

Abstract
A primary challenge for structural genomics is the automated functional characterization of protein structures. We have developed a sequence-independent method called S-BLEST (Structure-Based Local Environment Search Tool) for the annotation of previously uncharacterized protein structures. S-BLEST encodes the local environment of an amino acid as a vector of structural property values. It has been applied to all amino acids in a nonredundant database of protein structures to generate a searchable structural resource. Given a query amino acid from an experimentally determined or modeled structure, S-BLEST quickly identifies similar amino acid environments using a K-nearest neighbor search. In addition, the method gives an estimation of the statistical significance of each result. We validated S-BLEST on X-ray crystal structures from the ASTRAL 40 nonredundant dataset. We then applied it to 86 crystallographically determined proteins in the protein data bank (PDB) with unknown function and with no significant sequence neighbors in the PDB. S-BLEST was able to associate 20 proteins with at least one local structural neighbor and identify the amino acid environments that are most similar between those neighbors.
Keywords: protein structure, bioinformatics, similarity search, protein function, data mining
INTRODUCTION
Understanding the relationship between protein structure and chemical function is a problem of growing importance.1 In particular, structural genomics initiatives are determining the structures of targets without known or characterized function. Some of these initiatives have prioritized targets with potentially novel folds based on sequence with little similarity to known structure. Currently, conservation within a sequence alignment or phylogenetic tree remains the primary method for computationally identifying functional residues, and many experimental methods rely on site-directed mutagenesis in combination with other functional assays.2
Generally, function is inferred computationally by assessing similarity to proteins of known function. This guilt-by-association approach has proven to be valuable. In addition to sequence comparative methods, current structural methods for identifying function rely on one of the following:
Few clustering methods can identify functional residues automatically based on structural properties alone. Sequence-based methods for functional characterization rely on identifying conserved residues within protein structures. More sophisticated methods, such as the evolutionary trace method, use phylogenies combined with structure to define residues of functional importance.2 It is important to develop sequence-independent methods for identifying function to complement sequence-based methods when they are limited by lack of sequence similarity or small datasets.
Methods for identifying key functional residues, or molecular fingerprints, can classify function. These include Fuzzy Functional Forms,4 PROCAT,3 a neural network method developed by Stawiski et al.,6 and FEATURE.7 FEATURE describes a local environment around an arbitrary three-dimensional point in space by building a vector of property values that lie within several radial shells centered about the point. The properties are discrete structural property values for each atom within a shell. These values contain the number of atoms associated with a given residue type, secondary structure, van der Waals volume, and solvent accessibility. Given two sets of vectors, one set associated with some common functional or structural attribute and the other set lacking that attribute, FEATURE uses supervised machine learning to predict new positions within a protein structure that share the common attribute.
SCOP has proven to be a powerful tool for studying known protein structures.8 By maintaining a complete, annotated classification of all known proteins based on sequence, structure, and functional information, the structural components that classify a family can be determined. SCOP is a manually curated database and often is used as a gold standard for structural classification of proteins.
Sequence-independent structure-based methods for function assignment are challenging for several reasons. First, aligning local structure is a difficult computational task.9 Second, estimating the statistical significance of the results is challenging.10 Third, scanning through the entire protein data bank (PDB)11 can be computationally demanding. Finally, and perhaps vexing, structural similarity and functional similarity are not always well correlated.12
We have developed a method for unsupervised mining of structural datasets and automatically identifying local regions within protein structures that are statistically associated with a given annotation. Methods exist for unsupervised mining of structural topology. These include VAST,13 DALI,14 the method of Singh and Saha,15 Dubey et al.,16 and PINTS.17 Our method is complementary to these methods by defining the most structurally significant residue environments for given a classification, based on the structural environments represented in that database. S-BLEST (Structure-Based Local Environment Search Tool) is based on the FEATURE representation of a local environment, and rapidly searches databases of vectors of local structure properties. This method is a structural analog to sequence-based similarity search methods such as BLAST.18 We parameterized and evaluated the method by evaluating how well selected residue environments in the ASTRAL 40 dataset are associated with their annotated SCOP family.
MATERIALS AND METHODS
Residue-Based K Nearest Neighbor (KNN) Searches Against Structural Databases
A residue is encoded as a vector of properties using the following procedure, similar to others.7,19 To describe the local environment for each residue, a vector of properties is taken from a set of concentric shells extending outward from the position of the residue’s beta-carbon (Cβ) atom. Cβ atom positions for glycine residues were estimated by determining the average position of a Cβ (relative to the Cα, N, and C atoms) from serine protease 1DSU. Each shell contains 66 properties, which include the number of atoms associated with a given residue type, the number of positively and negatively charged ions, the van der Waals volume of the shell, and the solvent accessibility. Each vector contains three shells with the following radial boundaries: 1.875, 3.75, 5.625, and 7.5 Å. With three shells each having 66 properties, the resulting vector that describes the local environment of a residue has 264 dimensions. The properties are identical to the ones used by Bagley and Altman.7 This representation is orientation independent and can be used on arbitrary coordinates within a protein structure.
To encode the entire PDB, the Cβ of each amino acid from each structure in the ASTRAL 40 nonredundant structure database was encoded as a vector as described above. We use a nonredundant database so that features derived from recent common descent are minimized. All hetero-atoms were removed before encoding, as was all atomic information not associated with the chain the input residue is associated with. A vector set containing the entire PDB with other chains included was also built and tested. Each component of the vectors was normalized as integers from 0 to 255 with the formula:
where mini and maxi are the minimum and maximum value of the ith component across the entire vector set. In addition, the minimum and maximum are capped at a maximum of 18 standard deviations from the mean, in order to prevent odd outliers from skewing the results. Only X-ray crystal structures were used in the analysis. Approximately one million vectors were in the ASTRAL 40 v1.65 vector set. This set was stored in a binary file that contains the normalization factors, mini and maxi, for each dimension and the vector data. Each vector is encoded with the PDB id and chain (5 bytes), the residue type (1 byte), the residue number (1 byte), the insertion code (1 byte), and the vector data (264 bytes).
The S-BLEST method relies on nearest-neighbor searches using a Manhattan distance metric. Manhattan distance was chosen because it is inexpensive to calculate and the most derivative statistics are easy to determine. The closest vector from each chain in the dataset is determined, sorted, and output. A significance score (z-score) is calculated by estimating the mean and variance of all distances between the query residue and the residues in the dataset using the following formula:
Given a query residue, S-BLEST can find the most similar residue in each chain in the dataset and provide a score for the similarity using the z-score.
Identification of Residue Environments Associated With a Structural or Functional Annotation
If a query protein is a member of a known class (such as SCOP family), the residue environments most associated with that family can be readily determined by performing an S-BLEST query on each residue and performing the following protocol. The performance of each residue can be determined by creating a receiver operator characteristics (ROC) plot of the ranking, where the true-positive rate is plotted against the false-positive rate. A true positive is a protein structure that belongs to the same SCOP family as the query protein with a z-score of greater magnitude than the threshold. A false positive is a protein structure that does not belong to the same SCOP family but has a z-score of greater magnitude than the threshold. Each point on the plot represents the true-positive rate and false-positive rate of the ranking at a given z-score threshold. The ROC plot can be summarized by calculating the area under the curve (AUC). The AUC of a residue in a query structure of known function indicates how well the residue environment classifies the SCOP family of the structure and can range from 0.0 (perfect reverse classification) to 1.0 (perfect classification).
Congruence Approach for Combining S-BLEST Searches
Congruence approaches are a useful way to combine several searches to increase statistical significance.20 When given a query with multiple residues, such as all the residues in a query chain, S-BLEST can identify chains in the dataset that are most similar to the query chain and pinpoint the residues between the query chain and the dataset chain that are similar. The score for the dataset chain is the average z-score of the k most similar residues in the chain.
The following procedure is used to identify and score the most similar chain in the dataset to a query chain. For each residue in the query chain, the most similar residue in each dataset chain is identified and scored (using the above z-score). If there were n residues in the query chain, there would be n residues (possibly redundant) in the dataset chain that are identified as most similar to each of the n residues in the query each with a z-score. The score for the chain is the average of the top k z-scores. Each chain can then be ranked according to this averaged z-score, and the top k residues are reported as the residues bringing the query chain and database chain together. Because of the large computational task of building and ranking a table, z-scores of less than -2.5 are filtered out. Although it is possible that filtering out low-scoring hits may affect the results, we did not observe any significant differences in the test cases (data not shown).
We empirically determined the z-score threshold for search results by taking 100 random SCOP families in ASTRAL 40. We then calculated the best cutoff by balancing a high positive predictive value and a large number of true positive hits. This analysis is displayed in Figure 1.
Fig. 1.
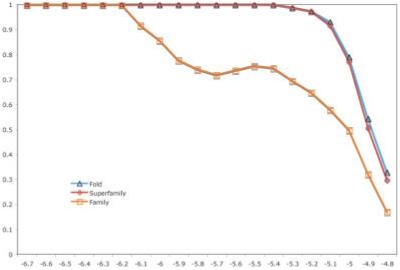
The relationship between average PPV and a given threshold S-BLEST z-score. The proteins used were 100 random members of random SCOP families in ASTRAL 40 v1.65.
RESULTS
Identification of Structurally Similar Residue Environments in ASTRAL 40 v1.65
ASTRAL 40 v1.65 encoded 4,129 crystallographically determined structures. Each search takes approximately 2 s to encode and query as single vector on an Intel Xeon 2.8-GHz processor. Figure 2 shows example background distributions used to calculate the z-scores. These distributions are generally not Gaussian and often contain shoulders or evidence of higher complexity.
Fig. 2.
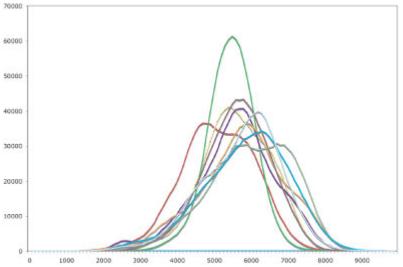
Illustration of the background distributions used to calculate z-score. The distance histogram distribution of the first nine residue environments of pdb 2TRX:A with respect to the ASTRAL 40 v1.65 dataset.
Identification of the Residue Environments Associated With a Structural Class
To illustrate the utility of using the AUC of an ROC plot, we determined how well each residue environment in a protein was associated with the protein’s annotated SCOP family. We looked in detail at the S-BLEST search results for residues in P38 mitogen-activated protein kinase from Homo sapiens 1DI9 chain A (1DI9:A)21 and found that S-BLEST identifies residues near functional regions of the structure as being associated with the protein’s SCOP family of protein kinases. The functional environments were considered to be the adenosine 5′-triphosphate (ATP) binding site, the peptide binding channel residues, and residues known to be phosphorylated. Figure 3 illustrates how the top scoring residues discriminate function. The functional residues were identified by ranking the AUC for S-BLEST search of each residue in the protein. The top 10 residues are shown in Figure 3. The residues that are good at classification form a core that is close to all three of the functionally interesting regions of the enzyme, the peptide binding channel, the ATP binding site, and the activating phosphorylated residues.
Fig. 3.
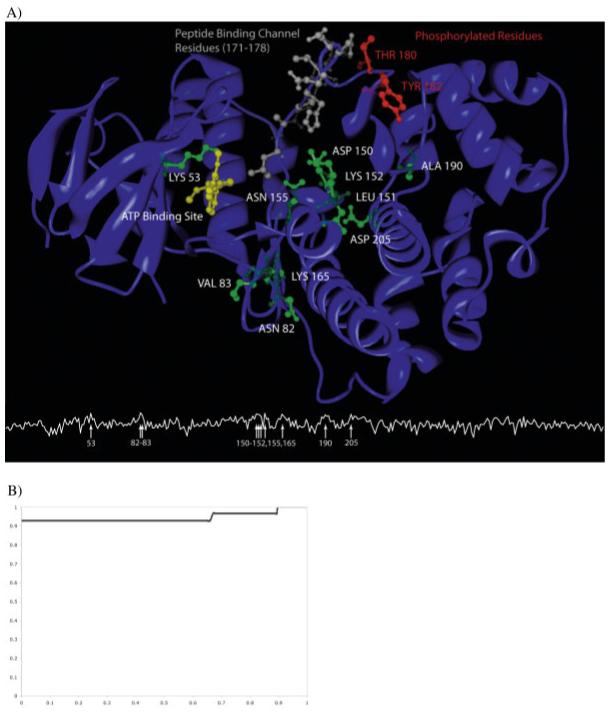
Illustration of classifying residues within map kinase, 1DI9. A: The line plot below indicates the AUC at each position along the chain. The arrows indicate the locations in the sequence with AUC above 0.90. These classifying residues are shown in green on the structure. 1DI9 illustrates the underlying reason for classification. The good classifiers form a core that is surrounded by the ATP binding site (in yellow), the peptide binding channel (in gray), and the residues that, when phosphorylated, activate the enzyme (in red). Additionally, LYS53 directly interacts with the ATP ligand. Interestingly, residues ASN82, VAL83, and LYS165 form another environment that classifies the function well. They are directly behind the peptide binding channel and are in close proximity to the ATP binding site. B: ROC of the ranked chains outputted from the congruence approach. Of the 27 members in our dataset, the first 25 chains ranked were true positives, whereas the method failed to recognize 1KOA and 1FMK as structurally similar (AUC is 0.935).
Congruence Approach to Characterize Protein Structures
Our goal is to show that S-BLEST finds structurally similar environments with potential implications for fold, family, and function. To do this, we selected 100 random SCOP families in ASTRAL 40. For each protein structure, an S-BLEST search was performed for every amino acid in the structure. The result of each search is a list of residue environments from a database of protein structures ranked by their similarity to the query residue based on a significance score (z-score). Only one residue, the one with highest similarity, is selected from each structure in the dataset. The datasets developed include all analyzable X-ray crystallographic structures from the ASTRAL 40 nonredundant dataset.22 To evaluate how well the environmental similarity of a residue from each structure can be used to assign the SCOP family of the structure, we examined the rankings of the members of the SCOP family associated with each structure using the procedure described in Materials and Methods.
A z-score threshold for each protein was offset at -5.5. The positive predicted value (PPV) of the search can be defined as the number of true positives above the threshold divided by the total number of hits above the threshold.
Analysis of Uncharacterized PDB Structures
We next applied S-BLEST to crystal structures of proteins with unknown function. Eighty-six of these structures had no significant hits when searched against the PDB using BLAST with e-value cutoff of 1e-4 (Table I). These proteins were selected from the PDB by searching for the phrase “unknown function.” Because the search phrase “unknown function” can have several intended meanings, these proteins represent a broad spectrum of proteins whose function is understood to variable degrees of precision. Table I lists the 86 structures and highlights the structures that were returned for each query protein with an average z-score better than the threshold of -5.00. With this procedure, we have identified residues in the PDB that have similar local environments as those in the query structure with potential structural significance.
TABLE I.
Eighty-Six Proteins With Partially Uncharacterized Function Used in the Analysis†
| 1G2R:A 1HY2:A 1I36:A 1IHN:A 1IN0:A 1IUJ:A 1IXL:A 1IZM:A 1J27:A 1J6R:A 1J6O:A 1JOP:A 1JOV:A 1JOG:A 1JRM:A |
| 1JSX:A 1JX7:A 1JYH:A 1JZT:A 1K4N:A 1K77:A 1KJN:A 1KUU:A 1KYH:A 1LPL:A 1M33:A 1M98:A 1MK4:A 1ML8:A |
| 1MOG:A 1MWW:A 1MWQ:A 1N81:A 1NE2:A 1NG6:A 1NI9:A 1NIG:A 1NIJ:A 1NJH:A 1NNW:A 1NNX:A 1NO5:A 1NRI:A |
| 1NXH:A 1NY1:A 1O4W:A 1O5H:A 1ORU:A 1OYZ:A 1OZ9:A 1P9I:A 1PBJ:A 1PC6:A 1PD3:A 1PG6:A 1PM3:A 1PV5:A |
| 1PVM:A 1Q2Y:A 1Q7H:A 1Q8B:A 1Q77:A 1Q8C:A 1Q9U:A 1QYI:A 1QZ4:A 1R4V:A 1R75:A 1RC6:A 1RFZ:A 1RI6:A |
| 1RU8:A 1SQH:A 1SQS:A 1UCR:A 1UFA:A 1VHN:A 1VHS:A 1VIZ:A 1VI7:A 1VI1:A 1VJG:A 1VJI:A 1VJN:A |
This set was obtained by selecting proteins from the PDB that were returned with the search “unknown function” and only proteins that were crystallographically determined with a minimum BLAST e-value of 1e-4. The structures with hits in the ASTRAL 40 v1.65 dataset with a z-score above -5.0 are shown in bold.
Among all the proteins of unknown function, we chose several interesting results for detailed analysis. Succinyl diaminopimelate desuccinylase from Neisseria meningitides, 1VGY:A, illustrates S-BLEST’s effectiveness in identifying statistically interesting residues from an uncharacterized protein structure. S-BLEST found that 1VGY:A shared highly significant residues with a dinuclear zinc aminopeptidase Pepv from Lactobacillis delbrueckii, 1LFW:A, with a z-score of -6.36. BLAST matches these proteins with an e-value of 3 × 10-4. The matching top five residues from 1VGY:A paired with 1LFW:A are ARG97 with ARG115, HIS68 with HIS87, ASP70 with ASP89, GLY98 with GLY112, and GLU136 with GLU154. As illustrated in Figure 4(A), the best matched residues in 1LFW flank the active site of the protein and are in close contact to the AEP ligand that was crystallized with the structure. This suggests that the corresponding residues in 1VGY are likely in this region as well. To further test S-BLEST, we assigned 1VGY to a SCOP family based on the top hit. 1LFW:A the protein with multiple similar residues is associated with SCOP family c.56.5.4, bacterial dinuclear zinc exopeptidases. We hypothesized that 1VGY:A is a member of this SCOP family and, in a process analogous to the one described earlier, we performed an S-BLEST search on each residue and determined the residues most associated with that SCOP family. There are five structures with less than 40% sequence identity (according to ASTRAL 1.65) thatbelong to the SCOP family of bacterial dinuclear zinc exopeptidases. We find that residues HIS68, GLU135, ASP70, ASP134, and HIS350 from 1VGY:A all can be used to annotate the structure very well (Fig. 5). Each of these residues is localized to one region of the structure, and the corresponding residues in 1LFW:A are near the active site.
Fig. 4.
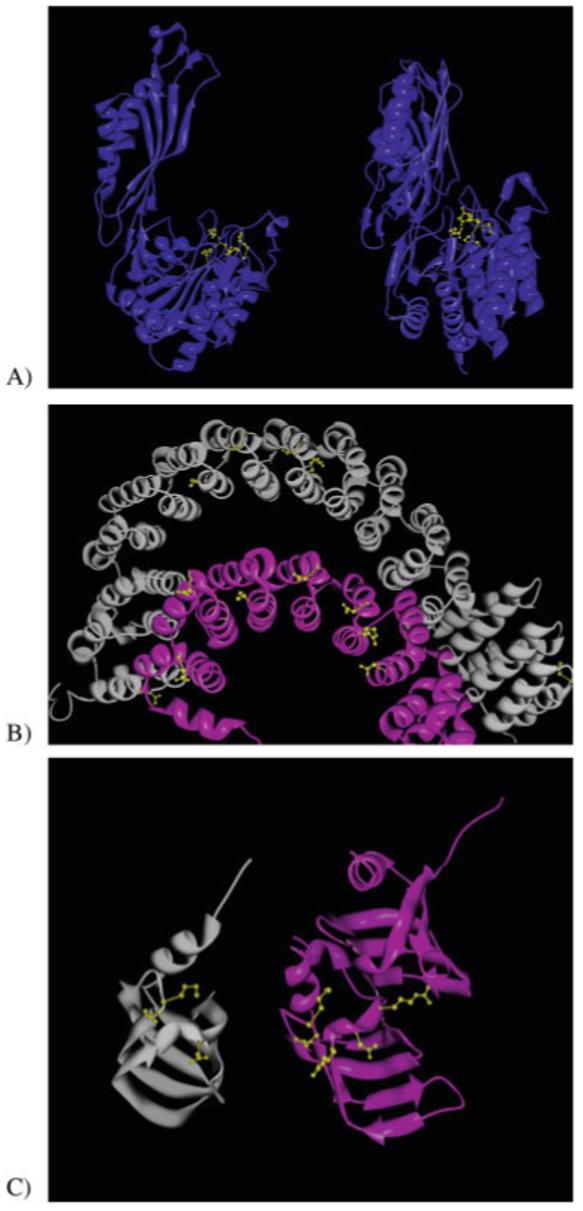
Illustration of the hit results from the 86 structures with unknown function. A: As an example of hit that is a true positive, 1VGY:A is matched with 1LFW:A with a z-score of -6.36. The best matching residues are ARG97 from 1VGY paired with ARG115 from 1LFW, HIS68 with HIS87, ASP70 with ASP89, GLY98 with GLY112, and GLU136 with GLU154. These residues are highlighted in yellow in the figure. B: An interesting hit that is of questionable significance is 1B3U:A, which is matched to the query of 1OYZ:A with a slightly below threshold z-score of -5.21. It is an interesting hit, because the proteins are clearly structurally related, and the best residue matches occur between secondary structural elements, and are often observed “bridging” the structural elements. C: An example of a possible unknown hit, between 1LJO:A and 1B34:A with a z-score of -5.64. Although the proteins share the same fold, their functional relationship is not known.
Fig. 5.
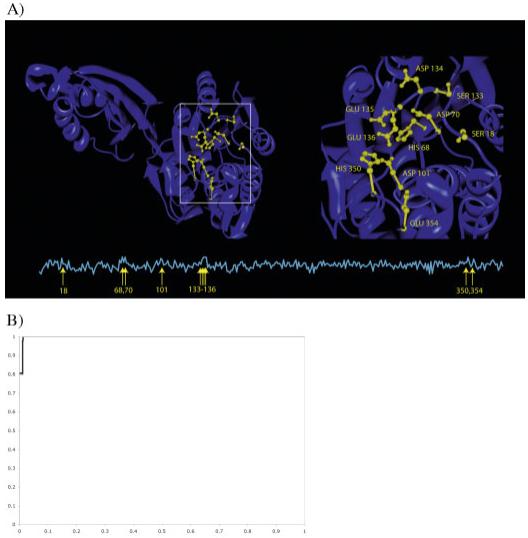
Characterization of a hit (1VGY:A). A: Top hits were associated with a common SCOP family. We then calculated the area under an ROC curve for each residue in that structure, quantifying how well each protein classifies the SCOP family the hits were associated with. The line plot below indicates the AUC at each position along the chain. The arrows indicate the locations in the sequence with AUC above 0.90. We highlight these locations in yellow on the structure. These hits fall into a predicted active site and are localized to a single region. B: The ROC for the congruence approach is shown. Of the five true positives in our dataset, three were the top hits, the fourth was in position five, and the fifth was ranked 65th overall (AUC is 0.995).
Sometimes residues sit in an environment that is sufficiently unique to give several hits, but those hits are based on unique structural properties, and not necessarily the protein’s function. For example, the hypothetical gene product from Escherichia coli, 1OYZ:A, is matched with protein phosphatase PP2A from H. sapiens, 1B3U:A, with a z-score of -5.21. We observe that residues found at the helix-loop interface and are oriented toward another secondary structural element are often identified as being good matches between structures [Fig. 4(B)]. For the strict purposes of this article, these should be considered as false positives, although the underlying reason for their uniqueness and any functional relationship between these seemingly unrelated proteins is intriguing and may deserve follow-up.
Another interesting hit, an archael SM-like protein AF-SM2 from Archeoglobus fulgidus, 1LJO:A, is matched with a small nuclear ribonucleoprotein SM D1 from H. sapiens, 1B34:A, with a z-score of -5.64. BLAST matches these proteins with an e-value of 1e-7. The top five matching residues all are close in space and all are close in sequence between the query and the match [Fig. 4(C)]. The proteins share the same fold, and several matching environments are identified.
DISCUSSION
The characterization of proteins from their structure is an important goal for the high throughput structural genomics pipeline. S-BLEST provides a method for quickly identifying similar local structures and the corresponding residue environments. Furthermore, it does not rely on fold recognition or the pre-identification of evolutionarily conserved residues. This method is intended to identify statistically significant environments in protein structures and will be complementary to both sequence-based methods such as BLAST or HMMs and fold recognition methods.
S-BLEST can be easily combined with BLAST for a sequence-structure analysis of a query protein. This allows for identification of highly conserved structural sites, as well as highly conserved sequence neighbors. For example, with the analysis of a random member from each of 100 random families, S-BLEST (threshold of -5.1) finds 28 SCOP family members that BLAST (threshold of 1e-5) does not find, and BLAST finds 89 family members that S-BLEST does not find, because of local structural variability between the proteins. There is a cost, however, of 66 false-family positives, all but 13 of which share the superfamily of the query. Additionally, for each BLAST hit, the degree of structural conservation of each residue environment can be easily determined using S-BLEST.
We were surprised to find that many residues that were annotated as being important for enzyme chemistry are not the ones that are most useful for recognizing structural similarities. The method sometimes does not select the critical residues (such as the catalytic triad) likely because the environments around those residues are structurally variable between members. The residue environments that are chosen, however, are those environments that are structurally conserved across a family. There are several possible explanations for why apparently critical residue environments are not conserved. These residue environments may adopt different structures in the presence of different ligands, crystallizing conditions, or in the presence of mutation. Methods that take into account protein may uncover similar ensembles of important residues that appear different in static structures.
The computational requirements of our method are relatively modest. For a single residue search, a 3.06-GHz Intel Xeon CPU can complete the search in less than 30 s. Querying with a 300-residue protein against the PDB can take as long as 4 h and requires a relatively large amount of memory (1-2 GB), whereas the same protein takes less than one half of an hour with ASTRAL 40. The vector data for the entire PDB is currently split between two files, each around 1.2 GB in size.
CONCLUSION
We developed S-BLEST to meet a need for rapidly identifying similar structures to a query protein using local structural features. To complement fold-recognition methods, we sought a method that could identify the local residue environments that correspond to that match. Our solution, S-BLEST, identifies constellations of structurally similar residues between the query protein and the full database of known protein structures. Moreover, we find that many of the structural environments in SCOP have statistically significant local environment neighbors.
ACKNOWLEDGMENTS
S.D.M. was funded by an American Cancer Society John Peter Hoffman Fellowship and NIH grant LM06244 (Russ Altman, PI) and is now funded by the INGEN grant from the Lilly Endowment. M.H.-P.L. is funded by NIH grants LM-05652, LM-07033, and GM-63495. The authors acknowledge Giselle Knudsen and David Konerding for helpful comments.
Grant sponsor: American Cancer Society; Grant sponsor: National Institutes of Health; Grant numbers: LM06244, LM-05652, LM-07033, and GM-63495; Grant sponsor: Lilly Endownment
REFERENCES
- 1.Thornton JM, Todd AE, Milburn D, Borkakoti N, Orengo CA. From structure to function: approaches and limitations. Nat Struct Biol. 2000;7(Suppl):991–994. doi: 10.1038/80784. [DOI] [PubMed] [Google Scholar]
- 2.Lichtarge O, Bourne H, Cohen F. An evolutionary trace method defines binding surfaces common to protein families. J Mol Biol. 1996;257(2):342–358. doi: 10.1006/jmbi.1996.0167. [DOI] [PubMed] [Google Scholar]
- 3.Wallace AC, Laskowski RA, Thornton JM. Derivation of 3D coordinate templates for searching structural databases: application to Ser-His-Asp catalytic triads in the serine proteinases and lipases. Protein Sci. 1996;5(6):1001–1013. doi: 10.1002/pro.5560050603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fetrow J, Skolnick J. Method for prediction of protein function from sequence using the sequence-to-structure-to-function paradigm with application to glutaredoxins/thioredoxins and T1 ribonucleases. J Mol Bioly. 1998;281:949–968. doi: 10.1006/jmbi.1998.1993. [DOI] [PubMed] [Google Scholar]
- 5.Holm L, Sander C. Dali/FSSP classification of three-dimensional protein folds. Nucleic Acids Res. 1997;25(1):231–234. doi: 10.1093/nar/25.1.231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stawiski EW, Baucom AE, Lohr SC, Gregoret LM. Predicting protein function from structure: unique structural features of proteases. Proc Natl Acad Sci USA. 2000;97(8):3954–3958. doi: 10.1073/pnas.070548997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bagley S, Altman R. Characterizing the microenvironments surrounding protein sites. Protein Sci. 1995;4(4):622–635. doi: 10.1002/pro.5560040404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Murzin A, Brenner S, Hubbard T, Chothia C. SCOP: a structural classification of proteins database for the investigation of sequences and structures. J Mol Biol. 1995;247:536–540. doi: 10.1006/jmbi.1995.0159. [DOI] [PubMed] [Google Scholar]
- 9.Jewett AI, Huang CC, Ferrin TE. MINRMS: an efficient algorithm for determining protein structure similarity using root-mean-squared-distance. Bioinformatics. 2003;19(5):625–634. doi: 10.1093/bioinformatics/btg035. [DOI] [PubMed] [Google Scholar]
- 10.Stark A, Russell RB. Annotation in three dimensions. PINTS: patterns in non-homologous tertiary structures. Nucleic Acids Res. 2003;31(13):3341–3344. doi: 10.1093/nar/gkg506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Berman H, Westbrook J, Feng Z, et al. The protein data bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jones S, Thornton JM. Searching for functional sites in protein structures. Curr Opin Chem Biol. 2004;8(1):3–7. doi: 10.1016/j.cbpa.2003.11.001. [DOI] [PubMed] [Google Scholar]
- 13.Panchenko AR, Bryant SH. A comparison of position-specific score matrices based on sequence and structure alignments. Protein Sci. 2002;11(2):361–370. doi: 10.1110/ps.19902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Holm L, Sander C. Protein structure comparison by alignment of distance matrices. J Mol Biol. 1993;233(1):123–138. doi: 10.1006/jmbi.1993.1489. [DOI] [PubMed] [Google Scholar]
- 15.Singh R, Saha M. Identifying structural motifs in proteins. Pac Symp Biocomput. 2003:228–239. [PubMed] [Google Scholar]
- 16.Dubey A, Hwang S, Rangel C, Rasmussen CE, Ghahramani Z, Wild DL. Clustering protein sequence and structure space with infinite Gaussian mixture models. Pac Symp Biocomput. 2004:399–410. doi: 10.1142/9789812704856_0038. [DOI] [PubMed] [Google Scholar]
- 17.Stark A, Sunyaev S, Russell RB. A model for statistical significance of local similarities in structure. J Mol Biol. 2003;326(5):1307–1316. doi: 10.1016/s0022-2836(03)00045-7. [DOI] [PubMed] [Google Scholar]
- 18.Altschul S, Madden T, Schaffer A, et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search tools. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Grossman T, Farber R, Lapedes A. Neural net representations of empirical protein potentials. Proc Int Conf Intell Syst Mol Biol. 1995;3:154–161. [PubMed] [Google Scholar]
- 20.Pegg SC, Babbitt PC. Shotgun: getting more from sequence similarity searches. Bioinformatics. 1999;15(9):729–740. doi: 10.1093/bioinformatics/15.9.729. [DOI] [PubMed] [Google Scholar]
- 21.Shewchuk L, Hassell A, Wisely B, et al. Binding mode of the 4-anilinoquinazoline class of protein kinase inhibitor: X-ray crystallographic studies of 4-anilinoquinazolines bound to cyclin-dependent kinase 2 and p38 kinase. J Med Chem. 2000;43(1):133–138. doi: 10.1021/jm990401t. [DOI] [PubMed] [Google Scholar]
- 22.Chandonia JM, Hon G, Walker NS, et al. The ASTRAL compendium in 2004. Nucleic Acids Res. 2004;32(database issue):D189–192. doi: 10.1093/nar/gkh034. [DOI] [PMC free article] [PubMed] [Google Scholar]