processed with Metamorph (Ver. 6.3, Molecular Devices, Sunnyvale, CA) software.
BPE method
Lateral diffusion coefficients of mobile fractions of TR-DHPE in SPBs were determined by the BPE methods (24) by using a microscope (BX51WI; Olympus) with a rectangular field stop (U-RFSS; Olympus). After photobleaching for 15 s, fluorescence recovery was monitored by taking a sequence of images every 10 s. Individual intensity profiles after bleaching were nonlinearly fitted to the Gaussian error function
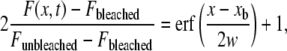 |
(1) |
where F(x, t) is the profile evolution with time; Fbleached and Funbleached are the fluorescence intensities inside and outside of the bleached region, respectively, immediately after the bleaching; xb is the position of the boundary between the bleached and unbleached areas; and (x-xb) is the distance to this boundary. The diffusion depth w is defined as
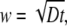 |
(2) |
where D is the diffusion coefficient and t is the elapsed time after bleaching. All measurements were conducted at room temperature.
RESULTS
Concentration dependence of ReLPS-DMPC lipid bilayer interaction
To examine the effect of the ReLPS concentration on its interaction with DMPC in lipid bilayers, the 31P NMR spectra of lipid bilayers in the presence and absence of ReLPS were measured under static conditions. Fig. 2 shows the 31P NMR spectra of DMPC lipid bilayers (Tm = 24°C) (25) containing various ratios of ReLPS at various temperatures. In the absence of ReLPS, an axially symmetric (principal axis of chemical shift anisotropy are motionally averaged) powder pattern was observed, indicating that the phospholipids were laterally diffusing in the lipid bilayers, i.e., they formed fluid bilayers. When bilayers were prepared from mixtures at a molar ratio of ReLPS/phospholipid = 1:50 (M/M) (hereafter relative amounts of lipid components are expressed as molar ratios), a small isotropic signal was observed, being superimposed on the bilayer component. This isotropic component may be attributed to fast isotropic tumbling of the phospholipids which may be caused by the formation of micelles, small unilamellar vesicles, small discoidal bilayers (26), inverted micelles, or a cubic phase (27). As the molar ratio of ReLPS/phospholipid increased, this isotropic component became bigger. This indicates that the isotropic component is ascribed to the micellization of the ReLPS component. However, the molar ratio of the isotropic to the bilayer component was smaller than that of ReLPS to phospholipids: the ratios of isotropic to bilayer components of ReLPS/DMPC membranes at a molar ratio of 1:50, 1:10, and 1:4 at 30°C are 1:66.4, 1:20.0, and 1:5.6, respectively. This suggests that a substantial part of ReLPS forms bilayers presumably together with DMPC. Further, the isotropic component became larger with increasing temperature, implying that the micellization by ReLPS is related to the acyl-chain fluidity. In addition, from the line shapes of the bilayer components we found that the liquid crystal-gel phase transition temperature (Tm) of these bilayers increased with increasing molar ratio of ReLPS/DMPC. This fact also indicates that at least a part of ReLPS is incorporated in the bilayer as judged from the observation of Gutsmann et al. that the Tm of lipid A becomes higher as the numbers of acyl chains increased (13). The Tm of ReLPS containing the hexaacylated lipid A part reported by Seydel et al. is ∼33°C (ethanolamine-salt form) (28). The Tm of the DMPC bilayer containing ReLPS in this study is higher than that of the lipid bilayer consisting of DMPC alone (Tm = 24°C).
FIGURE 2.

ReLPS concentration dependence of the 31PNMR spectra of DMPC lipid bilayers at various temperatures.
ReLPS membrane interactions
Next, we examined the effects of incorporated ReLPS on the morphological changes of various lipid membranes by observing the 31P NMR spectra of various lipid membranes in the absence and presence of ReLPS at a molar ratio of ReLPS/phospholipid = 1:10 under static conditions at above Tm. Fig. 3 a shows 31P NMR spectra of ReLPS/egg-PC (1:10) membranes (Tm ∼ −5°C) (29) at 0 and 30°C. In the presence of ReLPS, an isotropic component was observed already at 0°C and its proportion became larger at 30°C, implying that the higher temperature enhances micellization of ReLPS owing to the high mobility of the acyl chains of egg-PC (Fig. 1). A similar isotropic peak was observed on egg-PC/POPG (9:1) membranes in the presence of ReLPS at 30°C (Fig. 3 b). On the other hand, in the case of ReLPS/DEPE membranes (the gel-lamellar phase transition temperature Tc of DEPE ≈ 35°C and the lamellar-inverted hexagonal phase transition temperature Th of DEPE ≈ 60°C) (30) at 40°C, the isotropic signal became much smaller in the presence of ReLPS than those of the former two seen in Fig. 3, a and b (Fig. 3 c).
FIGURE 3.

Effect of ReLPS on the 31P NMR spectra of phospholipids in lipid bilayers composed of (a) egg-PC, (b) egg-PC/POPG (9:1), (c) DEPE, (d) E. coli polar lipid extract, and (e) PLMφ. The spectra were taken at 30°C for panels a, b, c, and e, and 40°C for panel d at molar ratio of ReLPS/lipids = 1:10.
It is interesting to examine how ReLPS acts within a membrane reconstituted from components of bacterial cells, since the bacterial membrane includes a large amount of ReLPS molecules in the outer leaflet of the outer membrane. The behavior of ReLPS was thus investigated in a model membrane composed of a phospholipid mixture extracted from E. coli. 31P NMR spectra were obtained from bilayers prepared from E. coli total lipid extract in the presence of ReLPS (Fig. 3 d). As in the case with DEPE membranes mentioned above (Fig. 3 c), the observed isotropic signal superimposed on a bilayer component is very small in the presence of ReLPS. E. coli total lipid extract contains ∼57.5% phosphatidylethanolamine (PE), 15.1% phosphatidylglycerol (PG), 9.8% cardiolipin, and 17.6% other lipids (w/w) (31). Thus, these results imply that PE-rich membranes are hardly affected by ReLPS and that ReLPS forms bilayers together with these phospholipids. Finally, we examined how ReLPS behaves in the membrane mimicking that of the host macrophage cell composed of a molar phospholipid mixture (PLMφ), PLMφ-[egg-PC]/[E. coli-PE]/[Bovine-SM]/[Brain-PS] = 1:0.7:0.5:0.4 (32). Fig. 3 e shows 31P NMR spectra of a PLMφ membrane at 30°C. In the absence of ReLPS, the spectrum showed that the membrane adopts a lamellar phase, and signals from PC, PE, sphingomyelin (SM), and phosphatidylserine (PS) appeared separately at different chemical shift values. In contrast, in the presence of ReLPS in the PLMφ mixture, an isotropic peak appeared superimposed on a bilayer component. Owing to the presence of PE as a major component in PLMφ, the relative intensity of the isotropic peak to the bilayer component diminished compared with the spectra shown in Fig. 3, a and b. In addition, the phospholipid peaks observed in the absence of ReLPS (Fig. 3 e, left) were fused in the presence of ReLPS, indicating that all phospholipids were homogeneously mixed in the membrane (33). For spectra where ReLPS was absent, shown in Fig. 3, b and e, we assigned the individual signals by comparing the individually acquired egg-PC, egg-PG, E. coli-PE, Bovine-SM, or Brain-PS MLV signals.
ReLPS in phospholipid giant unilamellar vesicles
ReLPS incorporated in phospholipid bilayers of GUVs was observed by the techniques of dark-field and fluorescence microscopy; the former can visualize the lipid membrane morphologies of GUV, whereas the location of ReLPS is detected by using the fluorescence antibody staining technique. Thus, we examined 1), whether the ReLPS is incorporated in the phospholipid layer of the GUV surface; and, if this is the case, 2), where ReLPS distributes on the GUV with the same membrane samples. Fig. 4 a shows the dark-field and fluorescence images of typical ReLPS/egg-PC (1:10) GUVs. The GUVs showed spherical shapes with a diameter of 10 μm in the dark-field image. In the subsequently acquired fluorescence image from Alexa Fluor-555 after the antibody treatment, the fluorescent probes were localized on the same GUV surface shown on the dark-field images, indicating that LPS was likely to localize in the GUV surface almost uniformly. ReLPS/egg-PC/POPG (1:9:1) GUV images, by contrast, showed spherical shapes with small spots on the dark-field image (Fig. 4 b). Since there was no such spot observed in the dark-field image of egg-PC/POPG (9:1) GUVs (Fig. 4 c) in the absence of ReLPS, these spots were obviously caused by the presence of ReLPS, and their appearance suggests that ReLPS and/or ReLPS/phospholipid aggregates were formed and localized in the GUV surface layer. Furthermore, the subsequently acquired fluorescence image showed that fluorescent probes are located partially on the GUV surface and fluorescence-positive small spots were also located in the same places as detected in the dark-field image. Both ReLPS/egg-PC/POPG (1:9:1) GUV-preparations without treatments with mouse anti-ReLPS antibody A20 and without treatment with Alexa Fluor-555 goat anti-mouse IgM antibody (second antibody) showed no fluorescent spots either (data not shown), confirming that the detected fluorescent probes in Fig. 4 b made the location of ReLPS molecules selectively visible. Therefore, the aggregates shown on both microscopy images were concluded to be composed of either ReLPS alone or ReLPS/phospholipid. The observed nonuniform, partial location of ReLPS on the ReLPS/egg-PC/POPG GUVs (shown in the fluorescence image) may be attributed to the electrostatic repulsion between the negatively charged LPS and POPG. ReLPS contains four and POPG contains one negative charge (as shown in Fig. 1). To prove these fluorescent probes are actually due to ReLPS molecules, we acquired fluorescence microscopy images of egg-PC/POPG (9:1) GUVs in the absence of ReLPS (Fig. 4 c). The dark-field images showed spherical shapes of this GUV similar to those in Fig. 4, a and b, whereas the fluorescence microscopy image showed, as anticipated, no binding of antibodies.
FIGURE 4.

Dark-field (top) and fluorescence (bottom) images of GUVs composed of (a) ReLPS/egg-PC (1:10), (b) ReLPS/egg-PC/POPG (1:9:1), and (c) PC/PG (9:1) at room temperature. Bar = 10 μm.
Vesicle fusion on SiO2 plate
Although we demonstrated that ReLPS is incorporated into lipid bilayers of GUVs in the previous section, the images in Fig. 4 are not sufficient to understand the precise manner of distribution of ReLPS in the lipid bilayer, because the membrane surface is shown only as a circle. To obtain further information on the distribution of ReLPS in lipid bilayers, we analyzed ReLPS in SPBs by using a fluorescence microscope. Before that, we checked the kinetics of SPB formation of SUVs on a SiO2 surface by using QCM-D (34).
Fig. 5 shows the QCM-D resonant frequency shift (Δf) and dissipation shift (ΔD) as a function of time for the adsorption of SUVs on SiO2 surfaces. As a control, we observed the adsorption of SUVs composed of egg-PC and egg-PC/POPG (9:1) (Fig. 5, a and b). In both cases, the adsorption was completed within 5 min, and both final Δf values were ∼26 Hz. Since these values are consistent with that reported for the formation of the egg-PC bilayer (34), these results indicate that both SUVs form bilayers. Since Δf is proportional to the mass of any materials adsorbed on the SiO2 surface, we had to normalize the molecular weights of the phospholipids (see discussion below). The temporal minimum Δf and the temporal maximum ΔD shown in all figures give evidence for the initial intact vesicle adsorption process and the subsequent breaking and spreading process of the vesicles to form a bilayer (34).
FIGURE 5.

QCM-D resonant frequency and dissipation shifts as a function of time for the adsorption of SUV onto SiO2 surface. The timings of buffer rinses are shown with arrows in all figures. Lipid compositions of SUVs are (a) ReLPS/egg-PC (1:10), (b) egg-PC, (c) ReLPS/egg-PC/POPG (1:9:1), and (d) egg-PC/POPG (9:1).
The SUVs containing LPS, i.e., ReLPS/egg-PC (1:10) SUVs took a longer time to form an SPB than the egg-PC SUV controls without LPS (Fig. 5 c). The final Δf, however, was approximately the same as that of the latter SUVs (∼27 Hz), suggesting that almost the entire SiO2 surface was covered with a lipid bilayer. Further, ReLPS/egg-PC/POPG (1:9:1) SUVs took the longest time of all four SUVs tested to complete the adsorption (Fig. 5 d). The final Δf was ∼36 Hz, a value ∼10 Hz larger than those of the other three SUVs. The times required for the formation of the SPBs were in the following order: ReLPS/egg-PC/POPG (1:9:1) > ReLPS/egg-PC (1:10) > egg-PC/POPG (9:1) > egg-PC. The longer time required to form the SPBs for ReLPS and POPG containing SUVs might be due to the electrostatic repulsion between the negatively charged SiO2 surface and LPS and/or POPG. Further, LPS aggregates on ReLPS/egg-PC/POPG SUVs also might be another reason for the disturbance of SPB formation.
The molecular weights of egg-PC, POPG, and ReLPS are 760, 771, and 2238, respectively. Since ReLPS has six acyl chains and egg-PC and POPG have two acyl chains (Fig. 1), one molecule of ReLPS is expected to occupy an area on the SiO2 surface approximately three times larger than those required for egg-PC or POPC. According to the ratio of acyl groups, the area on the SiO2 surface occupied by ReLPS should be corrected in relation to that of egg-PC and POPG; the standardized molecular weight of ReLPS would be 746 (= 2238/3). Keller and Kasemo (34) have shown that SUVs composed of egg-PC form an SPB by a process of adsorption on the SiO2 surface and found final Δf and ΔD values of 26 Hz and zero, respectively. When SUVs composed of egg-PC/POPG (9:1), ReLPS/egg-PC (1:10), and ReLPS/egg-PC/POPG (1:9:1) form planar bilayers, the final Δf for all samples also should be 26 Hz; since Δf is proportional to the mass of any materials adsorbed on the SiO2 surface.
Among all four SUVs tested, only the ones from ReLPS/egg-PC/POPG (1:9:1) showed the large final ΔD value. This may suggest that these particular SUVs form an SPB having a higher energy dissipation as compared to those formed from the others.
Distribution of ReLPS in SPBs
We examined the distribution of ReLPS in SPBs by observing fluorescence microscopy images of planar bilayers using the LPS-containing SPBs prepared as described in the preceding section. Fig. 6 a shows the fluorescence images of the SPBs composed of ReLPS/egg-PC (1:10) and ReLPS/egg-PC/POPG (1:9:1) with the fluorescence signals originating from DiO. Both images show a homogeneous distribution of phospholipids, thus confirming that both SPBs formed planar, uniform bilayers.
FIGURE 6.

Fluorescence microscope observation of SPBs. (a and b) Fluorescence images of planar bilayers composed of ReLPS/egg-PC (1:10) and ReLPS/egg-PC/POPG (1:9:1). Fluorescence signal are from (a) DiO and (b) Alexa Fluor-555. Bar = 5 μm. (c) Intensity profiles along the lines A–D shown in panel b. (d) Fluorescence intensity distribution of the images of Fig. 6 b. The average fluorescence intensity of the ReLPS/egg-PC (1:10) SPBs image is 88.8 with a relative standard uncertainty of 22%. The average fluorescence intensity of the ReLPS/egg-PC/POPG (1:9:1) SPBs image is 39.7 with a relative standard uncertainty of 17%.
In the next step we observed the fluorescence staining images of both SPBs with anti-ReLPS mouse IgM (first antibody) followed by anti-mouse IgM antibody conjugated with Alexa Fluor-555 dye (second antibody), where fluorescence signals from Alexa Fluor-555 reflect the locations of ReLPS (Fig. 6 b). In the image of the ReLPS/egg-PC (1:10) bilayer, fluorescence signals were widely and rather uniformly distributed on the SPB. However, when observed more closely, they were inhomogeneously distributed. In contrast, in the image of the ReLPS/egg-PC/POPG (1:9:1) bilayer, the intensity of the fluorescence signals remarkably decreased as compared to that of the cross-linked aggregates of antibodies and ReLPS or multimeric aggregates formed only by ReLPS.
Fig. 6 c shows the intensity profiles along the lines A, B, C, and D shown in Fig. 6 b. In these profiles, fluorescence dots show sharp peaks. Both the number and intensities of these peaks are larger in ReLPS/egg-PC (1:10) SPBs than in ReLPS/egg-PC/POPG (1:9:1) SPBs. Fig. 6 d shows the histograms of fluorescence signal intensities per pixel in the images of Fig. 6 b to demonstrate the distribution of fluorescence signal intensities. The average fluorescence intensity of the ReLPS/egg-PC (1:10) SPBs image was 88.8 and that of the ReLPS/egg-PC/POPG (1:9:1) SPBs image was 39.7; the average fluorescence intensity of the image of ReLPS/egg-PC (1:10) SPBs was more than twice than that of the ReLPS/egg-PC/POPG (1:9:1) SPBs. Although the average fluorescence signal intensity was weak in the ReLPS/egg-PC/POPG (1:9:1) SPB image, it contained many strong dotlike signals, which might be from aggregates composed of either only ReLPS or ReLPS/phospholipid as observed in the GUVs of the same lipid composition (Fig. 4 b).
For negative controls, we observed antibody binding to egg-PC and egg-PC/POPG (9:1) SPBs in the absence of ReLPS. Neither SPB showed any binding of fluorescent antibody (data not shown), indicating that the fluorescence probes show the location of ReLPS molecules selectively.
Effect of ReLPS incorporation on lateral diffusion coefficient in lipid bilayer
To evaluate the effect of ReLPS incorporation on the lateral diffusion of lipids in SPBs on a SiO2 surface, we determined the lateral diffusion coefficients by the BPE method. Fig. 7 shows the dependence of the lipid lateral diffusion coefficient D on the ReLPS concentration and POPG/(egg-PC+POPG) mol % of SPBs observed at 25°C.
FIGURE 7.

Dependence of the lipid lateral diffusion coefficients D on the ReLPS concentration and POPG/(egg-PC+POPG) mol % of SPBs at 25°C. The ReLPS/lipids molar ratios were 0:100 (•), 1:50 (▴), 1:20 (□), and 1:10 (○), respectively. The error bars include the standard deviations originating from sample and spot variations.
In the absence of ReLPS and POPG, the lateral diffusion coefficient was 2.14 μm2/s, a value in good agreement with those previously reported (24,35,36). The lateral diffusion coefficients of phospholipids were reduced depending on the amount of ReLPS added. For the egg-PC phospholipid membrane, the value changed from 2.14 μm2/s (in the absence of ReLPS) to 1.58 μm2/s (in the presence of 10 mol % ReLPS). It should be noted that the reduction of the coefficient value became larger with increasing POPG concentration in the membrane at high ReLPS/phospholipid molar ratios: from 1.58 μm2/s (in the absence of POPG) to 1.13 μm2/s (in the presence of 10 mol % POPG) at an ReLPS/phospholipid molar ratio of 1:10; and less effective in the absence of ReLPS, from 2.14 μm2/s (in the absence of POPG) to 2.12 μm2/s (in the presence of 10 mol % POPG).
DISCUSSION
The aim of this study was to investigate the interaction between ReLPS and phospholipids in mixed membranes. We used solid-state 31P-NMR spectroscopy for the analysis of the ensemble average of the system and microscopy for direct observation of the lipid membranes.
A concentration-dependent behavior of ReLPS in DMPC bilayers was examined by solid-state 31P-NMR (Fig. 2). In the presence of ReLPS, an isotropic component appeared and it became larger as the LPS concentration increased. Schromm et al. (38) have observed by using x-ray diffraction measurements that ReLPS from E. coli adopted a inverted cubic structure (Q212) (37) at 85% water content and at 40°C (13,14). Considering their results, the isotropic component superimposed on a lamellar component observed in this study, where ReLPS coexisted with phospholipid, can probably be assigned to the inverted cubic phase. Fig. 2 also shows that, in the presence of ReLPS, the liquid-gel transition temperature of the bilayer component became higher and that a certain amount of ReLPS was incorporated in the lipid bilayer. Considering the results from several previous studies on the incorporation of LPS into membranes (13,38), our findings show the high mixing ability of ReLPS with phospholipids in the host cell and imply the possibility that ReLPS exists in the host cell membrane in the first step of LPS-induced cell activation. The 31P NMR spectra (Fig. 3) also demonstrated that ReLPS generated partial micellization of egg-PC-rich membranes, whereas this tendency is suppressed in PE-rich membranes. Ethanolamine headgroup of PE has known to have a high hydrogen-bonding capability than the choline headgroup of PC (39) due to the absence of three methyl groups. Owing to the high hydrogen-bonding capability of the headgroup in PE, carboxyl and phosphate groups in ReLPS is expected to access more readily to the PE headgroups in PE-rich membranes as compared to PC-rich membranes. As a consequence, the high miscibility between ReLPS and PE brings uniform distribution of ReLPS in the PE-rich membrane, while ReLPS might be restricted from mixing with the PC-rich membrane and then isolate and form the ReLPS-rich cubic structure. Urban et al. demonstrated by using small-angle and wide-angle x-ray scattering and calorimetry that LPS induced a mixed cubic/lamellar phase in the DEPE/1,2-dipalmitoyl-sn-glycero-3-phosphoglycerol (8:2) MLV and that the cubic structure domain in the membrane became larger with increasing LPS concentration (from 2 to 50 mol % LPS) (40). This result may be explained by a strong tendency for PG-containing phospholipid membrane to induce a cubic phase in the presence of LPS, even in a PE-rich membrane.
In addition to the NMR results, microscopy was used to visualize the interactions between ReLPS and phospholipid membrane directly and individually (Figs. 4 and 6). Combined use of dark-field and fluorescence microscopy for imaging GUVs (Fig. 4) demonstrated that LPS is incorporated in the ReLPS/egg-PC GUV surface uniformly, but the same LPS was found to be located partially on the ReLPS/egg-PC/POPG (1:9:1) GUV surface. We also observed that ReLPS or ReLPS/phospholipid aggregates are present on the latter GUV surface. The lower intensities of the fluorescence signals, and the presence of ReLPS aggregates in negatively-charged membrane containing POPG, were also observed in ReLPS/egg-PC/POPG (1:9:1) SPBs (Fig. 6 b). This could be attributed to the low relative amount of LPS incorporated into the membrane owing to the electrostatic repulsion between negatively charged ReLPS and POPG, since experiments with both GUV and SPB microscopy demonstrated similar tendencies of the behavior of ReLPS in the charged membranes. Roes et al. (15) have shown by atomic force microscopy experiments that LPS formed domain structures (with diameters up to 20 μm) on PS/LBP membranes but not on PS/LBP membranes with α-LBP antibodies. In our observation, such large domains were observed neither for ReLPS/egg-PC nor for ReLPS/egg-PC/POPG SPBs, and these results are in good agreement with their latter observation. To examine the size of LPS domain structure on phospholipid membranes in the absence of LBP, more precise analysis on a molecular level may be required.
The QCM-D measurement (Fig. 5) showed that the ReLPS/egg-PC (1:10) membranes form almost complete bilayers, but ReLPS/egg-PC/POPG (1:9:1) SUVs obtained a larger final Δf value than ReLPS/egg-PC (1:10) SUVs even after being washed twice by the buffer solution. The larger final Δf can be attributed to the presence of ReLPS or ReLPS/phospholipid aggregates on the planar bilayer on the SiO2 surface, as shown in the fluorescence microscopy images of both ReLPS/egg-PC/POPG (1:9:1) GUV (Fig. 4 b) and SPBs (Fig. 6 b). These aggregates do not simply attach onto the SPBs and behave as part of the SPBs because they are not removed even after washing twice with buffer solution. Mechler et al. (41) have also shown ∼10 Hz larger final Δf value for caerin/DMPC/DMPG bilayers than that for caerin/DMPC bilayers by QCM. Since caerin (GLLSVLGSVAKHVLPHVVPVIAEHL-NH2) is transmembrane type antimicrobial peptide with +3 net charges, the change of final Δf value could be due to the higher packing between caerin and negatively-charged DMPG. Though the reason is different, charged phospholipid, like DMPG, would have an ability to change a packing density or thickness of a bilayer. Furthermore, ReLPS/egg-PC/POPG (1:9:1) SUVs showed a larger final ΔD value than that of ReLPS/egg-PC (1:9) SUVs. The difference can be ascribed to the formation of planar bilayers with incorporated ReLPS or ReLPS/phospholipid aggregates in ReLPS/egg-PC/POPG (1:9:1) SPBs causing an increase in the energy dissipation but not to a rupture of vesicles. ReLPS/egg-PC (1:9) SUV formed almost completely planar bilayers lying close to the SiO2 surface.
The lipid lateral diffusion coefficient measurements (Fig. 7) at various molar ratios of ReLPS/egg-PC/POPG showed that the ReLPS incorporation reduces the lateral diffusion coefficient of the phospholipids. This reduction became more prominent with increasing POPG concentration in the membrane at high ReLPS/phospholipid molar ratio. The POPG-dependent reduction was negligible in the absence of ReLPS. The reduction in the lateral diffusion rate of phospholipids in the presence of ReLPS suggests slow movement of ReLPS in the lipid bilayer. The rate of diffusion is known to depend on the size of the molecules in the lipid membrane (42,43). For example, Lee et al. (42) have reported that aggregates of transmembrane molecules, which occupy approximately three times larger membrane surface area than phospholipids, move at 58% of the diffusion rate of phospholipids in phospholipid membranes. ReLPS requires an approximately three times larger membrane surface area than egg-PC and POPG. Because of its size alone, ReLPS is expected to laterally diffuse more slowly than phospholipids in the membrane. The presence of the six acyl chains in ReLPS is also likely to be an additional reason for its slow diffusion, because the strong hydrophobic interaction between acyl chains of LPS itself and of phospholipids might reduce the diffusion rate.
In the case of SPBs containing both POPG and ReLPS, even though LPS incorporation in the membrane is suppressed in the presence of POPG as a membrane component, the diffusion of phospholipids was much slower than in ReLPS/egg-PC SPBs. The lateral diffusion of the phospholipids could possibly be disturbed by the presence of the aforementioned ReLPS or ReLPS/phospholipid aggregates detected in the fluorescence microscopy images of ReLPS/egg-PC/POPG (1:9:1) GUVs (Fig. 4 b) and SPBs (Fig. 6 b), as well as in the QCM-D measurement (Fig. 5 c). The disturbance by the membrane-adsorbed ReLPS or ReLPS/phospholipid aggregates seems to be more effective than the rather uniformly distributed ReLPS in the membrane lacking the POPG component.
In conclusion, both NMR and microscopic studies showed that, when lipid bilayers are prepared from mixtures of ReLPS and phospholipids, the majority (but not all) ReLPS is incorporated in phospholipid bilayers and the remaining portion of ReLPS forms isotropic aggregates (presumably adopting an inverted cubic phase). The degree of mixing of ReLPS and phospholipid bilayers depends on the lipid composition. In particular, for egg-PC bilayers containing POPG, mixing was significantly suppressed as compared to the bilayers composed of egg-PC alone. This should be at least partially due to electrostatic repulsion between POPG and ReLPS, which increases the energy required for mixing. The hydrogen bonds and strong mutual lipophilic affinity among ReLPS molecules should also contribute to aggregate formation. Segregated ReLPS should form inverted aggregates as depicted in Fig. 8. On the other hand, in ReLPS/DEPE or ReLPS/E. coli total lipid extract membranes, almost all ReLPS molecules intercalate into the membrane. In this study, we have investigated how LPS behaves in phospholipid membranes in very simple model systems, where lipid bilayers were prepared in the presence of ReLPS to have sufficient LPS incorporated in the membrane. During naturally occurring immune cell activation, the LPS-binding protein (LBP) in serum is expected to facilitate the delivery of LPS via interaction with the cell membrane (11,12) to the membrane-bound acceptor proteins. LBP has a positively charged region in the N-terminal domain which is assumed to be the LPS-binding domain (44). In fact, Gutsman et al. demonstrated that LBP enhances the transfer of lipid A into model lipid membranes by fluorescence resonance energy transfer experiments. In the negatively charged membranes, such as those consisting mainly of PS or PG, LBP intercalates more readily than into neutral PC membranes. Subsequent LPS intercalation into negatively charged membranes in the presence of LBP is hence expected to be enhanced as well (12). Gutsman et al. also demonstrated that LBP induces two different mechanisms for LPS binding to membrane using electrical measurements (11) and a surface plasmon resonance experiment (12). LPS binds to PS/LBP membrane but not to PS membrane (activation mechanism). LPS/LBP complexes, however, bind neither to PS nor to PS/LBP membrane (neutralization mechanism).
FIGURE 8.

Schematic model of ReLPS intercalations into the various phospholipid membranes composed of (a) egg-PC and (b) egg-PC/POPG (9:1) at 30°C, and (c) DEPE or E. coli polar lipid extract at 40°C at molar ratio of ReLPS/phospholipids = 1:10. The inverted cubic phase in panels a and b is assumed to be composed mainly of ReLPS.
In this work, though no LBP was involved, we have provided a basic understanding of the individual interactions between LPS, as a key molecule in innate immune recognition, and target cell membranes. This represents a starting point for more precise characterization of the mutual interaction between LPS, target cell membrane, LBP, and the membrane-bound receptor proteins. The next important step will be to elucidate the manner by which ReLPS aggregates in phospholipid membranes, which should be approached by solid-state NMR analyses of 13C and/or 15N-enriched specimens incorporated into phospholipid membranes. This will provide deeper insight into the problem of whether ReLPS molecules in membranes exist as isolated monomers surrounded by phospholipids, or form oligo- or polymeric aggregates (Fig. 8) as has been observed (45) and anticipated in the outer leaflet of Gram-negative bacterial cells.
Acknowledgments
We thank Prof. Helmut Brade (Research Center Borstel, Borstel, Germany) for providing the mouse anti-ReLPS antibody, A20 (IgM). We also thank Ms. Saori Mori (NAIST, Osaka, Japan) for her assistance in the preparation of glass slides and Dr. Kazunori Kawasaki and Dr. Yoshiro Tatsu for supporting the research conducted at AIST.
This work was supported by Grants-in-Aid for Scientific Research (No. 19550174) from the Ministry of Education, Culture, Sports, Science and Technology of Japan.
Editor: Arthur G. Palmer III.
References
- 1.Rietschel, E. T., L. Brade, B. Lindner, and U. Zahringer. 1992. Biochemistry of lipopolysaccharides. In Bacterial Endotoxic Lipopolysaccharides. D. C. Morison, and J. L. Ryan, editors. CRC Press, Boca Raton, FL.
- 2.Brade, H., S. M. Opal, S. N. Vogel, and D. C. Morrison. 1999. Endotoxin in Health and Disease. Marcel Dekker, New York.
- 3.Tobias, P. S., K. Soldau, and R. J. Ulevitch. 1986. Isolation of a lipopolysaccharide-binding acute phase reactant from rabbit serum. J. Exp. Med. 164:777–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schumann, R. R. 1992. Function of lipopolysaccharide (LPS)-binding protein (LBP) and CD14, the receptor for LPS/LBP complexes: a short review. Res. Immunol. 143:11–15. [DOI] [PubMed] [Google Scholar]
- 5.Aderem, A., and R. J. Ulevitch. 2000. Toll-like receptors in the induction of the innate immune response. Nature. 406:782–787. [DOI] [PubMed] [Google Scholar]
- 6.Gangloff, M., and N. J. Gay. 2004. MD-2: the Toll “gatekeeper” in endotoxin signaling. Trends Biochem. Sci. 29:294–300. [DOI] [PubMed] [Google Scholar]
- 7.Shimazu, R., S. Akashi, H. Ogata, Y. Nagai, K. Fukudome, K. Miyake, and M. Kimoto. 1999. MD-2, a molecule that confers lipopolysaccharide responsiveness on Toll-like receptor 4. J. Exp. Med. 189:1777–1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Olsson, S., and R. Sundler. 2006. The role of lipid rafts in LPS-induced signaling in a macrophage cell line. Mol. Immunol. 43:607–612. [DOI] [PubMed] [Google Scholar]
- 9.Triantafilou, M., K. Miyake, D. T. Golenbock, and K. Triantafilou. 2002. Mediators of innate immune recognition of bacteria concentrate in lipid rafts and facilitate lipopolysaccharide-induced cell activation. J. Cell Sci. 115:2603–2611. [DOI] [PubMed] [Google Scholar]
- 10.Wu, E. S., K. Jacobson, and D. Papahadjopoulos. 1977. Lateral diffusion in phospholipid multibilayers measured by fluorescence recovery after photobleaching. Biochemistry. 16:3836–3841. [DOI] [PubMed] [Google Scholar]
- 11.Gutsmann, T., N. Haberer, S. F. Carroll, U. Seydel, and A. Wiese. 2001. Interaction between lipopolysaccharide (LPS), LPS-binding protein (LBP), and planar membranes. Biol. Chem. 382:425–434. [DOI] [PubMed] [Google Scholar]
- 12.Gutsmann, T., M. Muller, S. F. Carroll, R. C. MacKenzie, A. Wiese, and U. Seydel. 2001. Dual role of lipopolysaccharide (LPS)-binding protein in neutralization of LPS and enhancement of LPS-induced activation of mononuclear cells. Infect. Immun. 69:6942–6950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gutsmann, T., A. B. Schromm, M. H. Koch, S. Kusumoto, K. Fukase, M. Oikawa, U. Seydel, and K. Brandenburg. 2000. Lipopolysaccharide-binding protein-mediated interaction of lipid A from different origin with phospholipid membranes. Phys. Chem. Chem. Phys. 2:4521–4528. [Google Scholar]
- 14.Schromm, A. B., K. Brandenburg, H. Loppnow, U. Zahringer, E. T. Rietschel, S. F. Carroll, M. H. Koch, S. Kusumoto, and U. Seydel. 1998. The charge of endotoxin molecules influences their conformation and IL-6-inducing capacity. J. Immunol. 161:5464–5471. [PubMed] [Google Scholar]
- 15.Roes, S., F. Mumm, U. Seydel, and T. Gutsmann. 2006. Localization of the lipopolysaccharide-binding protein in phospholipid membranes by atomic force microscopy. J. Biol. Chem. 281:2757–2763. [DOI] [PubMed] [Google Scholar]
- 16.Brabetz, W., S. Muller-Loennies, O. Holst, and H. Brade. 1997. Deletion of the heptosyltransferase genes rfaC and rfaF in Escherichia coli K-12 results in an Re-type lipopolysaccharide with a high degree of 2-aminoethanol phosphate substitution. Eur. J. Biochem. 247:716–724. [DOI] [PubMed] [Google Scholar]
- 17.Raetz, C. R., T. A. Garrett, C. M. Reynolds, W. A. Shaw, J. D. Moore, D. C. Smith, Jr., A. A. Ribeiro, R. C. Murphy, R. J. Ulevitch, C. Fearns, D. Reichart, C. K. Glass, C. Benner, S. Subramaniam, R. Harkewicz, R. C. Bowers-Gentry, M. W. Buczynski, J. A. Cooper, R. A. Deems, and E. A. Dennis. 2006. Kdo2-Lipid A of Escherichia coli, a defined endotoxin that activates macrophages via TLR-4. J. Lipid Res. 47:1097–1111. [DOI] [PubMed] [Google Scholar]
- 18.Hotani, H. 1984. Transformation pathways of liposomes. J. Mol. Biol. 178:113–120. [DOI] [PubMed] [Google Scholar]
- 19.Nomura, F., M. Nagata, T. Inaba, H. Hiramatsu, H. Hotani, and K. Takiguchi. 2001. Capabilities of liposomes for topological transformation. Proc. Natl. Acad. Sci. USA. 98:2340–2345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nomura, F., T. Inaba, S. Ishikawa, M. Nagata, S. Takahashi, H. Hotani, and K. Takiguchi. 2004. Microscopic observations reveal that fusogenic peptides induce liposome shrinkage prior to membrane fusion. Proc. Natl. Acad. Sci. USA. 101:3420–3425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Alexander, C., and E. T. Rietschel. 2001. Bacterial lipopolysaccharides and innate immunity. J. Endotoxin Res. 7:167–202. [PubMed] [Google Scholar]
- 22.Brade, L., P. Kosma, B. J. Appelmelk, H. Paulsen, and H. Brade. 1987. Use of synthetic antigens to determine the epitope specificities of monoclonal antibodies against the 3-deoxy-D-manno-octulosonate region of bacterial lipopolysaccharide. Infect. Immun. 55:462–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Inaba, T., A. Ishijima, M. Honda, F. Nomura, K. Takiguchi, and H. Hotani. 2005. Formation and maintenance of tubular membrane projections require mechanical force, but their elongation and shortening do not require additional force. J. Mol. Biol. 348:325–333. [DOI] [PubMed] [Google Scholar]
- 24.Merzlyakov, M., E. Li, and K. Hristova. 2006. Directed assembly of surface-supported bilayers with transmembrane helices. Langmuir. 22:1247–1253. [DOI] [PubMed] [Google Scholar]
- 25.Lewis, B. A., and D. M. Engelman. 1983. Lipid bilayer thickness varies linearly with acyl chain length in fluid phosphatidylcholine vesicles. J. Mol. Biol. 166:211–217. [DOI] [PubMed] [Google Scholar]
- 26.Dufourc, E. J., J. F. Faucon, G. Fourche, J. Dufourcq, T. Gulik-krzywicki, and M. Le Maire. 1986. Reversible disk-to-vesicle transition of melittin-DPPC complexes triggered by the phospholipid acyl chain melting. FEBS Lett. 201:205–209. [Google Scholar]
- 27.Prenner, E. J., R. N. A. H. Lewis, K. C. Neuman, S. M. Gruner, L. H. Kondejewski, R. S. Hodges, and R. N. McElhaney. 1997. Nonlamellar phases induced by the interaction of gramicidin s with lipid bilayers. A possible relationship to membrane-disrupting activity. Biochemistry. 36:7906–7916. [DOI] [PubMed] [Google Scholar]
- 28.Seydel, U., H. Labischinski, M. Kastowsky, and K. Brandenburg. 1993. Phase behavior, supramolecular structure, and molecular conformation of lipopolysaccharide. Immunobiology. 187:191–211. [DOI] [PubMed] [Google Scholar]
- 29.Seelig, J., and N. Waespe-Sarcevic. 1978. Molecular order in cis and trans unsaturated phospholipid bilayers. Biochemistry. 17:3310–3315. [DOI] [PubMed] [Google Scholar]
- 30.Epand, R. M. 1985. Diacylglycerols, lysolecithin, or hydrocarbons markedly alter the bilayer to hexagonal phase transition temperature of phosphatidylethanolamines. Biochemistry. 24:7092–7095. [DOI] [PubMed] [Google Scholar]
- 31.Newman, M. J., and T. H. Wilson. 1980. Solubilization and reconstitution of the lactose transport system from Escherichia coli. J. Biol. Chem. 255:10583–10586. [PubMed] [Google Scholar]
- 32.Kroner, E. E., B. A. Peskar, H. Fischer, and E. Ferber. 1981. Control of arachidonic acid accumulation in bone marrow-derived macrophages by acyltransferases. J. Biol. Chem. 256:3690–3697. [PubMed] [Google Scholar]
- 33.Pukala, T. L., M. P. Boland, J. D. Gehman, L. Kuhn-Nentwig, F. Separovic, and J. H. Bowie. 2007. Solution structure and interaction of cupiennin 1a, a spider venom peptide, with phospholipid bilayers. Biochemistry. 46:3576–3585. [DOI] [PubMed] [Google Scholar]
- 34.Keller, C. A., and B. Kasemo. 1998. Surface specific kinetics of lipid vesicle adsorption measured with a quartz crystal microbalance. Biophys. J. 75:1397–1402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chan, P. Y., M. B. Lawrence, M. L. Dustin, L. M. Ferguson, D. E. Golan, and T. A. Springer. 1991. Influence of receptor lateral mobility on adhesion strengthening between membranes containing LFA-3 and CD2. J. Cell Biol. 115:245–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Weng, K. C., J. J. Stalgren, D. J. Duval, S. H. Risbud, and C. W. Frank. 2004. Fluid biomembranes supported on nanoporous aerogel/xerogel substrates. Langmuir. 20:7232–7239. [DOI] [PubMed] [Google Scholar]
- 37.Khoo, I. C. a. F. S. 1991. Physics of Liquid Crystalline Materials. Gordon and Breach Science Publishers, Philadelphia, PA.
- 38.Schromm, A. B., K. Brandenburg, E. T. Rietschel, and U. Seydel. 1995. Do endotoxin aggregates intercalate into phospholipid membranes in a nonspecific, hydrophobic manner? J. Endotoxin Res. 2:313–323. [Google Scholar]
- 39.Hakizimana, P., M. Masureel, B. Gbaguidi, J. M. Ruysschaert, and C. Govaerts. 2008. Interactions between phosphatidylethanolamine headgroup and LMRP, a multidrug transporter: a conserved mechanism for proton gradient sensing? J. Biol. Chem. 283:9369–9376. [DOI] [PubMed] [Google Scholar]
- 40.Urban, E., A. Bota, and B. Kocsis. 2006. Non-bilayer formation in the DPPE-DPPG vesicle system induced by deep rough mutant of Salmonella minnesota R595 lipopolysaccharide. Colloids Surf. B Biointerfaces. 48:106–111. [DOI] [PubMed] [Google Scholar]
- 41.Mechler, A., S. Praporski, K. Atmuri, M. Boland, F. Separovic, and L. L. Martin. 2007. Specific and selective peptide-membrane interactions revealed using quartz crystal microbalance. Biophys. J. 93:3907–3916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lee, C. C., M. Revington, S. D. Dunn, and N. O. Petersen. 2003. The lateral diffusion of selectively aggregated peptides in giant unilamellar vesicles. Biophys. J. 84:1756–1764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liu, C., A. Paprica, and N. O. Petersen. 1997. Effects of size of macrocyclic polyamides on their rate of diffusion in model membranes. Biophys. J. 73:2580–2587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lamping, N., A. Hoess, B. Yu, T. C. Park, C. J. Kirschning, D. Pfeil, D. Reuter, S. D. Wright, F. Herrmann, and R. R. Schumann. 1996. Effects of site-directed mutagenesis of basic residues (Arg 94, Lys 95, Lys 99) of lipopolysaccharide (LPS)-binding protein on binding and transfer of LPS and subsequent immune cell activation. J. Immunol. 157:4648–4656. [PubMed] [Google Scholar]
- 45.Oikawa, M., T. Shintaku, N. Fukuda, H. Sekljic, Y. Fukase, H. Yoshizaki, K. Fukase, and S. Kusumoto. 2004. NMR conformational analysis of biosynthetic precursor-type lipid A: monomolecular state and supramolecular assembly. Org. Biomol. Chem. 2:3557–3565. [DOI] [PubMed] [Google Scholar]