

Abstract
The principal difficulty in experimental exploration of the folding and stability of membrane proteins (MPs) is their aggregation outside of the native environment of the lipid bilayer. To circumvent this problem, we recently applied fluorinated nondetergent surfactants that act as chemical chaperones. The ideal chaperone surfactant would 1), maintain the MP in solution; 2), minimally perturb the MP's structure; 3), dissociate from the MP during membrane insertion; and 4), not partition into the lipid bilayer. Here, we compare how surfactants with hemifluorinated (HFTAC) and completely fluorinated (FTAC) hydrophobic chains of different length compare to this ideal. Using fluorescence correlation spectroscopy of dye-labeled FTAC and HFTAC, we demonstrate that neither type of surfactant will bind lipid vesicles. Thus, unlike detergents, fluorinated surfactants do not compromise vesicle integrity even at concentrations far in excess of their critical micelle concentration. We examined the interaction of surfactants with a model MP, DTT, using a variety of spectroscopic techniques. Site-selective labeling of DTT with fluorescent dyes indicates that the surfactants do not interact with DTT uniformly, instead concentrating in the most hydrophobic patches. Circular dichroism measurements suggest that the presence of surfactants does not alter the structure of DTT. However, the cooperativity of the thermal unfolding transition is reduced by the presence of surfactants, especially above the critical micelle concentration (a feature of regular detergents, too). The linear dependence of DTT's enthalpy of unfolding on the surfactant concentration is encouraging for future application of (H)FTACs to determine the stability of the membrane-competent conformations of other MPs. The observed reduction in the efficiency of Förster resonance energy transfer between donor-labeled (H)FTACs and acceptor-labeled DTT upon addition of lipid vesicles indicates that the protein sheds the layer of surfactant during its bilayer insertion. We discuss the advantages of fluorinated surfactants over other types of solubilizing agents, with a specific emphasis on their possible applications in thermodynamic measurements.
INTRODUCTION
The principal difficulty of structural and thermodynamic studies with membrane proteins (MPs) is related to their hydrophobic nature, which causes them to aggregate and precipitate outside of their native membrane environment. Detergent solubilization, which is a general way of handling MPs in vitro, very often makes them unstable. Several approaches have been suggested to try to circumvent this problem (see, e.g., (1,2)), among which is the use of new, milder nondetergent surfactants such as amphipols (3–5) or fluorinated nonionic surfactants (6–9). Fluorinated surfactants are comprised of a polar head and a hydrophobic moiety that features partially or completely fluorinated chains (Fig. 1 A). In the case of hemifluorinated compounds (e.g., HFTAC), the very tip of the hydrophobic chain is left unfluorinated, a design intended to promote interactions with hydrophobic surfaces of MPs. Due to poor packing of fluorinated and acetylated chains, these surfactants possess the unusual and useful qualities of being at the same time good solvents for proteins and poor solvents for lipids. It has been demonstsrated that substitution of detergents with HFTAC improves the biochemical stability of such detergent-sensitive proteins as bacteriorhodopsin and the cytochrome b6f complex (8). Recently, we demonstrated that HFTAC can chaperone the insertion of a model membrane protein (DTT) into preformed lipid bilayers by reducing nonproductive aggregation in the aqueous phase without compromising membrane insertion (9) (Fig. 1 B). Here, we use the same model system to demonstrate that this chaperonelike ability is shared by completely fluorinated surfactants (FTACs). We characterize the interactions of fluorinated surfactants with protein and lipid moieties, with an emphasis on potential applications in thermodynamic studies of MP folding and insertion.
FIGURE 1.

(A) The chemical structure of the fluorinated surfactants used in this study. Hemifluorinated surfactant (HFTAC) has an unfluorinated tip on its hydrophobic tail, whereas FTAC-C6 and FTAC-C8 contain completely fluorinated hydrophobic tails consisting of six and eight carbons, respectively (7). The average degree of polymerization of the polar head for the batches used in this study was n = 6–7. Because of the poor miscibility of fluorinated and hydrogenated chains, fluorinated surfactants have advantages over regular detergents: they don't dissolve lipid bilayers even at concentrations above their CMCs (see Fig. 2). (B) Schematic illustration of fluorinated surfactants chaperoning membrane insertion of diphtheria toxin T-domain by preventing its nonproductive aggregation in solution at low pH (adapted from (9)).
Membrane insertion of nonconstitutive MPs (e.g., bacterial toxins (10–13) and colicins (14–16)) is often achieved in response to changes in environment and occurs spontaneously without the help of any protein-translocating machinery. For example, acidification of the endosome causes a conformational change in endocytosed DTT, resulting in its insertion into the membrane and translocation of its own N-terminus with the attached catalytic domain into the cytoplasm (10). The insertion of constitutive membrane proteins, on the other hand, is managed by complex multiprotein assemblies, such as the endoplasmic reticulum translocon (17–19). Although neither translocon-assisted nor spontaneous membrane insertion of proteins is well understood on a molecular level, recent thermodynamic evidence indicates that the underlying physicochemical principles for these two processes are likely to be the same (20–22). Thus, deciphering these principles with the help of spontaneously inserting proteins is relevant to the larger problems of membrane protein folding and stability. Recently, we demonstrated that pH-triggered membrane insertion of diphtheria toxin T-domain (23) and annexin B12 (24) are reversible processes, opening the door to their use for thermodynamic characterization of transbilayer insertion, amended by the use of fluorinated surfactants (9).
DTT is a particularly useful model system for testing protein interactions with fluorinated surfactants, as it can be easily converted, by acidification, from a properly folded monomeric globular state with a known structure (25) to a membrane-competent molten globulelike conformation. Physiological function of the T-domain is associated with membrane insertion and terminus translocation accompanied by the formation of the pore. We use this pore-forming activity as a simplified functional/insertion assay conducted in the presence of fluorinated surfactants. We also use circular dichroism and various applications of fluorescence spectroscopy to characterize the interactions of these surfactants with the T-domain and to demonstrate their lack of interaction with lipid bilayers. We discuss the implications of our findings for the future use of fluorinated surfactants as a new tool for structural and thermodynamic studies of MPs.
MATERIALS AND METHODS
Materials
POPC, POPG, and lysophosphoethanolamine were purchased from Avanti Polar Lipids (Alabaster, AL). UniBlue A vinyl sulfone was purchased from Sigma (St. Louis, MO). N-((2-(iodoacetoxy)ethyl)-N-methyl)amino-NBD ester, Oregon Green isothiocyanate, Alexa-532 C5 meleimide, and Alexa-647 C2 maleimide were purchased from Molecular Probes (Eugene, OR). The acidic buffer (pH 4.6) was comprised of 10 mM sodium acetate and 50 mM NaCl, and the neutral buffer (pH 8.0) of either 50 mM phosphate (for CD studies) or 10 mM HEPES and 50 mM NaCl. (H)FTACs were synthesized as described in (7). DTT (amino acids 202–378) was cloned into NdeI-EcoRI-treated pET15b vector containing an N-terminal 6xHis-tag and a thrombin cleavage site and isolated as described in (26). Labeling of single-cysteine mutants of the T-domain (N235C, L350C, Q369C, and P378C) was performed using a standard procedure for the thiol-reactive maleimide derivatives, as described in (23) and (9) for NBD- and Alexa-labeling, respectively. The concentration of T-domain was 3 μM for samples used in CD experiments and 0.5–1 μM for samples used in fluorescence experiments, unless otherwise specified.
LUV preparation
LUVs of diameter 0.1 μm were prepared by extrusion (27,28) from 3:1 molar mixtures of POPC and POPG. For FCS studies, a 0.5% of OG-labeled lipid was added. For leakage studies, the vesicles were preloaded with 1 mM ANTS and 10 mM DPX as described in (29).
Syntheses of fluorescent surfactants FTAC-C6-OG, FTAC-C8-OG, and HFTAC-OG
Starting telomers FTAC-C8 or FTAC-C8 and HFTAC were synthesized according to the procedure previously described (7). On each telomer, Oregon Green isothiocyanate (O488) was grafted via a thiocarbamate bond to a hydroxyl pendant of Tris moities. This reaction was carried out in pyridine at 50°C in the presence of a catalytic amount of diazabicyclo[2,2,2]octane. After completion of the reaction, fluorescent organic telomers were purified by chromatography through a Sephadex G15 column and lyophilized. The abundance of Oregon Green grafted on each telomer was specified in 1H-NMR (solvent: DMSO-d6) by comparing the peak area of typical signals ascribed to Oregon Green and free Tris moieties. As regards FTAC-C6 and FTAC-C8 telomers, having an average degree of polymerization (DPn) of 5, i.e., five Tris moieties compose the polar head, the proportion of Tris residues endowed with Oregon Green was only 1.3–2% (molecular ratio), whereas for HFTAC telomer, having a DPn of 10, it was higher and equal to 20%.
Steady-state fluorescence measurements
Fluorescence was measured either using a SPEX Fluorolog FL3-22 steady-state fluorescence spectrometer (Jobin Yvon, Edison, NJ) equipped with double-grating excitation and emission monochromators (most of the measurements) or using an SLM 8100 steady-state fluorescence spectrometer (Jobin Yvon, Urbana, IL) equipped with double-grating excitation and single-grating emission monochromators (see data in Fig. 4), as described previously (9,24). The measurements were made in 4 × 10-mm cuvettes oriented perpendicular to the excitation beam and maintained at 25°C using a Peltier device from Quantum Northwest (Spokane, WA). Direct contribution of scattering was subtracted using a signal from a blank sample containing vesicles, but not labeled proteins or surfactants. Cross-orientation of polarizers was used to minimize the scattering contribution from vesicles, eliminate spectral polarization effects in monochromator transmittance (30), and enhance the sensitivity of FRET measurements (24). Fluorescence excitation spectra of a 1:2 mixture of DTT-donor/DTT-acceptor were obtained by averaging 5–10 scans collected over a 470–660 nm range using 1-nm steps. The emission monochromator was set at 680 nm. All measurements were done after the system was equilibrated for at least 2 h. For the purpose of clear presentation, the spectra were normalized to direct acceptor excitation intensity, which accounts for minor variation in sample concentration (see Figs. 4 and 10). NBD emission spectra were collected using 465-nm excitation. Leakage of ANTS/DPX was followed by kinetic measurements of intensity with excitation and emission wavelengths of 353 nm and 520 nm, respectively.
FIGURE 4.

Influence of surfactants on the formation of the T-domain aggregates using the FRET-based experimental scheme from our previous study (9). (A) Excitation spectra for a mixture of donor- and acceptor-labeled T-domain (DTT-Alexa532 and DTT-Alexa647, respectively, at pH 4.6 in buffer (solid line) and in the presence of 0.6 mM of surfactants HFTAC (dashed line), FTAC-C6 (dotted line), and FTAC-C8 (dash-dotted line). Disappearance of the donor excitation peak (large arrow) indicates elimination of aggregation by surfactants. (B) Dependence of the relative FRET signal on the surfactant concentration. Intensity of the FRET peak is measured at 530 nm and is normalized to 1 for the sample in the absence of surfactants (solid line in A) and to 0 for the sample in the absence of the donor-labeled T-domain. Note that fluorinated surfactants can prevent aggregation at concentrations below their CMCs.
FIGURE 10.

Excitation spectra of a mixture of 0.5 μM DTT labeled with Alexa647 (acceptor) and 0.4 mM FTAC-C6 containing ∼1% of FTAC-C6-OG (donor) in buffer (solid line) and upon addition of 0.5 or 2 mM LUV (dotted and dashed lines, respectively). The decrease in the FRET-associated donor peak indicates that the protein sheds the surfactant upon insertion into the lipid bilayer.
FCS measurements
The FCS experiment was conducted on a MicroTime 200 confocal microscope (PicoQuant, Berlin, Germany). The fluorescence was excited with a pulsed picosecond diode laser LDH-P-C-470 operated at 40 MHz. Narrow-band clean-up filters ensured that no parasitic light reached the sample. The fluorescence was detected confocally after passing through an emission bandpass filter (HQ 520/40, Chroma, McHenry, IL) blocking the excitation wavelength. To suppress influences from the afterpulsing typically observed with single-photon avalanche diodes, the fluorescence light was split with a 50/50 beam splitter cube onto two single-photon avalanche diodes (SPCM-AQR-14, Perkin Elmer, Wellesley, MA), and cross-correlation analysis was applied. The high numerical aperture apochromatic water immersion objective (60×, NA 1.2, Olympus, Melville, NY), together with the 50-μm confocal pinhole, resulted in a confocal detection volume of 0.5 fl. The system was calibrated with 5 nM Rhodamine 6G solution and the focus volume was found to be 0.4 fl. The fluorescence was detected by applying time-correlated single-photon counting with the TimeHarp 200 board. The data was stored in the time-tagged time-resolved mode, which allowed the recording of every detected photon with its individual timing and detection channel information. Concentration of fluorescent particles (micelles and vesicles) in the FCS samples was in the nanomolar range. Other details were the same as in (31).
CD measurements and analysis of thermal unfolding
CD measurements were performed using an upgraded Jasco-720 spectropolarimeter (Jasco, Tokyo, Japan). Normally, 40–80 scans were recorded between 190 and 260 nm with a 1-nm step at +20°C, using a 1-mm optical path cuvette. All spectra were corrected for background. Temperature dependencies of unfolding were measured at 222 nm with a 1-deg/min scan rate and analyzed as described by (32). The thermal unfolding was analyzed using thermodynamic equations for a reversible two-state, N↔U unfolding transition, where N and U are the native and the unfolded states of the protein, respectively. To obtain the transition temperature (Tm) and the enthalpy changes (ΔHu), raw data were fitted by applying nonlinear least-square analysis with six fitting parameters, YN, mN, YU, mU, ΔHu and Tm with the equations (31)
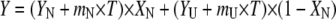 |
(1) |
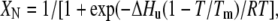 |
(2) |
where Y is the experimentally observed CD signal at a given temperature, YN and YU represent the signals of the pure N and U states at 0 K, and mN and mU are the temperature dependencies of these CD signals for the N and U states, respectively.
RESULTS
Membrane permeabilization experiments
Because fluorocarbons do not mix well with the hydrogenated acyl chains of lipids, fluorinated surfactants are not expected to readily solubilize membranes (they are not detergents). We test their membrane inertness in a vesicle-leakage assay by following the release of ANTS/DPX markers from LUVs after mixing them with (H)FTACs (Fig. 2). Both FTACs caused absolutely no leakage (dotted and dash-dotted lines), which is quite remarkable, as they were present at concentrations way above their CMCs and at a 10-fold molar excess over lipid. A small amount of leakage was detected for 1 mM HFTAC; however, decreasing surfactant concentration to 0.6 mM (Fig. 2) or increasing lipid concentration (9) eliminates the leakage. Much higher leakage levels are obtained with detergents, such as DM (solid line). (This work is not intended as a comprehensive study of detergents; thus, we limit our illustrations to DM, because it is considered to be a mild detergent and because its CMC value of 0.17 mM falls into the range of CMCs for fluorinated surfactants.) Because DM's molecular weight is smaller than that of (H)FTAC, this difference would have been even more pronounced if we had compared the leakage caused by surfactants and detergent on a per-weight, rather than a per-mole, basis.
FIGURE 2.

Permeabilization of lipid vesicles by various surfactants. Solutions of surfactants were mixed with LUV preloaded with ANTS/DPX markers at time zero. Release of markers was followed by changes in fluorescence and normalized to 100% after complete solubilization of LUV in 1% Triton. The final mixture contained 0.2 mM lipid and various amounts of surfactants or DM detergent (CMC 0.17 mM). Hemifluorinated surfactant HFTAC caused no release of vesicle content at 0.6 mM and a partial release at 1 mM, which was much smaller than that caused by DM. Completely fluorinated surfactants showed no signs of permeabilization despite relatively high concentrations and a 10-fold molar excess over lipid.
Next, we test the ability of fluorinated surfactants to insure efficient pore formation by the T-domain. Previously, in a similar experiment, we demonstrated that HFTAC has this chaperonelike ability (9) (Fig. 1 B). We incubate the T-domain in a membrane-competent form at low pH in the presence of various amounts of surfactants and then compare the pore-forming activity of the samples by adding them to marker-loaded LUVs. The percentage of the marker release, observed 20 min after leakage induction, is plotted as a function of the surfactant concentration in the stock in Fig. 3. (The baseline activity of the T-domain in this experiment is purposely reduced by acid-induced aggregation in the stock (9) to test the ability of the surfactants to rescue it. We emphasize that the data in Fig. 3 refer to the action of surfactants on the T-domain and not on the membranes themselves.) Final mixtures contained 50 nM T-domain and 0.2 mM lipid. The dose-response dependencies for FTAC-C6 and HFTAC are very similar, whereas the behavior for FTAC-C8 is somewhat different. The decrease in chaperonelike activity, observed at higher FTAC-C8 concentrations, can be explained, for example, by retention of the increasing amounts of the T-domain by surfactant micelles in the final solution (the concentration of surfactants in the final sample is ∼1% of that in the stock). This retention would not be affecting the other two surfactants, since their CMCs are an order of magnitude higher (Fig. 1 A). More important, however, is that all surfactants at any concentration enhance the activity of the T-domain above the baseline level, and thus all of them can chaperone its membrane insertion.
FIGURE 3.

Comparison of the chaperonelike ability of fluorinated surfactants to rescue pore-forming activity of the diphtheria toxin T-domain (see text for details). Incubation of the T-domain in concentrated stock solutions at low pH leads to aggregation-related deactivation, whereas it has been demonstrated that additions of HFTAC rescue T-domain activity by preventing nonproductive protein aggregation (9). Addition of surfactants to the T-domain stock leads to higher permeabilization of LUV preloaded with ANTS/DPX by the protein (compare data to the baseline). This chaperonelike ability of the HFTAC (squares) is shared by both FTAC-C6 (triangles) and FTAC-C8 (circles).
Testing interactions of surfactants with the T-domain in solution
First, we compare the ability of FTACs to prevent T-domain aggregation at low pH with the ability of HFTAC to do so, determined in our previous publication (9). We use the same experimental scheme: various amounts of fluorinated surfactants are added to the mixture of donor- and acceptor-labeled T-domain at neutral pH, and then an aliquot of concentrated acidic buffer is added to bring the pH to 4.6. In the absence of surfactants, such acidification results in aggregation, readily detectable by the appearance of the FRET-associated peak of the donor in the acceptor excitation spectrum (Fig. 4 A, solid line). This peak is not observed when either of the surfactants is present at 0.6 mM concentration (dashed and dotted lines), indicating an absence of aggregation. In Fig. 4 B, we have plotted the relative decrease of FRET-associated intensity in the presence of various amounts of different (H)FTACs. All of these are quite efficient at preventing T-domain aggregation, even at concentrations of 0.1–0.2 mM, which are below the CMCs for HFTAC and FTAC-C6.
We examined surfactant interactions of the T-domain at different pH by following the fluorescence of the environment-sensitive dye NBD. The probe was selectively attached to various protein sites by reacting with single-cysteine mutants, such as Q369C, with the labeling site on the hydrophobic helix 9. Addition of (H)FTACs to this dye-labeled mutant (designated as DTT-369NBD) results in fluorescence increase at either neutral or acidic pH (Fig. 5), indicating that surfactants interact with both globular and membrane-competent forms of the protein. However, the modes of interaction are different. At pH 8 (Fig. 5 A), addition of monomeric surfactants causes only a marginal change in fluorescence, whereas the presence of micelles leads to a strong increase of intensity and pronounced spectral shift, consistent with the transfer of the probe into a more hydrophobic environment. Acidification leads to conformational change, exposing the hydrophobic core and resulting in an increased fluorescence of DTT-369NBD (Fig. 5 B). It is not clear whether the possible aggregation of the T-domain contributes to the change of the signal. The presence of fluorinated surfactants leads to further increase in fluorescence, but the difference in increase caused by monomers and micelles is small. Similar changes in NBD intensity were observed with DTT-350NBD and DTT-378NBD (not shown), in which the probe is placed at the hydrophobic site of the T-domain, known to insert into the membrane (13). However, when NBD was attached to a polar site of the T-domain not expected to insert into the bilayer (e.g., DTT-235NBD), the intensity was low at either pH and addition of surfactants at any concentration had no effect (data not shown).
FIGURE 5.

Interactions of NBD-labeled single-cysteine mutant of diphtheria toxin T-domain (DTT-369NBD) with fluorinated surfactants at concentrations below (lighter lines) and above their CMCs (normal lines). NBD fluorescence was measured in samples containing 1 μM protein without any surfactants (solid lines) and in the presence of 0.1 mM or 0.6 mM HFTAC (dashed lines); 0.1 mM or 0.6 mM FTAC-C6 (dotted lines); 0.01 mM or 0.1 mM FTAC-C8 (dash-dotted lines). (A) For fully folded DTT at pH 8, the change in NBD emission depends strongly on whether surfactants are present at concentrations below or above their CMCs. (B) Acidification leads to conformational-change aggregation (see Fig. 4) causing an increase in NBD emission (the scales in both panels use the same intensity units). At this pH, 4.6, additional increase in emission caused by the surfactants no longer depends dramatically on concentrations in excess of the corresponding CMCs.
We checked the secondary structure of the folded T-domain at pH 8 in the presence of 0.6 mM surfactants by CD spectroscopy (Fig. 6 A). Although the surfactants are bound to the protein under these conditions (Fig. 5 A), they do not affect its secondary structure. On the other hand, addition of detergents causes certain structural rearrangements, as evidenced by CD in the case of the DM (thin solid line). A similarly altered CD spectrum was also observed in the presence of sodium dodecyl sulfate (not shown). At low pH, however, the CD appearance of the T-domain is affected by the presence of surfactants (Fig. 6 B). Exposure of hydrophobic patches and subsequent aggregation in the absence of membranes results in a lower CD signal, presumably due to protein precipitation and adhesion to the cuvette. Since it is rather difficult to quantitate the actual amount of the protein in the sample, we rely on the known concentration of the stock solution (kept at pH 8) to calculate the molar ellipticity. Thus, the resulting reduction in CD signal for DTT in the absence of surfactants (Fig. 6, A and B, heavy solid lines) is likely to be a combination of true unfolding and sample loss. The latter can be prevented by the presence of surfactants. Indeed, samples with 0.6 mM FTAC-C6 (Fig. 6 B, heavy dotted line) and 0.2 mM FTAC-C8 (dash-dotted line) have an identical CD signal higher than that obtained with no surfactants. It is of interest that the presence of FTAC-C6 at a sub-CMC of 0.2 mM has a similar effect (thin dotted line), which correlates with its ability to prevent aggregation (Fig. 4 B).
FIGURE 6.

Effects of surfactants on the far-UV CD spectrum of 5 μM DTT at pH 8 (A) and pH 4.6 (B). (A) DTT in buffer at pH 8 (solid heavy line) and in the presence or 0.6 mM surfactants HFTAC (dashed line), FTAC-C6 (dotted line), and FTAC-C8 (dash-dotted line) and DM detergent (solid light line). Even at concentrations above their CMCs, fluorinated surfactants do not cause changes in protein secondary structure at this pH. (B) DTT in buffer at pH 4.6 (solid line) and in the presence of 0.6 mM FTAC-C6 (heavy dotted line), 0.2 mM FTAC-C6 (thin dotted line), or 0.2 mM FTAC-C8 (dash-dotted line). Acidification triggers exposure of hydrophobic patches of the T-domain, which in the absence of membranes leads to aggregation (see Fig. 4 and (9)) and partial loss of the sample. The latter is seen here as a loss of CD signal (solid line), which can be rescued by the presence of surfactants.
In addition to CD measurements at 20°C, we monitored the effects of surfactants on T-domain thermal unfolding. Because the low-pH molten globule state does not undergo a cooperative melting transition, we examined the temperature dependence of the ellipticity at 222 nm for the globular T-domain only at pH 8 (Figs. 6 and 7). When present below CMCs (e.g., FTAC-C6 in Fig. 7 A, solid symbols), the surfactants did not change the transition temperature (∼77°C), but reduced the unfolding enthalpy ΔHu. At higher concentrations, they practically abolished the cooperative unfolding transition (Fig. 7 A, open symbols), which makes them similar to detergents (e.g., Fig. 7 B, DM). The concentration dependence of the ΔHu coincides for HFTAC and FTAC-C6, but is much steeper for FTAC-C8 (Fig. 8).
FIGURE 7.

Thermal unfolding of DTT monitored by changes in molar ellipticity at 222 nm in the presence of FTAC-C6 (upper) and DM (lower). Solid lines correspond to the least-squares analysis with Eqs. 1 and 2 to determine transition temperature Tm and enthalpy ΔHu. Increasing the concentration of FTAC-C6 leads to a progressive decrease in ΔHu (similar to that seen with other surfactants, Fig. 8) without the change in Tm, until the transition becomes undefined at 0.6 mM surfactant. Similar loss of cooperative transition is also observed in 0.6 mM DM (lower).
FIGURE 8.

Dependence of the enthalpy of thermal unfolding of DTT (ΔHu) on the concentration of fluorinated surfactants in the sample. Lines correspond to the linear approximations of the data: dashed line, HFTAC (squares); dotted line, FTAC-C6 (triangles); and dotted line, FTAC-C8 (circles).
Testing interactions of surfactants with the membranes and membrane-inserted T-domain
The lack of efficient membrane permeabilization by surfactants (Fig. 2) does not guarantee that they don't interact with the lipid bilayer. They can also potentially interact with the membrane-inserted protein. The latter scenario could not be tested by following fluorescence of the NBD-labeled T-domain, as was done in solution (Fig. 5), because membrane insertion of the labeled T-domain affects NBD fluorescence. The best way to test for membrane association of the surfactants is to place the probe on the surfactants. We have synthesized a fluorescent version of each fluorinated surfactant, wherein one polar headgroup is labeled with the Oregon Green dye. This substitution is not expected to change the nature of the molecular interactions, since polar groups are expected to interact predominantly with water and addition of a polar dye is not likely to affect that.
We examined the mobility of the dye-labeled surfactants by means of fluorescence correlation spectroscopy, which measures intensity fluctuations of a small number of fluorescent molecules diffusing through a small focal volume (32). The autocorrelation curves for HFTAC-OG are presented in Fig. 9 (solid lines). The sample contained 3 nM of OG-labeled surfactant mixed into 50 μM of an unlabeled surfactant. The decay time is fast and is not affected by the addition of either pure lipid LUV (0.5 mM) or vesicles with preinserted T-domain (0.5 mM lipid, 0.2 μM protein). This indicates that the surfactant does not associate with the membrane. If it did, the autocorrelation curve would have moved toward the curve for slow-moving LUV labeled with lipid containing the same OG fluorophore (dashed line). Also, the addition of 1 mM of unlabeled HFTAC to 50 μM LUV (dotted line) did not result in any change in vesicle mobility. The same results were observed for FTAC-C6-OG and FTAC-C8-OG (not shown). Thus, neither of the surfactants associates permanently with the membrane. Nor do any of them sequester the dye-labeled lipid from the bilayer, as that would have added a fast-moving component to OG-labeled LUV in the presence of unlabeled surfactants, which is not observed (note that the dashed and dotted lines in Fig. 9 coincide).
FIGURE 9.

Fluorescence correlation curves of OG-labeled surfactant HFTAC-OG (solid lines) and vesicles containing OG-labeled lipid. The fast mobility of the HFTAC-OG (heavy solid line) is not affected by additions of either LUV alone (normal solid line) or LUV with inserted DTT (light solid line), which indicates the absence of interaction of surfactant with the vesicles. In the case of surfactant interaction with LUV, the autocorrelation curve would have moved toward that for LUV labeled with OG-attached lipid (dashed line). Addition of the 20-fold molar excess of unlabeled HFTAC did not change vesicle mobility (dotted line). Similar results were obtained with the FTACs (not shown), indicating the absence of stable interaction of either surfactant with lipid vesicles.
If the surfactants bind the T-domain in solution, but don't associate with it when it is inserted into the membrane, one would expect them to dissociate from the protein surface during the insertion process. We have directly tested this hypothesis in the following FRET experiments. The T-domain was labeled with Alexa-647 and mixed with OG-labeled surfactants. Because these dyes form a donor/acceptor pair, a strong FRET-associated donor peak is observed in the excitation spectra of the acceptor (Fig. 10, solid line). Upon addition of the LUV, this peak was reduced (dotted and dashed lines), suggesting loss of association of the surfactant with the T-domain upon insertion.
DISCUSSION
Solubilizing MPs for various functional, structural, and thermodynamic studies is normally achieved with the help of detergents, which often makes them unstable (as discussed in (1–5)). Several classes of nondetergent surfactants have been successfully applied in recent years, most prominently amphipols (34–39) and hemifluorinated surfactants (8,9). Although they have been shown to have advantages over detergents in maintaining the active conformation of MPs, detailed understanding of the molecular interactions of fluorinated surfactants with MPs is lacking. Despite their useful properties, these new surfactants are not intended to totally replace detergents, as the latter are essential for membrane solubilization. Normally, detergents are exchanged for surfactants in the final stages of MP purification. In this study, we have avoided the need for a detergent purification step by choosing diphtheria toxin T-domain as a model protein. The advantage of the T-domain is that it exists as a soluble globular protein at neutral pH, yet is converted into a membrane-competent form by acidification and inserts into the lipid bilayer as a part of its physiological action (10,13). We use this pH-triggered conformational change to study the details of the T-domain's interactions with fluorinated surfactants in both states.
Previously, we demonstrated, by a combination of FRET and pore-formation experiments, that the efficiency of the T-domain's membrane insertion is pathway-dependent and is affected by nonproductive aggregation at low pH (9). We also established that the hemifluorinated surfactant HFTAC suppresses aggregation in solution, and, by doing so, facilitates the correct insertion/folding of the T-domain into lipid vesicles. In this study, we demonstrate that this chaperonelike ability is shared by totally fluorinated FTACs (Figs. 3 and 4). Complete fluorination of hydrophobic chains promotes their self-association; thus, the surfactant with the same number of carbons (8) in the tail will have a lower CMC than a hemifluorinated version: 0.0.3 mM for FTAC-C8 and 0.45 mM for HFTAC (Fig. 1 A). It is obvious that the strength of a hydrophobic interaction is reduced by shortening the tail; hence, the CMC for FTAC-C6 is 0.33 mM (7). The original rationale behind the design of a nonfluorinated tip in HFTAC was to amend its interaction with MPs (8). Our data suggest that, at least for the T-domain, this feature is not necessary and HFTAC acts in a manner similar to FTAC-C6 (e.g., Figs. 3, 4, and 8), a totally fluorinated analog with a comparable CMC. An FTAC-C8 surfactant, which has a longer hydrophobic chain than C6, exhibits a comparable effect on the T-domain's properties, although its dose-response dependencies are altered due to its lower CMC.
The ideal chaperone surfactant for MP studies should 1), maintain the MP in solution; 2), minimally perturb the MP's structure; 3), dissociate from the MP during membrane insertion; and 4), not partition into the lipid bilayer. Let us consider how various surfactants and detergents compare to this ideal. The first requirement is satisfied for either surfactants or detergents and is a prerequisite for their applicability. It is worth noting, however, that at least for the T-domain, fluorinated surfactants were able to prevent aggregation at concentrations below their CMC (Figs. 4 B and 6 B). Apparently, the formation of the micelle is not necessary and covering the exposed hydrophobic area of the T-domain by a few surfactant molecules is sufficient to keep it in solution at low pH. In fact, the ability to maintain their function over a wide pH range distinguishes (H)FTACs from amphipols, which are anionic compounds bearing many carboxylate groups and thus are unstable at acidic pH (3).
The need for gentle solubilization that does not alter MP conformation was the rationale behind the introduction of nondetergent surfactants in the first place. The use of our model protein allows direct comparison of the structural perturbation by surfactants, at least for the folded form at neutral pH. Our data indicate that (H)FTACs had no effect on the secondary structure of the T-domain, whereas at least some detergents affected the T-domain CD spectra (Fig. 6 A). This suggests that a membrane-competent conformation, formed at low pH, might also be affected less by the presence of surfactants than by the presence of detergents. However, the cooperative unfolding transition of the T-domain at pH 8 is affected by surfactants and detergents alike, especially when they are present at concentrations above the CMC (Fig. 7). At concentrations below the CMC, fluorinated surfactants did not alter the temperature of the unfolding, though they decreased its cooperativity. The latter is seen as a decrease in the unfolding enthalpy, which changes linearly with surfactant concentration (Fig. 8). Here, again, the behaviors of the two surfactants with similar CMCs (HFTAC and FTAC-C6) are almost indistinguishable, but FTAC-C8 behaves differently. The relatively shallow linear dependence for the former is encouraging for possible future application of these surfactants in unfolding studies of MPs. The fact that the variation in enthalpy is not accompanied by changes in Tm distinguishes this system from conventional unfolding systems, for which linear dependence between ΔHu and Tm gives the value of specific heat capacity. Thus, the change in ΔHu is likely to be the property of the surfactant, and the heat capacity of the complex is not maintained with increasing surfactant concentration.
LUV leakage experiments demonstrated that FTACs cause no permeabilization of lipid vesicles, whereas the ability of HFTAC to cause leakage is very small, especially as compared with that of regular detergents (Fig. 2). Moreover, application of fluorescently labeled surfactants and the FCS methodology indicated that neither of the surfactants associated stably with the lipid bilayer (Fig. 9). A partial leakage observed with HFTAC at high excess over lipid is likely to be due to transient binding and bilayer destabilization. FCS experiments also ruled out the sequestering of the lipid from LUVs into surfactant micelles. This membrane inertness, especially impressive in FTACs, distinguishes fluorinated surfactants from detergents, and even from amphipols, which, though they do bind to lipid bilayers (3,4,35), do not, as a rule, solubilize them (40,41). Moreover, we found that surfactants are not associated with vesicles with preinserted T-domain, or are even shed from the T-domain interface when it inserts into the lipid bilayer (Fig. 10).
Given the evidence discussed above, we conclude that fluorinated surfactants have important advantages over detergents or even amphipols as new media for studies of MPs. They combine efficiently gentle solubilization without structurally altering MPs in a broad pH range, with complete absence of association with the lipid bilayer. This makes them potentially useful for a variety of functional and structural applications in which detergents are currently utilized. In addition, the unique properties of fluorinated surfactants open up possibilities for totally novel approaches that have been unexplored so far—particularly in thermodynamic studies of MPs. Currently, exploration of MP stability by denaturation is hindered by irreversible aggregation of unfolded proteins, which may be prevented by the use of surfactants, provided they do not interact with the bilayer. Another possibility is to use the surfactants to chaperone membrane insertion of constitutive MPs to determine the free energy stabilizing their native conformations in the bilayer. Until now, this approach, based on thermodynamic measurements of membrane partitioning (42,43), has been limited to short peptides (44–47) and nonconstitutive proteins ((23), Posokhov, Rodnin, Lu, and Ladokhin, Biochemistry, 2008, in press). Note that dialysis-based reconstitution from detergents cannot be used for thermodynamic characterization of insertion because 1), it is not an equilibrium technique, and 2), membrane association of detergents will affect the energetics of the insertion process. The use of fluorinated surfactants circumvents these limitations and in principle allows observation of direct insertion of MPs into preformed bilayers under equilibrium conditions.
Acknowledgments
We are grateful to Dr. J.-L. Popot for his critical comments and to Mr. M. D. A. Myers for his editorial assistance.
This research was supported by National Institutes of Health grant GM-069783.
Dr. Palchevskyy's present address is Institute of Molecular Biology and Genetics, National Academy of Sciences of Ukraine, Kiev, Ukraine.
Abbreviations used: MP, membrane protein; HFTAC, hemifluorinated surfactant C2H5C6F12C2H4-S-poly-Tris-(hydroxymethyl)aminomethane with a critical micelle concentration (CMC) of 0.45 mM; FTAC-C6 and FTAC-C8, fluorinated surfactants C6F13C2H4-S-poly-Tris-(hydroxymethyl)aminomethane and C8F17C2H4-S-poly-Tris-(hydroxymethyl)aminomethane with CMCs of 0.33 mM and 0.03 mM, respectively; DM, detergent n-dodecyl-β-D-maltopyranoside with a CMC of 0.17 mM; DTT (or T-domain), diphtheria toxin T-domain; DTT-Donor (or DTT-Alexa532), DTT-Acceptor (or DTT-Alexa647), T-domain single-cysteine mutant N235C labeled with the FRET donor dye (Alexa-532) or acceptor dye (Alexa-647), respectively; DTT-235NBD, DTT-350NBD, DTT-369NBD, and DTT-378NBD, NBD-labeled single-cysteine mutants N235C, L350C, Q369C, and P378C of DTT; NBD, 7-nitrobenz-2-oxa-1,3-diazol-4-yl; FRET, Förster resonance energy transfer; ANTS, 8-aminonaphthalene-1,3,6 trisulfonic acid; DPX, p-xylene-bis-pyridinium bromide; LUV, extruded large unilamellar vesicles of 100 nm diameter; POPC, palmitoyloleoylphosphatidylcholine; POPG, palmitoyloleoylphosphatidylglycerol; CD, circular dichroism; OG, Oregon Green 488 dye; HFTAC-OG, FTAC-C6-OG, and FTAC-C8-OG, OG-labeled surfactants in which a single Oregon Green 488 dye is attached to one of the hydroxyl groups of the polar head; FCS, fluorescence correlation spectroscopy.
Editor: Mark Girvin.
References
- 1.Bowie, J. U. 2001. Stabilizing membrane proteins. Curr. Opin. Struct. Biol. 11:397–402. [DOI] [PubMed] [Google Scholar]
- 2.Gohon, Y., and J.-L. Popot. 2003. Membrane protein-surfactant complexes. Curr. Opin. Colloid Interface Sci. 8:15–22. [Google Scholar]
- 3.Popot, J.-L., E. A. Berry, D. Charvolin, C. Creuzenet, C. Ebel, D. M. Engelman, M. Flötenmeyer, F. Giusti, Y. Gohon, P. Hervé, Q. Hong, J. H. Lakey, K. Leonard, H. A. Shuman, P. Timmins, D. E. Warschawski, F. Zito, M. Zoonens, B. Pucci, and C. Tribet. 2003. Amphipols: polymeric surfactants for membrane biology research. Cell. Mol. Life Sci. 60:1559–1574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tribet, C., R. Audebert, and J.-L. Popot. 1996. Amphipols: polymers that keep membrane proteins soluble in aqueous solutions. Proc. Natl. Acad. Sci. USA. 93:15047–15050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sanders, C. R., A. Kuhn Hoffmann, D. N. Gray, M. H. Keyes, and C. D. Ellis. 2004. French swimwear for membrane proteins. ChemBioChem. 5:423–426. [DOI] [PubMed] [Google Scholar]
- 6.Chabaud, E., P. Barthelemy, N. Mora, J. L. Popot, and B. Pucci. 1998. Stabilization of integral membrane proteins in aqueous solution using fluorinated surfactants. Biochimie. 80:515–530. [DOI] [PubMed] [Google Scholar]
- 7.Barthelemy, P., B. Ameduri, E. Chabaud, J. L. Popot, and B. Pucci. 1999. Synthesis and preliminary assessments of ethyl-terminated perfluoroalkyl nonionic surfactants derived from tris(hydroxymethyl)acrylamidomethane. Org. Lett. 1:1689–1692. [DOI] [PubMed] [Google Scholar]
- 8.Breyton, C., E. Chabaud, Y. Chaudier, B. Pucci, and J.-L. Popot. 2004. Hemifluorinated surfactants: a non-dissociating environment for handling membrane proteins in aqueous solutions? FEBS Lett. 564:312–318. [DOI] [PubMed] [Google Scholar]
- 9.Palchevskyy, S. S., Y. O. Posokhov, B. Olivier, J. L. Popot, B. Pucci, and A. S. Ladokhin. 2006. Chaperoning of insertion of membrane proteins into lipid bilayers by hemifluorinated surfactants: application to diphtheria toxin. Biochemistry. 45:2629–2635. [DOI] [PubMed] [Google Scholar]
- 10.Oh, K. J., L. Senzel, R. J. Collier, and A. Finkelstein. 1999. Translocation of the catalytic domain of diphtheria toxin across planar phospholipid bilayers by its own T domain. Proc. Natl. Acad. Sci. USA. 96:8467–8470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Miller, C. J., J. L. Elliott, and R. J. Collier. 1999. Anthrax protective antigen: prepore-to-pore conversion. Biochemistry. 38:10432–10441. [DOI] [PubMed] [Google Scholar]
- 12.Shatursky, O., A. P. Heuck, L. A. Shepard, J. Rossjohn, M. W. Parker, A. E. Johnson, and R. K. Tweten. 1999. The mechanism of membrane insertion for a cholesterol-dependent cytolysin: a novel paradigm for pore-forming toxins. Cell. 99:293–299. [DOI] [PubMed] [Google Scholar]
- 13.Senzel, L., M. Gordon, R. O. Blaustein, K. J. Oh, R. J. Collier, and A. Finkelstein. 2000. Topography of diphtheria toxin's T domain in the open channel state. J. Gen. Physiol. 115:421–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zakharov, S. D., M. Lindeberg, and W. A. Cramer. 1999. Kinetic description of structural changes linked to membrane import of the colicin E1 channel protein. Biochemistry. 38:11325–11332. [DOI] [PubMed] [Google Scholar]
- 15.Tory, M. C., and A. R. Merrill. 1999. Adventures in membrane protein topology: a study of the membrane-bound state of colicin E1. J. Biol. Chem. 274:24539–24549. [DOI] [PubMed] [Google Scholar]
- 16.Parker, M. W., A. D. Tucker, D. Tsernoglou, and F. Pattus. 1990. Insights into membrane insertion based on studies of colicins. Trends Biochem. Sci. 15:126–129. [DOI] [PubMed] [Google Scholar]
- 17.Johnson, A. E., and M. A. van Waes. 1999. The translocon: a dynamic gateway at the ER membrane. Annu. Rev. Cell Dev. Biol. 15:799–842. [DOI] [PubMed] [Google Scholar]
- 18.Alder, N. N., and A. E. Johnson. 2004. Cotranslational membrane protein biogenesis at the endoplasmic reticulum. J. Biol. Chem. 279:22787–22790. [DOI] [PubMed] [Google Scholar]
- 19.Rapoport, T. A., V. Goder, S. U. Heinrich, and K. E. S. Matlack. 2004. Membrane-protein integration and the role of the translocation channel. Trends Cell Biol. 14:568–575. [DOI] [PubMed] [Google Scholar]
- 20.Hessa, T., H. Kim, K. Bihlmaler, C. Lundin, J. Boekel, H. Andersson, I. Nilsson, S. H. White, and G. von Heijne. 2005. Recognition of transmembrane helices by the endoplasmic reticulum translocon. Nature. 433:377–381. [DOI] [PubMed] [Google Scholar]
- 21.White, S. H., and G. von Heijne. 2005. Do protein-lipid interactions determine the recognition of transmembrane helices at the ER translocon? Biochem. Soc. Trans. 33:1012–1015. [DOI] [PubMed] [Google Scholar]
- 22.Bowie, J. U. 2005. Border crossing. Nature. 433:367–369. [DOI] [PubMed] [Google Scholar]
- 23.Ladokhin, A. S., R. Legmann, R. J. Collier, and S. H. White. 2004. Reversible refolding of the diphtheria toxin T-domain on lipid membranes. Biochemistry. 43:7451–7458. [DOI] [PubMed] [Google Scholar]
- 24.Ladokhin, A. S., and H. T. Haigler. 2005. Reversible transition between the surface trimer and membrane-inserted monomer of annexin 12. Biochemistry. 44:3402–3409. [DOI] [PubMed] [Google Scholar]
- 25.Bennett, M. J., S. Choe, and D. Eisenberg. 1994. Refined structure of dimeric diphtheria toxin at 2.0 Å resolution. Protein Sci. 3:1444–1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhan, H., J. L. Elliott, W. H. Shen, P. D. Huynh, A. Finkelstein, and R. J. Collier. 1999. Effects of mutations in proline 345 on insertion of diphtheria toxin into model membranes. J. Membr. Biol. 167:173–181. [DOI] [PubMed] [Google Scholar]
- 27.Mayer, L. D., M. J. Hope, and P. R. Cullis. 1986. Vesicles of variable sizes produced by a rapid extrusion procedure. Biochim. Biophys. Acta. 858:161–168. [DOI] [PubMed] [Google Scholar]
- 28.Hope, M. J., M. B. Bally, L. D. Mayer, A. S. Janoff, and P. R. Cullis. 1986. Generation of multilamellar and unilamellar phospholipid vesicles. Chem. Phys. Lipids. 40:89–107. [Google Scholar]
- 29.Ladokhin, A. S., W. C. Wimley, K. Hristova, and S. H. White. 1997. Mechanism of leakage of contents of membrane vesicles determined by fluorescence requenching. Methods Enzymol. 278:474–486. [DOI] [PubMed] [Google Scholar]
- 30.Ladokhin, A. S., S. Jayasinghe, and S. H. White. 2000. How to measure and analyze tryptophan fluorescence in membranes properly, and why bother? Anal. Biochem. 285:235–245. [DOI] [PubMed] [Google Scholar]
- 31.Posokhov, Y. O., M. V. Rodnin, L. Lu, and A. S. Ladokhin. 2008. Membrane insertion pathway of Annexin B12: thermodynamic and kinetic characterization by fluorescence correlation spectroscopy and fluorescence quenching. Biochemistry. 47:5078–5087. [DOI] [PubMed] [Google Scholar]
- 32.Eftink, M. R. 1994. The use of fluorescence methods to monitor unfolding transitions in proteins. Biophys. J. 66:482–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Haustein, E., and P. Schwille. 2003. Ultrasensitive investigations of biological systems by fluorescence correlation spectroscopy. Methods. 29:153–166. [DOI] [PubMed] [Google Scholar]
- 34.Pocanschi, C. L., T. Dahmane, Y. Gohon, H.-J. Apell, J. H. Kleinschmidt, and J.-L. Popot. 2005. Amphipathic polymers: tools to fold integral membrane proteins to their active form. Biochemistry. 45:13954–13961. [DOI] [PubMed] [Google Scholar]
- 35.Zoonens, M., L. J. Catoire, F. Giusti, and J. L. Popot. 2005. NMR study of a membrane protein in detergent-free aqueous solution. Proc. Natl. Acad. Sci. USA. 102:8893–8898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pocanschi, C. L., T. Dahmane, Y. Gohon, F. Rappaport, H. J. Apell, J. H. Kleinschmidt, and J. L. Popot. 2006. Amphipathic polymers: tools to fold integral membrane proteins to their active form. Biochemistry. 45:13954–13961. [DOI] [PubMed] [Google Scholar]
- 37.Lebaupain, F., A. G. Salvay, B. Olivier, G. Durand, A. S. Fabiano, N. Michel, J. L. Popot, C. Ebel, C. Breyton, and B. Pucci. 2006. Lactobionamide surfactants with hydrogenated, perfluorinated or hemifluorinated tails: physical-chemical and biochemical characterization. Langmuir. 22:8881–8890. [DOI] [PubMed] [Google Scholar]
- 38.Zoonens, M., F. Giusti, F. Zito, and J. L. Popot. 2007. Dynamics of membrane protein/amphipol association studied by Förster resonance energy transfer: implications for in vitro studies of amphipol-stabilized membrane proteins. Biochemistry. 46:10392–10404. [DOI] [PubMed] [Google Scholar]
- 39.Gohon, Y., T. Dahmane, R. W. Ruigrok, P. Schuck, D. Charvolin, F. Rappaport, P. Timmins, D. M. Engelman, C. Tribet, J. L. Popot, and C. Ebel. 2008. Bacteriorhodopsin/amphipol complexes: structural and functional properties. Biophys. J. 94. In press. [DOI] [PMC free article] [PubMed]
- 40.Ladaviere, C., M. Toustou, T. Gulik-Krzywicki, and C. Tribet. 2001. Slow reorganization of small phosphatidylcholine vesicles upon adsorption of amphiphilic polymers. J. Colloid Interface Sci. 241:178–187. [DOI] [PubMed] [Google Scholar]
- 41.Nagy, J. K., A. Kuhn Hoffmann, M. H. Keyes, D. N. Gray, K. Oxenoid, and C. R. Sanders. 2001. Use of amphipathic polymers to deliver a membrane protein to lipid bilayers. FEBS Lett. 501:115–120. [DOI] [PubMed] [Google Scholar]
- 42.White, S. H., W. C. Wimley, A. S. Ladokhin, and K. Hristova. 1998. Protein folding in membranes: determining the energetics of peptide-bilayer interactions. Methods Enzymol. 295:62–87. [DOI] [PubMed] [Google Scholar]
- 43.White, S. H., A. S. Ladokhin, S. Jayasinghe, and K. Hristova. 2001. How membranes shape protein structure. J. Biol. Chem. 276:32395–32398. [DOI] [PubMed] [Google Scholar]
- 44.Wimley, W. C., and S. H. White. 1996. Experimentally determined hydrophobicity scale for proteins at membrane interfaces. Nat. Struct. Biol. 3:842–848. [DOI] [PubMed] [Google Scholar]
- 45.Ladokhin, A. S., and S. H. White. 2001. Protein chemistry at membrane interfaces: non-additivity of electrostatic and hydrophobic interactions. J. Mol. Biol. 309:543–552. [DOI] [PubMed] [Google Scholar]
- 46.Ladokhin, A. S., and S. H. White. 1999. Folding of amphipathic α-helices on membranes: energetics of helix formation by melittin. J. Mol. Biol. 285:1363–1369. [DOI] [PubMed] [Google Scholar]
- 47.Fernandez-Vidal, M., S. Jayasinghe, A. S. Ladokhin, and S. H. White. 2007. Folding amphipathic helices into membranes: amphiphilicity trumps hydrophobicity. J. Mol. Biol. 370:459–470. [DOI] [PMC free article] [PubMed] [Google Scholar]