

Summary
DNA wrapped in nucleosomes is sterically occluded from many protein complexes that must act on it; how such complexes gain access to nucleosomal DNA is not known. In vitro studies on isolated nucleosomes show that they undergo spontaneous partial unwrapping conformational transitions, which make the wrapped nucleosomal DNA transiently accessible. Thus, site exposure might provide a general mechanism allowing access of protein complexes to nucleosomal DNA. However, existing quantitative analyses of site exposure focused on single nucleosomes, while the presence of neighbor-nucleosomes, and concomitant chromatin folding, might significantly influence site exposure. Here, we carry out quantitative studies on the accessibility of nucleosomal DNA in homogeneous nucleosome arrays. Two striking findings emerge. Organization into chromatin fibers changes the accessibility of nucleosomal DNA only modestly, from ~3-fold decreases in accessibility to ~8-fold increases. This means that nucleosome arrays are intrinsically dynamic and accessible even when they are visibly condensed. In contrast, chromatin folding decreases the accessibility of linker DNA by as much as ~50-fold. Thus, nucleosome positioning dramatically influences the accessibility of target sites located inside nucleosomes, while chromatin folding dramatically regulates access to target sites in linker DNA.
Keywords: chromatin, nucleosome, higher-order structure, site exposure, gene regulation
Introduction
Eukaryotic genomes are organized into repeating arrays of nucleosomes, which occlude most of the genomic DNA from the many protein complexes required for genome function1. How such complexes gain access to their DNA target sites in vivo is not known. Genomes encode an intrinsic nucleosome organization in which transcription factor binding sites that need to be accessible have a relatively lower probability of being occluded inside a nucleosome2; but these are probabilistic biases only; they do not keep critical target sites nucleosome free 3; 4; 5; 6; 7; 8; 9. Nucleosomal DNA needs to be unwrapped at times to function 10; 11; 12; 13.
Two broad mechanisms have been characterized that provide access to buried nucleosomal target sites. One involves ATP-dependent nucleosome remodeling factors14; 15, which disassemble nucleosomes or allow nucleosomes to redistribute their locations in response to changing constellations of DNA binding proteins. These factors are recruited to particular chromatin regions by site specific DNA binding proteins14; 16; 17, raising the question of how those DNA binding proteins themselves gain access to their own target sites. One idea is that some ATP-dependent remodeling factors may act on nucleosomes ubiquitously, without a requirement for specific binding, thereby rendering chromatin inherently “fluid” for protein binding18. Such a ubiquitous activity has not yet been demonstrated in vivo.
We focus here on a second mechanism for site accessibility in nucleosomes, which is known to occur, is intrinsic to the nucleosomes themselves, and has been implicated in chromatin function in vivo. Nucleosomes spontaneously undergo large-scale conformational fluctuations (“site exposure”) in which stretches of their wrapped DNA partially unwrap off the histone protein surface, starting from one end. Site exposure provides spontaneous access to the entire nucleosomal DNA length. Access to the outer more stretches of the DNA is particularly rapid (spontaneous opening as often as every ~250 milliseconds)19 and efficient (the DNA ends are unwrapped as much as ~1–5% of the time)20; 21, depending on the DNA sequence22.
Two lines of evidence suggest that intrinsic nucleosomal site exposure is important for chromosome function in vivo. First, one consequence of site exposure is to confer a novel nucleosome-dependent positive cooperativity on the binding of pairs of arbitrary site specific DNA binding proteins when their DNA target sites are contained within the same nucleosome23. This phenomenon occurs in vivo24; 25; 26, and is now thought to contribute to cooperativity in transcription factor action, genome-wide27. Second, an analysis of rates of DNA repair by photolyase in vivo concludes that repair occurs too quickly to be explained by known ATP-dependent remodeling activities, and suggests instead that rapid repair is facilitated by intrinsic nucleosome site exposure28.
Existing studies characterized the site exposure process only in isolated nucleosomes; but nucleosomes in vivo occur in long arrays, which, in physiological conditions, compact into higher order structures that could hinder site exposure. Indeed, such chromatin compaction occurring in vivo has long been proposed to contribute to transcriptional gene silencing by sterically occluding DNA from access to activators or polymerases29; 30, although a recent study has challenged this interpretation31.
Here, we ask how the presence of nucleosome neighbors and concomitant chromatin folding influence site exposure. Two striking findings emerge. First, despite the visible compaction of a model 17-nucleosome chromatin fiber, access to target sites within the central nucleosome is only modestly affected relative to access to the same sites in an isolated nucleosome, and these modest changes range from ~3-fold decreases in accessibility to ~8-fold increases. This means that nucleosome arrays are intrinsically dynamic and accessible even when they are visibly condensed. This finding helps explain how upstream activators may have access to their DNA target sites even in transcriptionally silenced chromatin in vivo31. Second, in contrast to the modest changes in accessibility it causes within nucleosomes, chromatin folding greatly decreases the accessibility at sites in the linker DNA between nucleosomes, as much as ~50-fold. Thus, the genome’s intrinsic nucleosome positioning (“chromatin primary structure”) strongly influences the accessibility of target sites that, on average, are located inside nucleosomes, while chromatin fiber folding (“chromatin secondary structure”) regulates access to target sites in regions that, on average, are located in linker DNA.
Results
Reconstitution and characterization of nucleosome arrays
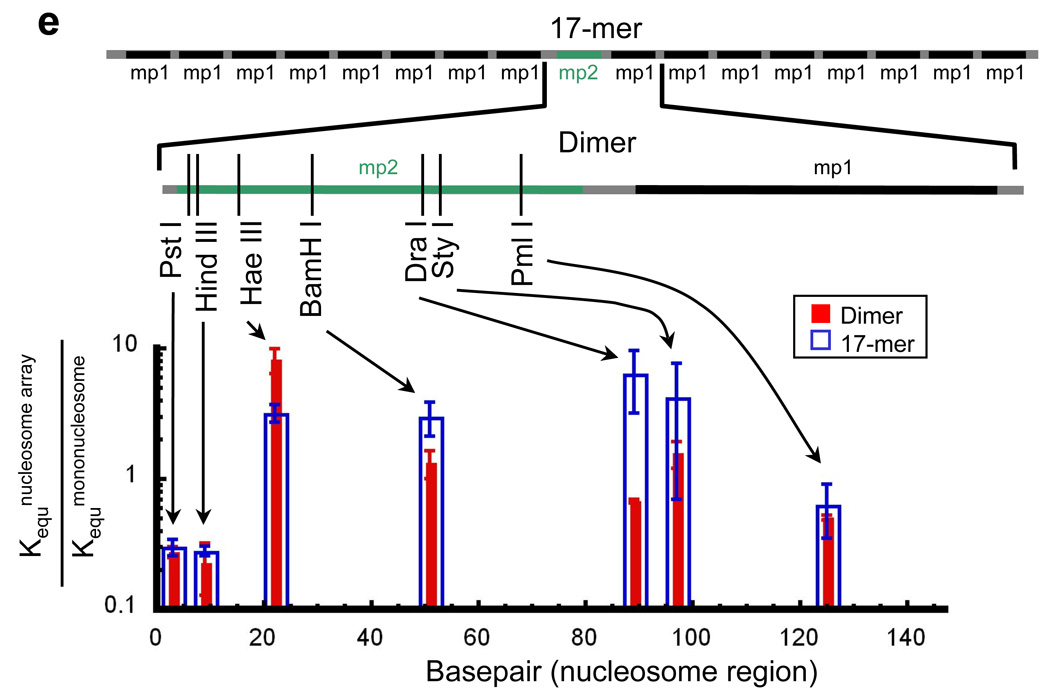
Restriction enzymes face the same steric problems for access to target sites inside nucleosomes as do many eukaryotic regulators and enzymes, and are convenient probes for quantitative analyses of nucleosomal DNA accessibility. Site exposure occurs as a rapid pre-equilibrium21, thus restriction enzymes digest nucleosomal DNA at a rate equal to their digestion rate on naked DNA multiplied by the (small) fraction of time that the nucleosomal target sites look like naked DNA, which is the equilibrium constant for exposure of the restriction site in the nucleosomes (Fig. 1). To assess how organization of nucleosomes into compact chromatin fibers influences DNA accessibility we created two new model systems, having one “test” nucleosome (“mp2”) that is flanked by one distinct “mp1” nucleosome on one side only (a dinucleosome), or one mp2 nucleosome flanked by 8 other mp1 nucleosomes on each side (a nucleosome 17-mer) (Fig. 2). In order that the resulting chromatin fibers have homogeneous nucleosome locations, each mp1 or mp2 nucleosome derives from the 147 bp nucleosome positioning region of sequence 60132, with an exactly repeating length of 30 bp of linker DNA separating consecutive nucleosome core particles. Thus our constructs reproduce essential features of the 177 bp nucleosome repeat systems used in recent biophysical and structural studies in the Richmond and Rhodes laboratories33; 34; 35; 36 and are novel only in their inclusion of a single distinguishable variant of 601, which allows us to probe accessibility within a particular central nucleosome embedded within a highly positioned nucleosome array.
Figure 1.
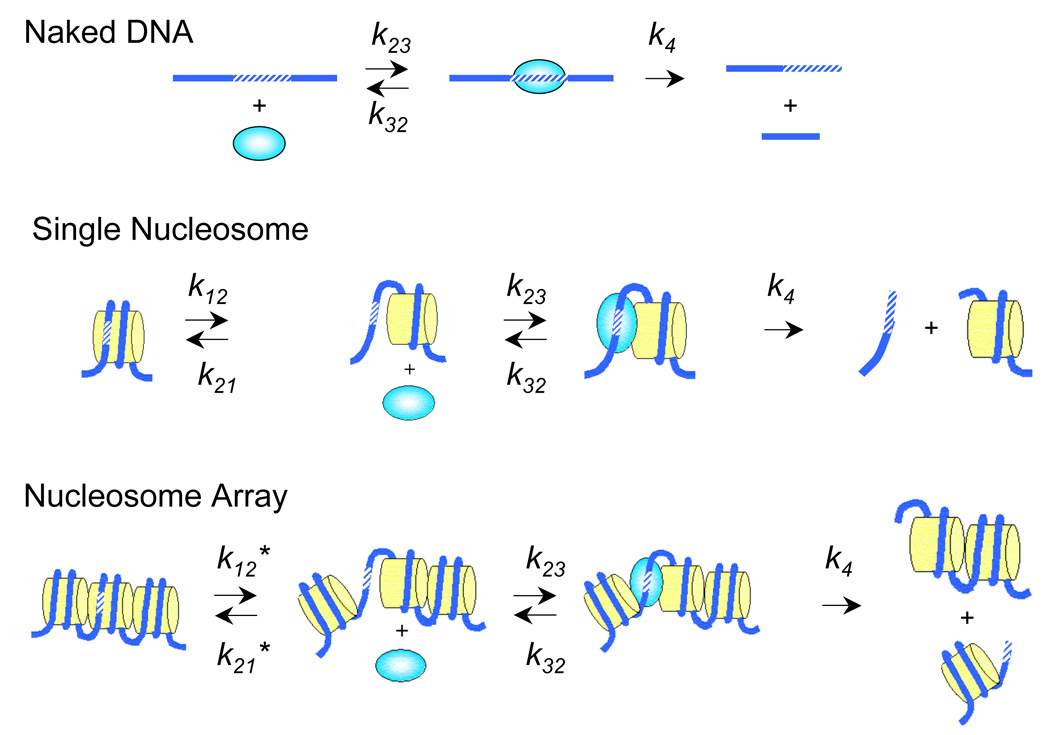
Restriction enzyme digestion assay for site exposure. k12 and k21 are the forward and reverse rates of site exposure of a unique DNA site in a single nucleosome; k12* and k21* are the corresponding rates for exposure of a unique site in a nucleosome array; k23 and k32 are the rates for restriction enzyme binding and unbinding; and k4 is the rate of restriction enzyme catalysis. The restriction enzyme kinetics method determines the equilibrium constant for site exposure, Kequ = k12 / k21 or = k12* / k21*.
Figure 2.
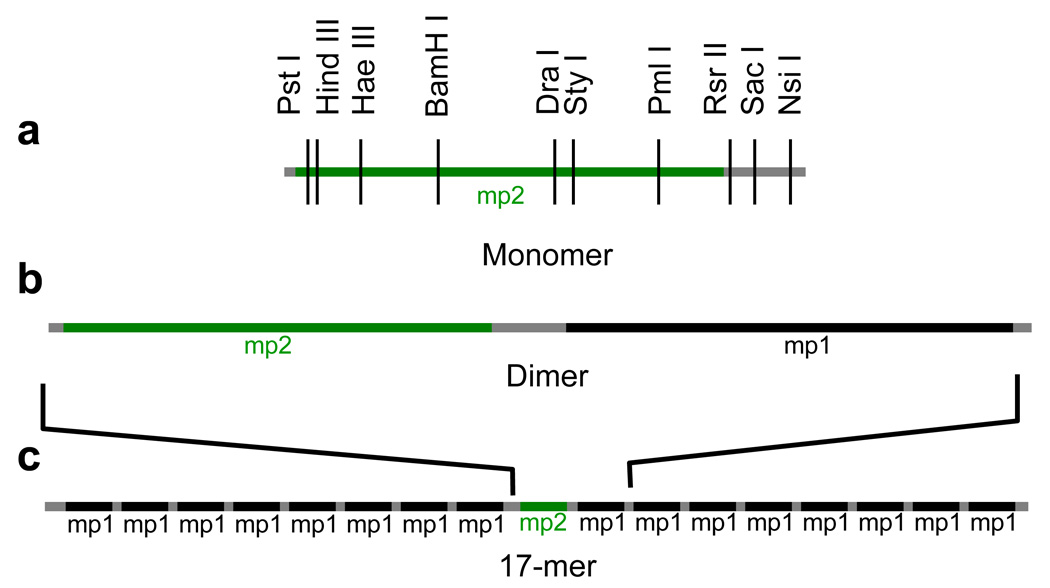
DNA templates used. The nucleosome positioning sequence mp1 is a variant of positioning sequence 60132, while mp2 is a variant of 601.222. mp2 contains many restriction enzyme recognition sites that are not present in mp1, allowing analysis of accessibility at unique sites within the dimer and 17-mer nucleosome arrays.
The nucleosome array reconstitutions were carried out similarly to (ref.33; 34; 35; 36), using excess nucleosome core particle DNA (cpDNA) as a histone buffer. When used together with high affinity nucleosome positioning arrays, the cpDNA allows the preparation of arrays in which every high affinity sequence is occupied by a nucleosome, without significant aggregation occurring. Optimal dimer reconstitution conditions were determined by titration with increasing concentrations of histone octamer (Fig. 3a). Native gel electrophoresis of the products reveals two distinct shifted single histone octamer-containing bands that converge to a single dinucleosome band by the highest histone octamer concentration used (0.8:1 mass of histone to mass of total DNA, 2.4 mol histone octamer per mol nucleosome positioning sequence). Products obtained with this highest histone concentration were purified by sucrose gradient centrifugation (Fig. 3b, lane 4).
Figure 3.
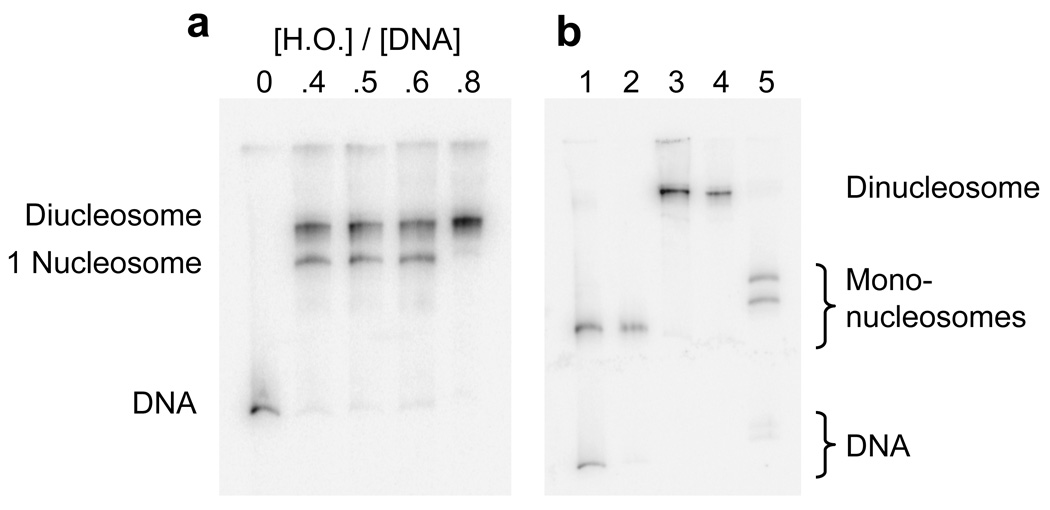
Reconstitution, purification, and characterization of mono- and di-nucleosomes. (a) Native 5% polyacrylamide gel of dinucleosome template DNA reconstituted with increasing concentrations of histone octamer. The shifts in DNA mobility are due to the formation of one and then two nucleosomes on each DNA. Numbers at the top (0–0.8) indicate the mass ratio (w/w) of histone octamer to total DNA used in the reconstitution reaction in that lane. Total DNA includes both the specific (high affinity) dinucleosome template DNA plus low affinity cpDNA competitor present as a histone buffer (see Methods). The specific template DNA saturates with histone octamer even though the amount of octamer is substoichiometric for the total amount of DNA. (b) Mononucleosome (lanes 1, 2) and dinucleosome (lanes 3, 4) reconstitutions before (lanes 1, 3) and after (lanes 2, 4) purification on sucrose gradients. Lane 5, purified dinucleosomes after digestion with 20 units ml−1 Sac I for 1 hour. Sac I cuts in the linker region, resulting in two bands having gel mobilities similar to that of mononucleosomes (lane 2), and two faint bands from digested naked template DNA (e.g., any template DNA that was not incorporated into nucleosomes). Quantification of these bands shows that greater than 95% of the dinucleosome’s positioning sequences are wrapped into nucleosomes.
To confirm that this product is dinucleosomes, and to verify the accuracy of nucleosome positioning within the dinucleosomes, the purified samples were digested in the linker DNA region with Sac I at low enzyme concentration (20 units ml−1) such that naked DNA was fully digested in 1 hour while there was essentially no digestion at sites within a nucleosome. As expected, Sac I fully cut the dinucleosomes into two distinct bands (Fig. 3b, lane 5) having mobilities appropriate for mononucleosomes (Fig. 3b, lane 2) containing extra linker DNA. This confirms that the original reconstitutes had two nucleosomes, and that essentially no dinucleosomes had mispositioned nucleosomes covering the Sac I site. Greater than 95% of all DNA was in the mononucleosomes, implying a near-complete saturation of the two nucleosome positioning sequences with histone octamer. Together these results confirm that the reconstitution procedure produces well-defined, homogeneous dinucleosomes.
We similarly optimized the heptadecamer (17-mer) reconstitutions by titration (Fig. 4a). Native gel electrophoresis reveals a range of mobility shifts that again converges to a distinct band by the highest concentration of histone octamer. This distinct band suggests that the reconstitutions produce a homogeneous population of nucleosome arrays; these were then purified by sucrose gradient centrifugation (Fig. 4b).
Figure 4.
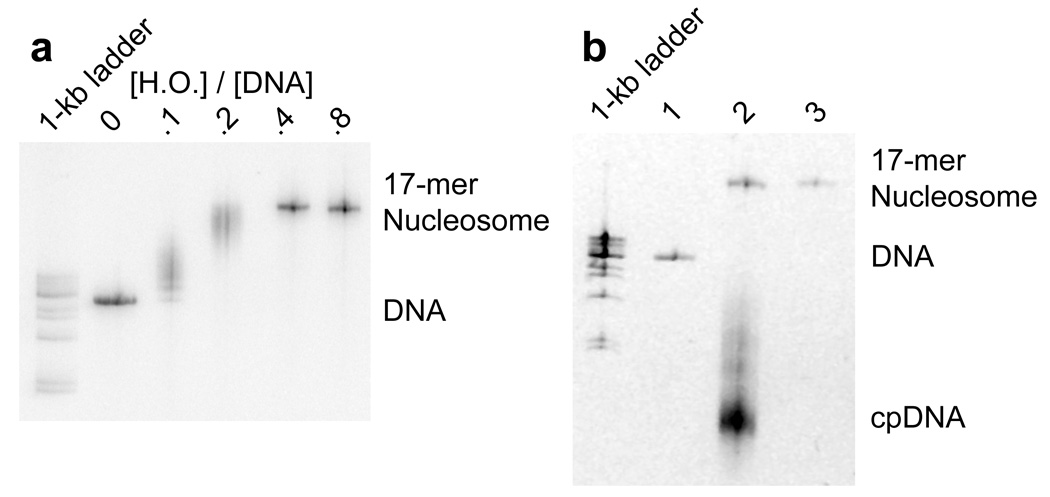

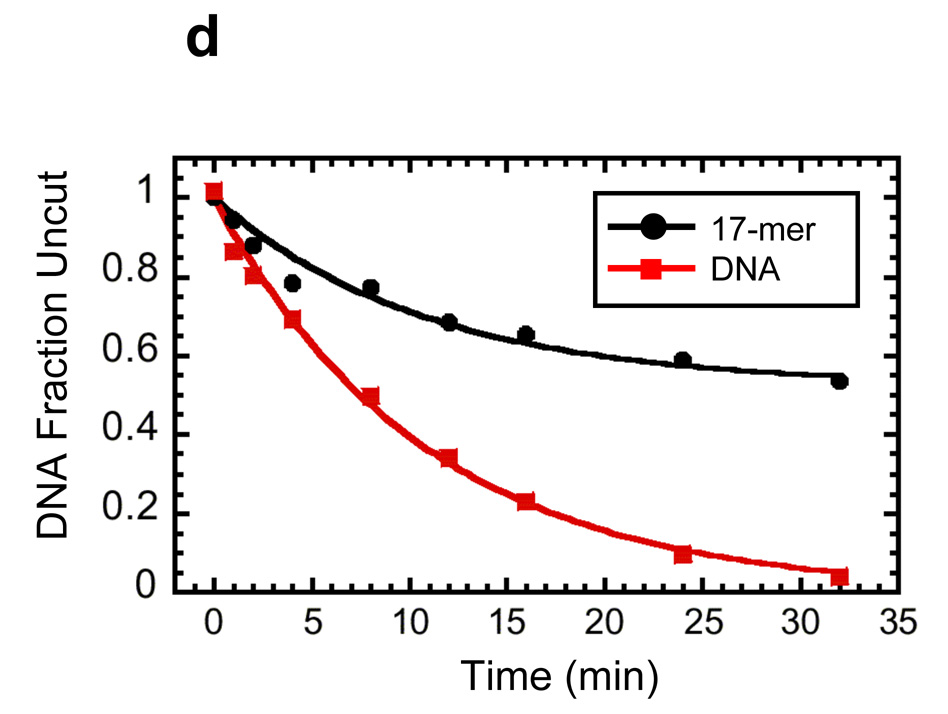
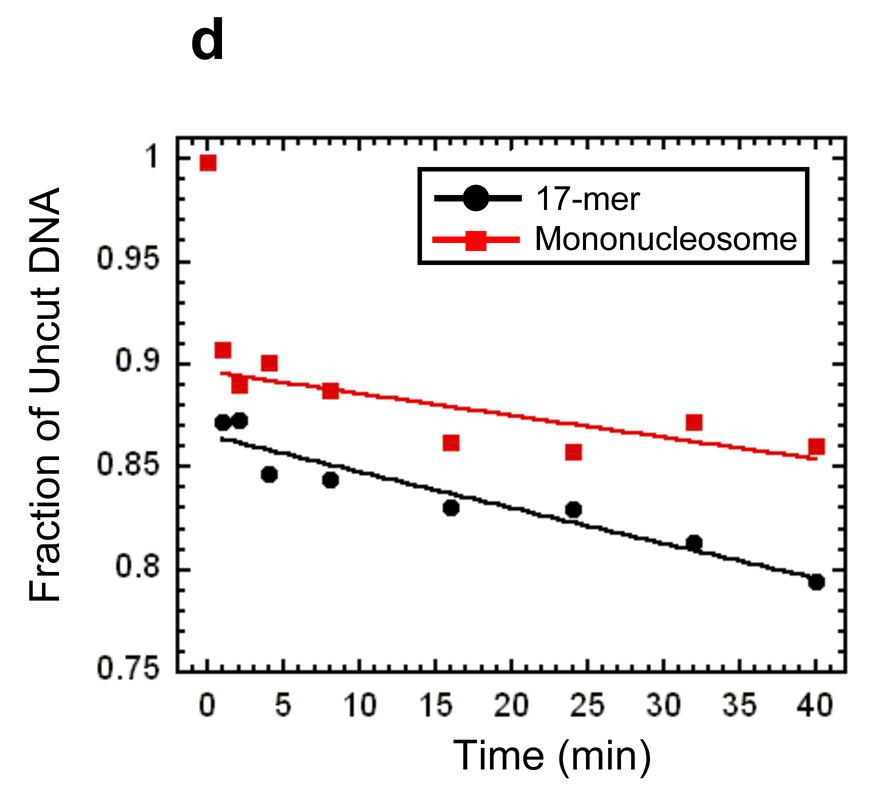
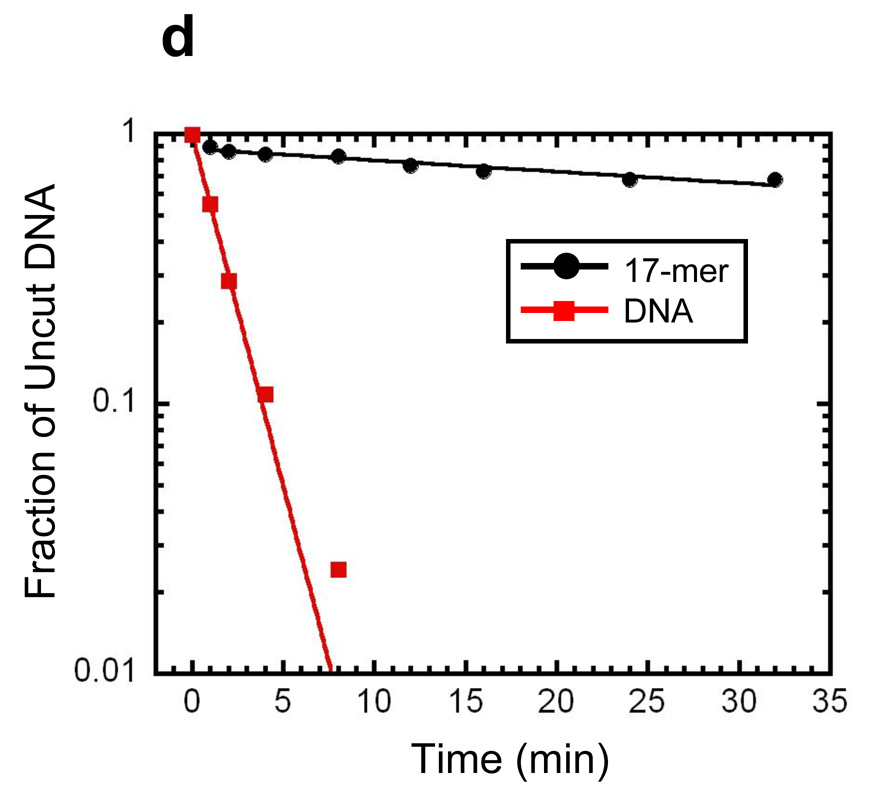
Reconstitution, purification, and characterization of nucleosome 17-mers. (a) Native gel analysis of 17-mer template DNA reconstituted with increasing concentrations of histone octamer. The mass ratio (w/w) of histone octamer to total DNA used in the reconstitution reaction in each lane is indicated. The decrease in mobility is due to nucleosome formation along the DNA template. The mass ratios 0.1 and 0.2 have wide range of gel mobilities owing to the inherent heterogeneity of DNA templates that are partially filled with nucleosomes. In contrast, the mass ratios of 0.4 and 0.8 yield a well-defined shift in gel mobility, suggesting that the DNA template is saturated with nucleosomes. (b) Ethidium-stained native gel analysis of the 17-mer DNA template (lane 1), and reconstituted nucleosome array (0.8 histone to DNA mass ratio) before (lane 2) and after purification (lane 3). The sucrose gradient removes the short low affinity competitor DNA and any aggregates. (c) Native 5% polyacrylamide gel analysis of DNA products from a Mlu I digestion of purified 17-mer nucleosomes mixed with naked DNA. The 17-mer contains 16 Mlu I sites and the naked DNA contains a single MluI site. (d) Quantification of results from (c) showing the fraction of naked DNA and the nucleosome 17-mer remaining uncut, as a function of time. The curves show best fits to the appropriate exponential decays (see Methods). These results show that 0.03 of the positioning sequences are not incorporated into nucleosomes, i.e., that 97 % of all positioning sequences in these nucleosome 17-mers are wrapped into nucleosomes.
To confirm that each mp1 nucleosome positioning region within the 17-mer contains a nucleosome, the array was digested with Mlu I, which cuts inside each mp1 nucleosome positioning region. A trace amount of a 400 bp naked DNA that contains one Mlu I site was added as an internal reference for the rate of naked DNA digestion. A low concentration of Mlu I (20 units ml−1) rapidly digested the naked DNA tracer and any naked DNA in the 17-mers, with negligible digestion internal to nucleosomes (Fig. 4c). Quantitative analysis of the digestion showed that the naked DNA digestion fits to a single exponential decay, with a decay rate of 6 min−1 (Fig. 4d). The fraction of uncut 17-mer decays with the same rate constant to a final value of 0.5. This means that at least 50% of the 17-mers have 100% of their Mlu I sites protected by being wrapped into nucleosomes. Since each array contains 16 mp1 sites, at most 0.5 of any of the 16 Mlu I sites (i.e. at most 3%) are unoccupied by a nucleosome. This in turn implies that at least 97% of all of the Mlu I sites (i.e., at least 97% of the mp1 nucleosome positioning regions) are wrapped into nucleosomes. A similar experiment using Sty I digestion showed that 100% of the Sty I sites in the mp2 nucleosome positioning sequences are incorporated into nucleosomes (results not shown).
We confirmed the biochemical quality of the 17-mers using atomic force microscopy (AFM) imaging. At low salt concentrations, nucleosome arrays adsorb to the substrate surface in an extended form, allowing individual nucleosomes to be resolved (Fig. 5a, b). AFM images of the 17-mers reveal distinct individual nucleosomes evenly spaced along the DNA template. We imaged 18 molecules; 13 of these had 17 well-resolved nucleosomes each; another three appeared to have 17 nucleosomes each, although in each case two putative nucleosomes were not well resolved; finally, the other 2 molecules contained 15 or 16 nucleosomes each. In summary, the 18 molecules together contain 306 nucleosome positioning sequences, and the images showed that 299–303 of these 306 positioning sequences were in fact wrapped into a nucleosome. This confirms the biochemical results that greater than 95% of all of the nucleosome positioning sequences in these 17-mers are incorporated into a nucleosome.
Figure 5.
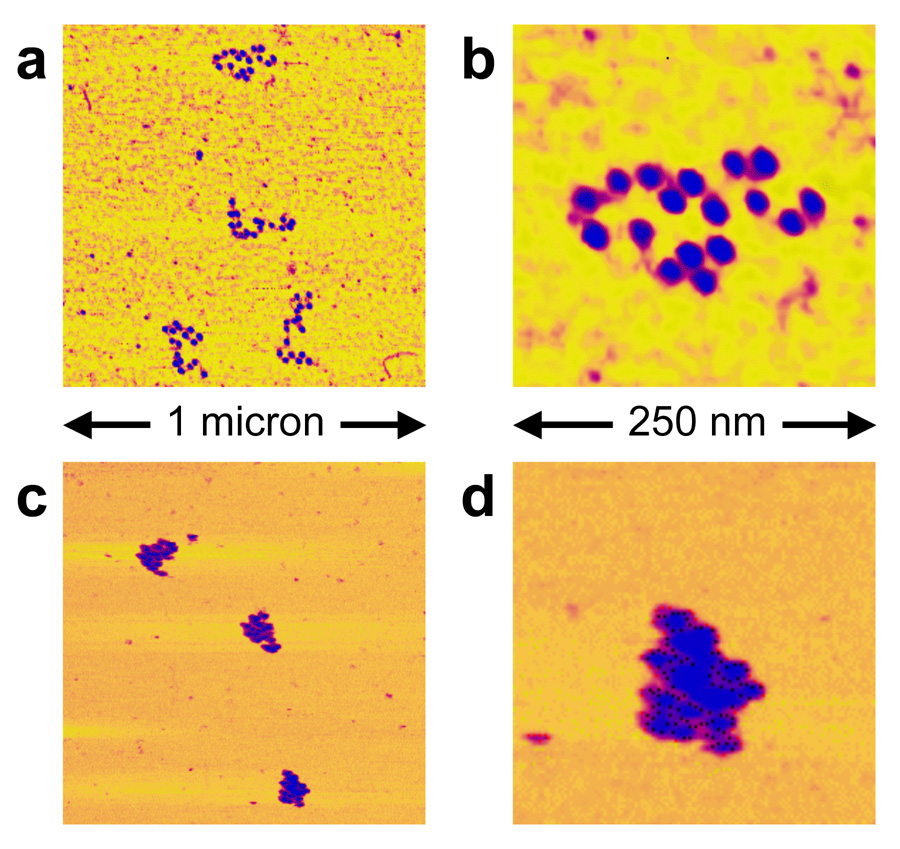
Atomic Force Microscopy images of nucleosome 17-mers. (a, b) purified nucleosome 17-mers adsorbed onto mica in 0.2 × TE (which causes them to adopt extended conformations) and imaged in air; (b) 4× zoom of the upper array in (a). These and additional images confirm the biochemical results showing that the 17-mers are greater than 95% saturated with nucleosomes. (c, d) 17-mers adsorbed in 0.5× TE plus 1 mM MgCl2 (which allows them to compact) and imaged in that buffer. (d) is a 4× zoom of the lower array in (c). As expected, the arrays fold into more compact structures in the presence of MgCl2.
Higher order folding of 17-mer nucleosome arrays
Chains of nucleosomes in physiological solution conditions fold into more compact higher order chromatin structures, even in the absence of the “linker histone” H1 (which is not essential for viability in at least some organisms37). In vitro biophysical studies on 12-mer nucleosome arrays containing the 601 nucleosome positioning sequence with a 177 bp repeat length — exactly the same as in our 17-mer design – show these arrays to undergo a reversible compaction in 1 mM Mg2+ that resembles the compaction of native chromatin fibers in vivo and remains essentially unchanged from 1–100 mM Mg2+ (ref. 33). Thus 12-mer arrays are already maximally compacted by 1 mM Mg2+, and they maintain this compaction in the 5–10 mM Mg2+ concentration range in which restriction enzymes are active. While it is conceivable that the nucleosome arrays could become even more highly compacted for Mg2+ concentrations greater than 100 mM, such conditions are too extreme to be of physiological relevance. Thus others, and we, focus our analyses on the quasi-physiological Mg2+ concentration range of 1–10 mM, in which the arrays are as compacted as they will ever be for any reasonable Mg2+ concentration. Because of the similarities in design, we expect that our 17-mer arrays would undergo a similar Mg2+-dependent folding. Indeed, AFM images of our 17-mers confirm that, like the 12-mer arrays, they are highly compacted already by 1 mM Mg2+ (Fig. 5c, d). Our restriction enzyme digestion assays are carried out in 5–10 mM Mg2+, and thus will probe the effects of both the presence of nucleosome neighbors together with this concomitant chromatin folding.
Site accessibility in folded nucleosome arrays
Having established a protocol for assembly of dinucleosomes and nucleosome 17-mers, we next used the restriction enzyme accessibility assay to quantify how the presence of nucleosome neighbors and concomitant chromatin folding influences accessibility inside the test nucleosome. For the restriction enzyme assay as originally developed, one measures the rate of digestion at a given nucleosomal DNA target site relative to the rate of digestion of the same site as naked DNA, scaled for differences in the enzyme concentrations used. The ratio of these scaled digestion rates yields the equilibrium constant for site exposure of that site in the nucleosome – i.e., the equilibrium fraction of time that a given nucleosomal DNA target site is as available to another protein as is the same site in naked DNA. The experiments are carried out in a rapid-preequilibrium regime (rapid re-wrapping of site-exposed DNA compared to the rate of restriction enzyme binding), where the restriction enzymes serve as neutral reporters of spontaneous nucleosomal site exposure 21; 38. Strictly speaking, the assay measures the “effective accessibility”, which could differ from true accessibility if the properties of DNA unwrapped transiently off the nucleosome surface differs from true naked DNA. Full experimental validation of the assumptions behind this assay have been published previously21; 38.
To apply this procedure for the present case, we would measure rates of digestion of a test nucleosome relative to the same sequence as naked DNA, for the test nucleosome present first in a mononucleosome, and then again in a dinucleosome or in a 17-mer. However, in pilot studies we observed that the rate of digestion at sites inside the test nucleosome in the arrays was similar to that for digestion of the same sites in mononucleosomes. This feature allowed us to improve our experimental design to more accurately measure the effects of nucleosome neighbors and concomitant chromatin folding. Rather than measure the mononucleosome and array sample independently relative to naked DNA, we instead add the mononucleosomes to the array sample together in the same tube, and measure their accessibility directly relative to each other, in the same reaction at the same time.
We used this approach to evaluate the equilibrium accessibility at seven sites spanning the length of the test (mp2) nucleosome (Fig. 6a–d). As shown previously21; 38, rapid mixing of the nucleosomes or nucleosome arrays into the restriction enzyme digestion buffer causes up to 10% of the nucleosomes to fall apart at the zero time-point of the digestion reaction; the resulting naked DNA is effectively instantly digested. We handle this in the kinetic analysis by omitting the first timepoint. With the nucleosome array constructs analyzed here, any nucleosome dissociation is dominated by loss of the test nucleosome itself, rather than the flanking nucleosomes, which, because of test nucleosome’s engineered sequence changes, is lower in affinity (stability) than are the flanking nucleosomes (for details, see Methods); and, as just explained, loss of the test nucleosome is accommodated in our analysis by omitting the first digestion timepoint. At most 3% of the nucleosomes flanking the test nucleosome dissociate (see Methods), thus, gaps in the nucleosome array surrounding the test nucleosome occur with negligible probability, and do not affect the measured accessibilities. Quantitative analysis of the reaction kinetics reveals that the presence of one or many nucleosome neighbors – with concomitant folding of the nucleosome array for the case of the 17-mer arrays – causes detectable but modest changes in equilibrium accessibility (Kequ) of target sites throughout the test nucleosome (Fig. 6e). These changes in equilibrium accessibility due to the nucleosome neighbors range from ~3-fold decreases to ~8-fold increases, which are small in comparison to the 103- to 106-fold quantitative effects on accessibility of wrapping 601 DNA into nucleosomes in the first place22.
Figure 6.

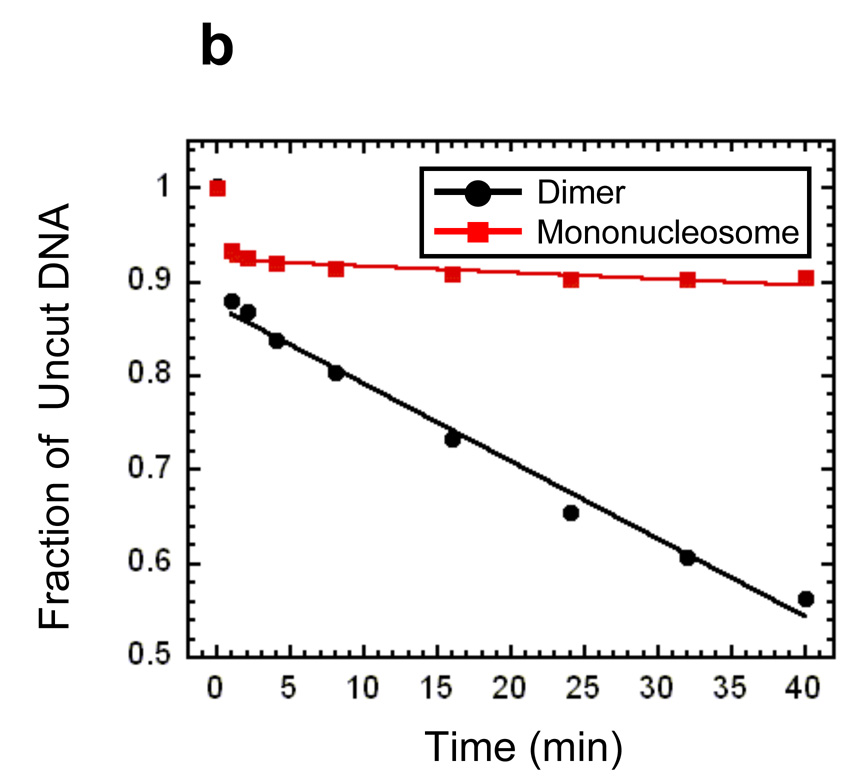
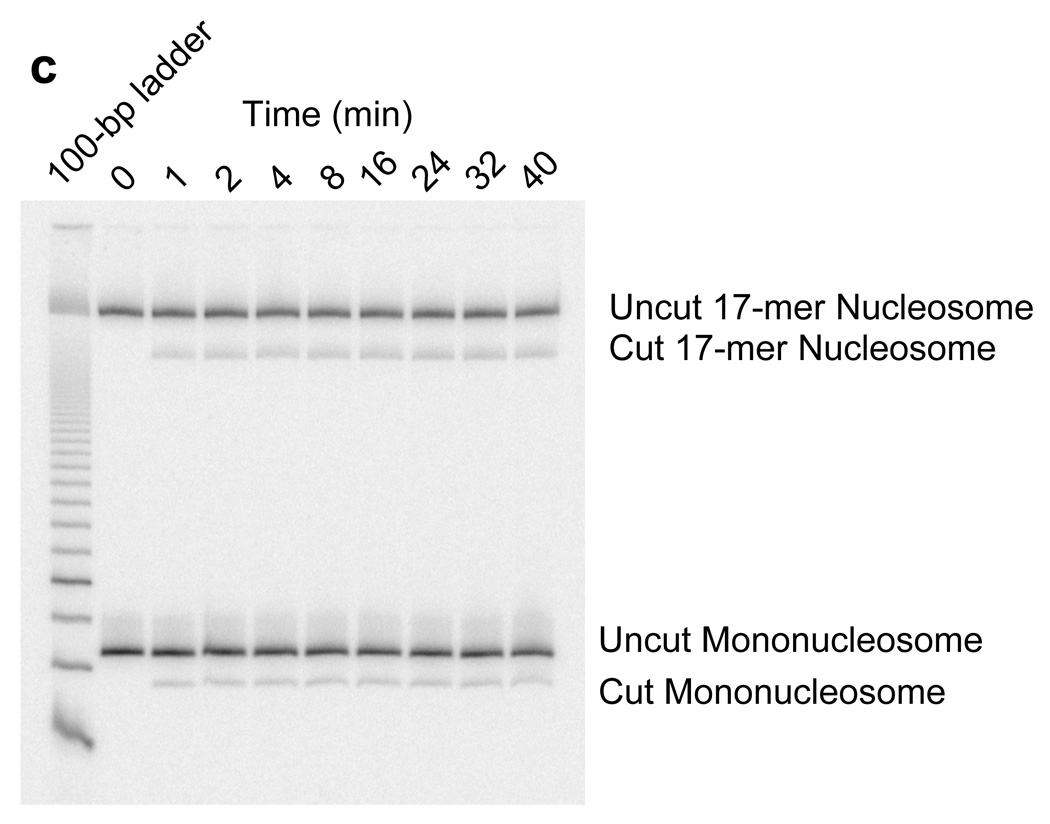
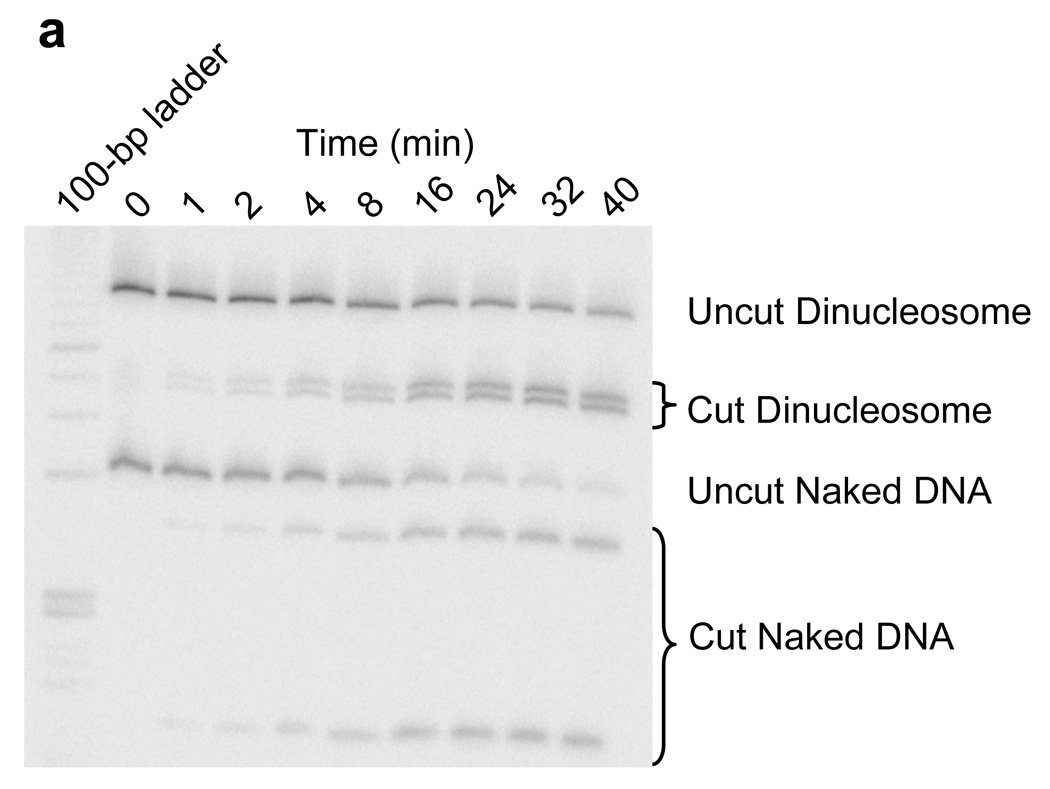
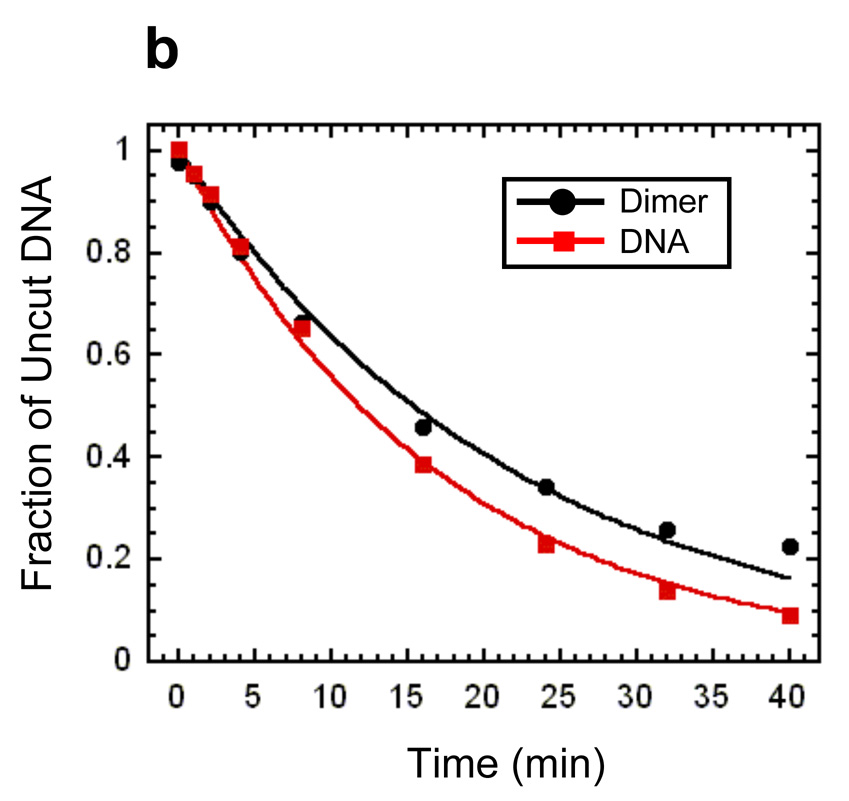
Site exposure in dinucleosomes and nucleosome 17-mers measured relative to mononucleosomes. (a) Native gel analysis of DNA products from Hae III digestions of a mixture of purified dinucleosomes and mononucleosomes; times of digestion (minutes) are indicated. (b) Quantification of the fraction of uncut DNA from (a), fit with an exponential decay (see Methods). (c, d) As in (a, b) except purified nucleosome 17-mers are used in place of dinucleosomes. (e) Summary of site accessibilities in dinucleosomes and nucleosome 17-mers, measured relative to accessibilities in mononucleosomes, for sites spanning the test nucleosome. Averages and standard deviations (n=2 or 3) are shown.
Site accessibility in linker DNA
We used the same approach to quantify the effects of nucleosome neighbors and concomitant chromatin folding on the accessibility of target sites in linker DNA, except in this case we used naked DNA as the internal reference, yielding linker accessibility relative to naked DNA. We made measurements at three sites spanning the 30 bp linker DNA (Fig. 7a–d). Strikingly, quantitative analysis of the reaction kinetics in this case reveals that, in conditions that stabilize higher order folding of the nucleosome arrays, the presence of one or many nucleosome neighbors strongly influences the accessibility of sites inside the linker: accessibility is reduced as much as ~50-fold, at a site in the middle of the linker in the 17-mer arrays (Fig. 7e).
Figure 7.

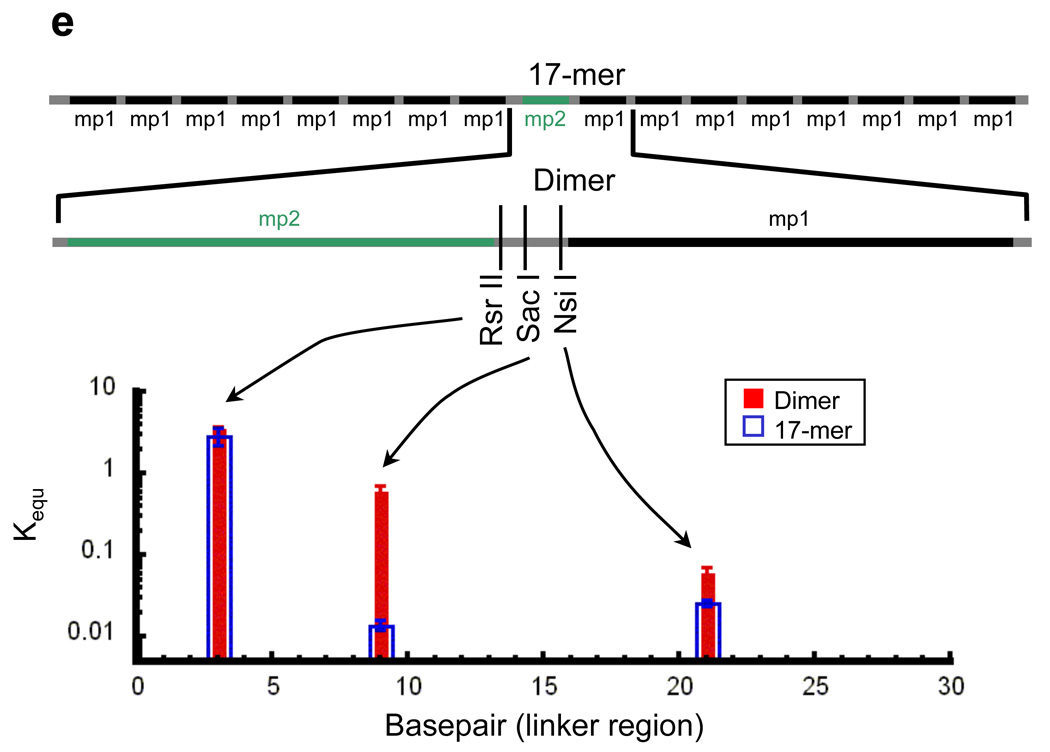
Site exposure in linker DNA within dinucleosomes and nucleosome 17-mers measured relative to naked DNA. (a) Native gel analysis of DNA products from Sac I digestions of a mixture of purified dinucleosomes and naked DNA; times of digestion (minutes) are indicated. (b) Quantification of the fraction of uncut DNA from (a), fit with an exponential decay. (c, d) As in (a, b) except purified nucleosome 17-mers are used in place of dinucleosomes. (e) Summary of site accessibilities in dinucleosome and nucleosome 17-mer linker DNA, measured relative to accessibilities in naked DNA, for sites spanning one linker DNA region. Averages and standard deviations (n=2–5) are shown.
These and other observations also imply that any self-association (aggregation) of the nucleosome arrays, which could be caused by the Mg2+ in the restriction enzyme buffers39, is not significantly influencing the observed site accessibilities. If aggregation significantly influenced the digestion kinetics, this should lead to non-single exponential behavior, with many nucleosomes resistant to digestion, and to comparably large decreases in accessibility for all sites in linker DNA or nucleosomes — all of which predictions are contrary to observation (above, and see Methods). Moreover, our results show that restriction enzymes digest nucleosomal DNA within arrays at rates that are similar to those for single nucleosomes, while in previous studies we showed that site exposure in single nucleosomes occurs to a similar quantitative extent regardless of whether the solutions contain Mg2+ or not20, and we proved that nucleosomes undergoing spontaneous site exposure in solutions lacking Mg2+ are not aggregated19. Based on all of these considerations, we conclude that any aggregation of the nucleosome arrays in our experiments negligibly influences the measured relative equilibrium accessibilities.
Discussion
Two results from this study stand out. First, surrounding a nucleosome with many neighbors on each side in a chromatin fiber, with concomitant higher order compaction of that fiber, causes only a modest change in accessibility of sites within the central nucleosome relative to the accessibility of the same sites in a single isolated nucleosome. Moreover, these modest changes in accessibility that do exist range from ~3-fold decreases in accessibility to ~8-fold increases. This means that folded nucleosome arrays are intrinsically dynamic to an extent that target sites even in the least-accessible locations in a nucleosome nevertheless are constantly but transiently accessible. This finding provides one explanation for how upstream activators have access to their DNA target sites even in transcriptionally silenced chromatin in vivo31.
Second, in contrast to the modest changes it causes in accessibility of nucleosomal sites, higher order compaction of the chromatin fiber dramatically influences the accessibility of certain sites in the linker DNA: the accessibility of the Sac I site in the middle of the linker DNA in a chromatin fiber, relative to the same site in naked DNA, is reduced by as much as ~50-fold. Accessibility at other sites in the linker is less-strongly decreased (Nsi I) or even slightly increased (Rsr II) by the existence of neighbor nucleosomes.
We emphasize that the restriction enzyme kinetics method quantifies the DNA accessibility for the specific restriction enzyme used in the experiment. Details of the size and shape of the enzyme and how it binds will differ between enzymes, and can in principle influence the apparent accessibilities. Nevertheless, for digestion inside the nucleosome, enzyme-specific differences appear to be small in comparison to the position-dependent protection against binding of any protein, which is conferred by the nucleosome: accessibilities decrease quite progressively from the ends in to the middle of the nucleosome, despite the use of many unrelated restriction enzymes and other site-specific DNA binding proteins22. Nevertheless, the distinct effects on accessibilities on nearby sites in linker DNA, observed here, may be attributable in some measure to enzyme-specific differences in the requirements for a site to become accessible. In that case, our findings would indicate that chromatin folding can specifically block certain DNA binding proteins while permitting others to bind in the same stretch of linker DNA.
Structural determinants of DNA target site accessibility in chromatin
Many nucleosomes in vivo are relatively well positioned: they have a high probability of occupying particular genomic locations2; 22; 40. It is now understood that these preferred locations are in part encoded directly in the genomic DNA sequence, and are also in part a consequence of competition with other site specific DNA binding proteins2; 27; 41; 42; 43; 44. In this context, what is the meaning of our new findings about relative accessibility of nucleosomal and linker DNA in chromatin fibers?
One conclusion is that the detailed location of a nucleosome along the genome, at a given point in time, is a dominant determinant of DNA site accessibility. DNA sites that are located far inside a nucleosome, e.g., near the middle, are as much as 103–105 times less accessible than are sites near the ends of the nucleosome or in linker DNA. For typical site specific DNA binding proteins, the resulting effective dissociation constant for binding to a target site in the middle of the nucleosome is likely to greatly exceed the actual free concentration of the protein. In this case (i.e., whenever free concentrations are low compared to the effective dissociation constant), a 103 - to 105-fold decrease in accessibility for a site in the middle of the nucleosome translates directly to a comparable decrease in expected occupancy of a typical regulatory protein at that target site. Compared to that very large potential decrease in occupancy attributable to the detailed location of a nucleosome itself, an additional ~3- to 8-fold (negative or positive) changes in accessibility attributable to organization of a nucleosome into a compacted chromatin array is plainly of more-modest significance.
In contrast, for sites in the middle of linker DNA regions, the opposite situation obtains. Here, accessibility is intrinsically high, yet proximity to neighboring nucleosomes coupled to chromatin folding can decrease that accessibility dramatically, by at least ~50-fold. If, as seems plausible, in vivo concentrations and DNA-binding affinities of a regulatory protein are tuned for significant but partial occupancy on naked DNA, then the occupancy of such a protein on a target site in linker DNA in chromatin will sensitively depend on the folded state of the chromatin fiber and on the presence or absence of other nucleosomes immediately nearby. Large effects on accessibility and occupancy at such genomic locations are to be expected.
Finally, for sites that are located near the ends of the nucleosome, the situation is more complex. The intrinsic accessibility of such sites in an isolated nucleosome is reduced relative to naked DNA, but is much less reduced than for sites near the middle of the nucleosome. The extent of reduction of accessibility depends sensitively on the particular DNA sequence, and may be as little as ~20-fold for sites near the end of an intrinsically less-stable nucleosome21. Few-fold effects of changing accessibility on top of that, due to placement in a chromatin fiber and concomitant chromatin folding, may lead to few-fold changes in occupancy. Whereas few-fold changes in occupancy for sites near the middle of the nucleosome may be of modest consequence, because the occupancy may be so low to begin with, comparable changes when occupancy is higher may be of great significance. The phenomena of dosage compensation and haplo-insufficiency diseases remind us of the potential significance of even 2-fold effects on gene expression.
In summary, for chromatin fibers composed of a repeated array of nucleosome core particles connected by typical-length segments of linker DNA, the detailed positioning of nucleosomes along the DNA is a dominant determinant of accessibility, while the presence of nucleosome neighbors and concomitant chromatin folding will subtly but significantly affect the accessibility of sites near the periphery of a nucleosome (especially for nucleosomes whose DNA end is not-too-stably wrapped), and will greatly affect the accessibility of regions in linker DNA.
Regulated DNA accessibility in chromatin fibers
Earlier studies showed that the accessibility of target sites within individual nucleosomes is quantitatively influenced by the acetylation patterns of the core histone tail domains45; 46. Here we are concerned with influences on target site accessibility arising from the organization of isolated nucleosomes into a repeating array in which each nucleosome is flanked with many neighbors, and in which the entire chain of nucleosomes then folds into a more compact higher order structure. Like other recent studies33; 34; 51; 53, our work has addressed chromatin folding as it occurs in the absence of other chromatin associated proteins. The “linker histone”, H1, is a chromatin associated protein of particular interest since it can occur in nearly stoichiometric amounts compared to nucleosomes. Even though histone H1 has been implicated in stabilizing higher order chromatin folding54, studying the accessibility of chromatin fibers lacking histone H1 is justified since the chromatin fibers visibly compact even without histone H1, and the single yeast gene for histone H1, which has significant homology to higher cell histone H1, is not essential for viability37. In higher cells, a regulated higher order chromatin folding, which might be facilitated by the regulated binding and unbinding of histone H1, might play more important roles, which are not revealed in our study. Although questions concerning the nature and biological roles of higher order chromatin folding remain unresolved, useful conclusions may already be drawn.
First, a dominant role for nucleosome positioning on accessibility of DNA target sites in vivo is supported by many recent studies41; 47; 48; 49. Importantly, however, nucleosome positions themselves are regulated. While nucleosome positions in vivo are influenced by the underlying genomic sequence2, they are also influenced by competition with the constellation of competing site specific DNA binding proteins2; 41; 43; 44. When this constellation changes, nucleosomes redistribute their locations in a manner that depends both on the concentrations and affinities of the competing proteins, and on the underlying intrinsic landscape for nucleosome positioning. The action of remodeling factors and existence of linker DNA can be required to facilitate the redistribution of nucleosome positions14; 15; 50.
Beyond regulated nucleosome positioning, our results suggest that regulated chromatin folding, too, may meaningfully influence target site accessibility in vivo. A recent study of chromatin folding in vitro established that acetylation of histone H4 lysine 16 (K16) specifically destabilizes the higher order folding of chromatin fibers51. Our studies utilized histone octamers purified from chicken erythrocytes, which are transcriptionally inert and have minimally-acetylated histones52. Thus, our results reflect a more stable form of the chromatin fiber45. Any destabilization of the fiber caused by specific acetylation of histone H4 K16 would only reduce the magnitudes of the already-modest changes in accessibility that we observed. Thus, we expect that regulated folding or unfolding of the chromatin fiber may have little role in regulating access to sites in the middle of a nucleosome, where accessibility is always strongly reduced. Nevertheless, regulated folding may play an important biological role by modulating access to sites near the ends of a nucleosome, or in linker DNA, where accessibility is intrinsically much greater.
Materials and Methods
DNA constructs
The DNA templates used are tandem repeats of variants of the 601 high affinity nucleosome positioning sequences (NPS), 60132 and 601.222 with exactly 30 bp of linker DNA length between each 147 bp-long nucleosome, yielding a repeat length of 177 bp. Eight base changes were made within 601 to give the sequence mp1 and two base changes were made within 601.2 to give the sequence mp2. With these sequence changes, there were eight restriction enzyme sites within mp2 across the nucleosome which are not present within mp1. The mp1 sequence was prepared by three rounds of PCR amplification. The first PCR amplification used the 601 sequence as template, with primers: AGCCGCTCAATTGGTCGTAGCAAGCTCTACCACCGCTTAAACGCAC-GTAAGGGCTGTCCCCCGCG and TACATGCACAGGATGTATATATCTGACGCGTGCCTGGAGACTGGGGAGTAATC-CTCTTGGCGGTT. The product was gel purified and used as the template for the second round of PCR amplification with primers: GCATCCCGCCCT-GGAGAATCTTGGTGCCGA-AGCCGCTCAATTGGTCGTAGCAA and CTTTGATCAAGA-TCTCCCGAGTTCA-ATACATGCACAGGATGTATATATCTGACGCG. This product too was gel purified and used as the template for the third round of PCR amplification with primers: TTGACGACCGGAATTCAGAGCTCCTCGGGATGCATCCCGCCCTGGAGAATC and CTCCTTATCCCAAGCTTTGATCAAGATCTCCCGAGTTCAATAC. This final product was gel purified and cloned into the multiple cloning site of pUC19 between the EcoR I and Hind III sites, yielding plasmid pMP1. The mp1 sequence is flanked on both sides by the asymmetric Ava I site CTCGGG, and by the EcoR I and Sac I sites on one side and by Bcl I and Bgl II on the other side.
A tandem repeat of 8 mp1 sites was prepared by ligation of Ava I-cut mp1 fragment. The single mp1 DNA insert was cleaved out of pMP1 with Ava I; the mp1 fragment and the linearized vector were gel purified. The mp1 fragment was ligated for one minute with Quick Ligase (Qiagen) and then the linearized vector was added and allowed to ligate for an additional 5 minutes. The resulting plasmid was transformed into DH5α cells. Repeats of up to 12 were cloned. The cloned mp1 repeats were sequenced; the longest insert that could be completely sequenced was the octamer of mp1.
The mp2 sequence too was made by three rounds of PCR amplification. The first PCR amplification used the 601.2 sequence as the template with primers: GACGAGGTGCGG-GGATGATCACTGCAGAAGCTTGGTGCCGGGGCCGC and TGTATATATCTGACACG-TGCCTGGAGACTAGGGAGTAATCCCCTTGGCGTTTAAAACGCG. The PCR product was gel purified and used as the template for the second round of PCR amplification with primers: AGGCGCCCCGGAATTCCGGTTGACGAGGTGCGGGGATGATCA and CTCGTCCTTA-TCGAGCTCGGTCCGACAGGATGTATATATCTGACACGTGCCTGGAGACTAG. This PCR product was gel purified and used as the template for the third round of PCR amplification with primers: AGGCGCCCCGGAATTCCGGTTGACGAGGTGCGGGGATGATCA and CGCCACACATGCATGCAGATCTATGTCGGGCTCGTCCTTATCGAGCTCGGTC. This final product was cloned into the multiple cloning site of pUC19 between the EcoR I and Sph I sites, yielding plasmid pMP2. The mp2 sequence is flanked on one side by EcoR I and Bcl I sites and on the other side by Sac I and Bgl II sites.
The dinucleosome DNA template was made by cloning a single mp1 sequence adjacent to the mp2 sequence between the Sac I and Bgl II sites. This strategy yields a 30 bp long linker DNA between the two positioning sequences, in plasmid pMP2_MP1. The heptadecamer (17-mer) DNA tandem repeat was made by cloning an mp1 8-mer between both the EcoR I and Bcl I sites and (separately) between the Sac I and Bgl II sites. This created a tandem repeat of 17 nucleosome positioning sequences with 8 mp1 sequences then 1 mp2 sequence and then 8 mp1 sequences, all spaced by exactly 30 bp of linker DNA, in plasmid pMP17.
Histones
Histone octamer was purified from chicken erythrocytes as described in ref.55.
Mononucleosome reconstitutions
Mononucleosomes were reconstituted and purified as described in22. Briefly, the DNA template used for reconstitutions was PCR amplified from plasmid pMP2 with primers: TGATCACTGCAGAAGCTTGGTGC and GAGCTCGGTCCGACAGGATGT. The PCR product was purified by reverse phase HPLC, 5’ end-labeled with 32P using T7 kinase, which was then heat denatured. Reconstitutions were done by double dialysis56 with 1 µg of radiolabel mp2 DNA, 4 µg of cold (unlabeled) mp2 DNA and 4 µg of chicken erythrocyte histone octamer. This incorporated about 80% of the mp2 DNA into nucleosomes (Fig.3b, lane1). The reconstituted mononucleosomes were then purified on a sucrose gradient32 to remove the free DNA and aggregates (Fig. 3b, lane2).
Nucleosome array reconstitutions
The dimer DNA template was prepared for reconstitution by PCR amplification using pMP2_MP1 template DNA with primers: GAATTCCGGTTG-ACGAGGTGC and GCTTGCATGCAGATCTCCCG. The product was purified by reverse phase HPLC and 32P end-labeled using T7 kinase. The 17-mer DNA template was cut out of pMP17 with EcoR I and Hind III. The vector was cut into many pieces with Dde I and then the 17mer was purified away from these pieces by agarose gel electrophoresis. The purified 17-mer DNA was dephosphorylated using Antarctic Phosphatase (NEB), which was then heat killed, and then the DNA 32P end-labeled using T7 kinase.
Nucleosome arrays were reconstituted onto these DNAs by double dialysis using radiolabeled dimer or 17-mer DNA templates, chicken erythrocyte histone octamer, and core particle DNA (cpDNA), which is used to buffer the reconstitutions so that the arrays saturate with positioned nucleosomes without substantial aggregation33; 35. The reconstitutions were done in a volume of 50 µl in lab-made dialysis buttons56. The mass ratio of histone octamer to total amount of DNA used to full reconstitute the arrays was 0.8. The buttons were inserted into a large dialysis tubing filled with 80 ml of 0.5 × (v/v) TE (TE is 10 mM Tris pH 8.0, 1 mM EDTA), 2 M NaCl, 1 mM benzamidine hydrochloride (BZA) and 0.5 mM phenylmethylsulfonyl fluoride (PMSF). The large dialysis tubing was placed into 4 liters of 0.5 × TE, 1 mM BZA and 0.5 mM PMSF. This was dialyzed for 6 hours at 4 C, and then the 4 liters were changed and dialyzed overnight at 4 °C. The reconstituted nucleosome arrays were then collected from the dialysis buttons and purified on sucrose gradients.
Biochemical characterization of nucleosome arrays
The dimer reconstitutions were assayed by gel shifts on 4% polyacrylamide gels in 0.3 × (v/v) TBE (TBE is 90 mM Tris-Borate, 2 mM EDTA). The dinucleosome reconstitutions display 2 distinct gel shifts, which correspond to the formation of a first followed by a second nucleosome on each template DNA. This allows us to determine the reconstitution conditions which incorporate ~100% of the positioning sequences into nucleosomes. To confirm this, we digested the dinucleosomes with Sac I at a concentration of 20 units ml−1, which cuts in the linker region between the mp1 and mp2 positioning sequences. This converts dinucleosomes into two mononucleosomes with little free DNA (Fig. 3b, lane 5). The difference in gel mobility of the two single nucleosomes is due to the difference in length of DNA that extends out from the nucleosome.
The 17-mer (Fig. 4a) reconstitutions were assayed by gel shifts on mixed 2% polyacrylamide plus 1% agarose gels in 0.2×TB (TB is 90 mM Tris-Borate). Unlike the dimer reconstitutions, the partially reconstituted heptadecamers gel shift bands were not all distinguishable and instead appeared as a smear (Fig. 4a, lanes “.1” and “.2”). It is therefore difficult from the gel shift assay alone to be sure the arrays are saturated with nucleosomes. However, the gel shift at higher concentrations of histone octamer is a tight, well-defined band, suggesting that those arrays are saturated. Mlu I digestions of the reconstituted 17-mers relative to naked DNA having a single Mlu I site were used to quantify the number of positioning sequences that were incorporated into nucleosomes in the 17-mer reconstitutes (Fig. 4b). The digestion kinetics of the 17-mer reconstitutes and naked DNA were measured in the same reaction volume. Time points were taken by quenching 10 µl of the digestion with 20 mM EDTA. Subsequently, the histone octamers were removed with 1 mg/ml Proteinase K and 0.02% SDS. Aliquots were analyzed on a 5% polyacrylamide gel using a PhosphorImager (Molecular Dynamics). Image Quant software was used to determine the amount of uncut 17-mer and naked DNA (Fig. 4c). The naked DNA digestion (Fig. 4d, red) is fit with a normalized exponential, exp (−k0*t), where k0 = 6 min−1. The 17-mer nucleosome digestion is fit with the function C + (1 − C) exp (− t * 6 min−1), where C = 0.5. The value C is the fraction of arrays that have all 16 Mlu I sites protected from digestion. It follows that 0.03 (3%) of the positioning sequences are not incorporated into nucleosomes and, therefore, that 97 % of the positioning sequences in the 17-mer reconstitutes are wrapped into nucleosomes.
Array imaging by atomic force microscopy
Atomic force microscopy (AFM) imaging was used to confirm that the arrays were saturated and to determine the folded state in various conditions. Imaging was done in air and in liquid using a Multimode (Digital Instruments) AFM operated in tapping mode. Freshly cleaved mica was pre-treated with 30 µl of an aqueous solution of poly-L-lysine (PL, Sigma) at a concentration of 10 µg ml−1. Following a 30 second incubation, unbound PL was removed by rinsing the mica disc with 3 ml of Millipore purified water followed by drying under a nitrogen stream. The arrays were diluted at a concentration of ~ 0.15 nM in 0.5 × TE or 0.2 × TE, pH 7.5, either with or without additional salt (see text). 30 µl of the array solution were placed on the PL-mica. For scanning directly in the adsorption buffer, silicon nitride probes (type NP-S20, Veeco Instruments) were used at a drive frequency of ~9.0 kHz and set point of 0.3 – 0.4 V. This method was chosen to verify the condensed state of the arrays in the presence of MgCl2, in order to circumvent the need of rinsing with water. The arrays were also scanned in air to reliably distinguish individual nucleosomes in extended array conformations at low salt conditions. For this, the mica disc was washed carefully with 2.0 ml of Millipore water after a one minute incubation with the array solution. The disc was then dried with nitrogen and subsequently scanned using etched silicon probes (type NHC, Nanosensors) at drive frequencies of 280 to 320 kHz and set point of 2.0–2.2 V. The images were recorded both in solution and in air at a scan diameter of 2×2 µm, a scan rate of 1–2 Hz and a resolution of 512×512 pixels. Using the Nanoscope IIIa software (version 5.12r3, Digital Instruments), recorded images were flattened and areas of the imaged fields were zoomed for counting nucleosomes and for presentation.
Restriction enzyme kinetics method for nucleosomal DNA
The restriction enzyme kinetics method21; 38 was used to quantify DNA accessibility, defined as the equilibrium constants of site exposure, Kequ, = k12/k21, the equilibrium fraction of time that a nucleosomal DNA target site acts as though it is naked DNA (see Fig. 1). The original method determined Kequ by determining the initial rates of cleavage of nucleosomal DNA relative to cleavage of naked DNA. The digestions occur in a rapid pre-equilibrium regime, in which
where kmonucleosome and knaked DNA are observed pseudo-first order rate constants for digestion of mononucleosomes and naked DNA, respectively, and [R.E.]mononucleosome and [R.E.]naked DNA are the concentrations of restriction enzyme used in those digestions.
In the present study we seek to quantify the effects of adding nucleosome neighbors, with concomitant folding of the resulting chromatin fiber. Thus, we measure Kequ of site exposure for a unique target site in a nucleosome array relative to exposure of the same site in mononucleosomes, which we defined as:
.
We assume here that the rates, k23 and k4, (Fig. 1) are the same for a test nucleosome in the middle of an array and for a single nucleosome. In reality, nucleosomal DNA that partially unwraps in the middle of an array is constrained at both ends, while in single nucleosomes it is constrained at only one end. This constraint could alter the rate of binding k23 and / or the rate of cleavage, k4. Given the lengths of unwrapped DNA involved (unwrapping of ~80 bp suffices to provide unhindered access even to the middle of the nucleosome, with shorter unwrapping lengths required for sites nearer the ends of the nucleosome), this is not likely to be a dramatic effect, since the DNA is relatively stiff on that length scale. In fact, the modest differences in relative equilibrium constants that we do detect may, in part, be due to this difference. This does not change the overall interpretation of our results because the effects on accessibility of nucleosomal DNA from adding neighbors to a single nucleosome are very small in comparison to the large effects of wrapping the DNA into a nucleosome in the first place.
We determine Krelative-equ by measuring the observed pseudo-first order rates of cleavage of a unique restriction enzyme target sites in nucleosome arrays and in mononucleosomes, karray and kmononucleosome, by six separate restriction enzymes (REs): Pst I, Hind III, Hae III, Bam HI, Sty I and Pml I. The reactions were done by digesting both the purified nucleosome arrays and the mononucleosomes at 1 nM concentration in the appropriate enzyme buffer in a volume of 100 µl in the recommended NEB buffer. NEBuffer 1 contains 10 mM Bis-Tris-Propane-HCl, 10 mM MgCl2, 1 mM Dithiothreitol NEBuffer 2 contains 50 mM NaCl, 10 mM Tris-HCl, 10 mM MgCl2, 1 mM Dithiothreitol. NEBuffer 3 contains 100 mM NaCl, 50 mM Tris-HCl, 10 mM MgCl2, 1 mM Dithiothreitol. NEBuffer 4 contains 50 mM potassium acetate, 20 mM Tris-acetate, 10 mM Magnesium Acetate, 1 mM Dithiothreitol. BamHI Buffer contains 150 mM NaCl, 10 mM Tris-HCl, 10 mM MgCl2, 1 mM Dithiothreitol. BSA was added when recommended by NEB. The concentration of RE was used so that the glycerol concentration was at 5%, which maximized enzyme concentration without introducing “star” activity and are first order in enzyme concentration. In preliminary studies we found that the cleavage rates of DNA within a nucleosome array and a mononucleosome are similar. Thus, we could improve the quantitative accuracy of the data by simultaneous measurement of relative cleavage rates on nucleosome arrays and mononucleosomes, in the same reaction at the same time. This ensured that [R.E.]mononucleosome / [R.E.]array = 1.
Relative rates of cleavage were determined by quenching 10 µl aliquots taken at 1, 2, 4, 8, 16, 24, 32 and 40 minutes of digestion, by addition of EDTA to 20 mM final concentration. We separately confirmed the adequacy of the quench procedure by reactions in which we added EDTA prior to the RE. The quenched reaction aliquots were digested with 1 mg ml−1 of proteinase K plus 0.02% (w/v) SDS for 20 minutes at 50 °C to remove the histone proteins. The resulting DNA was then run on a 5% native polyacrylamide gel, dried and quantified using a PhosphorImager and Image Quant software. There is an initial drop in undigested DNA following the first time point. This is due to the 5–10% of nucleosomes that fall apart during the rapid mixing at the beginning of the digestions (see below). We ignore this initial time point, as was done in previous studies21; 22, since the rapid drop in undigested DNA is due to digestion of naked DNA.
Restriction enzyme kinetics method for linker DNA
The restriction enzyme kinetics method was also used to determine the equilibrium constant for site exposure in the linker region, in this case measured relative to cleavage of naked DNA. The equilibrium constant, Kequ is defined as:
. The observed rates of cleavage, klinker DNA and knaked DNA, were determined as described above. The rates of digestion in the linker region of nucleosome arrays, and in naked DNA, were similar enough that they were done in the same reaction at the same time (and thus [R.E.]naked DNA / [R.E.]linker DNA = 1).
Nucleosome association
The restriction enzyme digests are carried out in buffers that contain 10 mM Mg2+, which can induce a reversible self-association (aggregation) of individual nucleosomes and of nucleosome arrays. This fact raises a question of whether such association influenced the observed site exposure equilibrium constants. Our results show that linker DNA sites in the nucleosome arrays are digested to completion in single exponential processes (Fig. 7), and that accessibility at some sites in the linker DNA is strongly influenced by the presence of neighboring nucleosomes, while accessibility at other sites is not. Moreover, we find that nucleosome arrays are digested inside the test nucleosome with overall rates that are close to those for digestion of individual nucleosomes. Each of these observations implies that most of the arrays must be participating in the restriction enzyme digestions, since, if association rendered many of the nucleosome arrays inaccessible to restriction enzyme digestion, the remaining fraction of the arrays would need to be digested at a correspondingly faster rate to give an effective rate that appears similar to single nucleosomes, which is not plausible. Together, these facts exclude the possibility that self-association creates bulk precipitation or heterogeneity in the sample, or that association significantly influenced the measured relative equilibrium accessibilities.
Histone octamer disassociation
The rapid mixing of the nucleosome arrays with the restriction enzyme causes up to 10% of the nucleosomes to fall apart21; 38, which is responsible for a rapid initial drop in uncut DNA as is seen for both dimer and 17-mer arrays (Fig. 6). We correct for this behavior by omitting the first digestion time point from the kinetic analysis. Several facts argue that this loss of nucleosomes does not alter our measured equilibrium measurements. First, the nucleosome that is lost is almost always the test nucleosome (mp2) itself, because it has more sequence changes deviating from the selected high affinity sequence (601) at key locations2 than do the flanking (mp1) nucleosomes: mp1 has 8 base pair changes while mp2 has 15, and two of the base pair changes in mp2 remove key TA dinucleotide steps, which are known to be critical for histone octamer affinity and nucleosome positioning. Therefore, the mp1 site is more stable and should have less histone octamers dissociation during the rapid mixing with the restriction enzyme. This is confirmed by digesting with Mlu I, which cuts in the mp1 site but not the mp2 site. We find that only 3% of the mp1 sites lose their histone octamer during the digestions (Fig. 4c–d) as compared to 10% for the mp2 site. In summary, the most likely nucleosome to be lost upon initial mixing of the reaction is mp2. With the enzyme concentrations used in these experiments, the naked DNA thus created is digested instantly; and we correct for this by eliminating the first digestion timepoint from the kinetic analysis.
A second way in which nucleosome loss could influence the measured relative accessibilities is if the lost nucleosome happened to be adjacent to the test nucleosome, in which case we would be looking at accessibility of a test nucleosome that did not, in fact, have an immediate neighbor on at least one side. Since only ~3% of flanking nucleosomes are lost, yet nucleosomes in arrays are digested at total rates that are similar to those for single nucleosomes, it follows that all of the nucleosome arrays are participating in the digestion, and that the measurements are overwhelmingly dominated by complete nucleosome arrays. If we assumed instead that the digestion is due arrays with a missing nucleosome neighboring the test nucleosome, the rates we report would not be normalized correctly, since only 6% of the arrays are missing one or the other flanking nucleosome. The actual digestion rates for the nucleosome arrays would need to be >16 (1/0.06) times faster than for single nucleosomes, for the total digestion rates to be similar. This would imply that the lack of a neighboring nucleosome increases the DNA accessibility by over 16-fold as compared to single nucleosomes – which is not plausible. For these reasons, we conclude that our results properly measure the actual relative equilibrium constants for DNA site exposure for nucleosomes surrounded by nucleosome neighbors.
Supplementary Material
ACKNOWLEDGEMENTS
We thank members of the Widom and Langowski labs for useful discussions, and the Keck Biophysics Facility at Northwestern University for the use of instruments. M.G.P. acknowledges support from US National Institutes of Health postdoctoral fellowship F32 GM072306 and a Career Award in the Biomedical Sciences from the Burroughs-Wellcome Fund. J.W. acknowledges research support from US National Institutes of Health grants R01 GM54692 and R01 GM58617.
COMPETING INTERESTS STATEMENT
The authors declare that they have no competing financial interests.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Richmond TJ, Davey CA. The structure of DNA in the nucleosome core. Nature. 2003;423:145–150. doi: 10.1038/nature01595. [DOI] [PubMed] [Google Scholar]
- 2.Segal E, Fondufe-Mittendorf Y, Chen L, Thastrom A, Field Y, Moore IK, Wang JP, Widom J. A genomic code for nucleosome positioning. Nature. 2006;442:772–778. doi: 10.1038/nature04979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Almer A, Rudolph H, Hinnen A, Horz W. Removal of positioned nucleosomes from the yeast PHO5 promoter upon PHO5 induction releases additional upstream activating DNA elements. EMBO J. 1986;5:2689–2696. doi: 10.1002/j.1460-2075.1986.tb04552.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Belikov S, Gelius B, Almouzni G, Wrange O. Hormone activation induces nucleosome positioning in vivo. Embo J. 2000;19:1023–1033. doi: 10.1093/emboj/19.5.1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Boeger H, Griesenbeck J, Strattan JS, Kornberg RD. Nucleosomes unfold completely at a transcriptionally active promoter. Mol Cell. 2003;11:1587–1598. doi: 10.1016/s1097-2765(03)00231-4. [DOI] [PubMed] [Google Scholar]
- 6.Fragoso G, John S, Roberts MS, Hager GL. Nucleosome Positioning on the MMTV LTR Results from the Frequency-Biased Occupancy of Multiple Frames. Genes & Dev. 1995;9:1933–1947. doi: 10.1101/gad.9.15.1933. [DOI] [PubMed] [Google Scholar]
- 7.Jack RS, Eggert H. Restriction enzymes have limited access to DNA sequences in Drosophila chromosomes. Embo J. 1990;9:2603–2609. doi: 10.1002/j.1460-2075.1990.tb07442.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lee W, Tillo D, Bray N, Morse RH, Davis RW, Hughes TR, Nislow C. A high-resolution atlas of nucleosome occupancy in yeast. Nat Genet. 2007;39:1235–1244. doi: 10.1038/ng2117. [DOI] [PubMed] [Google Scholar]
- 9.Sekinger EA, Moqtaderi Z, Struhl K. Intrinsic histone-DNA interactions and low nucleosome density are important for preferential accessibility of promoter regions in yeast. Mol Cell. 2005;18:735–748. doi: 10.1016/j.molcel.2005.05.003. [DOI] [PubMed] [Google Scholar]
- 10.Felsenfeld G. Chromatin Unfolds. Cell. 1996;86:13–19. doi: 10.1016/s0092-8674(00)80073-2. [DOI] [PubMed] [Google Scholar]
- 11.Felsenfeld G, Groudine M. Controlling the double helix. Nature. 2003;421:448–453. doi: 10.1038/nature01411. [DOI] [PubMed] [Google Scholar]
- 12.Kornberg RD, Lorch Y. Chromatin-modifying and -remodeling complexes. Curr Opin Genet Dev. 1999;9:148–151. doi: 10.1016/S0959-437X(99)80022-7. [DOI] [PubMed] [Google Scholar]
- 13.Kornberg RD, Lorch Y. Twenty-Five Years of the Nucleosome, Fundamental Paricle of the Eukaryote Chromosome. Cell. 1999;98:285–294. doi: 10.1016/s0092-8674(00)81958-3. [DOI] [PubMed] [Google Scholar]
- 14.Saha A, Wittmeyer J, Cairns BR. Chromatin remodelling: the industrial revolution of DNA around histones. Nat Rev Mol Cell Biol. 2006;7:437–447. doi: 10.1038/nrm1945. [DOI] [PubMed] [Google Scholar]
- 15.Smith CL, Peterson CL. ATP-dependent chromatin remodeling. Curr Top Dev Biol. 2005;65:115–148. doi: 10.1016/S0070-2153(04)65004-6. [DOI] [PubMed] [Google Scholar]
- 16.Narlikar GJ, Fan HY, Kingston RE. Cooperation between complexes that regulate chromatin structure and transcription. Cell. 2002;108:475–487. doi: 10.1016/s0092-8674(02)00654-2. [DOI] [PubMed] [Google Scholar]
- 17.Peterson CL, Logie C. Recruitment of Chromatin Remodeling Machines. J. Cell. Biochem. 2000;78:179–185. doi: 10.1002/(sici)1097-4644(20000801)78:2<179::aid-jcb1>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- 18.Kingston RE, Narlikar GJ. ATP-dependent remodeling and acetylation as regulators of chromatin fluidity. Genes Dev. 1999;13:2339–2352. doi: 10.1101/gad.13.18.2339. [DOI] [PubMed] [Google Scholar]
- 19.Li G, Levitus M, Bustamante C, Widom J. Rapid spontaneous accessibility of nucleosomal DNA. Nat. Struct. Mol. Biol. 2005;12:46–53. doi: 10.1038/nsmb869. [DOI] [PubMed] [Google Scholar]
- 20.Li G, Widom J. Nucleosomes facilitate their own invasion. Nat Struct Mol Biol. 2004;11:763–769. doi: 10.1038/nsmb801. [DOI] [PubMed] [Google Scholar]
- 21.Polach KJ, Widom J. Mechanism of Protein Access to Specific DNA Sequences in Chromatin: A Dynamic Equilibrium Model for Gene Regulation. J. Mol. Biol. 1995;254:130–149. doi: 10.1006/jmbi.1995.0606. [DOI] [PubMed] [Google Scholar]
- 22.Anderson JD, Widom J. Sequence- and Position-dependence of the Equilibrium Accessibility of Nucleosomal DNA Target Sites. J. Mol. Biol. 2000;296:979–987. doi: 10.1006/jmbi.2000.3531. [DOI] [PubMed] [Google Scholar]
- 23.Polach KJ, Widom J. A Model for the Cooperative Binding of Eukaryotic Regulatory Proteins to Nucleosomal Target Sites. J. Mol. Biol. 1996;258:800–812. doi: 10.1006/jmbi.1996.0288. [DOI] [PubMed] [Google Scholar]
- 24.Miller JA, Widom J. Collaborative Competition Mechanism for Gene Activation In Vivo. Mol. Cell. Biol. 2003;23:1623–1632. doi: 10.1128/MCB.23.5.1623-1632.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vashee S, Melcher K, Ding WV, Johnston SA, Kodadek T. Evidence for Two Modes of Cooperative DNA Binding in Vivo That Do Not Involve Direct Protein-Protein Interactions. Curr. Biol. 1998;8:452–458. doi: 10.1016/s0960-9822(98)70179-4. [DOI] [PubMed] [Google Scholar]
- 26.Vashee S, Willie J, Kodadek T. Synergistic Activation of Transciption by Physiologically Unrelated Transcription Factors through Cooperative DNA-Binding. Biochem. Biophys. Res. Com. 1998;247:530–535. doi: 10.1006/bbrc.1998.8820. [DOI] [PubMed] [Google Scholar]
- 27.Bernstein BE, Liu CL, Humphrey EL, Perlstein EO, Schreiber SL. Global nucleosome occupancy in yeast. Genome Biol. 2004;5:R62. doi: 10.1186/gb-2004-5-9-r62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bucceri A, Kapitza K, Thoma F. Rapid accessibility of nucleosomal DNA in yeast on a second time scale. Embo J. 2006;25:3123–3132. doi: 10.1038/sj.emboj.7601196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gottschling DE. Telomere-proximal DNA in Saccharomyces cerevisiae is refractory to methyltransferase activity in vivo. Proc Natl Acad Sci U S A. 1992;89:4062–4065. doi: 10.1073/pnas.89.9.4062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Loo S, Rine J. Silencers and Domains of Generalized Repression. Science. 1994;Vol. 264:1768–1771. doi: 10.1126/science.8209257. [DOI] [PubMed] [Google Scholar]
- 31.Chen L, Widom J. Mechanism of Transcriptional Silencing in Yeast. Cell. 2005;120:37–48. doi: 10.1016/j.cell.2004.11.030. [DOI] [PubMed] [Google Scholar]
- 32.Lowary PT, Widom J. New DNA Sequence Rules for High Affinity Binding to Histone Octamer and Sequence-Directed Nucleosome Positioning. J. Mol. Biol. 1998;276:19–42. doi: 10.1006/jmbi.1997.1494. [DOI] [PubMed] [Google Scholar]
- 33.Dorigo B, Schalch T, Bystricky K, Richmond TJ. Chromatin fiber folding: requirement for the histone H4 N-terminal tail. J Mol Biol. 2003;327:85–96. doi: 10.1016/s0022-2836(03)00025-1. [DOI] [PubMed] [Google Scholar]
- 34.Dorigo B, Schalch T, Kulangara A, Duda S, Schroeder RR, Richmond TJ. Nucleosome arrays reveal the two-start organization of the chromatin fiber. Science. 2004;306:1571–1573. doi: 10.1126/science.1103124. [DOI] [PubMed] [Google Scholar]
- 35.Huynh VA, Robinson PJ, Rhodes D. A method for the in vitro reconstitution of a defined "30 nm" chromatin fibre containing stoichiometric amounts of the linker histone. J Mol Biol. 2005;345:957–968. doi: 10.1016/j.jmb.2004.10.075. [DOI] [PubMed] [Google Scholar]
- 36.Robinson PJ, Fairall L, Huynh VA, Rhodes D. EM measurements define the dimensions of the "30-nm" chromatin fiber: evidence for a compact, interdigitated structure. Proc Natl Acad Sci U S A. 2006;103:6506–6511. doi: 10.1073/pnas.0601212103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ushinsky SC, Bussey H, Ahmed AA, Wang Y, Friesen J, Williams BA, Storms RK. Histone H1 in Saccharomyces cerevisiae. Yeast. 1997;13:151–161. doi: 10.1002/(SICI)1097-0061(199702)13:2<151::AID-YEA94>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 38.Polach KJ, Widom J. Restriction Enzymes as Probes of Nucleosome Stability. Meth. Enzymol. 1999;304:278–298. doi: 10.1016/s0076-6879(99)04017-3. [DOI] [PubMed] [Google Scholar]
- 39.Jason LJ, Moore SC, Ausio J, Lindsey G. Magnesium-dependent association and folding of oligonucleosomes reconstituted with ubiquitinated H2A. J Biol Chem. 2001;276:14597–14601. doi: 10.1074/jbc.M011153200. [DOI] [PubMed] [Google Scholar]
- 40.Yuan GC, Liu YJ, Dion MF, Slack MD, Wu LF, Altschuler SJ, Rando OJ. Genome-scale identification of nucleosome positions in S. cerevisiae. Science. 2005;309:626–630. doi: 10.1126/science.1112178. [DOI] [PubMed] [Google Scholar]
- 41.Belikov S, Holmqvist PH, Astrand C, Wrange O. Nuclear factor 1 and octamer transcription factor 1 binding preset the chromatin structure of the mouse mammary tumor virus promoter for hormone induction. J Biol Chem. 2004;279:49857–49867. doi: 10.1074/jbc.M409713200. [DOI] [PubMed] [Google Scholar]
- 42.Lee CK, Shibata Y, Rao B, Strahl BD, Lieb JD. Evidence for nucleosome depletion at active regulatory regions genome-wide. Nat Genet. 2004;36:900–905. doi: 10.1038/ng1400. [DOI] [PubMed] [Google Scholar]
- 43.Lomvardas S, Thanos D. Nucleosome Sliding by TBP Binding In Vivo. Cell. 2001;106:685–696. doi: 10.1016/s0092-8674(01)00490-1. [DOI] [PubMed] [Google Scholar]
- 44.Roth SY, Dean A, Simpson RT. Yeast alpha 2 repressor positions nucleosomes in TRP1/ARS1 chromatin. Mol Cell Biol. 1990;10:2247–2260. doi: 10.1128/mcb.10.5.2247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Anderson JD, Lowary PT, Widom J. Effects of Histone Acetylation on the Dynamic Equilibrium Accessibility of Nucleosomal DNA Target Sites. J. Mol. Biol. 2001;307:977–985. doi: 10.1006/jmbi.2001.4528. [DOI] [PubMed] [Google Scholar]
- 46.Polach KJ, Lowary PT, Widom J. Effects of Core Histone Tail Domains on the Equilibrium Constants for Dynamic DNA Site Accessibility in Nucleosomes. J. Mol. Biol. 2000;298:211–223. doi: 10.1006/jmbi.2000.3644. [DOI] [PubMed] [Google Scholar]
- 47.Goldmark JP, Fazzio TG, Estep PW, Church GM, Tsukiyama T. The Isw2 chromatin remodeling complex represses early meiotic genes upon recruitment by Ume6p. Cell. 2000;103:423–433. doi: 10.1016/s0092-8674(00)00134-3. [DOI] [PubMed] [Google Scholar]
- 48.Lomvardas S, Thanos D. Modifying Gene Expression Programs by Altering Core Promoter Architecture. Cell. 2002;110:261–271. doi: 10.1016/s0092-8674(02)00822-x. [DOI] [PubMed] [Google Scholar]
- 49.Martinez-Campa C, Politis P, Moreau JL, Kent N, Goodall J, Mellor J, Goding CR. Precise nucleosome positioning and the TATA box dictate requirements for the histone H4 tail and the bromodomain factor Bdf1. Mol Cell. 2004;15:69–81. doi: 10.1016/j.molcel.2004.05.022. [DOI] [PubMed] [Google Scholar]
- 50.Kagalwala MN, Glaus BJ, Dang W, Zofall M, Bartholomew B. Topography of the ISW2-nucleosome complex: insights into nucleosome spacing and chromatin remodeling. Embo J. 2004;23:2092–2104. doi: 10.1038/sj.emboj.7600220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shogren-Knaak M, Ishii H, Sun JM, Pazin MJ, Davie JR, Peterson CL. Histone H4-K16 acetylation controls chromatin structure and protein interactions. Science. 2006;311:844–847. doi: 10.1126/science.1124000. [DOI] [PubMed] [Google Scholar]
- 52.Mutskov V, Gerber D, Angelov D, Ausio J, Workman J, Dimitrov S. Persistent interactions of core histone tails with nucleosomal DNA following acetylation and transcription factor binding. Mol. Cell. Biol. 1998;18:6293–6304. doi: 10.1128/mcb.18.11.6293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Schalch T, Duda S, Sargent DF, Richmond TJ. X-ray structure of a tetranucleosome and its implications for the chromatin fibre. Nature. 2005;436:138–141. doi: 10.1038/nature03686. [DOI] [PubMed] [Google Scholar]
- 54.Van Holde KE. Chromatin. New York: Springer-Verlag; 1989. [Google Scholar]
- 55.Feng H-P, Scherl DS, Widom J. Lifetime of the Histone Octamer Studied by Continuous-flow Quasielastic Light Scattering: Test of a Model for Nucleosome Transcription. Biochem. 1993;32:7824–7831. doi: 10.1021/bi00081a030. [DOI] [PubMed] [Google Scholar]
- 56.Thåström A, Lowary PT, Widom J. Measurement of histone-DNA interaction free energy in nucleosomes. Methods. 2004;33:33–44. doi: 10.1016/j.ymeth.2003.10.018. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.