

Summary
The high immunogenicity of peptides displayed in dense repetitive arrays on virus-like particles makes recombinant VLPs promising vaccine carriers. Here we describe a platform for vaccine development based on the VLPs of RNA bacteriophage MS2. It serves for the engineered display of specific peptide sequences, but will also allow the construction of random peptide libraries from which specific binding activities can be recovered by affinity selection. Peptides representing the V3 loop of HIV gp120 and the ECL2 loop of the HIV coreceptor, CCR5, were inserted into a surface loop of MS2 coat protein. Both insertions disrupted coat VLP assembly, apparently by interfering with protein folding, but these defects were efficiently suppressed by genetically fusing coat protein's two identical polypeptides into a single-chain dimer. The resulting VLPs displayed the V3 and ECL2 peptides on their surfaces where they showed the potent immunogenicity that is the hallmark of VLP-displayed antigens. Experiments with random-sequence peptide libraries show the single-chain dimer to be highly tolerant of 6-, 8- and 10-amino acid insertions. Not only do MS2 VLPs support the display of a wide diversity of peptides in a highly immunogenic format, but they also encapsidate the mRNAs that direct their synthesis, thus establishing the genotype/phenotype linkage necessary for recovery of affinity selected sequences. The single-chain MS2 VLP therefore unites in a single structural platform the selective power of phage display with the high immunogenicity of VLPs.
Keywords: virus-like particle, phage display, epitope vaccine
Introduction
Many viral structural proteins have the intrinsic ability to self-assemble into virus-like particles (VLPs) that closely resemble authentic virions. VLPs make good vaccines because the regularity of capsid structure presents viral epitopes as dense repetitive arrays, which are highly stimulatory to B-cells 1; 2. Foreign epitopes displayed on VLPs by chemical conjugation or genetic fusion also show high immunogenicity. In fact, antigens presented in these formats are so immunogenic that antibody responses can be elicited even against self-antigens, thus overcoming the tolerance mechanisms that normally prevent the induction of autoantibodies 3; 4; 5; 6; 7, and raising the possibility that, in addition to the obvious infectious disease applications, recombinant VLP-based vaccines could replace therapeutic monoclonal antibodies as treatments for some diseases.
For maximum utility as a vaccine development platform, we think a VLP-display system should possess the following characteristics. First, to ensure high immunogenicity, including the ability to elicit antibodies to self-antigens, foreign peptides should be displayed as dense repetitive arrays on the VLP surface. Second, since display will normally be accomplished by genetic insertion of foreign sequences into a viral structural protein, a display site must be identified that is highly tolerant of diverse peptide insertions. Unfortunately, the effects on protein folding of such insertions, even in surface loops, are largely unpredictable and result frequently in folding failures. Third, the VLP should encapsidate the nucleic acid that encodes the viral protein-peptide fusion, thus enabling recovery of affinity selected sequences from complex peptide or antigen fragment libraries. This would allow a single VLP structural platform to serve both as an immunogen, and for epitope selection and evolution.
Most existing display platforms exhibit limitations with respect to these features. For example, the filamentous phages that form the basis of the most widely used display methods permit affinity selection from peptide libraries, but do not readily support the presentation of foreign peptides at the high densities required for potent immunogenicity 8. On the other hand, VLP systems able to present peptides at high density - Hepatitis B Virus core particles, for example - do not tolerate diverse peptide insertions without extensive engineering 9; 10, and have not yet been made to encapsidate their coding sequences. Other viral systems, including adeno-associated virus 11 and bacteriophage λ 12, have been adapted for high density display, but the immunogenicities of the particles have not been reported.
VLPs of the RNA bacteriophage MS2 offer a number of advantages for peptide display. The icosahedral shell can assemble spontaneously from a single coat protein expressed from a plasmid in E. coli. It is highly amenable to genetic manipulation 13; 14, is easily purified in large amounts 14, and the existence of detailed structural information facilitates genetic engineering 15. Moreover, it is a normal function of coat protein to encapsidate its own mRNA, which can be extracted from the assembled particle and amplified by reverse transcription and polymerase chain reaction. Coat protein's so-called AB-loop is a logical site for peptide display, because it protrudes prominently from the VLP surface, but despite some successes 16; 17; 18; 19; 20, insertions here frequently disrupt the ability of coat protein to correctly fold, leading to aggregation or proteolytic degradation 16. We previously produced a version of coat protein in which the subunits of the dimer are genetically fused 16; 21. The so-called “single-chain dimer” possesses greatly increased resistance to chemical denaturation and to the destabilizing effects of mutations 21; 22. Here we show that the single-chain dimer tolerates the insertion of a wide variety of peptides, that they are highly immunogenic when presented in the MS2 VLP format, and that the VLPs efficiently package the RNAs that direct their synthesis.
Results
Insertion of the ECL2 and V3 peptides in the coat protein AB-loop
The surface accessibility and regular geometric spacing of the AB-loop in the MS2 VLP make it an attractive site for the display of foreign peptides (Figure 1). We inserted two model peptides that in their natural environments are found in exposed loops. One was derived from the V3 loop of the HIV envelope protein of the lab-adapted strain, HIV-1LAI. Its core sequence is relatively conserved among HIV isolates, and this epitope is a target of neutralizing antibodies 23. The other peptide comes from the second extracellular loop (ECL2) of the macaque chemokine receptor, CCR5. In addition to its role in immune chemotaxis, CCR5 is a major HIV coreceptor. This particular sequence represents a region of ECL2 referred to as the undecapeptidyl arch (UPA) and is involved in HIV entry into cells 24.
Figure 1.
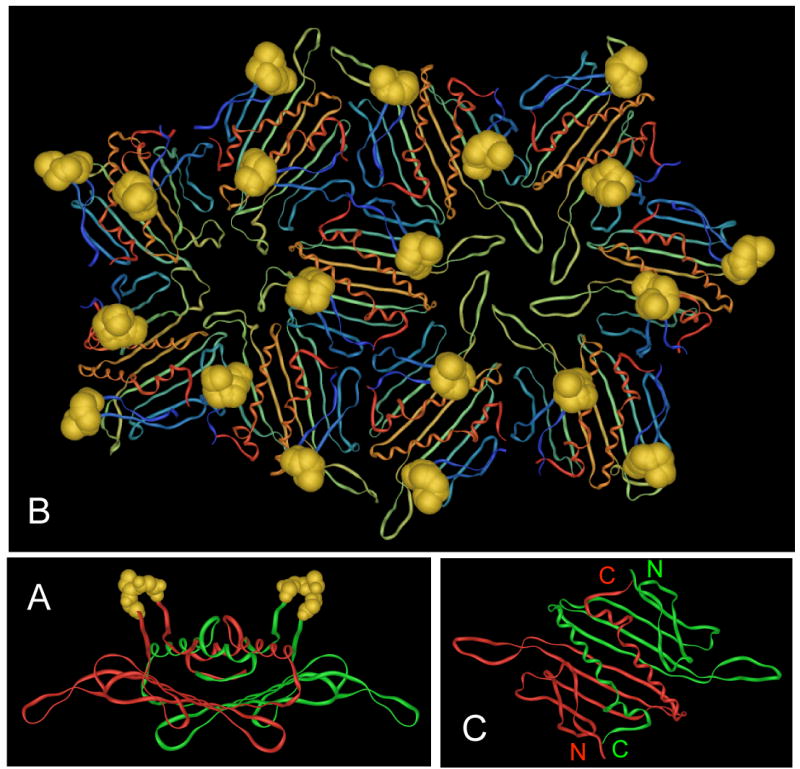
(A) Structure of the MS2 coat protein dimer seen edge-on, with its polypeptide chains in red and green and the AB-loops in gold. (B) A portion of the MS2 VLP showing the arrangment of subunits around the 5-fold and quasi-6-fold symmetry axes and the arrangement of the AB-loops (in gold). (C) Structure of the MS2 coat protein dimer as viewed from outside the VLP. Note the proximity of the N- and C-termini of the two polypeptides. Images were produced using imol software, available at http://www.pirx.com/iMol/.
To facilitate peptide insertion we constructed plasmid pMCTK2. It is similar to our previously described pCT119 14, but, following the example of Mastico et al. 17, was engineered to contain a Kpn I site in the AB-loop-encoding sequence (Figure 2A). We then inserted duplex oligonucleotides (Figure 2B) encoding the 10-amino acid V3 and ECL2 peptides. Because insertion at Kpn I results in duplication of codons 14 and 15, the length of coat protein was actually increased by a total of twelve amino acids. The resulting plasmids, pMCTK2-ECL2 and pMCTK2-V3 express the recombinant coat proteins from the E. coli lac promoter. A western blot of the soluble and insoluble fractions of crude cell lysates (Figure 3A) shows that the wild-type protein was abundantly produced in predominantly soluble form, but neither of the recombinant proteins was present in detectable quantities. It should be noted that if the AB-loop represented a single immunodominant epitope on wild-type MS2, our failure to detect these proteins could reflect insertional inactivation of the epitope, and consequent inability of our antiserum to bind. This cannot be case, however, since we show below that single-chain dimers with peptide insertions in both AB-loops are readily visualized by Western Blot. It seems likely, therefore, that the absence of these proteins is due to proteolytic degradation as a secondary consequence of a severe folding defect.
Figure 2.


(A) Arrangements of the coat protein reading frames on the plasmids used in this experiment. All express coat protein from the lac promoter. pMCTK is similar to the previously described pCT119 (5), but has silent mutations in codons 14 and 15 that introduce the Kpn I site (indicated by the arrow). p2MCTK3 expresses a single-chain dimer version of the protein with the Kpn I site in the C-terminal half of the single-chain dimer. Black boxes represent the ECL2 or V3 peptide insertions in the various plasmid derivatives. (B) Amino acid sequences (one-letter code) of the ECL2 and V3 peptides (top lines) and of the annealed oligonucleotides that encode them.
Figure 3.
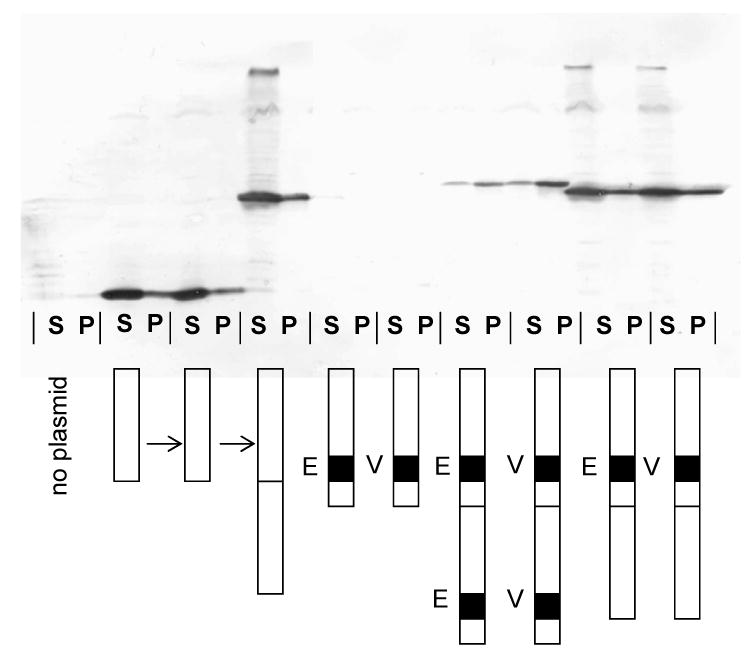

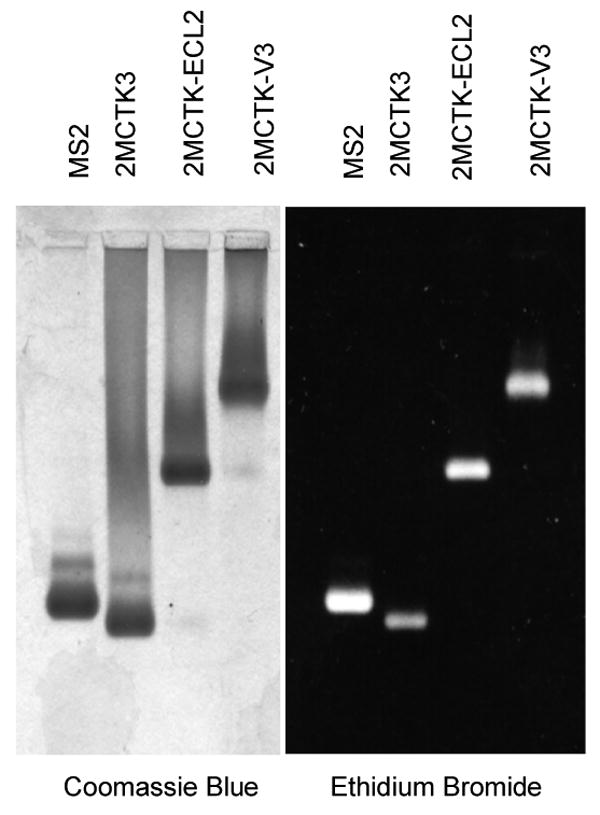
(A) Western blot analysis of the proteins produced by the various constructs described in the text and illustrated schematically here. In each case, a cell lysate was produced by sonication and then segregated into soluble (S) and insoluble (or pellet, P) fractions by centrifugation. Coat proteins were visualized using rabbit anti-MS2 serum and an alkaline phosphatase-labeled second antibody. (B) Elution of coat proteins from Sepharose CL-4B. Cell extracts were applied to the column and the coat protein content of individual fractions was determined by SDS-polyacrylamide gel electrophoresis. Proteins were visualized in the gel both by Coomassie Blue staining and by Western Blot, and the quantity of coat in each fraction was determined by densitometry. Authentic MS2 virus (not shown) co-elutes with VLPs. (C) Agarose gel electrophoresis of purified bacteriophage MS2 and the VLPs produced by the single-chain dimer construct and by the ECL2 and V3 recombinant VLPs. Protein was stained with Coomassie Blue R250 (left). Because the VLPs contain RNA they can also be visualized with ethidium bromide (at right).
Functional tests confirmed that the V3 and ECL2 recombinants are defective. Coat protein normally serves as a translational repressor, shutting off synthesis of the viral replicase by binding an 20-nucleotide RNA hairpin containing its ribosome binding site (the so-called translational operator). Fusing this sequence to the E. coli lacZ gene on the plasmid called pRZ5 14 provides a simple means of assessing translational repressor activity of coat protein variants. Cells containing pRZ5 form white colonies on x-gal plates when they express functional coat protein, but non-functional coat protein variants yield blue colonies. Neither the ECL2- nor V3-containing recombinant proteins inhibited β-galactosidase synthesis at all, indicating a complete failure to repress translation (Table I).
Table I.
Translational repressor activities of the various V3 and ECL2 recombinants as reflected in the relative blueness of colonies on X-Gal plates. Controls are pUCter3, which produces no coat protein and fails to repress, and pCT119 and p2MCTK3, which respectively produce wild-type coat protein and the single-chain dimer, and repress efficiently.
| Blueness on XGal | |
|---|---|
| pUCter3 | +++ |
| pCT119 | - |
| p2MCTK3 | - |
| pCT-ECL2 | +++ |
| pCT-V3 | +++ |
| p2M-ECL2-2 | +++ |
| p2M-V3-2 | +++ |
| p2M-ECL2-1 | - |
| p2M-V3-1 | - |
The defects are corrected in single-chain dimers
Figure 1C shows the structure of the coat protein dimer and illustrates the structural basis for construction of a single-chain dimer. Note the physical proximity of the C-terminus of one subunit to the N-terminus of the other. We previously demonstrated that genetic fusion of the two chains into single-chain dimers greatly protects the protein against the destabilizing effects of amino acid substitutions and chemical denaturants 16; 17; 21. In an effort to suppress the defects imparted by the ECL2 and V3 insertions, we constructed two types of single-chain dimer; one has a foreign peptide in both AB-loops, and the other contains the peptide in only its C-terminal half (Figure 2A).
We assessed the expression of the single-chain proteins by western blot (Figure 3A). When present in both AB-loops, the single-chain dimer seemed to revert partially the defects caused by the foreign peptides in the conventional dimer. The recombinant proteins were now detectable, but they were found predominantly as insoluble aggregates, suggesting that they were mostly misfolded. A folding failure is also consistent with an inability to repress translation (Table I). However, when the foreign peptides were incorporated into only the downstream copy of the single-chain dimer's two AB-loops the defect was fully corrected. The proteins were produced in normal amounts, were found mostly in the soluble fraction of the cell, and they repressed translation just like wild-type (Table I).
The elution of the ECL2 and V3 single-chain proteins from Sepharose CL-4B at the same position as authentic MS2 virus is another indication that the recombinant proteins assemble normally into VLPs (Figure 3B), and also provides a means to purify the ECL2 and V3 recombinant VLPs 14. Analysis by SDS-polyacrylamide gel electrophoresis shows that the VLPs contain only traces of contaminating cellular proteins (not shown). Electrophoresis of the VLPs in an agarose gel under native conditions is shown in Figure 3C. As expected of a properly assembled VLP, each contains RNA (it stains with ethidium bromide) and exhibits an altered electrophoretic mobility due to the charge differences conferred by the ECL2 and V3 peptides (Figure 2B). Staining of the gel with the protein stain Coomassie Brilliant Blue shows the same pattern. The mobility of the capsid produced by the unmodified single-chain dimer (p2MCTK3) is slightly greater than that shown by the wild-type VLP produced from pMCTK, perhaps because subunit fusion reduces by half the number of positively charged N-termini, which happen to reside near the VLP surface where they can influence mobility.
Recombinant VLPs were further characterized using a monoclonal antibody (mAb) (MAbIIIB-V3-13) that recognizes the V3 epitope and polyclonal sera raised against the ECL2 peptide. MAbIIIB-V3-13 specifically bound to purified V3-VLPs immobilized on an ELISA plate, but not to wild-type MS2 VLPs or ECL2-VLPs (Figure 4A), showing that the inserted V3 sequence is exposed on the surface of recombinant VLPs in a form competent for recognition by the monoclonal antibody. Similarly, polyclonal anti-ECL2 sera bound specifically to the ECL2 VLPs, but not to MS2 VLPs (Figure 4B).
Figure 4.


Analysis of immune responses to recombinant VLPs. (A) An anti-V3 mAb binds to V3-VLPs, but not ECL2-VLPs or wild-type MS2 VLPs. Dilutions of MAbIIIB-V3-13 were reacted with 500 ng/well of V3-VLPs, wild-type MS2 VLPs, or ECL2-VLPs. Binding was detected using a horseradish peroxidase-labeled goat anti-mouse IgG secondary followed by development with ABTS. Reactivity was measured by optical density at 405 nm (OD405). (B) Polyclonal sera raised against a cyclic ECL2 peptide binds to ECL2-VLPs, but not MS2 VLPs. Dilutions of sera from a mouse immunized with the cyclic ECL2 peptide (chemically conjugated to Qβ bacteriophage) were reacted with immobilized VLPs on an ELISA plate. Binding was detected as described above. (C) IgG antibody responses in C57Bl/6 mice immunized with wild-type MS2 VLPs, V3-VLPs, or ECL2-VLPs. End-point dilution ELISA titers against a peptide representing a portion of the V3 loop from HIVLAI (left panel), or a peptide representing the CCR5 ECL2 undecapeptidyl arch (UPA) (right panel) in serum from mice immunized three times with each VLP type. VLPs were administered either in the presence of complete Freund's adjuvant (CFA) or without adjuvant (NA). Results are from sera obtained 7 days after the third vaccination. Each data point represents the antibody titer from an individual mouse. Lines represent the geometric mean titer for each group. (D) Neutralization of HIV-1LAI infection of the MAGI-CCR5 indicator cell line using sera from mice immunized with V3-VLPs. Approximately 100 infectious virus particles were incubated with dilutions of pooled sera from mice immunized with wild-type MS2 VLPs, pooled sera from mice immunized with V3-VLPs, or the HIV neutralizing mAbs V3-13 or b12 (a potent neutralizing monoclonal antibody that recognizes an epitope outside of the V3 domain) for 1h and then added to target cells. Two days after infection, infected cells were scored by counting the number of blue cells in each well. Inhibition of HIV infection was determined by comparing the number of blue (infected) nuclei in the presence of antibody versus the number of blue nuclei in the absence of antibody. Data represents the average of two different experiments; error bars show the standard error of the mean. (E) Flow cytometric analysis of antibody binding to transiently transfected 293T cells. Cells were mock-transfected (shaded histogram) or transfected with pc.Rh-CCR5 (thick solid line) and then incubated with (upper left) a PE-labeled anti-CCR5 mAb (3A9), (upper right) secondary antibody alone, (lower left) sera from a mouse immunized with wild-type MS2 VLPs, or (lower right) sera from a mouse immunized with ECL2-VLPs.
Immunogenicity of the ECL2 and V3 recombinants
The abilities of purified recombinant ECL2- and V3-VLPs to induce antibodies against the target sequences were assessed by immunization of C57Bl/6 mice. Sera were tested for IgG antibodies specific for either the V3 or ECL2 peptides by end-point dilution ELISA. Mice immunized with V3-VLPs or ECL2-VLPs developed high titer (>104) IgG responses against the corresponding peptide, but not against the heterologous peptide (Figure 4C). No peptide-reactive antibodies were detected in sera from wild-type MS2 VLP-immunized mice. As with other VLP-based immunogens, high titer antibodies were induced without the use of exogenous adjuvants; coadministration of FA boosted IgG levels only slightly.
We next examined whether induced anti-V3 antibodies bound to full-length native protein. Because monoclonal antibodies that bind to this region of V3 have HIV neutralizing activity 23, we tested whether sera from V3-VLP immunized mice could inhibit HIV infection. Pooled sera from V3-VLP immunized mice, control sera, or two different HIV neutralizing monoclonal antibodies were preincubated with approximately 100 infectious HIV-1LAI particles, which were then used to infect an HIV indicator cell line (MAGI cells). Control sera from mice immunized with wild-type MS2 VLPs had no HIV neutralizing activity whereas sera from V3-VLP immunized mice neutralized HIV (∼75% neutralization at a 1:10 sera dilution) (Figure 4D). The more potent neutralizing activity displayed by the anti-V3 mAb (MAbIIIB V3-13) is consistent with the ∼10-fold higher V3-peptide ELISA binding activity of this mAb relative to the V3-VLP sera (data not shown).
The ability of ECL2-VLPs to elicit antibodies that bind native CCR5 was tested by flow cytometry. Macaque CCR5 was expressed on 293T cells by transient transfection with a rhesus macaque CCR5 expression vector (pc.Rh.CCR5), and the binding of mouse IgG was measured relative to mock-transfected cells. As shown in Figure 4E, sera from ECL2-VLP immunized mice bound to CCR5-transfected cells (relative to mock-transfected cells) whereas sera from control mice did not, demonstrating that anti-ECL2 antibodies bind native CCR5.
Testing the single-chain dimer's tolerance of random peptide insertions
We created a library of random 6-amino acid insertions in the AB-loop of pMCTK2 (wild-type coat) and of 6-, 8-, and 10-amino acids in the second AB-loop of p2MCTK3 (the single-chain dimer). This was accomplished by insertion of 6, 8 or 10 copies of the sequence NNY, where N=any nucleotide, and Y=C or T. Random NNY triplets produce codons for 15 of the 20 amino acids, and although such libraries cannot encode lys, glu, gln, trp and met, they create considerable diversity while avoiding the introduction of stop codons. The libraries were introduced into strain CSH41F-/pRZ5 and plated on X-Gal plates for the blue/white translational repression test. The results dramatically illustrated the importance of subunit fusion. In the case of the pMCTK2 6-mer library only 2% of colonies were white, showing that only rarely is a 6-mer insertion tolerated in the AB-loop of the conventional dimer. On the other hand, in the single-chain dimer 96% of 6-mer, 94% of 8-mer, and 92% of 10-mer insertions gave functional translational repressors. From each of these libraries we randomly selected 12 blue recombinants for sequence analysis, and, in the 6-mer and 8-mer libraries, found that about half the defective clones (5/12 and 6/12, respectively) had frameshift mutations (presumably caused by occasional errors during synthesis of the NNY primers), or were the products of anomolous ligation events. One quarter (3/12) of the defective 10-mer clones also had frameshift mutations. Thus, in each library a significant percentage of repressor-defective clones were not the results of failure to tolerate peptide insertions, but had other defects.
Visual inspection of the small percentage of sequences that resulted in folding failures immediately led to the impression that they are enriched in hydrophobic amino acids. This intuition was confirmed when all the peptide sequences were joined together (on paper) into a single 662-amino polypeptide, with the tolerated sequences (residues 1-482) and non-tolerated sequences (residues 483-662) grouped together. A Kyte-Doolittle hydrophobicity plot 25; 26 shows a distinct transition to higher average hydrophobicity at the white/blue junction (Figure 5).
Figure 5.

A hydrophobicity plot by the method of Kyte and Doolittle of a hypothetical protein produced by arbitrarily joining into a single long sequence all the random peptide sequences that resulted in a repressor-competent coat protein (residues 1-482) and those that interfered with repressor function (residues 483-662). Note the transition at about amino acid 483, indicating a substantially higher average hydrophobicity of peptides that interfere with coat protein folding.
White clones were picked from each of the 6-mer, 8-mer and 10-mer libraries and subjected to analysis of their abilities to support the synthesis of properly assembled VLPs. Sonicated cell lysates were subjected to agarose gel electrophoresis, and VLPs were visualized by ethidium bromide staining (upper half of each set in Figure 6) and by a Western Blot probed stained with anti-MS2 serum and alkaline phosphatase-conjugated anti-rabbit IgG (lower half of each set). The mobilities of the recombinant VLPs are diverse, because, as sequence analysis shows, most of the inserted peptides contain at least one charged amino acid. Importantly, tolerance of insertions is high: 100% (21/21) of 6-mer clones, and 87% (20/23) 8-mer clones contained a detectable VLP, while the frequency of successful 10-mers was 80% (16/20).
Figure 6.

Electrophoresis on agarose gel of VLPs found in crude lysates of cells producing a coat proteins with different random 6-, 8- or 10-amino acid insertions. The first two lanes of each gel are controls: pUCter3 produces no coat protein, while p2MCTK3 is the single-chain dimer construct without a peptide insertion. Ethidium bromide-stained gels (upper half of each set) and blots probed with anti-MS2 serum (lower half of each set) are shown.
MS2 VLPs encapsidate their mRNAs
Plasmids called pETCT and pET2CTdl-13 use the T7 promoter and transcription terminator to express wild-type coat protein and the single-chain dimer, respectively, from transcripts of predicted lengths of about 580 and 970 nucleotides. We purified their VLPs from bacteria, extracted the RNAs and subjected them to denaturing agarose gel electrophoresis 27. The ethidium-bromide-stained gel (Figure 7, left panel) shows that each VLP contains a dominant species comigrating with RNA markers produced by T7 transcription in vitro of the same pETCT and pET2CTdl-13 plasmids. Three other RNAs were also run as molecular weight markers and as hybridization controls. Two were genome RNAs extracted from the purified MS2 (3,569 nucleotides) and Qβ phages (4,220 nucleotides), and another, about 650 nucleotides long, was produced by transcription in vitro of a plasmid containing HCV core sequences. A Northern Blot probed with a 32P-labeled synthetic coat sense strand-specific oligonucleotide verifies the identities of the encapsidated RNAs; only RNAs expected to contain the coat coding sequence hybridize with the probe (Figure 7A, right). Thus, MS2 VLPs efficiently encapsidate RNAs whose characteristics suggest they represent the same RNAs that direct coat protein synthesis.
Figure 7.
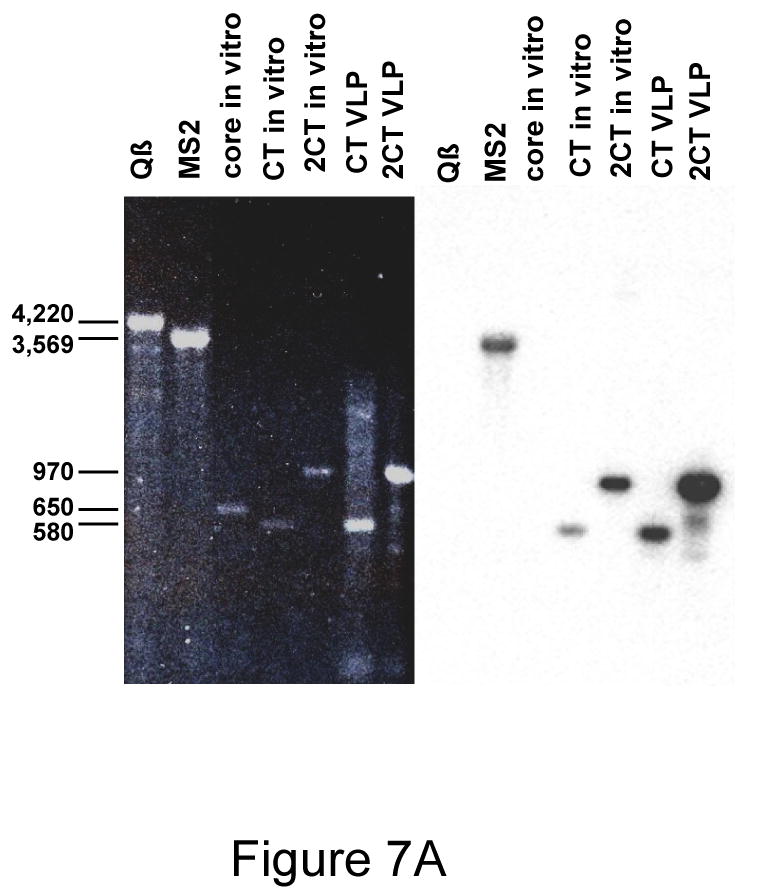
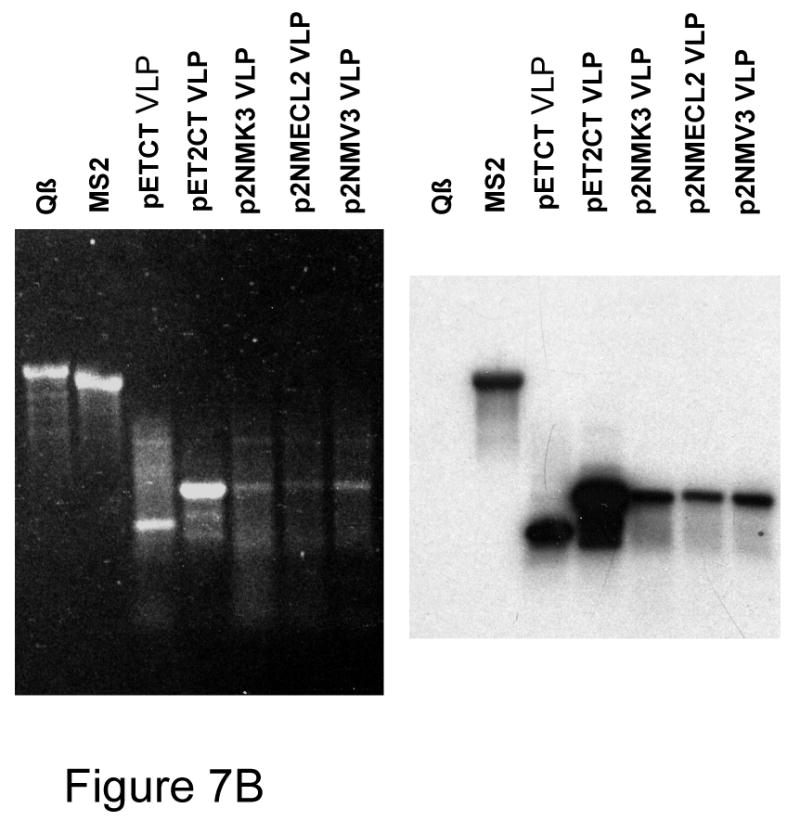
(A) Agarose gel electrophoresis of VLP-encapsdated RNAs. The left panel shows the ethidium bromide stained gel. A Northern Blot probed with a labeled MS2 coat-specfic oligonucleotide is shown in the right panel. “CT VLP” and “2CT VLP” refer to the RNAs extracted from conventional and single-chain dimer VLPs respectively. “CT in vitro” and “2CT in vitro” refer to the products of transcription in vitro of the same plasmids that produced the VLPs in bacteria. The in vitro transcription product of a similar plasmid containing HCV core sequences (“core in vitro”) was also run for comparison. MS2 and Qβ RNAs were obtained by extraction from the purified phages. (B) Agarose gel electorphoresis and Northern Blot of RNAs encapsidated in VLPs expressed from pET2NMK and its V3- and ECL2-dieplaying derivatives. Some of the same RNAs in (A) are also shown for comparision.
We wanted to verify that peptide-displaying recombinant VLPs also encapsidated their mRNAs. Using constructs similar to pETCT and pET2CT we synthesized in bacteria VLPs corresponding to the wild-type single-chain dimer with Kpn I in its second AB-loop (pET2NMK3), and its V3 and ECL2 derivatives (pET2NM-V3 and pET2NM-ECL2). The VLPs were purified and their RNAs extracted as described in Materials and Methods. Figure 7B shows agarose gel electrophoresis of these RNAs. Some of the RNAs shown in Figure 7A, namely those extracted from Qβ and MS2 virions, and from the pETCT and pET2CT VLPs, were run in this gel for comparision. The ethidium bromide-stained gel (left panel) shows that the V3 and ECL2 recombinants encapsidated the same coat-specific RNA species as pETCT and p2CT. Note also the small decrease in mobility of the V3 and ECL2 RNAs, consistent with the insertion of peptide-encoding sequences. Although the presence of a variable quantity of non-coat RNA is apparent in most samples, the most abundant single species is the coat-specific RNA in each case. A Northern Blot of a duplicate gel probed with the coat sense-strand-specific oligonucleotide (right panel) shows that the encapsidated RNAs are indeed coat-specific. Furthermore, RNA extracted from a peptide-displaying VLP was subjected to reverse transcription and polymerase chain reaction, and the nucleotide sequence of the product was determined. It was found to contain the peptide-encoding nucleotide sequence. Thus, VLPs faithfully encapsidate their mRNAs.
Discussion
The AB-loop is a natural location for peptide display, because it protrudes prominently from the MS2 VLP surface. Although correct folding of wild-type coat protein is usually prevented by AB-loop insertions, the single-chain dimer tolerates them. Insertion of either the V3 or ECL2 peptide into the AB-loop of wild-type coat protein resulted in a failure even to synthesize detectable amounts of the protein, but insertion of the same peptides into one AB-loop of the single-chain dimer yielded a protein that folded and assembled in a manner virtually indistinguishable from wild-type. The importance of subunit fusion was confirmed by the functional consequences of inserting random-sequence peptides. In the conventional dimer only 2% of random 6-mer insertions were functional for translational repression, but in the single-chain dimer between 93% and 98% (corrected for frameshift errors) of 6-mer, 8-mer or 10-mer insertions were fully competent repressors. Of these, a high proportion (80-100% depending on peptide insertion length) also produced a VLP.
The increased stability of the single-chain coat protein dimer is one example of a general approach to protein stabilization. In several other cases it was demonstrated that the covalent joining of subunits can stabilize the folded structures of oligomeric proteins 28; 29; 30. In coat protein the folding unit appears to be the dimer itself, since inspection of its three-dimensional structure (Figure 1) shows that each monomer's native conformation depends on association with its companion subunit. The covalent tethering of the two dimer halves likely reduces the entropy of the unfolded state, thus favoring the native structure. Another way of looking at it is to realize that when unfolded, the effective concentration of the two “monomers” of a single-chain dimer is high compared to that available to the conventional dimer at any normally attainable protein concentration, thus perturbing the folding-unfolding equilibrium in favor of the folded state.
Of course, a perfect platform for peptide library construction would show universal tolerance of insertions. While the single-chain coat protein does not quite achieve that objective, it does accommodate impressive peptide diversity. It is notable that many of the non-tolerated sequences are highly enriched in hydrophobic amino acids. The fact that most protein epitopes are themselves surface loops and naturally show a reduced incidence of hydrophobic residues, ameliorates concerns over the under-representation of such sequences in our VLP libraries. The use of an oligonucleotide synthesis method that controls codon frequencies to better mimic the amino acid compositions of natural proteins should result in an even higher frequency of tolerated insertions 31; 32. Furthermore, we suspect it is possible to produce additional engineered or randomly generated mutational variants of coat protein that, like the single-chain dimer, globally suppress the disruptive effects of the relatively few intolerable peptides. Even while we work toward the goal of a truly universal peptide display platform, however, we note that existing phage display systems are themselves subject to so-called library censorship issues; they also eliminate peptides whose sequences are inconsistent with viral morphogenesis 33; 34.
The necessity of displaying peptides in only one of the two halves of the coat protein dimer means that display density on our VLP is reduced by half, from 180 copies to 90. Since the high immunogenicity of recombinant VLPs is apparently due to the presentation of epitopes as dense repetitive arrays, we worried they might show reduced immunogenicity. However, the V3 and ECL2 VLPs induced specific high titer antibody responses, similar to those obtained using peptides displayed at even higher densities by chemical conjugation to RNA phage and Human Papilloma Virus (HPV) VLPs 35. These results are consistent with immunogenicity studies in which the density of epitope display was systematically varied on HPV VLPs; a 50% reduction of target density had minimal effects on immunogenicity 4.
One of coat protein's normal functions is to package the viral genome RNA that encodes it. This is normally assumed to occur through the specific interaction of coat protein with its RNA target, the translational operator of the replicase cistron. In this view, the same RNA-protein interaction responsible for translational repression also initiates RNA encapsidation and virus assembly. Consistent with this notion, the operator (or pac site) confers enhanced packagability to a heterologous RNA 36. Others have taken advantage of coat protein's encapsidation activity to incorporate specific RNAs into VLPs where they are resistant to RNase degradation 37. Some may find it surprising that pETCT and pET2CTdl-13 produce VLPs that efficiently encapsidate their own RNAs even though the translational operator sequence is not present. However, even before conducting the experiments described here, there were already indications that packaging does not absolutely require the coat protein-translational operator interaction. For example, MS2 viral variants whose mutant operators show no detectable ability to bind coat protein are viable 38. Moreover, a plasmid expressing a portion of the MS2 genome containing only maturase and coat sequences, and lacking the putative pac site produces a VLP that packages its mRNA (unpublished observations). Recently Bundy et al. also showed that MS2 coat protein expressed from plasmids similar to ours efficiently encapsidated its own RNA even though no translational operator was present 39. Perhaps the coat sequence itself contains an unidentified pac signal, or maybe our VLP RNAs are preferentially encapsidated through their sheer abundance, rather than by a specific RNA-protein recognition event. Alternatively, a high local concentration of newly synthesized coat protein in the vicinity of its mRNA on the ribosome might result in cis encapsidation. It should be noted that although coat-specific RNA is the most prevalent single species, VLPs also contain variable quantities of host RNAs. Experiments are underway to determine whether the presence of the translational operator can further enhance the specificity of encapsidation.
Whatever the packaging mechanism, it is clear that each individual member of a VLP library can be expected to encapsidate the mRNA that encodes it and its guest peptide, thus providing the phenotype-genotype linkage essential to any phage display-like technology, and making possible the affinity-selection of ligands for specific antibodies (or other receptors) from libraries of random-sequence peptides or antigen fragments. Reverse transcription and amplification by polymerase chain reaction of RNAs extracted from affinity selected VLPs will allow the recovery of the selected sequences, which can then be subjected to additional cycles of synthesis, assembly and selection as needed. Therefore, MS2 VLPs should provide the means both to identify epitopes and to present them directly to the immune system at high density on a single structural platform.
Materials and Methods
Plasmid construction
A PCR overlap extension method 40 introduced two silent nucleotide changes in codons 14 and 15 of the coat sequence and a unique KpnI site into the MS2 coat gene of pMCT, a plasmid nearly identical to the previously described pCT119 14. The new construct is called pMCTK2. Synthetic duplex oligonucleotides (from Integrated DNA Technologies, see Figure 2B) encoding the ECL2 and V3 peptides were inserted into the Kpn I site. DNA sequence analysis of the resulting recombinants confirmed the presence of the designed sequences. We call these plasmids pMCTK-ECL2 and pMCTK-V3 (Figure 2A).
The various single-chain dimer versions of the pMCTK-ECL2 and -V3 recombinants (Figure 2A) were produced by duplication of the wild-type and recombinant coat sequences. Briefly, the upstream half was produced by PCR amplification using a 5′ primer that anneals to plasmid sequences upstream of the coat sequence, and a downstream primer that creates a Bgl I site at the 3′-end of the coding sequence. This fragment was digested at the new Bgl I site and at the Hind III site in the upstream plasmid sequence. The downstream half was synthesized using a primer that creates a Bgl I site at the 5′-end of the coding sequence, and a 3′-primer that anneals to plasmid sequences downstream of coat. This fragment was digested at the Bgl I site at the 5′-end of this PCR fragment and at a Bam HI site present in plasmid sequences downstream of coat. These two DNAs were then joined by ligation to a vector fragment derived by Hind III - Bam HI cleavage of pMCT and introduced into E. coli by transformation. The resulting plasmids contain a duplication of the coat sequence, with the C-terminal amino acid of the upstream copy fused to amino acid 2 of the downstream sequence. This arrangement is identical to that found in the previously constructed p2CT-dl13 21, but with two conservative amino acid substitutions to accommodate the introduction of the Bgl I site, whose presence at the junction simplifies single-chain dimer construction. The plasmid p2MCTK3 was constructed by a similar process. It provides a single-chain dimer with a unique Kpn I site in the AB-loop of its downstream half.
Protein Expression, Purification and Functional Assays
To test the recombinant proteins for translational repressor activity, each plasmid was introduced into E. coli strain CSH41F- containing the translational repression reporter plasmid called pRZ5 14 and plated on LB medium containing the β-galactosidase chromogenic substrate, 5--bromo-4-chloro-3-indolyl-β-D-galactoside (X-gal). To determine the expression levels of recombinant proteins and their solubilities, cell lysates from 1 ml overnight cultures were separated into soluble and insoluble fractions and subjected to SDS-gel electrophoresis (see ref. 16 for details). Contents of the gel were transferred to a nitrocellulose membrane and probed with rabbit anti-MS2 serum and alkaline phosphatase-conjugated goat anti-rabbit IgG antibodies. The coat proteins encoded in p2MCTK3, p2MCTK-ECL2 and p2MCTK-V3 were purified to greater than 90% purity by chromatography in Sepharose CL-4B using methods described previously 14.
Rapid assessment of a recombinant protein's ability to assemble into a VLP was performed by electrophoresis of sonicated cell lysates (from 1ml overnight cultures) in gels of 1% agarose in 50mM potassium phosphate, pH7.5 13. Gels were stained with ethidium bromide to reveal the presence of VLPs, which contain host RNAs. The identity of the VLPs was then confirmed by transferring the contents of the gel to nitrocellulose and probing with rabbit anti-MS2 serum and an alkaline phosphatase-labeled second antibody.
Libraries of random sequence peptides
To insert random DNA sequences encoding 6-, 8- and 10-amino acid peptides into the AB-loop, the primers described below were used to amplify a coat fragment from pMCT in three different PCR reactions. Three different 5′-primers [called (NNY)6, (NNY)8 and (NNY)10] attach at codon 14 a Kpn I site and 6, 8 or 10 randomized codons of sequence NNY (where N=A,C,G, or T and Y=T or C). Each reaction employed a single 3′-primer that annealed downstream of a Bam HI site in the plasmid vector. The resulting PCR products were digested with Kpn I and Bam HI, gel purified and ligated to the similarly digested vector fragments of p2MCTK3 or pMCTK2. These were introduced by transformation into strain CSH41F- containing plasmid pRZ5 14 and plated on LB medium containing X-gal. Control ligations containing only vector DNA gave rise to at least 1000-fold fewer colonies than those that contained an insert fragment. After overnight incubation at 37°C the relative numbers of blue and white colonies obtained were determined. Properly folded coat proteins repress translation of β-galactosidase and yield white colonies. From each library 24 white and 12 blue colonies were picked to two different 1 ml cultures in LB medium and grown overnight with shaking at 37°C. Note that after sequence analysis, a few white clones in were dropped from the analysis. A few either lacked a peptide insertion or contained mutations at secondary sites which might complicate interpretation of results. One set of cultures was used for plasmid isolation and DNA sequence analysis. The other was lysed by sonication and subjected to agarose gel electrophoresis as described above. VLPs were visualized by ethidium bromide staining and by blotting to nitrocellulose and probing with rabbit anti-MS2 serum and alkaline phosphatase-conjugated goat anti-rabbit IgG.
Packaging of coat-specific RNAs
The Xba I - Bam HI fragments of plasmids pCT119 13 and p2CTdl-13 21 were inserted into the T7 expression vector, pET3d 41. Coat protein expression was induced by IPTG in bacterial strain BL21(DE3)/pLysS using standard methods, and VLPs were extracted and purified by Sepharose CL-4B chromatography 14 followed by centrifugation to equilibrium in CsCl gradients (1.40g/cc starting density) at 40,000 rpm in the SW50.1 rotor. RNAs were extracted from VLPs using phenol/chloroform and applied to a 1.5% agarose gel containing formaldehyde 27. The gel was blotted to nitrocellulose and probed with a coat-specific synthetic oligonucleotide (5′-CGAGTTAGAGCTGATCCATTCAGCGACCCC-3′) complementary to the sense-strand and labeled at its 5′-end with 32P. Control RNAs were produced by transcription of pETCT and pET2CTdl-13 in vitro using T7 RNA polymerase.
Immunization and characterization of antisera
Antisera were prepared by inoculating C57Bl/6 mice with 15 μg wild-type MS2 VLPs, 15 μg MS2-V3 VLPs, or 15 μg MS2-ECL2 VLPs. Mice were inoculated intramuscularly three times at 2-week intervals. Sera were collected prior to each injection and 2 weeks after the final boost. When adjuvant was used, antigen was diluted 1:1 in complete Freund's adjuvant (CFA; initial injection) or incomplete Freund's adjuvant (IFA; subsequent boosts) immediately prior to the injection. All animal care was in accordance with the National Institutes of Health and University of New Mexico guidelines. Antibody titers were determined by ELISA using peptides corresponding to the target sequences. A V3 peptide (RIQRGPGRAFVTGK; synthesized by Commonwealth Biotechnologies, Chantilly, VA) was conjugated to KLH using a carbodiimide crosslinker (Pierce). A cyclic peptide corresponding to macaque CCR5 ECL2 (C1D2R3S4Q5R6E7G8L9H10Y11T12G13, in which Gly13 was linked to Asp2 through a dipeptide spacer; synthesized by Celtek Peptides, Nashville TN) was conjugated to avidin using a heterobifunctional crosslinker (SMPH; Pierce). Conjugated peptides were immobilized (at 200 ng/well) onto Immulon II ELISA plates (Dynex Technologies, Chantilly, VA) overnight at 4°C and then wells were blocked with PBS plus 0.5% non-fat dry milk for 2h at room temperature. Mouse serum was serially diluted in PBS-0.5% milk and applied to wells for 2.5h at room temperature. Reactivity to antigen was determined using horseradish peroxidase (HRP)-labeled goat anti-mouse IgG (Jackson Immunoresearch, West Grove, PA) at a 1:2000 dilution in blocking buffer as a secondary antibody. Upon development, optical densities were read at 405 nm using an OpSys MR plate reader (Thermo Labsystems, Waltham, MA). OD405 values that were greater than twice background (usually >0.080) were considered positive.
HIV neutralization was measured using the MAGI-CCR5 indicator cell line. These cells and the MAGI-CCR5 assay are described in more detail by Chackerian et al. 42. One hour prior to infection, dilutions of mouse sera were incubated with approximately 100 infectious HIV-1LAI virus particles in a total volume of 50 μL at 37°C. The virus-antibody mixture was then added to wells in a total volume of 200 μL in the presence of 10 μg/mL DEAE-Dextran (Sigma-Aldrich, St. Louis, MO). After 2h at 37°C, virus and antibody were removed from each well and replaced with 0.5 mL of media. Two days after infection, cells were fixed, washed, and stained for β-galactosidase activity, as described previously 43.
Binding of mouse serum IgG to native CCR5 was tested by flow cytometry. 293T cells were transiently transfected with a rhesus macaque CCR5-encoding expression vector [pc.Rh-CCR5; 44]. Cells were detached from the monolayer using 5 mM EDTA and then washed three times in staining buffer (PBS plus 0.5% BSA). To remove antibodies that bound non-specifically to cells, sera was preincubated with untransfected 293T cells (105 cells for every 5 μL of sera) for 45 min at 4°C. Sera was removed from cells and then incubated with CCR5- or mock-transfected cells. Approximately 105 cells were resuspended in 50 μl of staining buffer plus 10 μL of mouse sera for 30 min at 4°C. After washing three times with staining buffer, cells were resuspended in 50 μl of staining buffer plus 250 ng of fluorescein isothiocyanate (FITC)-labeled goat anti-mouse IgG (Jackson Immunoresearch) and then incubated for 30 min at 4°C. As a control, cells were stained with secondary antibody alone or with a phycoerythrin (PE)-labeled anti-CCR5 monoclonal antibody (3A9; BD Pharmingen). Before analysis, cells were washed twice more with staining buffer and resuspended in 0.5 ml of staining buffer. Specific binding was measured relative to mock-transfected cells.
Table II.
The percentages of 6-mer, 8-mer and 10-mer AB-loop insertions that result in functional translational repressors (white colonies on X-Gal plates). Plasmid pMCTK2 produces a conventional coat protein dimer, while p2MCTK3 incorporates the insertions in the second AB-loop of a single-chain dimer. (nd = not determined).
| Percent White Colonies | ||
|---|---|---|
| pMCTK2 | p2MCTK3 | |
| (NNY)6 | 2 | 96 |
| (NNY)8 | nd | 94 |
| (NNY)10 | nd | 92 |
Acknowledgments
We thank John Schiller for comments on the manuscript and Paul Durfee for technical assistance. The following reagents were obtained through the AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH: HIV-1 V3 Monoclonal Antibody (IIIB-V3-13) from Dr. Jon Laman, pc.Rh-CCR5 from Dr. Preston Marx. This work was supported by NIH grants 5RO1 GM042901 (to D.S.P.) and R01 AI065240 (to B.C.)
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
This PDF receipt will only be used as the basis for generating PubMed Central (PMC) documents. PMC documents will be made available for review after conversion (approx. 2-3 weeks time). Any corrections that need to be made will be done at that time. No materials will be released to PMC without the approval of an author. Only the PMC documents will appear on PubMed Central -- this PDF Receipt will not appear on PubMed Central.
References
- 1.Bachmann MF, Rohrer UH, Kundig TM, Burki K, Hengartner H, Zinkernagel RM. The influence of antigen organization on B cell responsiveness. Science. 1993;262:1448–1451. doi: 10.1126/science.8248784. [DOI] [PubMed] [Google Scholar]
- 2.Milich DR, Chen M, Schodel F, Peterson DL, Jones JE, Hughes JL. Role of B cells in antigen presentation of the hepatitis B core. Proc Natl Acad Sci U S A. 1997;94:14648–53. doi: 10.1073/pnas.94.26.14648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chackerian B, Lowy DR, Schiller JT. Induction of autoantibodies to mouse CCR5 with recombinant papillomavirus particles. Proc Natl Acad Sci USA. 1999;96:2373–2378. doi: 10.1073/pnas.96.5.2373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chackerian B, Lenz P, Lowy DR, Schiller JT. Determinants of autoantibody induction by conjugated papillomavirus virus-like particles. J Immunol. 2002;169:6120–6. doi: 10.4049/jimmunol.169.11.6120. [DOI] [PubMed] [Google Scholar]
- 5.Chackerian B, Lowy DR, Schiller JT. Conjugation of a self-antigen to papillomavirus-like particles allows for efficient induction of protective autoantibodies. J Clin Invest. 2001;108:415–23. doi: 10.1172/JCI11849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fehr T, Bachmann MF, Bucher E, Kalinke U, Di Padova FE, Lang AB, Hengartner H, Zinkernagel RM. Role of repetitive antigen patterns for induction of antibodies against antibodies. J Exp Med. 1997;185:1785–92. doi: 10.1084/jem.185.10.1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Spohn G, Schwarz K, Maurer P, Illges H, Rajasekaran N, Choi Y, Jennings GT, Bachmann MF. Protection against osteoporosis by active immunization with TRANCE/RANKL displayed on virus-like particles. J Immunol. 2005;175:6211–8. doi: 10.4049/jimmunol.175.9.6211. [DOI] [PubMed] [Google Scholar]
- 8.Van Houten NE, Scott JK. Phage Libraries for Developing Antibody-Targeted Diagnostics and Vaccines. In: Sidhu S, editor. Phage Display in Biotechnology and Drug Discovery. Taylor and Francis Group; 2005. pp. 165–254. [Google Scholar]
- 9.Jegerlehner A, Tissot A, Lechner F, Sebbel P, Erdmann I, Kundig T, Bachi T, Storni T, Jennings G, Pumpens P, Renner WA, Bachmann MF. A molecular assembly system that renders antigens of choice highly repetitive for induction of protective B cell responses. Vaccine. 2002;20:3104–12. doi: 10.1016/s0264-410x(02)00266-9. [DOI] [PubMed] [Google Scholar]
- 10.Billaud JN, Peterson D, Barr M, Chen A, Sallberg M, Garduno F, Goldstein P, McDowell W, Hughes J, Jones J, Milich D. Combinatorial approach to hepadnavirus-like particle vaccine design. J Virol. 2005;79:13656–66. doi: 10.1128/JVI.79.21.13656-13666.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Muller OJ, Kaul F, Weitsman MD, Pasqualini R, Arap W, Kleinschmidt JA, Trepel M. Random peptide libraries displayed on adeno-associated virus to select for targeted gene therapy vectors. Nature Biotechnology. 2003;21:1040–1046. doi: 10.1038/nbt856. [DOI] [PubMed] [Google Scholar]
- 12.Gupta A, Onda M, Pastan I, Adhya S, Chaudhary VK. High-density Functional Display of Proteins on Bacteriophage Lambda. Journal of Molecular Biology. 2003;334:241–254. doi: 10.1016/j.jmb.2003.09.033. [DOI] [PubMed] [Google Scholar]
- 13.Peabody DS. The RNA binding site of bacteriophage MS2 coat protein. Embo J. 1993;12:595–600. doi: 10.1002/j.1460-2075.1993.tb05691.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Peabody DS. Translational repression by bacteriophage MS2 coat protein expressed from a plasmid. A system for genetic analysis of a protein-RNA interaction. J Biol Chem. 1990;265:5684–9. [PubMed] [Google Scholar]
- 15.Golmohammadi R, Valegard K, Fridborg K, Liljas L. The refined structure of bacteriophage MS2 at 2.8 A resolution. J Mol Biol. 1993;234:620–39. doi: 10.1006/jmbi.1993.1616. [DOI] [PubMed] [Google Scholar]
- 16.Peabody DS. Subunit fusion confers tolerance to peptide insertions in a virus coat protein. Arch Biochem Biophys. 1997;347:85–92. doi: 10.1006/abbi.1997.0312. [DOI] [PubMed] [Google Scholar]
- 17.Mastico RA, Talbot SJ, Stockley PG. Multiple presentation of foreign peptides on the surface of an RNA-free spherical bacteriophage capsid. J Gen Virol. 1993;74:541–8. doi: 10.1099/0022-1317-74-4-541. [DOI] [PubMed] [Google Scholar]
- 18.Stockley PG, Mastico RA. Use of fusions to viral coat proteins as antigenic carriers for vaccine development. Methods Enzymol. 2000;326:551–69. doi: 10.1016/s0076-6879(00)26076-x. [DOI] [PubMed] [Google Scholar]
- 19.Brown WL, Mastico RA, Wu M, Heal KG, Adams CJ, Murray JB, Simpson JC, Lord JM, Taylor-Robinson AW, Stockley PG. RNA bacteriophage capsid-mediated drug delivery and epitope presentation. Intervirology. 2002;45:371–80. doi: 10.1159/000067930. [DOI] [PubMed] [Google Scholar]
- 20.Kozlovskaya TM, Pushko PM, Stankevich EI, Dreimane AY, Sniker DY, Grinshtein EE, Greilinya DE. Genetically engineered mutants of the coat protein of an RNA-containing bacteriophage. Molecular Biology (USSR) 1988;22:584–593. [PubMed] [Google Scholar]
- 21.Peabody DS, Lim F. Complementation of RNA binding site mutations in MS2 coat protein heterodimers. Nucleic Acids Res. 1996;24:2352–9. doi: 10.1093/nar/24.12.2352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Peabody DS, Chakerian A. Asymmetric contributions to RNA binding by the Thr(45) residues of the MS2 coat protein dimer. J Biol Chem. 1999;274:25403–10. doi: 10.1074/jbc.274.36.25403. [DOI] [PubMed] [Google Scholar]
- 23.Laman JD, Schellekens MM, Abacioglu YH, Lewis GK, Tersmette M, Fouchier RA, Langedijk JP, Claassen E, Boersma WJ. Variant-specific monoclonal and group-specific polyclonal human immunodeficiency virus type 1 neutralizing antibodies raised with synthetic peptides from the gp120 third variable domain. J Virol. 1992;66:1823–31. doi: 10.1128/jvi.66.3.1823-1831.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Misumi S, Nakajima R, Takamune N, Shoji S. A cyclic dodecapeptide-multiple-antigen peptide conjugate from the undecapeptidyl arch (from Arg(168) to Cys(178)) of extracellular loop 2 in CCR5 as a novel human immunodeficiency virus type 1 vaccine. J Virol. 2001;75:11614–20. doi: 10.1128/JVI.75.23.11614-11620.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gasteiger E, H C, Gattiker A, Duvaud S, Wilkins MRm, Appel RD, Barioch A. Protein Identification and Analysis Tools on the ExPASy Server. In: Walker JM, editor. The Proteomics Protocols Handbook. Hum and Press; 2005. [Google Scholar]
- 26.Kyte J, Doolittle RF. A simple method for displaying the hydropathic character of a protein. Journal of Molecular Biology. 1982;157:105–132. doi: 10.1016/0022-2836(82)90515-0. [DOI] [PubMed] [Google Scholar]
- 27.Lehrach H, Diamond D, Wozney JM. RNA molecular weight determinations by gel electrophoresis under denaturing conditions: a critical reexamination. Biochemistry. 1977;16:4743–4751. doi: 10.1021/bi00640a033. [DOI] [PubMed] [Google Scholar]
- 28.Looker D, Abbott-Brown D, Cozart P, Durfee S, Hoffman S, Mathews AJ, Miller-Roehrich J, Shoemaker S, Trimble S, Fermi G, Komiyama G, Nagai K, Stetler GL. A human recombinant haemoglobin designed for use as a blood substitute. Nature. 1992;356:258–60. doi: 10.1038/356258a0. [DOI] [PubMed] [Google Scholar]
- 29.Liang H, Sandberg WS, Terwilliger TC. Genetic fusion of subunits of a dimeric protein substantially enhances its stability and rate of folding. Proc Natl Acad Sci U S A. 1993;90:7010–4. doi: 10.1073/pnas.90.15.7010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yang T, Olsen KW. Thermal stability of hemoglobin crosslinked in the T-state by bis(3,5-dibromosalicyl) fumarate. Biochem Biophys Res Commun. 1991;174:518–23. doi: 10.1016/0006-291x(91)91447-k. [DOI] [PubMed] [Google Scholar]
- 31.Ono A, Matsuda A, Zhao J, Santi DV. The synthesis of blocked triplet-phosphoramidites and their use in mutagenesis. Nucleic Acids Res. 1995;23:4677–82. doi: 10.1093/nar/23.22.4677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Virnekas B, Ge L, Pluckthun A, Schneider KC, Wellnhofer G, Moroney SE. Trinucleotide phosphoramidites: ideal reagents for the synthesis of mixed oligonucleotides for random mutagenesis. Nucleic Acids Res. 1994;22:5600–7. doi: 10.1093/nar/22.25.5600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rodi DJ, Mandava S, Makowski L. Filamentous bacteriophage structure and biology. In: Sidhu S, editor. Phage Display in Biotechnology and Drug Discovery. CRC Press; 2005. pp. 1–62. [Google Scholar]
- 34.Rodi DJ, Soares AS, Makowski L. Quantitative assessment of peptide sequence diversity in M13 combinatorial peptide phage display libraries. J Mol Biol. 2002;322:1039–52. doi: 10.1016/s0022-2836(02)00844-6. [DOI] [PubMed] [Google Scholar]
- 35.Chackerian B, Rangel M, Hunter Z, Peabody DS. Virus and virus-like particle-based immunogens for Alzheimer's disease induce antibody responses against amyloid-beta without concomitant T cell responses. Vaccine. 2006;24:6321–31. doi: 10.1016/j.vaccine.2006.05.059. [DOI] [PubMed] [Google Scholar]
- 36.Pickett GG, Peabody DS. Encapsidation of heterologous RNAs by bacteriophage MS2 coat protein. Nucleic Acids Res. 1993;21:4621–6. doi: 10.1093/nar/21.19.4621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.WalkerPeach CR, Winkler M, DuBois DB, Pasloske BL. Ribonuclease-resistant RNA Controls (Armored RNA) for Reverse Transcription-PCR, Branched DNA, and Genotyping Assays for Hepatitis C Virus. Clinical Chemistry. 1999;45:2079–2085. [PubMed] [Google Scholar]
- 38.Peabody DS. Role of the coat protein-RNA interaction in the life cycle of bacteriophage MS2. Mol Gen Genet. 1997;254:358–64. doi: 10.1007/s004380050427. [DOI] [PubMed] [Google Scholar]
- 39.Bundy B, Franciszkowicz M, Swartz J. Escherichia coli-based Cell-free Synthesis of Virusl-like Particles. Botechnology and Bioengineering. 2008;100:28–37. doi: 10.1002/bit.21716. [DOI] [PubMed] [Google Scholar]
- 40.Higuchi R, Krummel B, Saiki RK. A general method of in vitro preparation and specific mutagenesis of DNA fragments: study of protein and DNA interactions. Nucleic Acids Res. 1988;16:7351–67. doi: 10.1093/nar/16.15.7351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Studier FW, Rosenberg AH, Dunn JJ, Dubenforff JW. Use of T7 RNA polymerase to direct expression of cloned genes. Methods in Enzymology. 1990;185:60–89. doi: 10.1016/0076-6879(90)85008-c. [DOI] [PubMed] [Google Scholar]
- 42.Chackerian B, Long EM, Luciw PA, Overbaugh J. Human immunodeficiency virus type 1 coreceptors participate in postentry stages in the virus replication cycle and function in simian immunodeficiency virus infection. J Virol. 1997;71:3932–3939. doi: 10.1128/jvi.71.5.3932-3939.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kimpton J, Emerman M. Detection of replication-competent and pseudotyped human immunodeficiency virus with a sensitive cell line on the basis of activation of an integrated beta-galactosidase gene. J Virol. 1992;66:2232–2239. doi: 10.1128/jvi.66.4.2232-2239.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chen Z, Zhou P, Ho DD, Landau NR, Marx PA. Genetically divergent strains of simian immunodeficiency virus use CCR5 as a coreceptor for entry. J Virol. 1997;71:2705–14. doi: 10.1128/jvi.71.4.2705-2714.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]